 Bin Wang
Bin Wang Xiaoqing Shao1,2†
Xiaoqing Shao1,2†- 1Department of Endocrinology, Jinshan Hospital of Fudan University, Shanghai, China
- 2Department of Rheumatology and Immunology, Jinshan Hospital of Fudan University, Shanghai, China
- 3Department of Rheumatology and Immunology, The Affiliated Hospital of Weifang Medical University, Weifang, China
Autoimmune thyroid diseases (AITD) are a group of both B cell- and T cell-mediated organ-specific autoimmune diseases. Graves’ disease and Hashimoto thyroiditis are the two main clinical presentations of AITD. Both genetic and environmental factors have important roles in the development of AITD. Epigenetics have been considered to exert key roles in integrating those genetic and environmental factors, and epigenetic modifications caused by environmental factors may drive genetically susceptibility individuals to develop AITD. Recent studies on the epigenetics of AITD have provided some novel insights into the pathogenesis of AITD. The aim of this review is to provide an overview of recent advances in the epigenetic mechanisms of AITD, such as DNA methylation, histone modifications, and non-coding RNAs. This review highlights the key roles of epigenetics in the pathogenesis of AITD and potential clinical utility. However, the epigenetic roles in AITD are still not fully elucidated, and more researches are needed to provide further deeper insights into the roles of epigenetics in AITD and to uncover new therapeutic targets. Although there are many studies assessing the epigenetic modifications in AITD patients, the clinical utility of epigenetics in AITD remains poorly defined. More studies are needed to identify the underlying epigenetic modifications that can contribute to accurate diagnosis of AITD, adequate choice of treatment approach, and precise prediction of treatment outcomes.
Introduction
Autoimmune thyroid diseases (AITD) are a group of both B cell- and T cell-mediated autoimmune diseases, which are caused by loss of immune tolerance and autoimmune attack to thyroid tissues (1, 2). The prevalence of AITD including Graves’ disease (GD) and Hashimoto’s thyroiditis (HT) is more than 5%, but the prevalence of thyroid autoantibodies is more than 10% in general population (1, 3). Despite its high prevalence, the incidence and prevalence of AITD have increased obviously in recent years (1, 4, 5). GD is the main cause of clinical hyperthyroidism, and HT is the main cause of clinical hypothyroidism (1, 6). Besides, AITD can also increase the risk of non-thyroid diseases, such as cardiovascular diseases, cancers, and adverse pregnancy outcomes (7–13). In addition, there is still lack of major breakthrough in the treatment of AITD (1, 2, 6, 14). Therefore, AITD have become a serious harm to public health, and more studies are urgently needed to explore the pathogenesis of AITD and develop new therapeutic strategies to effectively treat AITD.
Although the clinical manifestations of GD and HT are different, loss of immune tolerance caused by immune imbalance is believed to exert critical roles in the development of both diseases (15–17). Both GD and HT are characterized by the presence of autoantibodies against thyroid tissues, such as thyroid-stimulating hormone receptor antibody (TRAb), thyroid peroxidase antibody (TPOAb), and thyroglobulin antibody (TgAb) (15–17). TRAb is thyroid-stimulating hormone receptor (TSHR)-directed autoantibodies, and it can be classified as TSHR-stimulating autoantibodies, TSHR-blocking autoantibodies, or neutral depending on their respective abilities to induce cAMP generation and thyrocyte proliferation or bind to the receptor without impacting cAMP generation (18, 19). TPOAb and TgAb are the major autoantibodies in HT, but they also exist in some GD patients. The TRAb is the major autoantibody in GD patients, but it also usually occurs in some HT patients.
Both GD and HT are characterized by lymphocytic infiltration in the thyroid tissues, and T cells and B cells can infiltrate into the thyroid gland during the development AITD (17, 20–22). Autoantibodies and B cell dysfunction are thought to be the primary immune reactions in AITD, and aberrant functions of T cell subsets also exert important roles in breaking the immune homeostasis and causing autoimmunity against thyroid tissues (17, 20–22) (Figure 1). Besides, T cells may amplify autoimmunity against thyroid by secreting pro-inflammatory cytokines, promoting B cells to generate more autoantibodies, and maintaining the number of autoreactive memory T cells against thyroid tissues (17, 20, 21, 23). In GD, the CD4+ T helper (Th) cells are important in the onset of the disease, and the roles of Th1 and Th2 in the development of AITD have also long been recognized in previous studies (24–27). Recent studies have suggested that the balance between T effector cells and T regulatory cells are important to maintain the immune tolerance to thyroid, and its imbalance can result in the development of AITD (22, 28, 29). Besides, the roles of other T subsets in AITD, such as Th17, Th22, and follicular helper T (Tfh) cells, have also been found in numerous researches (25, 30–34). Th17 cells are characterized by the transcription factors of signal transducer and activator of transcription 3 and Rorγt, and they mainly secret interleukin (IL)-17 (35). Increased circulating Th17 cells have been found in AITD patients, and IL-17 has also been identified as an important cytokine in the pathogenesis of AITD (25, 32–34, 36). Tfh cells are characterized by the expression of CXC chemokine receptor 5 (CXCR5) and the production of IL-21 and are critical for the activation of B cells and germinal center formation (37). Th22 cells are characterized by the transcription factor of aryl hydrocarbon receptor and the production of IL-22 (38). Increased proportions of circulating Th22 cells and Tfh cells and elevated of related cytokines in the AITD patients have been found in several published studies, suggesting the pathogenic roles of Th22 and Tfh in the pathogenesis of AITD (30, 31, 39). However, the molecular mechanisms underling the abnormal functions of those immune cells and immune imbalance during the development of autoimmune attacks to thyroid tissues are still unclear.
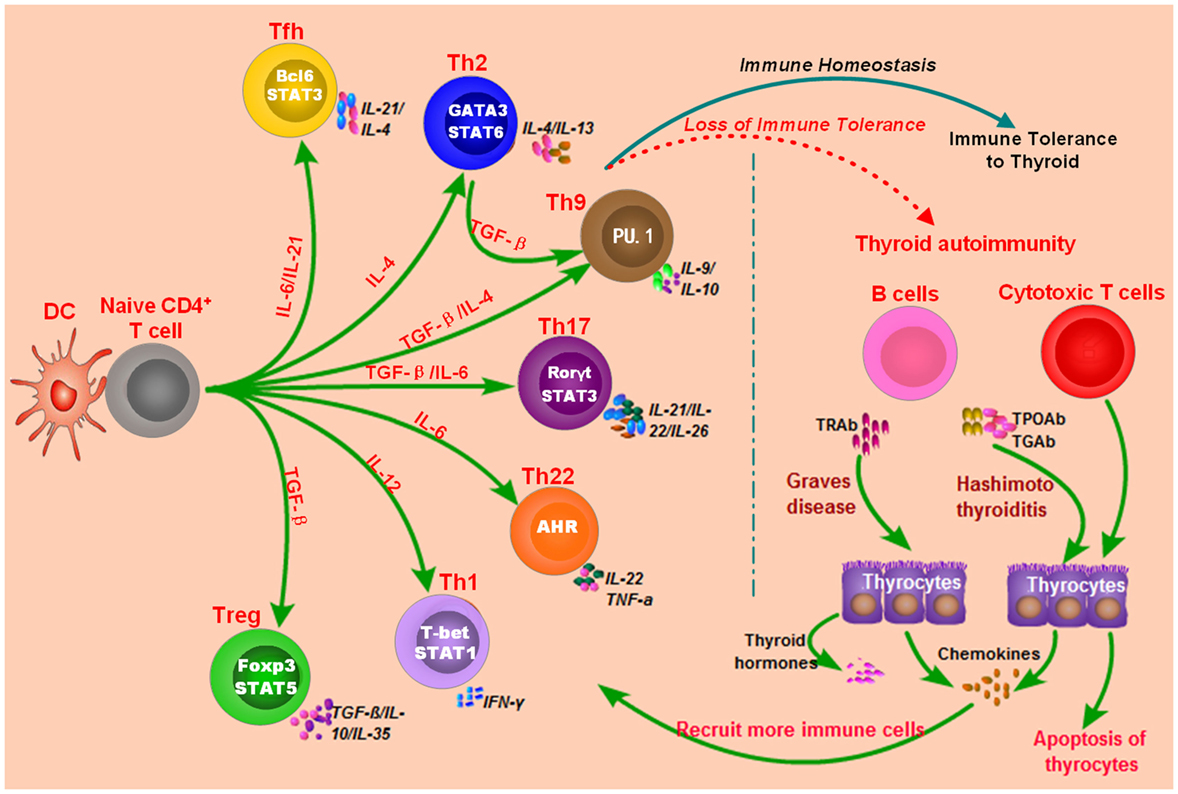
Figure 1. Loss of immune tolerance results in autoimmunity during the development of autoimmune thyroid diseases. Naive CD4+ T cells can be activated by dendritic cells (DC) or other antigen-presenting cells and they can differentiate into various subsets which are characterized by different cytokines and specific transcription factors. The balance of those immune cells is necessary for the maintenance of immune homeostasis. Under normal conditions, T cell subsets have normal functions, and there is immune homeostasis in human body, which can maintain the immune tolerance and avoid unwarranted immune attacks to thyroid tissues. Some genetic factors and environmental factors can result in the dysfunctions of these T cell subsets, B cells, and antigen-presenting cells, which may break up the immune homeostasis and cause thyroid autoimmunity.
