 Hua Su
Hua Su Chun-Tao Lei
Chun-Tao Lei Chun Zhang
Chun Zhang- Department of Nephrology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Interleukin-6 (IL-6) is a pleiotropic cytokine that not only regulates the immune and inflammatory response but also affects hematopoiesis, metabolism, and organ development. IL-6 can simultaneously elicit distinct or even contradictory physiopathological processes, which is likely discriminated by the cascades of signaling pathway, termed classic and trans-signaling. Besides playing several important physiological roles, dysregulated IL-6 has been demonstrated to underlie a number of autoimmune and inflammatory diseases, metabolic abnormalities, and malignancies. This review provides an overview of basic concept of IL-6 signaling pathway as well as the interplay between IL-6 and renal-resident cells, including podocytes, mesangial cells, endothelial cells, and tubular epithelial cells. Additionally, we summarize the roles of IL-6 in several renal diseases, such as IgA nephropathy, lupus nephritis, diabetic nephropathy, acute kidney injury, and chronic kidney disease.
Introduction
Interleukin-6 (IL-6) was discovered in 1986 as a B cell stimulatory factor initiating IgG production (1). Later, it was demonstrated to be a multifunctional cytokine that regulates numerous biological processes including the organ development, acute-phase responses, inflammation, and immune responses (2).
Up to date, ten IL-6 family cytokines have been identified: IL-6, oncostatin M (OSM), leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), cardiotropin-1 (CT-1), cardiotrophin-like cytokine (CLC), neuropoietin (NP), IL-11, IL-27, and IL-31 (3–5). All these cytokines share the membrane glycoprotein gp130 as a common receptor and signal transducer subunit in their receptor complexes except IL-31. The formation of the signal-transducing complex of each cytokine depends on specific ligand and receptor association. IL-6 and IL-11 bind to the specific membrane-bound α chain of IL-6 receptor (IL-6R) or IL-11R, respectively, and then connect to gp130, leading to the homodimerization of gp130. CLC, CT-1, OSM interact with gp130/LIFR or gp130/OSMR and then form heterodimeric receptor complexes to conduct signal. For IL-31, it utilizes the IL-31R and OSMR as signal-transducing receptor, and it is the only cytokine of IL-6 family that does not need gp130 for signal transmission [Figure S1 in Supplementary Material quoted from Garbers et al. (6)].
The IL-6 family cytokines show diverse physiological and pathological functions. Some of their functions are overlapping such as both IL-6 and IL-11 participate in promoting the synthesis of acute-phase protein in hepatocytes (7–9), and IL-11, IL-6, OSM as well as CT-1 are all involved in the bone metabolism by stimulating osteoclast formation (10, 11). However, more frequently, individual IL-6 family members have distinct functions by the respective specific receptors that may not present simultaneously or spatially restricted. IL-6 is the one that has been studied most, and we will mainly focused on it in this review.
Classic and Trans-Signaling Pathway of IL-6
On target cells, IL-6 first binds to the α-chain of non-signaling membrane-bound IL-6R (mbIL-6R, also named CD126 or gp80); subsequently, this complex connects to two molecules of gp130 and initiates signal transduction. JAK/STAT3 and SHP2/Gab/MAPK are the two major pathways involved in gp130 signaling which is activated via the YxxQ motif and Y759 of gp130, respectively (5, 12–14).
It is worthwhile to note that only a few cell types, such as macrophages, neutrophils, CD4+ T-cells, podocytes, and hepatocytes, express IL-6R on the cell surface, and therefore can directly respond to IL-6 (15, 16).
Soluble receptors have been identified for many cytokines and are pivotal regulators by acting as agonists or antagonists of cytokine signaling and inflammatory events. For example, the soluble receptors of TNF-α and IL-1α act as antagonists by neutralizing and inhibiting the activity of their ligands (17–19). A soluble form of the IL-6R (sIL-6R) has been detected in body fluids such as blood and urine (20). sIL-6R binds to IL-6 with comparable affinity as the mbIL-6R (21); consequently, the complex of IL-6/sIL-6R can activate gp130, a membrane protein is ubiquitously expressed (22–25). Activation of gp130 via the IL-6/sIL-6R complex is termed IL-6 trans-signaling pathway, whereas activation of gp130 via the mbIL-6R is named IL-6 classic signaling pathway (23, 26–28). IL-6 trans-signaling represents an alternative to classic IL-6 signaling and permits IL-6 to modulate a broad spectrum of target cells [Figure S2 in Supplementary Material, quoted from Ref. (29)].
Due to the ubiquitous expression of gp130 and the extensive involvement of IL-6 trans-signaling pathway in diverse physiologic and pathologic processes, to get a comprehensive understanding of the generation of sIL-6R is critical. In humans, the sIL-6R is generated via two distinct mechanisms. The first one implicates proteolytic cleavage of the mbIL-6R and relies on a metalloprotease activity, and the second mechanism is by the translation of a differentially spliced IL-6R mRNA lacking the transmembrane and cytosolic domains (30–32). It is believed that the generation of the sIL-6R is mainly regulated by metalloprotease cleavage rather than via differential mRNA splicing (33). In the mouse, only shedding via enzymatic digestion but no differential splicing of the IL-6R mRNA was observed (30). The shedding of the IL-6R is catalyzed by Zn2+-metalloproteases of the ADAM (a disintegrin and metalloprotease) family (34, 35), among which ADAM10 and ADAM17 are the most related enzymes that driving IL-6R proteolysis. It is documented that ADAM10 mediates the slow constitutive IL-6R cleavage, while ADAM17 is account for rapid IL-6R shedding upon diverse stimulations (36). Many factors can activate ADAM17, such as pro-inflammatory cytokines (IL-1β and TNF-α), bacterial toxins, cellular cholesterol depletion, PKC agonist, proteasome inhibitor, DNA damage, and so on (34, 36–40). Therefore, modulation of ADAM17 activity is essential for the IL-6 trans-signaling conduction.
Soluble form of gp130 (sgp130) is found naturally produced and it is detected in the circulation at relatively high concentration (100–400 ng/ml in human plasma) (4, 41–44). Unlike sIL-6R, sgp130 is predominantly generated by alternative splicing rather than proteolysis (45). Since sgp130 can interact with the IL-6/sIL-6R complex, it acts as a specific inhibitor of IL-6 trans-signaling pathway (46, 47), whereas it does not affect mbIL-6R-mediated classic signaling. Notably, sgp130 is specific for the IL-6/sIL-6R complex due to signaling of other IL-6 family cytokines like LIF and OSM were blocked at 100–1,000-folds higher concentrations and CNTF and IL-27 signaling were not affected at all (46, 48, 49). Experimentally, sgp130 can be utilized as molecular tool to discriminate between IL-6 classic signaling and trans-signaling because it competitively inhibits trans-signaling without affecting classic signaling.
Global IL-6 signaling can be halted by IL-6 or IL-6R neutralizing antibodies. In parallel experiments, IL-6 trans-signaling can be inhibited with the sgp130Fc protein, which could be provided by either injection of the recombinant protein or generation by sgp130Fc transgenic mice. Moreover, IL-6 trans-signaling can be also activated with hyper-IL-6 (recombinant soluble IL-6/sIL-6R fusion protein). The experimental design using these approaches could elucidate whether the IL-6-gp130-initiated effect is mediated by IL-6 classic or trans-signaling pathway (23, 50).
