 Annie Luo
Annie Luo Steven T. Leach
Steven T. Leach Romain Barres
Romain Barres Luke B. Hesson
Luke B. Hesson Michael C. Grimm1
Michael C. Grimm1 David Simar
David Simar- 1St George and Sutherland Clinical School, University of New South Wales, Sydney, NSW, Australia
- 2School of Women and Children’s Health, University of New South Wales, Sydney, NSW, Australia
- 3The Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 4Adult Cancer Program, Lowy Cancer Research Centre, Prince of Wales Clinical School, University of New South Wales, Sydney, NSW, Australia
- 5Mechanisms of Disease and Translational Research, School of Medical Sciences, University of New South Wales, Sydney, NSW, Australia
Immune cells not only affect tissue homeostasis at the site of inflammation but also exert systemic effects contributing to multiple chronic conditions. Recent evidence clearly supports an altered T helper 17/regulatory T cell (Th17/Treg) balance leading to the development and progression of inflammatory diseases that not only affect the gastrointestinal tract but also have whole-body manifestations, including insulin resistance. Epigenetic mechanisms are amenable to both environmental and circulating factors and contribute to determining the T cell landscape. The recently identified participation of the gut microbiota in the remodeling of the epigenome of immune cells has triggered a paradigm shift in our understanding of the etiology of various inflammatory diseases and opened new paths toward therapeutic strategies. In this review, we provide an overview of the contribution of the Th17/Treg balance in the development and progression of inflammatory bowel diseases and metabolic diseases. We discuss the involvement of epigenetic mechanisms in the regulation of T cell function in the particular context of dysbiosis. Finally, we examine the potential for nutritional interventions affecting the gut microbiota to reshape the T cell epigenome and address the inflammatory component of various diseases.
Introduction
Immune homeostasis is a complex process involving a wide variety of key immunological players. Failure to establish a balanced immune response contributes to an ever-growing list of chronic diseases. Over the last few decades, the involvement of T helper 17 (Th17) cells and regulatory T cells (Tregs) in the maintenance of immune homeostasis has received increasing attention. It is now widely accepted that an altered Th17/Treg balance is associated with a wide variety of inflammatory diseases [Figure 1; (1–7)]. Given the role of epigenetic mechanisms in the regulation of immune functions, notably in the control of Th17 cell and Treg differentiation, it is not surprising that inflammatory diseases are linked to a substantial remodeling of the host epigenome in various tissues [Figure 1; (8–10)]. There is growing evidence that the gut microbial community is critical for the maintenance of a healthy host, and clear links between the gut microbiota and the central nervous system, or the cardiovascular and metabolic functions have been established (11–14). Consequently, perturbations of the gut microbiota (or dysbiosis), encompassing reduced microbial diversity and changes in microbiota composition, have been associated with a large number of chronic conditions (15). The vast majority of diseases that develop in the face of dysbiosis are characterized by immune alterations, and in particular chronic inflammation, which affect not only the gastrointestinal (GI) tract but also spread to the systemic circulation and central or peripheral tissues (16, 17). Some of the key manifestations of these inflammatory conditions, that include inflammatory bowel disease (IBD) and metabolic diseases, involve modulation of the local and circulating cytokine profile, but also significant changes in the frequency, function, and trafficking pattern of immune cells, including major alterations in the Th17/Treg balance, affecting the “immunological environment” in the host tissues (18–20). How dysbiosis orchestrates the remodeling of the T-cell landscape and whether this could involve epigenetic mechanisms still remain unclear. Perhaps an even more important question that remains unanswered is whether the gut microbiota could be manipulated to reshape the T-cell epigenome in the context of inflammatory diseases providing novel strategies to limit the pandemic of IBD and metabolic diseases. Although further work is still needed to provide definitive answers to those questions, recent progress in the field has shed light on important mechanisms driving the Th17/Treg balance and that could be targeted to treat inflammatory diseases.