Autoimmune thyroid diseases are multifactorial and complex diseases, and multiple factors are involved in the development of AITD (1, 16). Studies in the past decade have proven the important role of genetic factors in the pathogenesis of AITD. In the past two decades, genetic studies in AITD have developed from candidate gene analyses, whole-genome linkage screening, genome-wide association study (GWAS), whole-genome sequencing, and epigenetic studies (16, 40–42). Some thyroid-specific genetic factors are found to be associated with AITD, such as polymorphisms in TSHR gene and thyroglobulin (TG) gene (43–45). Emerging evidence has suggested the important role of immunogenetics in the pathogenesis of AITD, and polymorphisms in these immune-modulating genes can impair immune tolerance and alter T cells’ interactions with antigen-presenting cells during the development of AITD (16, 46). Some immune-modulating genetic factors are also reported to be associated with AITD, such as polymorphisms in HLA-DR3, CTLA4, PTPN22, and FOXP3 (47–49). Of those AITD susceptibility genes, FOXP3 and CD25 play critical roles in the establishment of peripheral tolerance, and CD40, CTLA4, and the HLA genes are pivotal for T lymphocyte activation and antigen presentation (16, 42). Those immune-modulating genetic factors can cause dysfunction of immune cells and loss of immune homeostasis, which can further result in the development of AITD. However, those genetic factors cannot fully explain host’s predisposition to AITD, and environmental factors also have important roles in AITD (16, 50, 51). The lack of full concordance in monozygotic twins also proves the importance of environmental factors in AITD (52, 53). Several environmental factors such as high iodine intake and vitamin D deficiency are proven to be risk factors of AITD (54–58). The genetic and environmental factors may cooperate together and cause the dysfunctions of immune cells and thyroid autoimmunity, but the mechanisms involving the effects of genetic and environmental factors on the immune cells’ function and immune balance are still not well understood (59–61).
Recent studies propose that environmental factors can interact with susceptibility genes to produce a synergistic effect in triggering diseases through epigenetic modulation (62–64). Epigenetics aim to study how non-genetic factors regulate the gene expressions and phenotypes and their roles in the development of diseases without involving alterations of the DNA sequence (65). Major epigenetic mechanisms mainly include DNA methylation, histone modifications, and RNA interference through non-coding RNAs (65). For example, DNA methylation can cause inactivation of genes, and some histone modifications can lead to activation of genes, but these factors are usually dynamic and can be affected by environmental factors (65–67). In addition, non-coding RNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), can also regulate the expressions of targeted genes (68, 69). Therefore, genes involved in the immune system or thyroid can be regulated by epigenetic mechanisms, and dysfunctions of these genes caused by epigenetics can further result in autoimmune diseases. In the past decade, epigenetics have been considered to have key roles in integrating genetic and environmental factors in human complex diseases including autoimmune diseases (64, 70, 71). In the past decade, increasing evidence has suggested the critical roles of epigenetics in the pathogenesis of AITD, and epigenetic modifications caused by environmental factors may drive genetically susceptibility individuals to develop AITD (42, 60, 72–75). The aim of this review is to provide an overview of recent advances in the epigenetic mechanisms of AITD, to highlight the epigenetic roles in the pathogenesis of AITD, and to discuss the potential clinical utility of epigenetic modifications in AITD.
Epigenetics in AITD
DNA Methylation and AITD
DNA methylation is the most common type of DNA modifications, and it mainly occurs at the fifth carbon ring of cytosine in palindromic cytosine-phosphate-guanine dinucleotides (76–78). DNA methylation mainly results in transcriptional repression, especially when it occurs in the region of 5′ promoter regions with high density (79). In addition, methyl-binding domain family can recognize the methyl-CpG and result in transcriptional repression (79). Some important enzymes involved in DNA methylation have been found, such as DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) enzymes for demethylation (78). In addition, DNA methylation can be reversed by TET enzymes, and the dynamic turnover of DNA methylation may be modulated through the relative expressions of DNMTs and TET enzymes (77, 78). Resent researches have provided evidence for the critical roles of DNA methylation in many autoimmune diseases through regulating gene expressions (80–85). Some agents targeting DNA methylation also provide promising novel treatment strategies for human diseases (86–88).
In the past decade, increasing evidence has demonstrated the roles of epigenetic dysregulation in the pathogenesis of AITD. Several studies have shown that global DNA hypomethylation exists in AITD patients, which may cause the overexpression of some genes involved in immune function or the activation of immune cells and further result in autoimmune attack toward thyroid tissues (74, 75). We previously studied the genome-wide DNA methylation of GD patients and revealed more than 200 hypermethylated and hypomethylated genetic regions in GD patients, such as ICAM1, DNMT1, and MECP2 genes (74). Limbach et al. investigated the genome-wide DNA methylation of CD4+ and CD8+ T cells of GD patients and found more than 300 differentially methylated sites in CD4+ T cells and more than 3,000 differentially methylated sites in CD8+ T cells, and many of those genes were from T cell signaling (75). Many of those DNA methylations were immune-related modifications, such as hypermethylated sites in ICAM1, CD247, and CTLA4 (74, 75). Limbach et al. also found hypermethylation of the first intron area in TSHR gene was associated with GD, which suggested methylation in thyroid-specific genes could also be involved in the development of AITD. The above findings demonstrated the existence of aberrant DNA methylation at some genes during the development of AITD. However, the molecular mechanisms underlining the roles of those differentially methylated genes are still unclear.
Some genetic polymorphisms of DNA methylation-regulating genes can also cause dysfunction of these genes and aberrant DNA methylation, which further increases hosts’ susceptibility to diseases (89, 90). Arakawa et al. reported that several polymorphisms in methylation-regulating genes, such as DNMT1 and methionine synthase reductase (MTRR), were correlated with DNA hypomethylation levels and susceptibility to AITD (91). Our previous study also found evidence for a potential role of DNMT3B rs2424913 and DNMT1 rs2228611 in the susceptibility to AITD (92). 5,10-Methylenetetrahydrofolate reductase C677T polymorphism was also associated with GD and Graves’ ophthalmopathy (93, 94). The above findings also provide secondhand evidence for the important roles of DNA methylation in the pathogenesis of AITD.
The above findings suggest the emerging and important roles of DNA methylation in AITD, but current understanding on DNA methylation in AITD is still very limited. More studies in the future are necessary to further explore the possible key roles of DNA methylation in AITD and new promising treatment strategies targeting DNA methylation for AITD patients. In addition, the clinical utility of DNA methylation in AITD as biomarkers for disease diagnosis and predictors of treatment outcomes is still unclear, which need to be explored in future studies.
Histone Modifications and AITD
Histone has key roles in the compaction of DNA, interacting with DNA to form tightly packed chromatin, and it also has been suggested to have critical roles in many human diseases (95–98). The unstructured N-terminal amino acids in histones can be modified and then affect the chromatin structure and function directly or by binding some protein effectors (99). These modifications usually occur at the lysine or arginine of histones and are mediated by histone-modifying enzymes, which are intensively involved with chromosome remodeling (98). There are several main types of modifications in histones, such as methylation, acetylation, deacetylation, phosphorylation, ubiquitination, and sumoylation (99, 100). Histone modifications have important roles in controlling chromatin compaction, nucleosome dynamics, and DNA repair, and it can directly regulate transcription (101, 102). Like DNA methylation, histone modifications are highly dynamic and are regulated by “writer” and “eraser” enzymes (77, 100, 103). Recent researches have provided some evidence for the roles of histone modifications in modulating immune tolerance and autoimmune diseases (104–109). Some small-molecule inhibitors targeting histone-modifying enzymes, such as histone deacetylases (HDAC), also provide new treatment strategies for diseases such as cancers and autoimmune diseases (87, 110).
We found that the histone H4 acetylation level in the peripheral blood mononuclear cells of GD patients was significantly lower than that of healthy individuals, but the levels of HDAC1 and HDAC2 were obviously higher, which proved evidence for the aberrant histone modifications in GD patients (104). A genome-wide analysis by Limbach et al. found decreased levels of H3K4me3 and H3K27ac at several genes of T cell signaling in GD patients (75). In addition, phosphorylated histone protein H2A.X was also observed in the T cells and thyrocytes in the mice of AITD model (111).
Stefan et al. found that interferon-alpha (IFN)-α could induce alterations of TG gene expression and trigger AITD through enrichment of Lys-4 residue methylation in histone H3 at the promoter area of TG gene (112). IFN-α is a key cytokine secreted during viral infections, and it has been found to increase levels of H3K4me3 and H3K4me1 in thyroid cells (72). Another study by Kawashima et al. showed that DNA fragments released during thyroid injury could be recognized by histone H2B, which further resulted in the activation of genes of immune responses and triggered autoimmunity against thyroid tissue (113).
Genetic polymorphisms of histone-modifying genes can also cause dysfunction of these genes and aberrant histone modifications (114–116), which may further results in AITD. Sirtuin1 (SIRT1) is a class 3 nicotinamide adenine dinucleotide-dependent HDAC, which is intensively associated with immune response and autoimmune diseases (114–116). A case–control study by Sarumaru et al. reported that rs3758391 and rs4746720 in the SIRT1 gene were associated with higher levels of thyroid autoantibodies in AITD patients (117).
The findings above suggest the important role of histone modifications in AITD, but they are still not fully elucidated. More researches are needed to further elucidate the roles of histone modifications in AITD. Besides, the roles of histone modifications as diagnostic biomarker and predictors of treatment outcomes in AITD patients have also not been investigated, and future researches are recommended to study these roles of histone modifications in AITD.
miRNAs and AITD
microRNAs are endogenous small non-coding RNAs ranging from 18 to 25 nucleotides in length and have important roles in regulating gene expression (118). miRNAs can regulate about 60% of all mRNAs and are involved in many diseases, such as cancers, metabolic diseases, and inflammatory diseases (118–120). Some miRNAs also play important roles in regulating immune function and maintaining immune homeostasis, such as miR-223-3p and miR-155-5p (120–122). It is not surprising that abnormal expressions of miRNAs involving immune function can potentially contribute to the development of autoimmune diseases (123, 124). Recent studies have revealed that some miRNAs are also involved in the development of AITD, and most of them have been found to be intensively involved in modulating the differentiation or activation of immune cells and immune response (Table 1).