The property of IL-6 during inflammation process is complicated, in addition to its well-known pro-inflammatory properties, it also elicits anti-inflammatory effects under certain situations. Usually, it is believed that IL-6 classic signaling is anti-inflammatory whereas the trans-signaling is pro-inflammatory (51). In detail, IL-6 classic signaling is implicated in the synthesis of acute-phase proteins in hepatocyte, which have anti-inflammatory properties and are indispensable for immune defense (52). Whereas, in various inflammation and autoimmune diseases, as well as inflammation-associated cancer, IL-6 trans-signaling is pro-inflammatory and blockade of it is sufficient to alleviate the inflammatory reaction (53–61). But the controversy is existed. Several investigations showed that abrogating IL-6 trans-signaling cannot provide protective effects (28), even unexpectedly attenuated the recovery processes of the disease under certain conditions. For example, IL-6 trans-signaling is critical to regulate the transition from neutrophil to monocyte and prevent excessive tissue damage (62). Therefore, it appears in different diseases that the pathogenetic roles of IL-6 classic and trans-signaling are distinct.
IL-6 and Immune and Inflammatory Cells
T Lymphocytes
Growing data conclusively confirmed that IL-6 is involved in the regulation of T cell differentiation between two critical CD4+ T cell populations: regulatory T (Treg) cells and T helper 17 (Th17) cells. Specifically, IL-6 triggers the differentiation of Th17 cells together with TGF-β by enhancing RORγt expression (63–66), while it dampens the generation of Treg cells via STAT3 (67–70). Th17 cells secrete a number of pro-inflammatory cytokines, such as IL-17, and initiate various inflammatory responses. Meanwhile, diminished Treg further worsen the situation.
Besides that, it is well documented that IL-6 signaling promotes T cells proliferation and resists its apoptosis by producing IL-2 and activating STAT3 (53, 71, 72), and it also activates Th2 cytokine generation through the transcription factor C/EBP (73). Furthermore, follicular helper T cells (TFH), a newly identified CD4+ T helper subpopulation, which is differentiated from naive CD4+ T cells in the presence of IL-6, and is a strong inducer for B cell activation (74).
Therefore, IL-6 is clearly implicated in CD4+ T cells differentiation and expansion and plays a key role in the T-cell-mediated immune response. Additionally, IL-6 is indirectly involved in B cell-induced inflammation via TFH. Targeting IL-6 signaling is promising for the treatment of autoimmune and inflammatory Diseases.
Monocyte and Macrophage
It is well known that IL-6 modulates monocytes differentiation between macrophages and dendritic cells (DCs). Macrophages play an essential role in inflammatory response by secreting cytokines, chemokines, and matrix metalloproteinases. Local macrophages are differentiated from peripheral blood, with the exposure of GM-CSF, and IL-4 peripheral blood monocytes differentiate into DCs, whereas the additional IL-6 switches the differentiation of monocytes from DCs to macrophages. This switch is caused by IL-6-mediated upregulation of M-CSF receptors on monocytes (75, 76). However, this effect is not seen with the addition of other IL-6 family members, including IL-11, LIF, and OSM (77).
Interleukin-6 can trigger myeloid leukemia cell to express complement receptors and Fc receptors along with F4/80 (a marker for mature macrophages) (78–80). In addition, IL-6 stimulates various macrophages typical gene expression, including early response genes c-Jun, Jun B, Jun D, JAK3, Egr-1, and late response genes, such as lysozyme and ferritin light-chain (78, 80). Consistently, upon IL-6 stimulation, monocytic cells upregulate MCP-1 mRNA and protein, a chemokine more strongly expressed in macrophages rather than monocytes (81).
Moreover, gp130 and downstream STAT3 activation are required for IL-6-induced macrophage differentiation, because gp130 mutation and dominant-negative form of STAT3 can block it (82, 83). Thus, IL-6-mediated gp130-JAK/STAT3 signaling drives macrophage differentiation from monocyte.
In contrast to abundant evidence in vitro, there is little data confirmed that IL-6 plays a role in macrophage differentiation in vivo. Macrophages from IL-6-deficient and wild-type mice are similar, except for the decreased capability to defend microorganism (84–86). Hence, other factors compensating for IL-6 may induce macrophage differentiation. So IL-6 appears to be sufficient, but not necessary for macrophage differentiation in vivo.
Local Generation and Signaling of IL-6 in Kidney
Emerging data showed that local activation of IL-6 classic and trans-signaling pathway is implicated in renal autoimmune and inflammatory diseases. Kidney resident cells, including podocytes, endothelial cells, mesangial cells, and tubular epithelial cells (TECs) can secrete IL-6 under certain milieu. Podocyte is the only resident cell that expresses IL-6R, while others do not express IL-6R and not employ classic IL-6 signaling (87, 88) (Table 1).
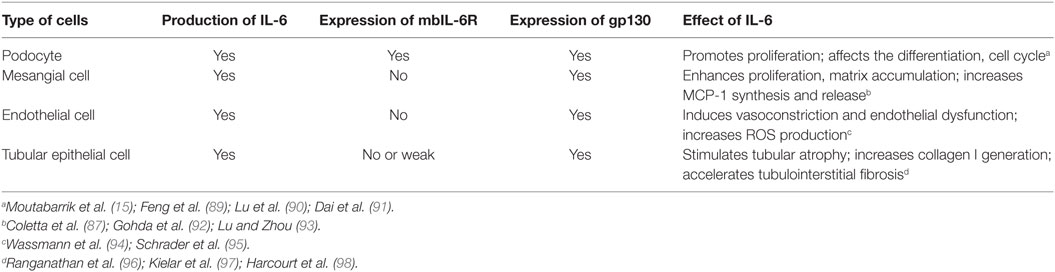
Table 1. Effect of IL-6 on kidney cells.
IL-6 and Podocytes
Podocyte is a well-known source of IL-6, under serum-deprived condition, there is no detectable IL-6 in the supernate of cultured podocytes, but with the exposure to pro-inflammatory mediators, such as LPS, TNF-α, and IL-1β, IL-6 was detected and its concentration was increased in a dose- and time-dependent manner (99–102). It was reported that dexamethasone and vitamin D can suppress IL-6 expression, which partially accounts for their anti-inflammation effects (103–105). Interestingly, podocyte is one of the limited number of cells that can express IL-6R and its abundance was also upregulated by pro-inflammatory stimulation. However, the soluble form of IL-6R (sIL-6R) was not identified in the culture supernatants collected from unstimulated or cytokine-stimulated cells. As we mentioned before, ADAM10 and ADAM17 are the key enzymes cleaving mbIL-6R and generating sIL-6R, thus maybe upregulation of ADAM activity could induce sIL-6R production; however, it requires further investigation. In addition, IL-6 promotes podocyte proliferation in an autocrine fashion (15).
Besides pro-inflammatory factors, high glucose also increased IL-6 secretion and triggered IL-6 signal transduction in podocytes which was abrogated by IL-6-neutralizing antibody or IL-6 siRNA (100). The author suggested that blocking IL-6 and its downstream mediators such as IL-6R and gp130 may attenuate the progression of diabetic nephropathy (DN).
More recently, Abkhezr (106) observed that upon 100 nM angiotensin II (Ang II) exposure for 6–24 h, the STAT3 was phosphorylated via gp130-mediated signaling in podocytes; however, it seems not by classic IL-6 pathway. Intriguingly, these effects were abrogated by TRPC6 gene depletion as well as by inhibitors of the Ca2+-dependent downstream enzymes calcineurin and Ca2+-calmodulin-dependent protein kinase II (CaMKII). It is well known that the STAT3 activity is positively correlated with glomerular cell proliferation in glomerular disease (107). Conditional deletion of STAT3 in podocyte ameliorated the glomerular and tubulointerstitial insults in several animal models (89–91). Taken together, it is suggested that IL-6 trans-signaling initiated by Ang II could activate STAT3 in podocytes in a TRPC6-dependent manner which consequently affect the differentiation, cell cycle, and other physiopathologic processes of podocytes.