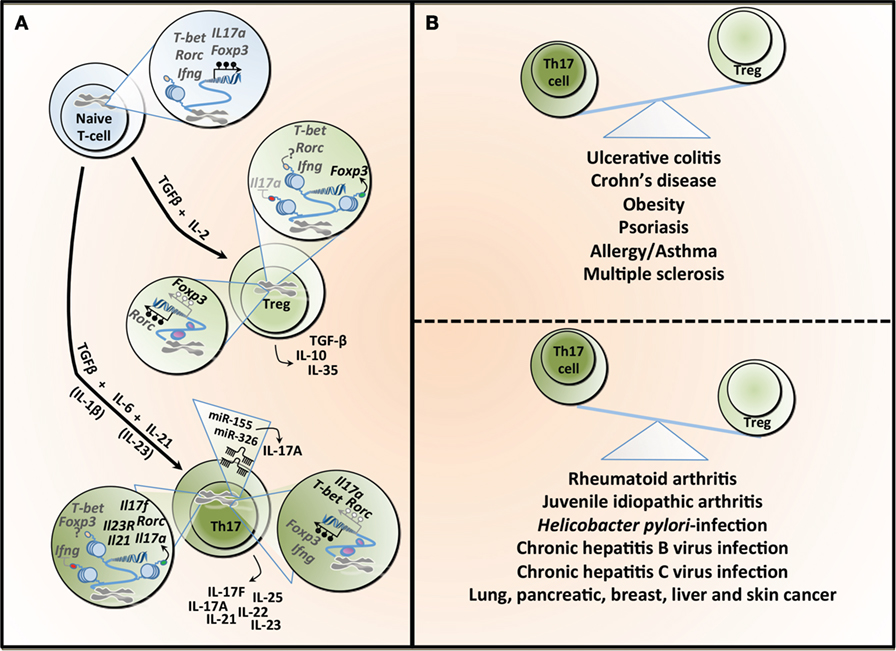
Figure 1. T helper 17 (Th17) and regulatory T cell (Treg) lineage determination and the Th17/Treg balance in health and diseases. (A) The differentiation of naïve T-cells into different lineages is regulated by specific cytokines. TGF-β in combination with IL-2 induces the expression of the transcription factor Foxp3 and Treg differentiation, which are characterized by the secretion of TGF-β, IL-10, and IL-33. TGF-β combined with IL-6 leads to the differentiation into Th17, which is characterized by the transcription factor Rorc (or RORγt). This process is amplified under the influence of IL-21 and both IL-1β and IL-23 have been reported to potentially contributing to Th17 differentiation. Th17 can secrete different cytokines, including IL-17A, IL-17F, IL-21, IL-22, IL-23, and IL-25. The differentiation into Treg or Th17 is a tightly controlled process regulated, at least partly, by epigenetic mechanisms. In naïve T-cell, the T-bet (or Tbx1) locus is characterized by both H3K4me3 (a permissive histone mark) and H3k27me3 (a repressive histone mark), whereas the Rorc, interferon gamma (Ifng), Foxp3, and Il17a loci show a total absence of such marks, making those five loci susceptible to both repression or expression of these genes. All five genes are hypermethylated in naïve T-cell contributing to gene silencing. Tregs show H3K4me3 on Foxp3 and H3K27me3 on Il17a, supporting the expression of Foxp3 while silencing Il17a. The demethylation of Foxp3 and the hypermethylation of Rorc further contribute to Foxp3 expression. In the same cells, T-bet and Rorc are marked by both histone modifications, whereas Ifng is devoid of these, rendering these loci more unstable. Th17 cells show H3K27me3 on Ifng and H3K4me3 on Rorc, Il17a, Il17f, Il21, and Il23, facilitating the expression of Rorc and supporting the secretion of the signature cytokines by these cells. This process is further supported by the hypermethylation observed in the Ifng and Foxp3 loci, whereas the Il17a, T-bet, and Rorc are characterized by a demethylation, supporting gene expression. microRNAs can also contribute to the differentiation and function of Th17, and both miR-155 and miR-326 contribute to the production of IL-17A. TGF-β, transforming growth factor-beta; IL-2, interleukin-2; Foxp3, forkhead box P3; Rorc (or RORγt), retinoic acid receptor-related orphan receptor; T-bet, T-box1; H3K4me3, trimethylation of histone H3 on lysine 4 (green dot); H3K4me27, trimethylation of histone H3 on lysine 27 (red dot); IFN-γ, interferon gamma. (B) An imbalance between the number and activity of Th17 and Treg has been linked to several diseases. The predominance of Th17 is associated with inflammatory bowel diseases, obesity, and some allergic conditions, whereas Tregs have been linked to different forms of arthritis as well as several types of infections.