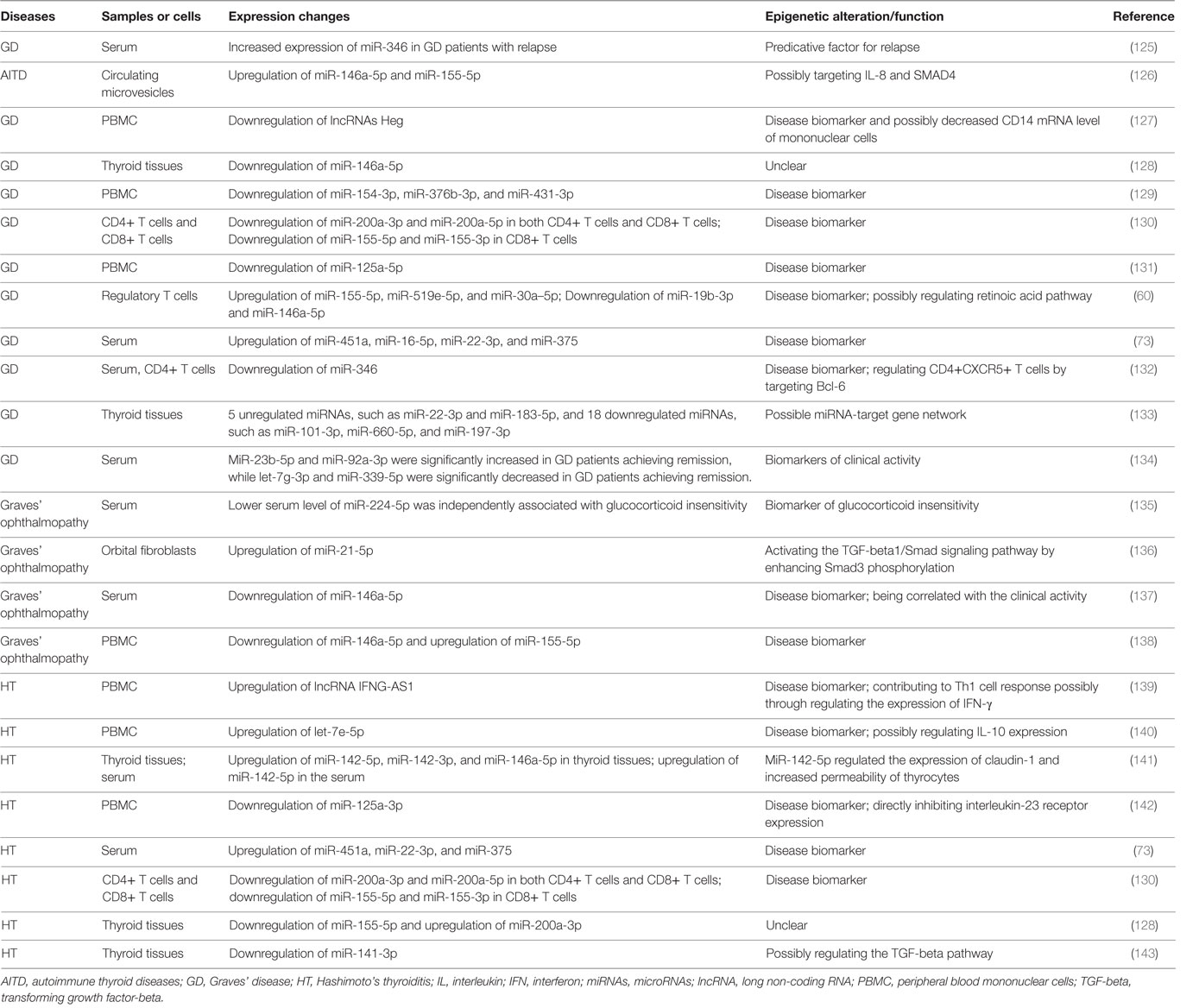
Table 1. Aberrant expressions of non-coding RNAs in AITD patients.
MiR-155-5p and miR-146a-5p are two widely studied miRNAs and have important roles in modulating immune response (122, 144–146). MiR-146a-5p could repress IL-1R-associated kinase 1 and TNF-receptor-associated factor 6, and its downexpression would increase the activation and antigen presentation of dendritic cells (144, 147). MiR-155-5p also could regulate the immune functions of Th cells or dendritic cells by targeting transcription factors or key molecules involved in immune response (122, 148, 149). Abnormal expressions of miR-155-5p and miRNA-146a-5p can contribute to the development of autoimmune diseases by breaking immune homeostasis and immune tolerance. There are several studies providing evidence for the abnormal expression of miR-155-5p and miR-146a-5p in AITD patients (Figure 2; Table 1). Bernecker et al. found that miR-146a-5p and miR-155-5p were differently expressed in the thyroid tissues of AITD (128). GD and HT patients had significantly lower levels of miR-146a-5p and miR-155-5p in the thyroid tissues, respectively (128). A subsequent study by Bernecker et al. found that GD and HT patients had lower level of miR-155-5p in HT in CD8+ T cells than controls (130). Wei et al. reported that Graves’ ophthalmopathy patients had significantly lower levels of miR-146a-5p than controls, and miR-146a-5p was negatively correlated with serum level of IL-17, which had been suggested to be an important pathogenic cytokine in the development of Graves’ ophthalmopathy (33, 34, 137). However, few studies have explored their clinical utility as diagnostic biomarkers or predictors of treatment outcomes. Besides no study has been performed to explore the feasibility of treatment strategies targeting those two miRNAs in AITD, especially for those with intractable GD.
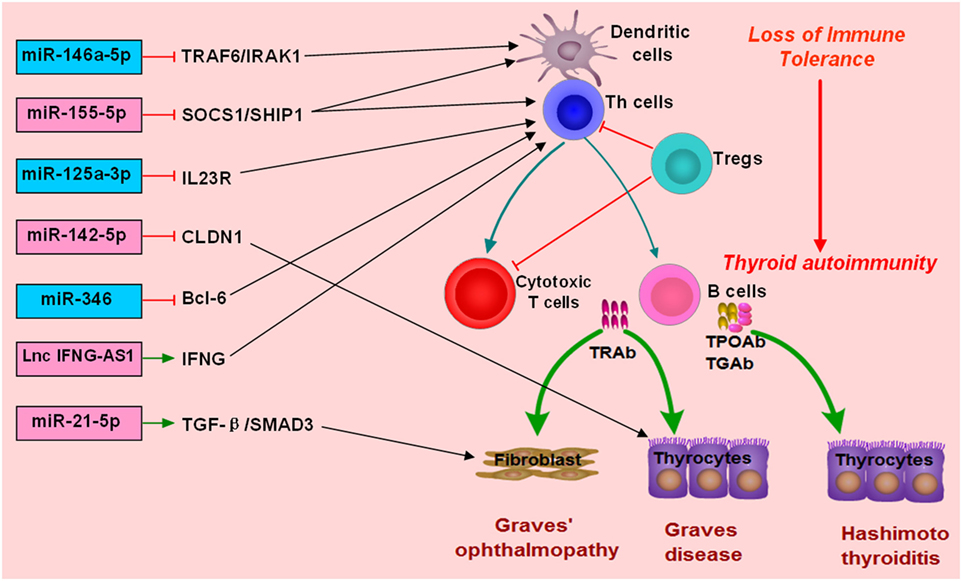
Figure 2. Roles of non-coding RNAs in the development of autoimmune thyroid diseases (AITD). microRNAs (miRNAs) or long non-coding RNAs (lncRNAs) can target some genes involved in immune response or the function of immune cells. The altered expression of miRNAs or lncRNAs can alter the normal function of immune cells, break immune homeostasis, and result in immune attacks toward thyroid tissues during the development of AITD. MiR-146a-5p can repress IL-1R-associated kinase 1 (IRAK1) and TNF-receptor-associated factor 6 (TRAF6), and its downexpression will increase the activation and antigen presentation of dendritic cells. Other miRNAs, such as miR-125-3p and miR-346 and miR-155-5p, can also regulate the immune functions of Th cells or dendritic cells by targeting transcription factors or key molecules. Lnc IFNG-AS1 can increase the expression of IFNG and increase the activation Th1 cell, and increased level of lncRNA IFNG-AS1 thus contributes to Th1 cell response in HT patients. MiR-21-5p can promote collagen I expression and total collagen production induced by TGF-beta1 in orbital fibroblasts, and increased expression of miR-21-5p thus can contribute to Graves’ ophthalmopathy. MiR-142-5p can target CLDN1, and its overexpression in thyrocytes can result in reduced expression of claudin-1 and increased permeability of thyrocytes monolayer.
There are also some other miRNAs found to be associated with AITD (Table 1). Chen et al. found downregulation of miR-346 in GD patients, and miR-346 could inhibit Bcl-6 expression and regulate the activation of CD4+CXCR5+ T cells (132). Zhu et al. reported that miR-142-5p was highly expressed in HT patients and was positively correlated with TgAb (141). Overexpression of miR-142-5p in thyrocytes resulted in reduced expression of claudin-1 and increased permeability of thyrocytes monolayer (141). Tong et al. found that the expression of miR-21-5p in orbital fibroblasts from Graves’ ophthalmopathy was higher than that in the controls, and miR-21-5p could promote collagen I expression and total collagen production induced by TGF-beta1 in orbital fibroblasts (136). Peng et al. found that miR-125a-3p could target IL-23 receptor (IL-23R), and its decreased expression of miR-125a-3p could upregulate IL-23R expression in HT patients (142). Our previous study simultaneously detected the expression profiles of miRNAs and mRNAs in the thyroid tissues of GD patients (133). We found five unregulated miRNAs, such as miR-22-3p and miR-183-5p, and 18 downregulated miRNAs in the thyroid tissues of GD patients, such as miR-101-3p, miR-660-5p, and miR-197-3p (133). The finding from our study highlighted a miRNA-target gene network in the pathogenesis of GD (133). Other abnormally expressed miRNAs identified in AITD patients could be found in Table 1.
Some studies also studied the clinical utility of miRNAs in AITD. Hiratsuka et al. used miRNA array to identify circulating miRNAs in relation to disease activity of GD by recruiting seven intractable GD patients, seven GD patients in remission, and seven healthy controls, and found that miR-23b-5p and miR-92a-3p were significantly increased in GD patients achieving remission, but let-7g-3p and miR-339-5p were significantly lower in GD patients achieving remission than intractable GD patients, demonstrating that some miRNAs could be as biomarkers of intractable GD and treatment response (134). A recent study by Li et al. found that patients with higher miR-346 level at diagnosis were at a higher risk of relapse during follow-up (125). A study by Shen et al. found that lower serum level of miR-224-5p was independently associated with glucocorticoid insensitivity in Graves’ ophthalmopathy, and in vitro overexpression of miR-224-5p could restore glucocorticoid sensitivity via targeting GSK-3beta (135). However, apart from those three studies above, no other studies assessing the clinical utility of miRNAs in AITD are available.
microRNAs are important regulators of gene expression, while genetic variants in miRNAs have been associated with many diseases (150–152). Inoue et al. found that MIR125A rs12976445 C/T was significantly associated with HD and intractable GD (131). Our recent study also proved that rs3746444 of miR-499a and rs12976445 of miR-125a-5p were associated with AITD susceptibility (153). The above study also indicated the roles of miRNAs in the pathogenesis of AITD. In addition, Dicer is an important ribonuclease involved in the biogenesis of miRNAs (154). Frezzetti et al. reported that the development of the thyroid gland was not affected by the absence of Dicer through using thyrocyte-specific Dicer knockout mice, but Dicer knockout resulted in severe hypothyroidism (155). In addition, Dicer inactivation also increased the expressions of Tg and decreased the expressions of cell adhesion proteins in the thyroid cells, such as Cdh16 and Cdh1 (155). Saeki et al. found that AITD patients had lower expression of Dicer and DROSHA compared with healthy controls, and DICER rs1057035 and DROSHA rs644236 were obviously associated with susceptibility to GD (156).