Notably, apart from above detrimental role in glomerular diseases, soluble IL-6 generated by podocytes also has anti-inflammation function which was demonstrated in co-culture system of podocytes and endothelial cells. Specifically, TNF-α stimulated podocyte to secrete IL-6 which upregulated the expression of suppressor of cytokine signaling 3 in glomerular endothelium and elicited the immunosuppressive action of IL-6, consequently reduced the recruitment of neutrophils to endothelium (108); however, this issue remains debatable.
Collectively, podocyte is a vigorous origin of IL-6, the local excessive expression of IL-6, and its receptor may actively involve in the process of diverse glomerular diseases in an autocrine and/or paracrine fashion. However, its detailed roles in the development and resolution of the insults require further investigation. The discrepancy between reports may explain by the different experiment setting and different downstream signal pathway involved. Debate about IL-6 function also exists in other inflammatory diseases, as data propose that it serves as a marker rather than a mediator of inflammation (109).
IL-6 and Mesangial Cells
Under certain situation, mesangial cells also can secrete IL-6 and activate inflammatory cells which play an essential role in immune and metabolism-mediated injury of kidney (110–112). As we know, mesangial cells only express the gp130 and not the IL-6R. Exposure to IL-6 and sIL-6R together strongly promoted mesangial cells to synthesize and release monocyte chemoattractant protein 1 and subsequently enhance monocyte recruitment (87). Whereas IL-6 or sIL-6R alone was ineffective to induce cytokine or chemokine secrete from mesangial cells.
Additionally, IL-6 involves in pathological abnormalities of mesangium by enhancing its proliferation, matrix accumulation, and sclerosis (92, 93).
IL-6 and Endothelial Cells
Inflammatory stimuli, specifically, IL-1, LPS, TNFα, and IL-4 are the common inducers for endothelial cells to generate IL-6. IL-6 promotes Ang II type 1 receptor (ATR1) gene expression and leads to Ang II-induced vasoconstriction and ROS production which ultimately results in endothelial dysfunction (94). Consistently, IL-6 deficiency mice are protected against Ang II-mediated endothelial dysfunction (95).
Endothelial cell does not express transmembrane IL-6R, thus IL-6 merely via trans-signaling pathway regulates endothelial function (113). Interplay between IL-6 and endothelial cells modulates leukocytes recruitment and chemokine secretion. IL-6 knockout mice showed reduced leukocyte aggregation at inflammatory sites, which is associated with diminished endothelial surface adhesion molecules expression and chemokines production (114).
IL-6 and Tubular Epithelial Cells
Numerous systemic or local insults, including hypoxemia, nephrotoxin, oxidized lipid, advanced glycation end products, immune complexes, cytokines, and chemokines, could initiate TEC to synthesize and secrete IL-6. For example, anti-dsDNA antibodies can induce IL-6 de novo synthesis in proximal TEC. Interesting, for IL-6 secretion, TEC is more sensitive to anti-dsDNA antibodies compared to mesangial cells (115). Notably, glomerular injury is a potent inducer for IL-6 generation in TEC which presents one aspect of glomeruli-tubules cross talk.
It is shown that IL-6 can trigger proximal TEC to generate the collagen I and accelerate tubulointerstitial fibrosis, which was associated with enhanced STAT3 phosphorylation. Suppression of IL-6 expression in the TEC hampered the interstitial fibrosis and tubular atrophy whereas chronic administration of IL-6 enhanced the fibrotic process (96). Although many studies favor that IL-6 contributes to acute and chronic kidney injury and fibrosis, opposing opinions still exist (97, 98).
IL-6 and Renal Disease
IL-6 and IgA Nephropathy (IgAN)
Local deposited high-molecular polymeric IgA1 can promote mesangial cells proliferation and secretion of the pro-inflammatory cytokine IL-6 (111, 116, 117). Moreover, MAPK/ERK signaling, apparently relying on AT1R activation, is implicated in mesangial cells IL-6 secretion. Because losartan, an AT1R blocker can block MAPK/ERK signaling and IL-6 production from mesangial cells. Thus, utilizing RAS blockers to treat IgAN is rationale and promising (118). Upon complement system is activated by immune complex, sublytic C5b-9 can stimulate IL-6 and TGF-β1 secretion in mesangial cells in a p300-C/EBPβ-dependent manner which was demonstrated in mesangioproliferative glomerulonephritis (MPG) model. In parallel, the Th17-associated cytokines in serum and urine were elevated and correlated with renal pathological change. Additionally, locally, blockage of IL-6 generation by p300 and C/EBPβ gene silence protected MPG model from renal injury, including the reduced Th17 cytokines, ameliorated mesangial cells proliferation, ECM accumulation, and diminished proteinuria (119). Therefore, in IgAN, immune complex and complement component both can stimulate mesangial cell to secrete IL-6 and ensuing itself proliferation and inflammatory cell recruitment.
IL-6 and Lupus Nephritis (LN)
In systemic lupus erythematosus (SLE) patients and mice, the concentration of IL-6 is elevated in serum, urine, and glomeruli, which positively associated with disease activity (120–126). B lymphocytes of SLE subjects spontaneously produced anti-dsDNA autoantibody which was halted by IL-6 inhibition meanwhile it was regained with exogenous supply of IL-6 (120). As we know, CD5 is a suppressor of B cell receptor signaling and negatively regulated B cells activity, meantime IL-6 can dampen CD5 expression via DNA methylation and consequently initiate the activation and expansion of autoreactive B cell lineage (127).
Interleukin-6 gene polymorphism (IL-6 174G>C) on the promoter region was found to convey the susceptibility of SLE in certain ethnical groups, such as in Caucasians. However, it is not identified in Asian population (128–130).
Besides its systemic effects, IL-6 was proved to have a strong correlation with the activity of LN. It was shown that elevated urinary IL-6 excretion was associated with a higher activity of LN (131, 132). In addition, local IL-6 expression within the glomeruli and tubules was evidently increased in LN (133). In SLE animal model, IL-6-deficient MRL-Faslpr mice were resistant to immune- and inflammatory-mediated tissue injury accompanying with delayed onset of proteinuria and hematuria. The lack of IL-6 led to marked reduction of macrophages, CD4+ and CD8+T lymphocytes infiltrates in the kidney, a reduction of IgG deposition, and C3 fixation in kidney (122). In another lupus-prone mice, Lyn−/− mice, inhibition of IL-6 trans-signaling by sgp130Fc exerted relatively little effect on abnormal immune processes along with unchanged pathogenic autoantibodies and renal immune complexes deposition, whereas sgp130Fc indeed ameliorated glomerulonephritis and preserved renal function by hampering complement fixation, leukocytes infiltration, and macrophage expansion in this model (134).
Collectively, IL-6 is a vigorous player in LN and will be a promising therapeutic target.
IL-6 and DN
High IL-6 level was identified in early stage of type 1 diabetes proposing IL-6 is involved in islet cells impairment. Additionally, an IL-6-inducible autoimmunity-related gene (HIP/PAP) was found to be expressed in the pancreas in patients with type-1 diabetes further indicating a potential correlation between IL-6 and autoimmune diabetes (135). In type 2 diabetes, IL-6 level was increased and associated with atherosclerosis development (136). In vitro IL-6 induced insulin resistance which support its role in type 2 diabetes occurrence (137, 138).