Th17/Treg Balance in IBDs
Th17 cells and Tregs are T-cell subsets that have essential roles in various immune processes, including inflammation, as well as disease progression. The Th17 lineage mediates effector functions and is under the transcriptional regulation of retinoic acid-related orphan receptor (ROR) γt (21–23). Th17 cells are further characterized by the production of their signature cytokines, including interleukin (IL)-17, IL-21, IL-22, IL-23, and IL-25, which promote tissue inflammation via the induction of other pro-inflammatory cytokines and chemokines [Figure 1; (23–25)]. At the other end of the immune regulation spectrum, natural Tregs and inducible Tregs differentiate in the thymus and periphery, respectively, and play an indispensable role in the suppressive control of both innate and adaptive immunity in vivo (26, 27). Developmentally, Tregs are regulated by the transcription factor forkhead box P3 (Foxp3) and express high levels of the IL-2-α chain receptor, CD25. Both Foxp3 and CD25 play pivotal roles in Treg differentiation, induction, and stabilization of their regulatory phenotype and immunosuppressive functions, including the secretion of IL-10, IL-35, and transforming growth factor (TGF)-β [Figure 1; (28–34)]. Tregs can be further divided into naturally occurring and peripherally induced Tregs. Phenotypically, these Treg cell subsets are indistinguishable from each other and utilize similar mechanisms to suppress immune responses including (1) cell-to-cell contact, (2) regulation of the effects of dendritic cells on effector cells, (3) secretion of inhibitory cytokines including IL-10, IL-35, and TGF-β, and (4) cytolysis of target cells (35). Interestingly, some studies have demonstrated that the development and function of a subset of peripherally derived Tregs are independent of Foxp3 expression and are characterized by their ability to produce high levels of the immunosuppressive IL-10 and TGF-β to suppress both adaptive and innate immune responses (36).
Despite these distinct functional differences, Th17 and Tregs are linked by their shared requirement for the cytokine TGF-β during development (21–23). This represents a close, fundamental relationship between the two cell types and further suggests shared developmental pathways during the progression of immune-mediated inflammatory conditions (22, 37). This is supported by data suggesting that the balance of Th17/Treg plays a role in the regulation of intestinal inflammation in IBD (38–40). A balance between the effector and regulatory functions (respectively mediated by Th17 cells and Tregs) is particularly important in the GI tract as it represents a critical barrier between the internal and external environments. The GI tract is under constant challenge from microbial and food antigens, which can generate both beneficial and harmful effects; hence, the normal intestinal immune system is constantly activated and tightly regulated. When this delicate balance between effector and regulatory responses is breached, the development of IBD ensues (38, 39). IBD are chronic inflammatory diseases of the GI tract, with unknown etiology, consisting of two major forms called ulcerative colitis (UC) and Crohn’s disease (CD), characterized by excessive inflammation of the intestine. The current hypothesis regarding the pathophysiology of IBD postulates that inflammation in these conditions is a result of the interaction of multiple factors including the intestinal microbiota, abnormalities of mucosal permeability, environmental and genetic factors, and a dysregulated host-immune response (41–44). Nonetheless, it still remains unclear whether chronic inflammation is responsible for the development of dysbiosis in IBD, or if disturbance in the gut microbiota is the trigger leading to inflammation in the GI tract. Despite the fact that animal models have suggested the possibility of dysbiosis preceding chronic inflammation (45), similar evidence is yet to be clearly demonstrated in human studies.