Although there are many studies investigating the differently expressed miRNAs in AITD patients, few studies have explored their clinical utility. The diagnostic values of those miRNAs and their roles in the risk stratification of AITD patients have not been clearly defined. More studies are needed to investigate the clinical significance of miRNAs and assess whether miRNAs can help to promote advances in the personalized therapeutics for AITD patients. Besides, no study has been performed to explore the feasibility of treatment strategies targeting miRNAs in AITD, especially for the treatment of intractable GD. Therefore, more studies in the future are needed to find more AITD-related miRNAs, to explore their molecular roles in the pathogenesis of AITD, and to investigate their clinical utility in AITD.
LncRNAs and AITD
Long non-coding RNAs are a class of non-coding RNAs with a length of more than 200 nt, which are also involved in autoimmune diseases (157, 158). LncRNAs are found to be involved in a variety of biological processes, and lncRNAs may regulate gene expressions at various levels, such as epigenetic regulation, transcriptional regulation, posttranscriptional regulation, and regulating miRNAs (159–161). Many studies have confirmed that lncRNAs play critical roles in immune system development and function regulation (162, 163). LncRNAs are involved in regulating T cell production and differentiation, and different types of T cells may express certain specific lncRNAs (162, 164–166). Previous studies have reported aberrant expressions of many lncRNAs in various autoimmune diseases, but the precise mechanisms underling the roles of lncRNAs in autoimmune diseases are still largely unknown (167–170). There are also several studies suggesting the possible important roles of lncRNAs in AITD (127, 139, 171, 172).
Christensen et al. first reported one lncRNA associated with GD, namely lncRNA Heg (127). LncRNA Heg was correlated with the levels of TRAb and CD14 mRNA in the mononuclear cells of GD patients (127). In vitro studies suggested that lncRNA Heg could significantly decrease the level of CD14 mRNA of mononuclear cells (127). However, antithyroid treatment was unable to change the level of lncRNA Heg in GD patients (171). Peng et al. found that lncRNA-IFNG-AS1 was upregulated in HT patients, and it was associated with the frequency of circulating Th1 cells and IFN-γ expression (139). In addition, lncRNA-IFNG-AS1 could regulate IFN-γ expression in human CD4+ T cells and may promote Th1 response in the development of HT (139). A GWAS by Zhao et al. found a susceptibility locus of GD at an intergenic region harboring two non-coding RNAs at 14q32.2, and two lncRNAs including C14orf64 and GDCG14q32.2 were reported to be potentially involved in the pathogenesis of GD (172).
Researches on the lncRNAs associated with AITD will also provide potential drug targets and help us to find some novel treatment strategies for AITD (42, 173). Currently, there is still lack of a good understanding on the regulatory network of lncRNAs/miRNAs/mRNAs in the molecular mechanisms of AITD. More studies in the future are needed to find more AITD-related lncRNAs, to explore their roles in AITD in details, and to investigate the feasibility of non-coding RNA-based therapeutic agents for AITD (174). In addition, the clinical utility of lncRNAs in AITD patients is also poorly studied, and more studies are needed to identify those lncRNAs associated with different types of AITD and to assess their roles in predicting treatment responsiveness, guiding the choice of treatment approach for GD patients, and predicting relapse risk during follow-up.
X Chromosome Inactivation (XCI) in AITD
X chromosomes are randomly inactivated in females, which can result in a mosaic pattern of cells expressing genes from either chromosome (175). XCI is a major epigenetic feature in which one X chromosome is transcriptionally silenced, and the X-inactive-specific transcript has major roles in the silencing (176, 177). Currently, histone modifications, DNA methylation, and non-coding RNAs are all considered to be involved in the formation of XCI (178). Skewed XCI occurs when the inactivation of one X chromosome is silenced more than the other one (178, 179). Skewed XCI can also result in loss of imbalance of gene products and immune tolerance and thus is involved in many autoimmune diseases (180–183).
Autoimmune thyroid diseases occur more often in females, suggesting a key role for the XCI in AITD (176). Skewed XCI has been proposed as a potential mechanism explaining the female preponderance of AITD (184). Previous studies have proven the increased frequency of skewed XCI in AITD patients (182, 184–187). Brix et al. first conducted a case–control study and found that the frequencies of skewed XCI in female twins with GD and HT were both significantly higher than the controls (11%) (185). Ozcelik et al. reported that extreme skewing of XCI was present in 19% of AITD patients, but only in 2.4% of controls (P < 0.0001) (182). Yin et al. also found that XCI skewing was significantly associated with AITD (OR = 4.0, P = 0.004) (186). However, a recent study by Ishido at al. reported that there was no obvious difference of skewed XCI between AITD cases and controls, but it was significantly higher in intractable GD patients (66.7%) than those with GD remission (25.0%), which suggested that skewed XCI was related to the GD progression (188).
Summary
The important roles of epigenetics in AITD have been increasingly recognized, but many of them are still largely unknown, which need to be elucidated in more epigenetic researches in the future. Specially, more studies are needed to find more AITD-related epigenetic modifications, to explore deeper complex interactions of epigenetic factors in the pathogenesis of AITD, and to investigate the feasibility of epigenetic-based therapeutic strategies for the treatment of AITD. In addition, it is still unclear whether some thyroid-specific lncRNAs and miRNAs could have roles in the pathogenesis of AITD, and future studies are recommended to explore it. Currently, there are still great challenges in providing effective healthcare for AITD patients, such as adequate choice of treatment protocols for GD, precise prediction of treatment response, and appropriate personalized therapeutics for AITD. Epigenetics undoubtedly provide opportunities of a better understanding of the mechanism of AITD and may help to solve these challenges and promote advances in the personalized therapeutics of AITD. However, although there are many studies assessing the epigenetic modifications in AITD patients, few studies have explored those epigenetic modifications that are associated with types of AITD, treatment outcomes, and risk of relapse during follow-up, and the clinical utility of epigenetics in AITD remains poorly defined. A better understanding of those epigenetic modifications can contribute to accurate diagnosis of AITD, adequate choice of treatment approach, and precise prediction of treatment outcomes, and it’s recommended in future researches. Besides most studies available now focus on one type of epigenetic modifications (e.g., DNA methylation) in isolation, which is unlikely to fully explain the etiology of AITD. Hence, to get a better understanding of the etiology of AITD and improve the clinical utility of epigenetics in AITD, it is essential to integrate analyses of multiple epigenetic modifications together in future studies. The increasing use of bioinformatics and high-throughput sequencing will provide much help in interpreting analyses of multiple epigenetic modifications in AITD. Finally, clinical observational studies with large number of AITD patients are also needed, which will provide essential evidence for the clinical utility of those epigenetic modifications in AITD.
Author Contributions
BW and JZ designed the study. BW, XS, RS, DX, and JZ contributed to the literature search, interpretation, writing, and proofreading of the manuscript. BW generated the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The present work was supported by grants from the National Natural Science Foundation of China (No. 81670722 and 81471004).