Chronic inflammatory process participates in the development of microvascular complications of diabetes. DN patients showed an elevated serum level of inflammatory cytokines, including IL-6, which positively correlated with the extent of proteinuria (139, 140). Meantime, hyperglycemia can trigger podocytes, mesangial cells, interstitial tissue, and tubules to generate IL-6 which contribute to local and systemic inflammatory process in DN (110, 141).
Apart from its involvement in LN, IL-6 gene polymorphism also conveys the susceptibility to cancer (142), lipid metabolic abnormalities, and inflammatory disorders (143). Recently, it was found that IL-6 gene 174G>C polymorphism is an independent risk factor for DN in Turkish and Greek type 2 diabetic mellitus patients (144, 145). Therefore, it again suggested that IL-6 is a cardinal player in DN.
IL-6 and Acute Kidney Injury (AKI)
Recent studies have shown a close correlation between IL-6 expression and AKI. In ischemic AKI animal model, it was found that IL-6 transcription and signaling are elevated locally and systemically after 60 min bilateral kidney ischemia. This finding indicated that IL-6 signaling connected local and systemic inflammation and can be employed as a biomarker and therapeutic target in ischemic AKI (146).
Similarly, in nephrotoxin-induced AKI, IL-6 expression was dramatically enhanced in kidney (113-fold), predominantly in renal TECs, and strongly correlated with the damage of kidney. While IL-6 deficiency attenuated neutrophil accumulation and caused mice relatively resistant to the insult. Moreover, neutrophil depletion in wild-type mice markedly reduced nephrotoxin-induced injury as well. Thus, IL-6-mediated neutrophil activation is one of the central mechanisms for AKI. Intriguingly, stimulation of IL-6 trans-signaling significantly mitigated renal damage and preserved renal function via underlying anti-oxidative stress mechanism (88). The similar observation was reported in ischemia-reperfusion-induced AKI model which proposed that IL-6 trans-signaling may play a protective role by promoting repair process (147, 148).
IL-6 and Chronic Kidney Disease (CKD)
The elevated plasma IL-6 level is commonly observed in CKD patients (149), which is largely caused by the increased generation resulting from oxidative stress, chronic inflammation, and fluid overload. Meanwhile, the reduced clearance of IL-6 due to the impaired renal function also contributes to its accumulation. In the end stage renal disease (ESRD) patients, the therapeutic hemodialysis and peritoneal dialysis per se further stimulate inflammatory responses and increase IL-6 production (150, 151).
Interleukin-6 accelerates the progression of CKD not only by aggravating kidney injury as described above but also by initiating its complications, especially the chronic vascular disease (CVD). It is demonstrated that IL-6 initiates the endothelial injury mainly via reducing endothelial nitric oxide synthase (eNOS) and adiponectin (an anti-atherogenic adipokine) expression (152), and the injection of recombinant IL-6 exacerbates atherosclerosis (153); these findings suggest that IL-6 also contributes to the increased incidence of CVD in CKD patients.
Taken together, elevated IL-6 level is not only a consequence of CKD, more importantly, it also acts as a trigger for the progression of CKD and its related complications.
Conclusion and Perspective
In brief, IL-6 could be produced by renal resident cells, including podocytes, mesangial cells, endothelial cells, and TECs. Meantime, all these cells, as well as immune and inflammatory cells will actively respond to IL-6 via classic or/and trans-signaling pathway. It has already been evidently elucidated that IL-6 participates in renal instinct cell injury and repair process, as well as a variety of immune, metabolic, ischemic, and toxemic-mediated renal diseases.
More importantly, IL-6R-neutralizing mAb tocilizumab (Actemra) has already been approved for treatment in patients with certain autoimmune diseases, such as rheumatoid arthritis in more than 100 countries, including European Union, United State, Brazil, and India. And in Japan, it was authorized to be used in patients with juvenile idiopathic arthritis and Castleman’s disease (154). Several second generation of IL-6 inhibitors, including anti-IL-6 antibodies, is under development.
Intriguingly, recently it was found that cardiotrophin-like cytokine-1 (CLC-1), a member of the IL-6 family and transmitting signal via gp130, is almost probable permeability factor of FSGS. Strikingly, the concentration of CLC-1 is up to 100 times higher in the circulation of FSGS patients compared to normal subjects (155). In vitro CLC-1 can mimic the effects of plasma from FSGS patients on albumin permeability, and it reduces nephrin expression in glomeruli and cultured podocytes which can be prevented by CLC-1 monoclonal antibody.
Thus, further investigation of IL-6 and its family members as well as its signaling pathway are imperative for getting a full view of kidney disease and developing more effective drugs.
Author Contributions
Dr. CZ organized and approved the final version of this review; Dr. HS organized and drafted this review; Dr. C-TL drafted this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81522010, No. 81570671, No. 81470964, No.31200872, No.81570657, and No.81400720), a grant from Wuhan Science and Technology Bureau (No. 2015060101010039), a Doctoral Fund from the Ministry of Education of China (No. 20130142110064), and the Academic Frontier Youth Team Program of Huazhong University of Science and Technology (2016).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00405/full#supplementary-material.
References
1. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature (1986) 324:73–6. doi: 10.1038/324073a0
2. Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol (1998) 16:249–84. doi:10.3109/08830189809042997
3. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J (2003) 374:1–20. doi:10.1042/BJ20030407
4. Scheller J, Grötzinger J, Rose-John S. Updating interleukin-6 classic- and trans-signaling. Signal Transduct (2006) 6:240–59. doi:10.1002/sita.200600086
5. Costa-Pereira AP. Regulation of IL-6-type cytokine responses by MAPKs. Biochem Soc Trans (2014) 42:59–62. doi:10.1042/BST20130267
6. Garbers C, Hermanns HM, Schaper F, Muller-Newen G, Grotzinger J, Rose-John S, et al. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev (2012) 23:85–97. doi:10.1016/j.cytogfr.2012.04.001
7. Dorner AJ, Goldman SJ, Keith JC Jr. Interleukin-11. BioDrugs (1997) 8:418–29. doi:10.2165/00063030-199708060-00002
8. Peters M, Odenthal M, Schirmacher P, Blessing M, Fattori E, Ciliberto G, et al. Soluble IL-6 receptor leads to a paracrine modulation of the IL-6-induced hepatic acute phase response in double transgenic mice. J Immunol (1997) 159:1474–81.
9. Chodorowska G, Glowacka A, Tomczyk M. Leukemia inhibitory factor (LIF) and its biological activity. Ann Univ Mariae Curie Sklodowska Med (2004) 59:189–93.
10. Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci U S A (1993) 90:11924–8. doi:10.1073/pnas.90.24.11924
11. Richards CD, Langdon C, Deschamps P, Pennica D, Shaughnessy SG. Stimulation of osteoclast differentiation in vitro by mouse oncostatin M, leukaemia inhibitory factor, cardiotrophin-1 and interleukin 6: synergy with dexamethasone. Cytokine (2000) 12:613–21. doi:10.1006/cyto.1999.0635
12. Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, et al. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity (1996) 5:449–60. doi:10.1016/S1074-7613(00)80501-4
13. Ohtani T, Ishihara K, Atsumi T, Nishida K, Kaneko Y, Miyata T, et al. Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity (2000) 12:95–105. doi:10.1016/S1074-7613(00)80162-4
14. Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol (2003) 149:1–38. doi:10.1007/s10254-003-0012-2
15. Moutabarrik A, Nakanishi I, Ishibashi M. Interleukin-6 and interleukin-6 receptor are expressed by cultured glomerular epithelial cells. Scand J Immunol (1994) 40:181–6. doi:10.1111/j.1365-3083.1994.tb03448.x
16. Oberg HH, Wesch D, Grussel S, Rose-John S, Kabelitz D. Differential expression of CD126 and CD130 mediates different STAT-3 phosphorylation in CD4+CD25- and CD25high regulatory T cells. Int Immunol (2006) 18:555–63. doi:10.1093/intimm/dxh396
17. Orlando S, Sironi M, Bianchi G, Drummond AH, Boraschi D, Yabes D, et al. Role of metalloproteases in the release of the IL-1 type II decoy receptor. J Biol Chem (1997) 272:31764–9. doi:10.1074/jbc.272.50.31764
18. Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. An essential role for ectodomain shedding in mammalian development. Science (1998) 282:1281–4. doi:10.1126/science.282.5392.1281
19. Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, et al. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem (2000) 275:14608–14. doi:10.1074/jbc.275.19.14608
20. Novick D, Engelmann H, Wallach D, Rubinstein M. Soluble cytokine receptors are present in normal human urine. J Exp Med (1989) 170:1409–14. doi:10.1084/jem.170.4.1409
21. Atsumi T, Ishihara K, Kamimura D, Ikushima H, Ohtani T, Hirota S, et al. A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med (2002) 196:979–90. doi:10.1084/jem.20020619
22. Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol (2006) 195:173–83. doi:10.1007/s00430-006-0019-9
23. Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets (2007) 11:613–24. doi:10.1517/14728222.11.5.613
24. Li H, Rokavec M, Hermeking H. Soluble IL6R represents a miR-34a target: potential implications for the recently identified IL-6R/STAT3/miR-34a feed-back loop. Oncotarget (2015) 6:14026–32. doi:10.18632/oncotarget.4334
25. Zhuang PY, Wang JD, Tang ZH, Zhou XP, Quan ZW, Liu YB, et al. Higher proliferation of peritumoral endothelial cells to IL-6/sIL-6R than tumoral endothelial cells in hepatocellular carcinoma. BMC Cancer (2015) 15:830. doi:10.1186/s12885-015-1763-2
26. Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J (1994) 300(Pt 2):281–90. doi:10.1042/bj3000281
27. Ebihara N, Matsuda A, Nakamura S, Matsuda H, Murakami A. Role of the IL-6 classic- and trans-signaling pathways in corneal sterile inflammation and wound healing. Invest Ophthalmol Vis Sci (2011) 52:8549–57. doi:10.1167/iovs.11-7956
28. Malchow S, Thaiss W, Janner N, Waetzig GH, Gewiese-Rabsch J, Garbers C, et al. Essential role of neutrophil mobilization in concanavalin A-induced hepatitis is based on classic IL-6 signaling but not on IL-6 trans-signaling. Biochim Biophys Acta (2011) 1812:290–301. doi:10.1016/j.bbadis.2010.11.009
29. Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta (2016) 1863:1218–27. doi:10.1016/j.bbamcr.2016.03.018
30. Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine (1992) 4:96–100. doi:10.1016/1043-4666(92)90043-Q
31. Mullberg J, Schooltink H, Stoyan T, Gunther M, Graeve L, Buse G, et al. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol (1993) 23:473–80. doi:10.1002/eji.1830230226
32. Lamas JR, Rodriguez-Rodriguez L, Tornero-Esteban P, Villafuertes E, Hoyas J, Abasolo L, et al. Alternative splicing and proteolytic rupture contribute to the generation of soluble IL-6 receptors (sIL-6R) in rheumatoid arthritis. Cytokine (2013) 61:720–3. doi:10.1016/j.cyto.2012.12.025
33. Dimitrov S, Lange T, Benedict C, Nowell MA, Jones SA, Scheller J, et al. Sleep enhances IL-6 trans-signaling in humans. FASEB J (2006) 20:2174–6. doi:10.1096/fj.06-5754fje
34. Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, et al. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood (2007) 110:1748–55. doi:10.1182/blood-2007-01-067918
35. Chalaris A, Gewiese J, Paliga K, Fleig L, Schneede A, Krieger K, et al. ADAM17-mediated shedding of the IL6R induces cleavage of the membrane stub by gamma-secretase. Biochim Biophys Acta (2010) 1803:234–45. doi:10.1016/j.bbamcr.2009.12.001
36. Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J Biol Chem (2003) 278:38829–39. doi:10.1074/jbc.M210584200
37. Walev I, Vollmer P, Palmer M, Bhakdi S, Rose-John S. Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc Natl Acad Sci U S A (1996) 93:7882–7. doi:10.1073/pnas.93.15.7882
38. Franchimont N, Lambert C, Huynen P, Ribbens C, Relic B, Chariot A, et al. Interleukin-6 receptor shedding is enhanced by interleukin-1beta and tumor necrosis factor alpha and is partially mediated by tumor necrosis factor alpha-converting enzyme in osteoblast-like cells. Arthritis Rheum (2005) 52:84–93. doi:10.1002/art.20727
39. Vahdat AM, Reiners KS, Simhadri VL, Eichenauer DA, Boll B, Chalaris A, et al. TNF-alpha-converting enzyme (TACE/ADAM17)-dependent loss of CD30 induced by proteasome inhibition through reactive oxygen species. Leukemia (2010) 24:51–7. doi:10.1038/leu.2009.230
40. Schumacher N, Meyer D, Mauermann A, Von Der Heyde J, Wolf J, Schwarz J, et al. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J Biol Chem (2015) 290:26059–71. doi:10.1074/jbc.M115.649509
41. Narazaki M, Yasukawa K, Saito T, Ohsugi Y, Fukui H, Koishihara Y, et al. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood (1993) 82:1120–6.