References
1. Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmun Rev (2015) 14(2):174–80. doi: 10.1016/j.autrev.2014.10.016
2. Burch HB, Cooper DS. Management of Graves disease: a review. JAMA (2015) 314(23):2544–54. doi:10.1001/jama.2015.16535
4. Leese GP, Flynn RV, Jung RT, Macdonald TM, Murphy MJ, Morris AD. Increasing prevalence and incidence of thyroid disease in Tayside, Scotland: the Thyroid Epidemiology Audit and Research Study (TEARS). Clin Endocrinol (Oxf) (2008) 68(2):311–6. doi:10.1111/j.1365-2265.2007.03051.x
5. McLeod DS, Cooper DS. The incidence and prevalence of thyroid autoimmunity. Endocrine (2012) 42(2):252–65. doi:10.1007/s12020-012-9703-2
6. De Leo S, Lee SY, Braverman LE. Hyperthyroidism. Lancet (2016) 388(10047):906–18. doi:10.1016/S0140-6736(16)00278-6
7. Chen WH, Chen YK, Lin CL, Yeh JH, Kao CH. Hashimoto’s thyroiditis, risk of coronary heart disease, and L-thyroxine treatment: a nationwide cohort study. J Clin Endocrinol Metab (2015) 100(1):109–14. doi:10.1210/jc.2014-2990
8. Medici M, Korevaar TI, Schalekamp-Timmermans S, Gaillard R, de Rijke YB, Visser WE, et al. Maternal early-pregnancy thyroid function is associated with subsequent hypertensive disorders of pregnancy: the generation R study. J Clin Endocrinol Metab (2014) 99(12):E2591–8. doi:10.1210/jc.2014-1505
9. Karch A, Thomas SL. Autoimmune thyroiditis as a risk factor for stroke: a historical cohort study. Neurology (2014) 82(18):1643–52. doi:10.1212/WNL.0000000000000377
10. Fallah M, Liu X, Ji J, Forsti A, Sundquist K, Hemminki K. Autoimmune diseases associated with non-Hodgkin lymphoma: a nationwide cohort study. Ann Oncol (2014) 25(10):2025–30. doi:10.1093/annonc/mdu365
11. Liu CL, Cheng SP, Lin HW, Lai YL. Risk of thyroid cancer in patients with thyroiditis: a population-based cohort study. Ann Surg Oncol (2014) 21(3):843–9. doi:10.1245/s10434-013-3363-1
12. Gleicher N. Maternal autoimmunity and adverse pregnancy outcomes. J Autoimmun (2014) 50:83–6. doi:10.1016/j.jaut.2013.12.009
13. Chen YK, Lin CL, Cheng FT, Sung FC, Kao CH. Cancer risk in patients with Hashimoto’s thyroiditis: a nationwide cohort study. Br J Cancer (2013) 109(9):2496–501. doi:10.1038/bjc.2013.597
14. Franklyn JA, Boelaert K. Thyrotoxicosis. Lancet (2012) 379(9821):1155–66. doi:10.1016/S0140-6736(11)60782-4
15. Ramos-Levi AM, Marazuela M. Pathogenesis of thyroid autoimmune disease: the role of cellular mechanisms. Endocrinol Nutr (2016) 63(8):421–9. doi:10.1016/j.endonu.2016.04.003
16. Lee HJ, Li CW, Hammerstad SS, Stefan M, Tomer Y. Immunogenetics of autoimmune thyroid diseases: a comprehensive review. J Autoimmun (2015) 64:82–90. doi:10.1016/j.jaut.2015.07.009
17. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev (2014) 35(1):59–105. doi:10.1210/er.2013-1055
18. Prabhakar BS, Bahn RS, Smith TJ. Current perspective on the pathogenesis of Graves’ disease and ophthalmopathy. Endocr Rev (2003) 24(6):802–35. doi:10.1210/er.2002-0020
19. Gilbert JA, Gianoukakis AG, Salehi S, Moorhead J, Rao PV, Khan MZ, et al. Monoclonal pathogenic antibodies to the thyroid-stimulating hormone receptor in Graves’ disease with potent thyroid-stimulating activity but differential blocking activity activate multiple signaling pathways. J Immunol (2006) 176(8):5084–92. doi:10.4049/jimmunol.176.8.5084
20. Mikos H, Mikos M, Obara-Moszynska M, Niedziela M. The role of the immune system and cytokines involved in the pathogenesis of autoimmune thyroid disease (AITD). Endokrynol Pol (2014) 65(2):150–5. doi:10.5603/EP.2014.0021
21. Li H, Wang T. The autoimmunity in Graves’s disease. Front Biosci (Landmark Ed) (2013) 18:782–7. doi:10.2741/4141
22. Smith TJ, Hegedus L. Graves’ disease. N Engl J Med (2016) 375(16):1552–65. doi:10.1056/NEJMra1510030
23. Guan LJ, Wang X, Meng S, Shi LF, Jiang WJ, Xiao L, et al. Increased IL-21/IL-21R expression and its proinflammatory effects in autoimmune thyroid disease. Cytokine (2015) 72(2):160–5. doi:10.1016/j.cyto.2014.11.005
24. Guo J, Rapoport B, McLachlan SM. Balance of Th1/Th2 cytokines in thyroid autoantibody synthesis in vitro. Autoimmunity (1999) 30(1):1–9. doi:10.3109/08916939908994754
25. Nanba T, Watanabe M, Inoue N, Iwatani Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid (2009) 19(5):495–501. doi:10.1089/thy.2008.0423
26. Phenekos C, Vryonidou A, Gritzapis AD, Baxevanis CN, Goula M, Papamichail M. Th1 and Th2 serum cytokine profiles characterize patients with Hashimoto’s thyroiditis (Th1) and Graves’ disease (Th2). Neuroimmunomodulation (2004) 11(4):209–13. doi:10.1159/000078438
27. Rapoport B, McLachlan SM. Graves’ hyperthyroidism is antibody-mediated but is predominantly a Th1-type cytokine disease. J Clin Endocrinol Metab (2014) 99(11):4060–1. doi:10.1210/jc.2014-3011
28. Yuan Q, Zhao Y, Zhu X, Liu X. Low regulatory T cell and high IL-17 mRNA expression in a mouse Graves’ disease model. J Endocrinol Invest (2017) 40(4):397–407. doi:10.1007/s40618-016-0575-9
29. Li C, Yuan J, Zhu YF, Yang XJ, Wang Q, Xu J, et al. Imbalance of Th17/Treg in different subtypes of autoimmune thyroid diseases. Cell Physiol Biochem (2016) 40(1–2):245–52. doi:10.1159/000452541
30. Zhu C, Ma J, Liu Y, Tong J, Tian J, Chen J, et al. Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J Clin Endocrinol Metab (2012) 97(3):943–50. doi:10.1210/jc.2011-2003
31. Song RH, Yu ZY, Qin Q, Wang X, Muhali FS, Shi LF, et al. Different levels of circulating Th22 cell and its related molecules in Graves’ disease and Hashimoto’s thyroiditis. Int J Clin Exp Pathol (2014) 7(7):4024–31.
32. Xue H, Yu X, Ma L, Song S, Li Y, Zhang L, et al. The possible role of CD4(+)CD25(high)Foxp3(+)/CD4(+)IL-17A(+) cell imbalance in the autoimmunity of patients with Hashimoto thyroiditis. Endocrine (2015) 50(3):665–73. doi:10.1007/s12020-015-0569-y
33. Fang S, Huang Y, Wang S, Zhang Y, Luo X, Liu L, et al. IL-17A exacerbates fibrosis by promoting the proinflammatory and profibrotic function of orbital fibroblasts in TAO. J Clin Endocrinol Metab (2016) 101(8):2955–65. doi:10.1210/jc.2016-1882
34. Fang S, Huang Y, Zhong S, Zhang Y, Liu X, Wang Y, et al. IL-17A promotes RANTES expression, but not IL-16, in orbital fibroblasts via CD40-CD40L combination in thyroid-associated ophthalmopathy. Invest Ophthalmol Vis Sci (2016) 57(14):6123–33. doi:10.1167/iovs.16-20199
35. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13(6):668–77. doi:10.1016/j.autrev.2013.12.004
36. Klatka M, Grywalska E, Partyka M, Charytanowicz M, Kiszczak-Bochynska E, Rolinski J. Th17 and Treg cells in adolescents with Graves’ disease. Impact of treatment with methimazole on these cell subsets. Autoimmunity (2014) 47(3):201–11. doi:10.3109/08916934.2013.879862
37. Jogdand GM, Mohanty S, Devadas S. Regulators of Tfh cell differentiation. Front Immunol (2016) 7:520. doi:10.3389/fimmu.2016.00520
38. Jia L, Wu C. The biology and functions of Th22 cells. Adv Exp Med Biol (2014) 841:209–30. doi:10.1007/978-94-017-9487-9_8
39. Bai X, Sun J, Wang W, Shan Z, Zheng H, Li Y, et al. Increased differentiation of Th22 cells in Hashimoto’s thyroiditis. Endocr J (2014) 61(12):1181–90. doi:10.1507/endocrj.EJ14-0265
40. Wang Y, Smith TJ. Current concepts in the molecular pathogenesis of thyroid-associated ophthalmopathy. Invest Ophthalmol Vis Sci (2014) 55(3):1735–48. doi:10.1167/iovs.14-14002
41. Wong KH, Rong SS, Chong KK, Young AL, Pang CP, Chen LJ. Genetic associations of interleukin-related genes with Graves’ ophthalmopathy: a systematic review and meta-analysis. Sci Rep (2015) 5:16672. doi:10.1038/srep16672
42. Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annu Rev Pathol (2014) 9:147–56. doi:10.1146/annurev-pathol-012513-104713
43. Hsiao JY, Hsieh MC, Tien KJ, Hsu SC, Lin SR, Ke DS. Exon 33 T/T genotype of the thyroglobulin gene is a susceptibility gene for Graves’ disease in Taiwanese and exon 12 C/C genotype protects against it. Clin Exp Med (2008) 8(1):17–21. doi:10.1007/s10238-008-0151-5
44. Bufalo NE, Dos Santos RB, Marcello MA, Piai RP, Secolin R, Romaldini JH, et al. TSHR intronic polymorphisms (rs179247 and rs12885526) and their role in the susceptibility of the Brazilian population to Graves’ disease and Graves’ ophthalmopathy. J Endocrinol Invest (2015) 38(5):555–61. doi:10.1007/s40618-014-0228-9
45. Fujii A, Inoue N, Watanabe M, Kawakami C, Hidaka Y, Hayashizaki Y, et al. TSHR gene polymorphisms in the enhancer regions are most strongly associated with the development of Graves’ disease, especially intractable disease, and of Hashimoto’s disease. Thyroid (2017) 27(1):111–9. doi:10.1089/thy.2016.0345