42. Zhang JG, Zhang Y, Owczarek CM, Ward LD, Moritz RL, Simpson RJ, et al. Identification and characterization of two distinct truncated forms of gp130 and a soluble form of leukemia inhibitory factor receptor alpha-chain in normal human urine and plasma. J Biol Chem (1998) 273:10798–805. doi:10.1074/jbc.273.17.10798
43. Tanaka M, Kishimura M, Ozaki S, Osakada F, Hashimoto H, Okubo M, et al. Cloning of novel soluble gp130 and detection of its neutralizing autoantibodies in rheumatoid arthritis. J Clin Invest (2000) 106:137–44. doi:10.1172/JCI7479
44. Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, et al. Regulation of a lymphocyte-endothelial-IL-6 trans-signaling axis by fever-range thermal stress: hot spot of immune surveillance. Cytokine (2007) 39:84–96. doi:10.1016/j.cyto.2007.07.184
45. Mullberg J, Dittrich E, Graeve L, Gerhartz C, Yasukawa K, Taga T, et al. Differential shedding of the two subunits of the interleukin-6 receptor. FEBS Lett (1993) 332:174–8. doi:10.1016/0014-5793(93)80507-Q
46. Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem (2001) 268:160–7. doi:10.1046/j.1432-1327.2001.01867.x
47. Moreno Velasquez I, Golabkesh Z, Kallberg H, Leander K, De Faire U, Gigante B. Circulating levels of interleukin 6 soluble receptor and its natural antagonist, sgp130, and the risk of myocardial infarction. Atherosclerosis (2015) 240:477–81. doi:10.1016/j.atherosclerosis.2015.04.014
48. Hui W, Bell M, Carroll G. Soluble glycoprotein 130 (gp130) attenuates OSM- and LIF-induced cartilage proteoglycan catabolism. Cytokine (2000) 12:151–5. doi:10.1006/cyto.1999.0550
49. Scheller J, Schuster B, Holscher C, Yoshimoto T, Rose-John S. No inhibition of IL-27 signaling by soluble gp130. Biochem Biophys Res Commun (2005) 326:724–8. doi:10.1016/j.bbrc.2004.11.098
50. Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opin Ther Targets (2012) 16:225–36. doi:10.1517/14728222.2012.660307
51. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta (2011) 1813:878–88. doi:10.1016/j.bbamcr.2011.01.034
52. Tilg H, Dinarello CA, Mier JW. IL-6 and APPs: anti-inflammatory and immunosuppressive mediators. Immunol Today (1997) 18:428–32. doi:10.1016/S0167-5699(97)01103-1
53. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med (2000) 6:583–8. doi:10.1038/75068
54. Mitsuyama K, Matsumoto S, Rose-John S, Suzuki A, Hara T, Tomiyasu N, et al. STAT3 activation via interleukin 6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut (2006) 55:1263–9. doi:10.1136/gut.2005.079343
55. Finotto S, Eigenbrod T, Karwot R, Boross I, Doganci A, Ito H, et al. Local blockade of IL-6R signaling induces lung CD4+ T cell apoptosis in a murine model of asthma via regulatory T cells. Int Immunol (2007) 19:685–93. doi:10.1093/intimm/dxm037
56. Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, et al. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood (2008) 111:1021–8. doi:10.1182/blood-2007-07-102137
57. Matsumoto S, Hara T, Mitsuyama K, Yamamoto M, Tsuruta O, Sata M, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol (2010) 184:1543–51. doi:10.4049/jimmunol.0801217
58. Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ, et al. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol (2011) 186:1199–208. doi:10.4049/jimmunol.1002971
59. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell (2011) 19:456–69. doi:10.1016/j.ccr.2011.03.009
60. Lo CW, Chen MW, Hsiao M, Wang S, Chen CA, Hsiao SM, et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res (2011) 71:424–34. doi:10.1158/0008-5472
61. Jasiewicz M, Knapp M, Waszkiewicz E, Ptaszynska-Kopczynska K, Szpakowicz A, Sobkowicz B, et al. Enhanced IL-6 trans-signaling in pulmonary arterial hypertension and its potential role in disease-related systemic damage. Cytokine (2015) 76:187–92. doi:10.1016/j.cyto.2015.06.018
62. Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol (2003) 24:25–9. doi:10.1016/S1471-4906(02)00013-3
63. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
64. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi:10.1038/nature04754
65. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
66. Kimura A, Kishimoto T. Th17 cells in inflammation. Int Immunopharmacol (2011) 11:319–22. doi:10.1016/j.intimp.2010.10.004
67. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
68. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol (2004) 172:5149–53. doi:10.4049/jimmunol.172.9.5149
69. Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol (2007) 179:2041–5. doi:10.4049/jimmunol.179.4.2041
70. Fujimoto M, Nakano M, Terabe F, Kawahata H, Ohkawara T, Han Y, et al. The influence of excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J Immunol (2011) 186:32–40. doi:10.4049/jimmunol.0903314
71. Teague TK, Marrack P, Kappler JW, Vella AT. IL-6 rescues resting mouse T cells from apoptosis. J Immunol (1997) 158:5791–6.
72. Yoshida H, Hashizume M, Suzuki M, Mihara M. Anti-IL-6 receptor antibody suppressed T cell activation by inhibiting IL-2 production and inducing regulatory T cells. Eur J Pharmacol (2010) 634:178–83. doi:10.1016/j.ejphar.2010.02.026
73. Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell RA. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med (1997) 185:461–9. doi:10.1084/jem.185.3.461
74. Linterman MA, Vinuesa CG. Signals that influence T follicular helper cell differentiation and function. Semin Immunopathol (2010) 32:183–96. doi:10.1007/s00281-009-0194-z
75. Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol (2000) 20:2175–83. doi:10.1161/01.ATV.20.10.2175
76. Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol (2003) 91:7A–11A. doi:10.1016/S0002-9149(02)03144-2
77. Mitani H, Katayama N, Araki H, Ohishi K, Kobayashi K, Suzuki H, et al. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br J Haematol (2000) 109:288–95. doi:10.1046/j.1365-2141.2000.02020.x
78. Krishnaraju K, Hoffman B, Liebermann DA. The zinc finger transcription factor Egr-1 activates macrophage differentiation in M1 myeloblastic leukemia cells. Blood (1998) 92:1957–66.
79. Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol (2000) 1:510–4. doi:10.1038/82763
80. Tanaka H, Matsumura I, Nakajima K, Daino H, Sonoyama J, Yoshida H, et al. GATA-1 blocks IL-6-induced macrophage differentiation and apoptosis through the sustained expression of cyclin D1 and bcl-2 in a murine myeloid cell line M1. Blood (2000) 95:1264–73.
81. Biswas P, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, et al. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood (1998) 91:258–65.
82. Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J (1996) 15:3651–8.
83. Yamanaka Y, Nakajima K, Fukada T, Hibi M, Hirano T. Differentiation and growth arrest signals are generated through the cytoplasmic region of gp130 that is essential for Stat3 activation. EMBO J (1996) 15:1557–65.
84. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature (1994) 368:339–42. doi:10.1038/368339a0
85. Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, et al. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell (1995) 80:353–61. doi:10.1016/0092-8674(95)90418-2
86. Akira S, Kishimoto T. Role of interleukin-6 in macrophage function. Curr Opin Hematol (1996) 3:87–93. doi:10.1097/00062752-199603010-00013
87. Coletta I, Soldo L, Polentarutti N, Mancini F, Guglielmotti A, Pinza M, et al. Selective induction of MCP-1 in human mesangial cells by the IL-6/sIL-6R complex. Exp Nephrol (2000) 8:37–43. doi:10.1159/000059327
88. Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, et al. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol (2008) 19:1106–15. doi:10.1681/ASN.2007070744
89. Feng X, Lu TC, Chuang PY, Fang W, Ratnam K, Xiong H, et al. Reduction of Stat3 activity attenuates HIV-induced kidney injury. J Am Soc Nephrol (2009) 20:2138–46. doi:10.1681/ASN.2008080879
90. Lu TC, Wang ZH, Feng X, Chuang PY, Fang W, Shen Y, et al. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int (2009) 76:63–71. doi:10.1038/ki.2009.98
91. Dai Y, Gu L, Yuan W, Yu Q, Ni Z, Ross MJ, et al. Podocyte-specific deletion of signal transducer and activator of transcription 3 attenuates nephrotoxic serum-induced glomerulonephritis. Kidney Int (2013) 84:950–61. doi:10.1038/ki.2013.197
92. Gohda T, Makita Y, Shike T, Funabiki K, Shirato I, Tomino Y. Dilazep hydrochloride, an antiplatelet drug, inhibits lipopolysaccharide-induced mouse mesangial cell IL-6 secretion and proliferation. Kidney Blood Press Res (2001) 24:33–8. doi:10.1159/000054203
93. Lu H, Zhou J. HBV X gene transfection upregulates IL-1beta and IL-6 gene expression and induces rat glomerular mesangial cell proliferation. J Huazhong Univ Sci Technolog Med Sci (2008) 28:247–50. doi:10.1007/s11596-008-0304-5
94. Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Bohm M, et al. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res (2004) 94:534–41. doi:10.1161/01.RES.0000115557.25127.8D
95. Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol (2007) 27:2576–81. doi:10.1161/ATVBAHA.107.153080
96. Ranganathan P, Jayakumar C, Ramesh G. Proximal tubule-specific overexpression of netrin-1 suppresses acute kidney injury-induced interstitial fibrosis and glomerulosclerosis through suppression of IL-6/STAT3 signaling. Am J Physiol Renal Physiol (2013) 304:F1054–65. doi:10.1152/ajprenal.00650.2012
97. Kielar ML, John R, Bennett M, Richardson JA, Shelton JM, Chen L, et al. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol (2005) 16:3315–25. doi:10.1681/ASN.2003090757
98. Harcourt BE, Forbes JM, Matthews VB. Obesity-induced renal impairment is exacerbated in interleukin-6-knockout mice. Nephrology (Carlton) (2012) 17:257–62. doi:10.1111/j.1440-1797.2011.01547.x
99. Lee SJ, Borsting E, Decleves AE, Singh P, Cunard R. Podocytes express IL-6 and lipocalin 2/ neutrophil gelatinase-associated lipocalin in lipopolysaccharide-induced acute glomerular injury. Nephron Exp Nephrol (2012) 121:e86–96. doi:10.1159/000345151
100. Kim DI, Park SH. Sequential signaling cascade of IL-6 and PGC-1alpha is involved in high glucose-induced podocyte loss and growth arrest. Biochem Biophys Res Commun (2013) 435:702–7. doi:10.1016/j.bbrc.2013.05.046
101. Srivastava T, Sharma M, Yew KH, Sharma R, Duncan RS, Saleem MA, et al. LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease: changes in TLR profile. J Cell Commun Signal (2013) 7:49–60. doi:10.1007/s12079-012-0184-0
102. Abkhezr M, Dryer SE. Angiotensin II and canonical transient receptor potential-6 activation stimulate release of a signal transducer and activator of transcription 3-activating factor from mouse podocytes. Mol Pharmacol (2014) 86:150–8. doi:10.1124/mol.114.092536
103. Xing CY, Saleem MA, Coward RJ, Ni L, Witherden IR, Mathieson PW. Direct effects of dexamethasone on human podocytes. Kidney Int (2006) 70:1038–45. doi:10.1038/sj.ki.5001655
104. Matsumoto T, Hess S, Kajiyama H, Sakairi T, Saleem MA, Mathieson PW, et al. Proteomic analysis identifies insulin-like growth factor-binding protein-related protein-1 as a podocyte product. Am J Physiol Renal Physiol (2010) 299:F776–84. doi:10.1152/ajprenal.00597.2009
105. Sanchez-Nino MD, Bozic M, Cordoba-Lanus E, Valcheva P, Gracia O, Ibarz M, et al. Beyond proteinuria: VDR activation reduces renal inflammation in experimental diabetic nephropathy. Am J Physiol Renal Physiol (2012) 302:F647–57. doi:10.1152/ajprenal.00090.2011
106. Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol (2014) 229:434–42. doi:10.1002/jcp.24461
107. Arakawa T, Masaki T, Hirai T, Doi S, Kuratsune M, Arihiro K, et al. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol Dial Transplant (2008) 23:3418–26. doi:10.1093/ndt/gfn314
108. Kuravi SJ, Mcgettrick HM, Satchell SC, Saleem MA, Harper L, Williams JM, et al. Podocytes regulate neutrophil recruitment by glomerular endothelial cells via IL-6-mediated crosstalk. J Immunol (2014) 193:234–43. doi:10.4049/jimmunol.1300229
109. Song L, Schindler C. IL-6 and the acute phase response in murine atherosclerosis. Atherosclerosis (2004) 177:43–51. doi:10.1016/j.atherosclerosis.2004.06.018
110. Pfafflin A, Brodbeck K, Heilig CW, Haring HU, Schleicher ED, Weigert C. Increased glucose uptake and metabolism in mesangial cells overexpressing glucose transporter 1 increases interleukin-6 and vascular endothelial growth factor production: role of AP-1 and HIF-1alpha. Cell Physiol Biochem (2006) 18:199–210. doi:10.1159/000097667
111. Schmitt R, Stahl AL, Olin AI, Kristoffersson AC, Rebetz J, Novak J, et al. The combined role of galactose-deficient IgA1 and streptococcal IgA-binding M protein in inducing IL-6 and C3 secretion from human mesangial cells: implications for IgA nephropathy. J Immunol (2014) 193:317–26. doi:10.4049/jimmunol.1302249
112. Ji M, Lu Y, Zhao C, Gao W, He F, Zhang J, et al. C5a induces the synthesis of IL-6 and TNF-alpha in rat glomerular mesangial cells through MAPK signaling pathways. PLoS One (2016) 11:e0161867. doi:10.1371/journal.pone.0161867
113. Sironi M, Breviario F, Proserpio P, Biondi A, Vecchi A, Van Damme J, et al. IL-1 stimulates IL-6 production in endothelial cells. J Immunol (1989) 142:549–53.
114. Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity (1997) 6:315–25. doi:10.1016/S1074-7613(00)80334-9
115. Yung S, Tsang RC, Sun Y, Leung JK, Chan TM. Effect of human anti-DNA antibodies on proximal renal tubular epithelial cell cytokine expression: implications on tubulointerstitial inflammation in lupus nephritis. J Am Soc Nephrol (2005) 16:3281–94. doi:10.1681/ASN.2004110917
116. Gomez-Guerrero C, Lopez-Armada MJ, Gonzalez E, Egido J. Soluble IgA and IgG aggregates are catabolized by cultured rat mesangial cells and induce production of TNF-alpha and IL-6, and proliferation. J Immunol (1994) 153:5247–55.
117. Moura IC, Arcos-Fajardo M, Gdoura A, Leroy V, Sadaka C, Mahlaoui N, et al. Engagement of transferrin receptor by polymeric IgA1: evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J Am Soc Nephrol (2005) 16:2667–76. doi:10.1681/ASN.2004111006
118. Tamouza H, Chemouny JM, Raskova Kafkova L, Berthelot L, Flamant M, Demion M, et al. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int (2012) 82:1284–96. doi:10.1038/ki.2012.192
119. Zhang J, Li Y, Shan K, Wang L, Qiu W, Lu Y, et al. Sublytic C5b-9 induces IL-6 and TGF-beta1 production by glomerular mesangial cells in rat Thy-1 nephritis through p300-mediated C/EBPbeta acetylation. FASEB J (2014) 28:1511–25. doi:10.1096/fj.13-242693
120. Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol (1991) 147:117–23.
121. Peterson E, Robertson AD, Emlen W. Serum and urinary interleukin-6 in systemic lupus erythematosus. Lupus (1996) 5:571–5. doi:10.1177/096120339600500603
122. Cash H, Relle M, Menke J, Brochhausen C, Jones SA, Topley N, et al. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: the IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J Rheumatol (2010) 37:60–70. doi:10.3899/jrheum.090194
123. Ball EM, Gibson DS, Bell AL, Rooney MR. Plasma IL-6 levels correlate with clinical and ultrasound measures of arthritis in patients with systemic lupus erythematosus. Lupus (2014) 23:46–56. doi:10.1177/0961203313512882
124. Abdel Galil SM, Ezzeldin N, El-Boshy ME. The role of serum IL-17 and IL-6 as biomarkers of disease activity and predictors of remission in patients with lupus nephritis. Cytokine (2015) 76:280–7. doi:10.1007/BF02253829
125. Solus JF, Chung CP, Oeser A, Li C, Rho YH, Bradley KM, et al. Genetics of serum concentration of IL-6 and TNFalpha in systemic lupus erythematosus and rheumatoid arthritis: a candidate gene analysis. Clin Rheumatol (2015) 34:1375–82. doi:10.1007/s10067-015-2881-6
126. Wang JB, Li H, Wang LL, Liang HD, Zhao L, Dong J. Role of IL-1beta, IL-6, IL-8 and IFN-gamma in pathogenesis of central nervous system neuropsychiatric systemic lupus erythematous. Int J Clin Exp Med (2015) 8:16658–63.