46. Khalilzadeh O, Noshad S, Rashidi A, Amirzargar A. Graves’ ophthalmopathy: a review of immunogenetics. Curr Genomics (2011) 12(8):564–75. doi:10.2174/138920211798120844
47. Ban Y, Concepcion ES, Villanueva R, Greenberg DA, Davies TF, Tomer Y. Analysis of immune regulatory genes in familial and sporadic Graves’ disease. J Clin Endocrinol Metab (2004) 89(9):4562–8. doi:10.1210/jc.2003-031693
48. Gu LQ, Zhu W, Zhao SX, Zhao L, Zhang MJ, Cui B, et al. Clinical associations of the genetic variants of CTLA-4, Tg, TSHR, PTPN22, PTPN12 and FCRL3 in patients with Graves’ disease. Clin Endocrinol (Oxf) (2010) 72(2):248–55. doi:10.1111/j.1365-2265.2009.03617.x
49. Zheng L, Wang X, Xu L, Wang N, Cai P, Liang T, et al. Foxp3 gene polymorphisms and haplotypes associate with susceptibility of Graves’ disease in Chinese Han population. Int Immunopharmacol (2015) 25(2):425–31. doi:10.1016/j.intimp.2015.02.020
50. Hasham A, Tomer Y. Genetic and epigenetic mechanisms in thyroid autoimmunity. Immunol Res (2012) 54(1–3):204–13. doi:10.1007/s12026-012-8302-x
51. Yin X, Latif R, Bahn R, Davies TF. Genetic profiling in Graves’ disease: further evidence for lack of a distinct genetic contribution to Graves’ ophthalmopathy. Thyroid (2012) 22(7):730–6. doi:10.1089/thy.2012.0007
52. Brix TH, Hegedus L. Twins as a tool for evaluating the influence of genetic susceptibility in thyroid autoimmunity. Ann Endocrinol (Paris) (2011) 72(2):103–7. doi:10.1016/j.ando.2011.03.013
53. Brix TH, Hegedus L. Twin studies as a model for exploring the aetiology of autoimmune thyroid disease. Clin Endocrinol (Oxf) (2012) 76(4):457–64. doi:10.1111/j.1365-2265.2011.04318.x
54. Muscogiuri G, Mari D, Prolo S, Fatti LM, Cantone MC, Garagnani P, et al. 25 hydroxyvitamin D deficiency and its relationship to autoimmune thyroid disease in the elderly. Int J Environ Res Public Health (2016) 13(9):E850. doi:10.3390/ijerph13090850
55. Giovinazzo S, Vicchio TM, Certo R, Alibrandi A, Palmieri O, Campenni A, et al. Vitamin D receptor gene polymorphisms/haplotypes and serum 25(OH)D3 levels in Hashimoto’s thyroiditis. Endocrine (2016) 55(2):599–606. doi:10.1007/s12020-016-0942-5
56. Miranda DM, Massom JN, Catarino RM, Santos RT, Toyoda SS, Marone MM, et al. Impact of nutritional iodine optimization on rates of thyroid hypoechogenicity and autoimmune thyroiditis: a cross-sectional, comparative study. Thyroid (2015) 25(1):118–24. doi:10.1089/thy.2014.0182
57. Camargo RY, Tomimori EK, Neves SC, Rubio IGS, Galrao AL, Knobel M, et al. Thyroid and the environment: exposure to excessive nutritional iodine increases the prevalence of thyroid disorders in Sao Paulo, Brazil. Eur J Endocrinol (2008) 159(3):293–9. doi:10.1530/EJE-08-0192
58. Laurberg P, Jorgensen T, Perrild H, Ovesen L, Knudsen N, Pedersen IB, et al. The Danish investigation on iodine intake and thyroid disease, DanThyr: status and perspectives. Eur J Endocrinol (2006) 155(2):219–28. doi:10.1530/eje.1.02210
59. Effraimidis G, Wiersinga WM. Mechanisms in endocrinology: autoimmune thyroid disease: old and new players. Eur J Endocrinol (2014) 170(6):R241–52. doi:10.1530/EJE-14-0047
60. Wang Z, Fan X, Zhang R, Lin Z, Lu T, Bai X, et al. Integrative analysis of mRNA and miRNA array data reveals the suppression of retinoic acid pathway in regulatory T cells of Graves’ disease. J Clin Endocrinol Metab (2014) 99(12):E2620–7. doi:10.1210/jc.2014-1883
61. Ajjan RA, Weetman AP. The pathogenesis of Hashimoto’s thyroiditis: further developments in our understanding. Horm Metab Res (2015) 47(10):702–10. doi:10.1055/s-0035-1548832
62. Marsit CJ. Influence of environmental exposure on human epigenetic regulation. J Exp Biol (2015) 218(Pt 1):71–9. doi:10.1242/jeb.106971
63. Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol (2015) 16(10):593–610. doi:10.1038/nrm4048
64. Canas CA, Canas F, Bonilla-Abadia F, Ospina FE, Tobon GJ. Epigenetics changes associated to environmental triggers in autoimmunity. Autoimmunity (2016) 49(1):1–11. doi:10.3109/08916934.2015.1086996
65. Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet (2012) 13(2):97–109. doi:10.1038/nrg3142
66. Yet I, Tsai PC, Castillo-Fernandez JE, Carnero-Montoro E, Bell JT. Genetic and environmental impacts on DNA methylation levels in twins. Epigenomics (2016) 8(1):105–17. doi:10.2217/epi.15.90
67. Dik S, Scheepers PT, Godderis L. Effects of environmental stressors on histone modifications and their relevance to carcinogenesis: a systematic review. Crit Rev Toxicol (2012) 42(6):491–500. doi:10.3109/10408444.2012.684146
68. Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol (2014) 15(6):484–91. doi:10.1038/ni.2887
69. Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, Curti S, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol (2015) 16(3):318–25. doi:10.1038/ni.3093
70. Jeffries MA, Sawalha AH. Autoimmune disease in the epigenetic era: how has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev Clin Immunol (2015) 11(1):45–58. doi:10.1586/1744666X.2015.994507
71. Brown CC, Wedderburn LR. Genetics: mapping autoimmune disease epigenetics: what’s on the horizon? Nat Rev Rheumatol (2015) 11(3):131–2. doi:10.1038/nrrheum.2014.210
72. Stefan M, Wei C, Lombardi A, Li CW, Concepcion ES, Inabnet WB III, et al. Genetic-epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc Natl Acad Sci U S A (2014) 111(34):12562–7. doi:10.1073/pnas.1408821111
73. Yamada H, Itoh M, Hiratsuka I, Hashimoto S. Circulating microRNAs in autoimmune thyroid diseases. Clin Endocrinol (Oxf) (2014) 81(2):276–81. doi:10.1111/cen.12432
74. Cai TT, Muhali FS, Song RH, Qin Q, Wang X, Shi LF, et al. Genome-wide DNA methylation analysis in Graves’ disease. Genomics (2015) 105(4):204–10. doi:10.1016/j.ygeno.2015.01.001
75. Limbach M, Saare M, Tserel L, Kisand K, Eglit T, Sauer S, et al. Epigenetic profiling in CD4+ and CD8+ T cells from Graves’ disease patients reveals changes in genes associated with T cell receptor signaling. J Autoimmun (2016) 67:46–56. doi:10.1016/j.jaut.2015.09.006
76. Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol (2015) 16(9):519–32. doi:10.1038/nrm4043
77. Torres IO, Fujimori DG. Functional coupling between writers, erasers and readers of histone and DNA methylation. Curr Opin Struct Biol (2015) 35:68–75. doi:10.1016/j.sbi.2015.09.007
78. Schubeler D. Function and information content of DNA methylation. Nature (2015) 517(7534):321–6. doi:10.1038/nature14192
79. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet (2016) 17(9):551–65. doi:10.1038/nrg.2016.83
80. Richardson BC, Patel DR. Epigenetics in 2013. DNA methylation and miRNA: key roles in systemic autoimmunity. Nat Rev Rheumatol (2014) 10(2):72–4. doi:10.1038/nrrheum.2013.211
81. Ai R, Hammaker D, Boyle DL, Morgan R, Walsh AM, Fan S, et al. Joint-specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun (2016) 7:11849. doi:10.1038/ncomms11849
82. Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, Schones DE, et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A (2016) 113(21):E3002–11. doi:10.1073/pnas.1603712113
83. Hammaker D, Whitaker JW, Maeshima K, Boyle DL, Ekwall AH, Wang W, et al. LBH gene transcription regulation by the interplay of an enhancer risk allele and DNA methylation in rheumatoid arthritis. Arthritis Rheumatol (2016) 68(11):2637–45. doi:10.1002/art.39746
84. Paul DS, Teschendorff AE, Dang MA, Lowe R, Hawa MI, Ecker S, et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat Commun (2016) 7:13555. doi:10.1038/ncomms13555
85. Tahara T, Hirata I, Nakano N, Nagasaka M, Nakagawa Y, Shibata T, et al. Comprehensive DNA methylation profiling of inflammatory mucosa in ulcerative colitis. Inflamm Bowel Dis (2017) 23(1):165–73. doi:10.1097/MIB.0000000000000990
86. Licht JD. DNA methylation inhibitors in cancer therapy: the immunity dimension. Cell (2015) 162(5):938–9. doi:10.1016/j.cell.2015.08.005
87. Chistiakov DA, Orekhov AN, Bobryshev YV. Treatment of cardiovascular pathology with epigenetically active agents: focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int J Cardiol (2017) 227:66–82. doi:10.1016/j.ijcard.2016.11.204
88. Erdmann A, Halby L, Fahy J, Arimondo PB. Targeting DNA methylation with small molecules: what’s next? J Med Chem (2015) 58(6):2569–83. doi:10.1021/jm500843d
89. Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet (2015) 47(11):1282–93. doi:10.1038/ng.3405
90. Shin J, Bourdon C, Bernard M, Wilson MD, Reischl E, Waldenberger M, et al. Layered genetic control of DNA methylation and gene expression: a locus of multiple sclerosis in healthy individuals. Hum Mol Genet (2015) 24(20):5733–45. doi:10.1093/hmg/ddv294
91. Arakawa Y, Watanabe M, Inoue N, Sarumaru M, Hidaka Y, Iwatani Y. Association of polymorphisms in DNMT1, DNMT3A, DNMT3B, MTHFR and MTRR genes with global DNA methylation levels and prognosis of autoimmune thyroid disease. Clin Exp Immunol (2012) 170(2):194–201. doi:10.1111/j.1365-2249.2012.04646.x
92. Cai TT, Zhang J, Wang X, Song RH, Qin Q, Muhali FS, et al. Gene-gene and gene-sex epistatic interactions of DNMT1, DNMT3A and DNMT3B in autoimmune thyroid disease. Endocr J (2016) 63(7):643–53. doi:10.1507/endocrj.EJ15-0596
93. Mao R, Fan Y, Zuo L, Geng D, Meng F, Zhu J, et al. Association study between methylenetetrahydrofolate reductase gene polymorphisms and Graves’ disease. Cell Biochem Funct (2010) 28(7):585–90. doi:10.1002/cbf.1694
94. Lee JY, Kim NK, Cho YW, Lew H. Association between methylenetetrahydrofolate reductase (MTHFR) polymorphisms and susceptibility to Graves’ ophthalmopathy. Mol Med Rep (2016) 14(3):2276–82. doi:10.3892/mmr.2016.5458
95. Lan F, Shi Y. Histone H3.3 and cancer: a potential reader connection. Proc Natl Acad Sci U S A (2015) 112(22):6814–9. doi:10.1073/pnas.1418996111
96. Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell (2014) 56(2):298–310. doi:10.1016/j.molcel.2014.08.022
97. Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature (2014) 514(7523):513–7. doi:10.1038/nature13605
98. Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, et al. Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell (2016) 165(2):331–42. doi:10.1016/j.cell.2016.02.064
99. Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol (2017) 18(2):90–101. doi:10.1038/nrm.2016.140
100. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol (2014) 15(11):703–8. doi:10.1038/nrm3890
101. Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet (2016) 32(1):42–56. doi:10.1016/j.tig.2015.10.007
102. Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol (2015) 16(3):178–89. doi:10.1038/nrm3941
103. Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci (2010) 35(11):618–26. doi:10.1016/j.tibs.2010.05.006
104. Yan N, Zhou JZ, Zhang JA, Cai T, Zhang W, Wang Y, et al. Histone hypoacetylation and increased histone deacetylases in peripheral blood mononuclear cells from patients with Graves’ disease. Mol Cell Endocrinol (2015) 414:143–7. doi:10.1016/j.mce.2015.05.037
105. Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes (2014) 63(5):1748–62. doi:10.2337/db13-1251
106. Liu Y, Liao J, Zhao M, Wu H, Yung S, Chan TM, et al. Increased expression of TLR2 in CD4(+) T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur J Immunol (2015) 45(9):2683–93. doi:10.1002/eji.201445219
107. Araki Y, Tsuzuki Wada T, Aizaki Y, Sato K, Yokota K, Fujimoto K, et al. Histone methylation and STAT-3 differentially regulate interleukin-6-induced matrix metalloproteinase gene activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol (2016) 68(5):1111–23. doi:10.1002/art.39563
108. Angiolilli C, Kabala PA, Grabiec AM, Van Baarsen IM, Ferguson BS, Garcia S, et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann Rheum Dis (2017) 76(1):277–85. doi:10.1136/annrheumdis-2015-209064
109. Cheng F, Lienlaf M, Wang HW, Perez-Villarroel P, Lee C, Woan K, et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J Immunol (2014) 193(6):2850–62. doi:10.4049/jimmunol.1302778
110. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov (2014) 13(9):673–91. doi:10.1038/nrd4360
111. Ciornei RT, Hong SH, Fang Y, Zhu Z, Braley-Mullen H. Mechanisms and kinetics of proliferation and fibrosis development in a mouse model of thyrocyte hyperplasia. Cell Immunol (2016) 304-305:16–26. doi:10.1016/j.cellimm.2016.04.006
112. Stefan M, Jacobson EM, Huber AK, Greenberg DA, Li CW, Skrabanek L, et al. Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J Biol Chem (2011) 286(36):31168–79. doi:10.1074/jbc.M111.247510
113. Kawashima A, Tanigawa K, Akama T, Wu H, Sue M, Yoshihara A, et al. Fragments of genomic DNA released by injured cells activate innate immunity and suppress endocrine function in the thyroid. Endocrinology (2011) 152(4):1702–12. doi:10.1210/en.2010-1132
114. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity (2016) 44(6):1337–49. doi:10.1016/j.immuni.2016.05.009
115. Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest (2009) 119(10):3048–58. doi:10.1172/JCI38902
116. Yang H, Lee SM, Gao B, Zhang J, Fang D. Histone deacetylase sirtuin 1 deacetylates IRF1 protein and programs dendritic cells to control Th17 protein differentiation during autoimmune inflammation. J Biol Chem (2013) 288(52):37256–66. doi:10.1074/jbc.M113.527531
117. Sarumaru M, Watanabe M, Inoue N, Hisamoto Y, Morita E, Arakawa Y, et al. Association between functional SIRT1 polymorphisms and the clinical characteristics of patients with autoimmune thyroid disease. Autoimmunity (2016) 49(5):329–37. doi:10.3109/08916934.2015.1134506
118. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol (2016) 16(5):279–94. doi:10.1038/nri.2016.40
119. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet (2015) 16(7):421–33. doi:10.1038/nrg3965
120. Vicente R, Noel D, Pers YM, Apparailly F, Jorgensen C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat Rev Rheumatol (2016) 12(8):496. doi:10.1038/nrrheum.2016.119
121. Rothchild AC, Sissons JR, Shafiani S, Plaisier C, Min D, Mai D, et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2016) 113(41):E6172–81. doi:10.1073/pnas.1608255113
122. Seddiki N, Brezar V, Ruffin N, Levy Y, Swaminathan S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology (2014) 142(1):32–8. doi:10.1111/imm.12227
123. Chen JQ, Papp G, Szodoray P, Zeher M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev (2016) 15(12):1171–80. doi:10.1016/j.autrev.2016.09.003
124. Fedeli M, Riba M, Garcia Manteiga JM, Tian L, Vigano V, Rossetti G, et al. miR-17 approximately 92 family clusters control iNKT cell ontogenesis via modulation of TGF-beta signaling. Proc Natl Acad Sci U S A (2016) 113(51):E8286–95. doi:10.1073/pnas.1612024114
125. Li J, Cai Y, Sun X, Yao D, Xia J. MiR-346 and TRAb as predicative factors for relapse in Graves’ disease within one year. Horm Metab Res (2017) 49(3):180–4. doi:10.1055/s-0043-100116
126. Rodriguez-Munoz A, Martinez-Hernandez R, Ramos-Levi AM, Serrano-Somavilla A, Gonzalez-Amaro R, Sanchez-Madrid F, et al. Circulating microvesicles regulate Treg and Th17 differentiation in human autoimmune thyroid disorders. J Clin Endocrinol Metab (2015) 100(12):E1531–9. doi:10.1210/jc.2015-3146
127. Christensen NJ, Habekost G, Bratholm P. A RNA transcript (Heg) in mononuclear cells is negatively correlated with CD14 mRNA and TSH receptor autoantibodies. Clin Exp Immunol (2008) 154(2):209–15. doi:10.1111/j.1365-2249.2008.03744.x
128. Bernecker C, Lenz L, Ostapczuk MS, Schinner S, Willenberg H, Ehlers M, et al. MicroRNAs miR-146a1, miR-155_2, and miR-200a1 are regulated in autoimmune thyroid diseases. Thyroid (2012) 22(12):1294–5. doi:10.1089/thy.2012.0277
129. Liu R, Ma X, Xu L, Wang D, Jiang X, Zhu W, et al. Differential microRNA expression in peripheral blood mononuclear cells from Graves’ disease patients. J Clin Endocrinol Metab (2012) 97(6):E968–72. doi:10.1210/jc.2011-2982
130. Bernecker C, Halim F, Lenz L, Haase M, Nguyen T, Ehlers M, et al. microRNA expressions in CD4+ and CD8+ T-cell subsets in autoimmune thyroid diseases. Exp Clin Endocrinol Diabetes (2014) 122(2):107–12. doi:10.1055/s-0033-1361088
131. Inoue Y, Watanabe M, Inoue N, Kagawa T, Shibutani S, Otsu H, et al. Associations of single nucleotide polymorphisms in precursor-microRNA (miR)-125a and the expression of mature miR-125a with the development and prognosis of autoimmune thyroid diseases. Clin Exp Immunol (2014) 178(2):229–35. doi:10.1111/cei.12410
132. Chen J, Tian J, Tang X, Rui K, Ma J, Mao C, et al. MiR-346 regulates CD4(+)CXCR5(+) T cells in the pathogenesis of Graves’ disease. Endocrine (2015) 49(3):752–60. doi:10.1007/s12020-015-0546-5
133. Qin Q, Wang X, Yan N, Song RH, Cai TT, Zhang W, et al. Aberrant expression of miRNA and mRNAs in lesioned tissues of Graves’ disease. Cell Physiol Biochem (2015) 35(5):1934–42. doi:10.1159/000374002
134. Hiratsuka I, Yamada H, Munetsuna E, Hashimoto S, Itoh M. Circulating MicroRNAs in Graves’ disease in relation to clinical activity. Thyroid (2016) 26(10):1431–40. doi:10.1089/thy.2016.0062
135. Shen L, Huang F, Ye L, Zhu W, Zhang X, Wang S, et al. Circulating microRNA predicts insensitivity to glucocorticoid therapy in Graves’ ophthalmopathy. Endocrine (2015) 49(2):445–56. doi:10.1007/s12020-014-0487-4
136. Tong BD, Xiao MY, Zeng JX, Xiong W. MiRNA-21 promotes fibrosis in orbital fibroblasts from thyroid-associated ophthalmopathy. Mol Vis (2015) 21:324–34.