127. Garaud S, Le Dantec C, Jousse-Joulin S, Hanrotel-Saliou C, Saraux A, Mageed RA, et al. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J Immunol (2009) 182:5623–32. doi:10.4049/jimmunol.0802412
128. Huang CM, Huo AP, Tsai CH, Chen CL, Tsai FJ. Lack of association of interleukin-6 and interleukin-8 gene polymorphisms in Chinese patients with systemic lupus erythematosus. J Clin Lab Anal (2006) 20:255–9. doi:10.1002/jcla.20151
129. Santos MJ, Fernandes D, Capela S, Da Silva JC, Fonseca JE. Interleukin-6 promoter polymorphism -174 G/C is associated with nephritis in Portuguese Caucasian systemic lupus erythematosus patients. Clin Rheumatol (2011) 30:409–13. doi:10.1007/s10067-010-1640-y
130. Lee YH, Lee HS, Choi SJ, Ji JD, Song GG. The association between interleukin-6 polymorphisms and systemic lupus erythematosus: a meta-analysis. Lupus (2012) 21:60–7. doi:10.1177/0961203311422711
131. Iwano M, Dohi K, Hirata E, Kurumatani N, Horii Y, Shiiki H, et al. Urinary levels of IL-6 in patients with active lupus nephritis. Clin Nephrol (1993) 40:16–21.
132. Tsai CY, Wu TH, Yu CL, Lu JY, Tsai YY. Increased excretions of beta2-microglobulin, IL-6, and IL-8 and decreased excretion of Tamm-Horsfall glycoprotein in urine of patients with active lupus nephritis. Nephron (2000) 85:207–14. doi:10.1159/000045663
133. Herrera-Esparza R, Barbosa-Cisneros O, Villalobos-Hurtado R, Avalos-Diaz E. Renal expression of IL-6 and TNFalpha genes in lupus nephritis. Lupus (1998) 7:154–8. doi:10.1191/096120398678919949
134. Tsantikos E, Maxwell MJ, Putoczki T, Ernst M, Rose-John S, Tarlinton DM, et al. Interleukin-6 trans-signaling exacerbates inflammation and renal pathology in lupus-prone mice. Arthritis Rheum (2013) 65:2691–702. doi:10.1002/art.38061
135. Gurr W, Yavari R, Wen L, Shaw M, Mora C, Christa L, et al. A Reg family protein is overexpressed in islets from a patient with new-onset type 1 diabetes and acts as T-cell autoantigen in NOD mice. Diabetes (2002) 51:339–46. doi:10.2337/diabetes.51.2.339
136. Andriankaja OM, Barros SP, Moss K, Panagakos FS, Devizio W, Beck J, et al. Levels of serum interleukin (IL)-6 and gingival crevicular fluid of IL-1beta and prostaglandin E(2) among non-smoking subjects with gingivitis and type 2 diabetes. J Periodontol (2009) 80:307–16. doi:10.1902/jop.2009.080385
137. Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun (2003) 311:372–9. doi:10.1016/j.bbrc.2003.10.013
138. Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem (2003) 278:45777–84. doi:10.1074/jbc.M301977200
139. Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol (2005) 16(Suppl 1):S78–82. doi:10.1681/ASN.2004110961
140. Navarro JF, Milena FJ, Mora C, Leon C, Garcia J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol (2006) 26:562–70. doi:10.1159/000098004
141. Kaur H, Chien A, Jialal I. Hyperglycemia induces toll like receptor 4 expression and activity in mouse mesangial cells: relevance to diabetic nephropathy. Am J Physiol Renal Physiol (2012) 303:F1145–50. doi:10.1152/ajprenal.00319.2012
142. Liu RY, Song X, Chen P, Lei Z, Miao J, Yi N, et al. Association between IL6 -174G/C and cancer: a meta-analysis of 105,482 individuals. Exp Ther Med (2012) 3:655–64. doi:10.3892/etm.2012.454
143. Smith AJ, Humphries SE. Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine Growth Factor Rev (2009) 20:43–59. doi:10.1016/j.cytogfr.2008.11.006
144. Papaoikonomou S, Tentolouris N, Tousoulis D, Papadodiannis D, Miliou A, Papageorgiou N, et al. The association of the 174G>C polymorphism of interleukin 6 gene with diabetic nephropathy in patients with type 2 diabetes mellitus. J Diabetes Complications (2013) 27:576–9. doi:10.1016/j.jdiacomp.2013.06.006
145. Karadeniz M, Erdogan M, Berdeli A, Yilmaz C. Association of interleukin-6 -174 G>C promoter polymorphism with increased risk of type 2 diabetes mellitus patients with diabetic nephropathy in Turkey. Genet Test Mol Biomarkers (2014) 18:62–5. doi:10.1089/gtmb.2013.0357
146. Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol (2008) 19:547–58. doi:10.1681/ASN.2007040469
147. Lemay S, Rabb H, Postler G, Singh AK. Prominent and sustained up-regulation of gp130-signaling cytokines and the chemokine MIP-2 in murine renal ischemia-reperfusion injury. Transplantation (2000) 69:959–63. doi:10.1097/00007890-200003150-00049
148. Yoshino J, Monkawa T, Tsuji M, Hayashi M, Saruta T. Leukemia inhibitory factor is involved in tubular regeneration after experimental acute renal failure. J Am Soc Nephrol (2003) 14:3090–101. doi:10.1097/01.ASN.0000101180.96787.02
149. Pecoits-Filho R, Heimburger O, Barany P, Suliman M, Fehrman-Ekholm I, Lindholm B, et al. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am J Kidney Dis (2003) 41:1212–8. doi:10.1016/S0272-6386(03)00353-6
150. Takahashi T, Kubota M, Nakamura T, Ebihara I, Koide H. Interleukin-6 gene expression in peripheral blood mononuclear cells from patients undergoing hemodialysis or continuous ambulatory peritoneal dialysis. Ren Fail (2000) 22:345–54. doi:10.1081/JDI-100100878
151. Caglar K, Peng Y, Pupim LB, Flakoll PJ, Levenhagen D, Hakim RM, et al. Inflammatory signals associated with hemodialysis. Kidney Int (2002) 62:1408–16. doi:10.1111/j.1523-1755.2002.kid556.x
152. Bruun JM, Lihn AS, Verdich C, Pedersen SB, Toubro S, Astrup A, et al. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab (2003) 285:E527–33. doi:10.1152/ajpendo.00110.2003
153. Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol (1999) 19:2364–7. doi:10.1161/01.ATV.19.10.2364
154. Nishimoto N. Interleukin-6 as a therapeutic target in candidate inflammatory diseases. Clin Pharmacol Ther (2010) 87:483–7. doi:10.1038/clpt.2009.313
Keywords: IL-6 classic signaling, IL-6 trans-signaling, podocyte, mesangial cells, endothelial cells, tubular epithelial cells, renal disease
Citation: Su H, Lei C-T and Zhang C (2017) Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 8:405. doi: 10.3389/fimmu.2017.00405
Received: 21 January 2017; Accepted: 22 March 2017;
Published: 21 April 2017
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases, USAReviewed by:
Behdad Afzali, King’s College London, UK; National Institutes of Health, USAJames R. Whiteford, Queen Mary University of London, UK
Copyright: © 2017 Su, Lei and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun Zhang, ZHJ6aGFuZ2NodW5AaHVzdC5lZHUuY24=