137. Wei H, Guan M, Qin Y, Xie C, Fu X, Gao F, et al. Circulating levels of miR-146a and IL-17 are significantly correlated with the clinical activity of Graves’ ophthalmopathy. Endocr J (2014) 61(11):1087–92. doi:10.1507/endocrj.EJ14-0246
138. Li K, Du Y, Jiang BL, He JF. Increased microRNA-155 and decreased microRNA-146a may promote ocular inflammation and proliferation in Graves’ ophthalmopathy. Med Sci Monit (2014) 20:639–43. doi:10.12659/MSM.890686
139. Peng H, Liu Y, Tian J, Ma J, Tang X, Rui K, et al. The long noncoding RNA IFNG-AS1 promotes T helper type 1 cells response in patients with Hashimoto’s thyroiditis. Sci Rep (2015) 5:17702. doi:10.1038/srep17702
140. Kagawa T, Watanabe M, Inoue N, Otsu H, Saeki M, Katsumata Y, et al. Increases of microRNA let-7e in peripheral blood mononuclear cells in Hashimoto’s disease. Endocr J (2016) 63(4):375–80. doi:10.1507/endocrj.EJ15-0577
141. Zhu J, Zhang Y, Zhang W, Fan L, Wang L, Liu Y, et al. MicroRNA-142-5p contributes to Hashimoto’s thyroiditis by targeting CLDN1. J Transl Med (2016) 14(1):166. doi:10.1186/s12967-016-0917-6
142. Peng H, Liu Y, Tian J, Ma J, Tang X, Yang J, et al. Decreased expression of microRNA-125a-3p upregulates interleukin-23 receptor in patients with Hashimoto’s thyroiditis. Immunol Res (2015) 62(2):129–36. doi:10.1007/s12026-015-8643-3
143. Dorris ER, Smyth P, O’Leary JJ, Sheils O. MIR141 expression differentiates Hashimoto thyroiditis from PTC and benign thyrocytes in Irish archival thyroid tissues. Front Endocrinol (2012) 3:102. doi:10.3389/fendo.2012.00102
144. Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA. The role of microRNA-146a (miR-146a) and its target IL-1R-associated kinase (IRAK1) in psoriatic arthritis susceptibility. Scand J Immunol (2010) 71(5):382–5. doi:10.1111/j.1365-3083.2010.02381.x
145. Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell (2010) 142(6):914–29. doi:10.1016/j.cell.2010.08.012
146. Garo LP, Murugaiyan G. Contribution of MicroRNAs to autoimmune diseases. Cell Mol Life Sci (2016) 73(10):2041–51. doi:10.1007/s00018-016-2167-4
147. Park H, Huang X, Lu C, Cairo MS, Zhou X. MicroRNA-146a and microRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J Biol Chem (2015) 290(5):2831–41. doi:10.1074/jbc.M114.591420
148. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A (2009) 106(17):7113–8. doi:10.1073/pnas.0902636106
149. Lu C, Huang X, Zhang X, Roensch K, Cao Q, Nakayama KI, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood (2011) 117(16):4293–303. doi:10.1182/blood-2010-12-322503
150. Hashemi M, Bahari G, Naderi M, Sadeghi-Bojd S, Taheri M. Pri-miR-34b/c rs4938723 polymorphism is associated with the risk of childhood acute lymphoblastic leukemia. Cancer Genet (2016) 209(11):493–6. doi:10.1016/j.cancergen.2016.09.009
151. Morales S, Gulppi F, Gonzalez-Hormazabal P, Fernandez-Ramires R, Bravo T, Reyes JM, et al. Association of single nucleotide polymorphisms in Pre-miR-27a, Pre-miR-196a2, Pre-miR-423, miR-608 and Pre-miR-618 with breast cancer susceptibility in a South American population. BMC Genet (2016) 17(1):109. doi:10.1186/s12863-016-0415-0
152. Wright C, Gupta CN, Chen J, Patel V, Calhoun VD, Ehrlich S, et al. Polymorphisms in MIR137HG and microRNA-137-regulated genes influence gray matter structure in schizophrenia. Transl Psychiatry (2016) 6:e724. doi:10.1038/tp.2015.211
153. Cai T, Li J, An X, Yan N, Li D, Jiang Y, et al. Polymorphisms in MIR499A and MIR125A gene are associated with autoimmune thyroid diseases. Mol Cell Endocrinol (2016) 440:106–15. doi:10.1016/j.mce.2016.11.017
154. Kim YK, Kim B, Kim VN. Re-evaluation of the roles of DROSHA, export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci U S A (2016) 113(13):E1881–9. doi:10.1073/pnas.1602532113
155. Frezzetti D, Reale C, Cali G, Nitsch L, Fagman H, Nilsson O, et al. The microRNA-processing enzyme Dicer is essential for thyroid function. PLoS One (2011) 6(11):e27648. doi:10.1371/journal.pone.0027648
156. Saeki M, Watanabe M, Inoue N, Tokiyoshi E, Takuse Y, Arakawa Y, et al. DICER and DROSHA gene expression and polymorphisms in autoimmune thyroid diseases. Autoimmunity (2016) 49(8):514–22. doi:10.1080/08916934.2016.1230846
157. Stachurska A, Zorro MM, van der Sijde MR, Withoff S. Small and long regulatory RNAs in the immune system and immune diseases. Front Immunol (2014) 5:513. doi:10.3389/fimmu.2014.00513
158. Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PH, et al. A long noncoding RNA associated with susceptibility to celiac disease. Science (2016) 352(6281):91–5. doi:10.1126/science.aad0467
159. Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature (2016) 539(7629):452–5. doi:10.1038/nature20149
160. Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol (2016) 17(12):756–70. doi:10.1038/nrm.2016.126
161. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet (2016) 17(1):47–62. doi:10.1038/nrg.2015.10
162. Huang W, Thomas B, Flynn RA, Gavzy SJ, Wu L, Kim SV, et al. DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature (2015) 528(7583):517–22. doi:10.1038/nature16193
163. Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science (2014) 344(6181):310–3. doi:10.1126/science.1251456
164. Aune TM, Crooke PS III, Spurlock CF III. Long noncoding RNAs in T lymphocytes. J Leukoc Biol (2016) 99(1):31–44. doi:10.1189/jlb.1RI0815-389R
165. Casero D, Sandoval S, Seet CS, Scholes J, Zhu Y, Ha VL, et al. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat Immunol (2015) 16(12):1282–91. doi:10.1038/ni.3299
166. Ranzani V, Arrigoni A, Rossetti G, Panzeri I, Abrignani S, Bonnal RJ, et al. Next-generation sequencing analysis of long noncoding RNAs in CD4+ T cell differentiation. Methods Mol Biol (2017) 1514:173–85. doi:10.1007/978-1-4939-6548-9_14
167. Mayama T, Marr AK, Kino T. Differential expression of glucocorticoid receptor noncoding RNA repressor Gas5 in autoimmune and inflammatory diseases. Horm Metab Res (2016) 48(8):550–7. doi:10.1055/s-0042-106898
168. Mirza AH, Berthelsen CH, Seemann SE, Pan X, Frederiksen KS, Vilien M, et al. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Med (2015) 7(1):39. doi:10.1186/s13073-015-0162-2
169. Santoro M, Nociti V, Lucchini M, De Fino C, Losavio FA, Mirabella M. Expression profile of long non-coding RNAs in serum of patients with multiple sclerosis. J Mol Neurosci (2016) 59(1):18–23. doi:10.1007/s12031-016-0741-8
170. Zhang Y, Xu YZ, Sun N, Liu JH, Chen FF, Guan XL, et al. Long noncoding RNA expression profile in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res Ther (2016) 18(1):227. doi:10.1186/s13075-016-1129-4
171. Christensen NJ, Habekost G, Bratholm P. Decrease in TSH receptor autoantibodies during antithyroid treatment: relationship with a long noncoding Heg RNA and Cdk1 mRNA in mononuclear cells. ISRN Endocrinol (2011) 2011:287052. doi:10.5402/2011/287052
172. Zhao SX, Xue LQ, Liu W, Gu ZH, Pan CM, Yang SY, et al. Robust evidence for five new Graves’ disease risk loci from a staged genome-wide association analysis. Hum Mol Genet (2013) 22(16):3347–62. doi:10.1093/hmg/ddt183
173. Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov (2017) 16(3):167–79. doi:10.1038/nrd.2016.117
174. Sullenger BA, Nair S. From the RNA world to the clinic. Science (2016) 352(6292):1417–20. doi:10.1126/science.aad8709
175. Briggs SF, Reijo Pera RA. X chromosome inactivation: recent advances and a look forward. Curr Opin Genet Dev (2014) 28:78–82. doi:10.1016/j.gde.2014.09.010
176. Brooks WH, Renaudineau Y. Epigenetics and autoimmune diseases: the X chromosome-nucleolus nexus. Front Genet (2015) 6:22. doi:10.3389/fgene.2015.00022
177. Invernizzi P, Pasini S, Selmi C, Miozzo M, Podda M. Skewing of X chromosome inactivation in autoimmunity. Autoimmunity (2008) 41(4):272–7. doi:10.1080/08916930802024574
178. Gendrel AV, Heard E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Annu Rev Cell Dev Biol (2014) 30:561–80. doi:10.1146/annurev-cellbio-101512-122415
179. Minks J, Robinson WP, Brown CJ. A skewed view of X chromosome inactivation. J Clin Invest (2008) 118(1):20–3. doi:10.1172/JCI34470
180. Seidel MG, Rami B, Item C, Schober E, Zeitlhofer P, Huber WD, et al. Concurrent FOXP3- and CTLA4-associated genetic predisposition and skewed X chromosome inactivation in an autoimmune disease-prone family. Eur J Endocrinol (2012) 167(1):131–4. doi:10.1530/EJE-12-0197
181. Broen JC, Wolvers-Tettero IL, Geurts-van Bon L, Vonk MC, Coenen MJ, Lafyatis R, et al. Skewed X chromosomal inactivation impacts T regulatory cell function in systemic sclerosis. Ann Rheum Dis (2010) 69(12):2213–6. doi:10.1136/ard.2010.129999
182. Ozcelik T, Uz E, Akyerli CB, Bagislar S, Mustafa CA, Gursoy A, et al. Evidence from autoimmune thyroiditis of skewed X-chromosome inactivation in female predisposition to autoimmunity. Eur J Hum Genet (2006) 14(6):791–7. doi:10.1038/sj.ejhg.5201614
183. Uz E, Mustafa C, Topaloglu R, Bilginer Y, Dursun A, Kasapcopur O, et al. Increased frequency of extremely skewed X chromosome inactivation in juvenile idiopathic arthritis. Arthritis Rheum (2009) 60(11):3410–2. doi:10.1002/art.24956
184. Simmonds MJ, Kavvoura FK, Brand OJ, Newby PR, Jackson LE, Hargreaves CE, et al. Skewed X chromosome inactivation and female preponderance in autoimmune thyroid disease: an association study and meta-analysis. J Clin Endocrinol Metab (2014) 99(1):E127–31. doi:10.1210/jc.2013-2667
185. Brix TH, Knudsen GP, Kristiansen M, Kyvik KO, Orstavik KH, Hegedus L. High frequency of skewed X-chromosome inactivation in females with autoimmune thyroid disease: a possible explanation for the female predisposition to thyroid autoimmunity. J Clin Endocrinol Metab (2005) 90(11):5949–53. doi:10.1210/jc.2005-1366
186. Yin X, Latif R, Tomer Y, Davies TF. Thyroid epigenetics: X chromosome inactivation in patients with autoimmune thyroid disease. Ann N Y Acad Sci (2007) 1110:193–200. doi:10.1196/annals.1423.021
187. Chabchoub G, Uz E, Maalej A, Mustafa CA, Rebai A, Mnif M, et al. Analysis of skewed X-chromosome inactivation in females with rheumatoid arthritis and autoimmune thyroid diseases. Arthritis Res Ther (2009) 11(4):R106. doi:10.1186/ar2759
Keywords: epigenetics, autoimmune thyroid diseases, pathogenesis, DNA methylation, histone modifications, microRNAs, long non-coding RNAs
Citation: Wang B, Shao X, Song R, Xu D and Zhang J-a (2017) The Emerging Role of Epigenetics in Autoimmune Thyroid Diseases. Front. Immunol. 8:396. doi: 10.3389/fimmu.2017.00396
Received: 09 January 2017; Accepted: 21 March 2017;
Published: 07 April 2017
Edited by:
Anne Cooke, University of Cambridge, UKReviewed by:
Terry Francis Davies, Icahn School of Medicine at Mount Sinai, USABin Li, Shanghai Jiao Tong University School of Medicine, China
Copyright: © 2017 Wang, Shao, Song, Xu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-an Zhang, emhhbmdqaW5hbkBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work.