Changes in Th17 cells and Tregs numbers and functions are well established in both patients with IBD and murine colitis models. Elevated transcript levels of IL17 have been reported in the mucosa of both UC and CD patients compared to healthy controls [Table 1; (2, 3)]. The relationship between IL-17 and IBD genesis, however, is likely to be complex, given that anti-IL-17 monoclonal antibody therapy failed to improve outcomes in CD (46). Serum levels of IL-22, another Th17-related cytokine, are significantly increased in IBD patients, an observation further recapitulated in a mouse model of colitis (47, 48). This excessive pro-inflammatory response results from a decreased differentiation and accumulation, as well as functional defects in Tregs in the inflamed tissues (49) or in peripheral blood of IBD patients with active disease (50). The upregulation of these pro-inflammatory cytokines is correlated with a significant reduction in the levels of several immunosuppressive cytokines including TGF-β, IL-10, and IL-33 (41, 49). All these events favor the establishment of a local effector immune response, leading to a disturbed intestinal immune system, resulting in chronic tissue injury. Accordingly, the modulation of the immunological balance between Th17 cells and Tregs could be exploited to revert the pro-inflammatory intestinal environment. Indeed, several studies have demonstrated that the re-tuning of the Th17/Treg balance could dampen down intestinal inflammation resulting in the re-establishment of intestinal immune homeostasis (40, 51). For instance, all-trans retinoic acid (ATRA) downregulated pro-inflammatory responses in both murine and human colitis by shifting the Th17/Treg profile (51). In this study, the upregulation of Foxp3 post treatment with ATRA and elevated levels of regulatory cytokines including IL-10 and TGF-β were associated with the downregulation of IL-17. Furthermore, metformin was able to attenuate IBD severity and reduce inflammation through the regulation of Il17 and Foxp3 expression, demonstrating the importance of the Th17/Treg balance in the regulation of intestinal inflammation [Table 1; (40)]. Taken together, these data support the notion that the attenuation of chronic intestinal inflammation in IBD is achieved through the restoration of a fine balance between pro- and anti-inflammatory responses (40, 51–54).
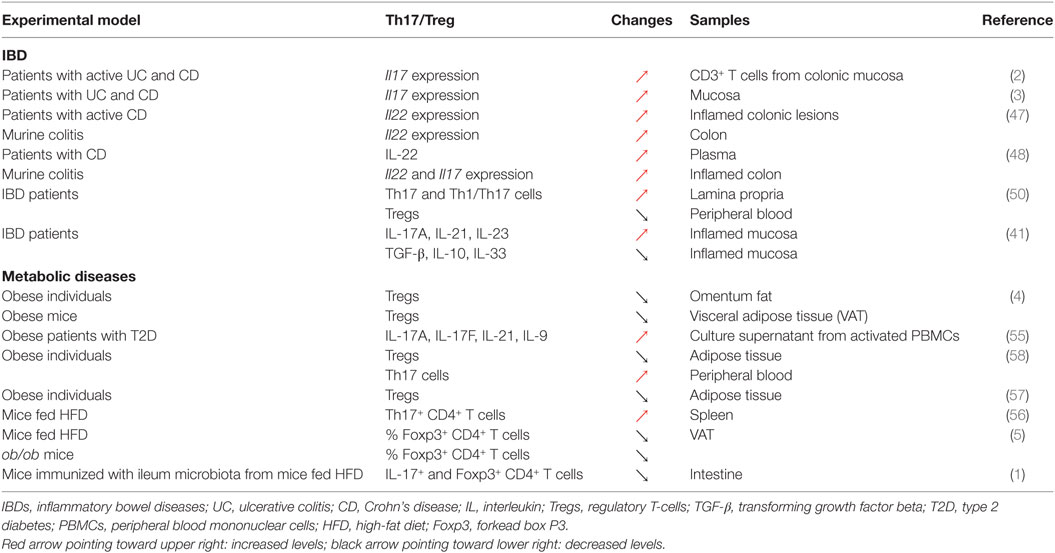
Table 1. The T helper 17/regulatory T cell (Th17/Treg) balance in IBD and metabolic disease.
Th17/Treg Balance in Metabolic Diseases
The Th17/Treg balance has also been linked with the development of obesity-associated diseases such as type 2 diabetes (T2D) [Table 2; (1, 4, 5, 7, 55–57)]. Immunologically, obesity is characterized by low-grade chronic inflammation with abnormal cytokine production and the activation of various inflammatory pathways, associated with adverse clinical outcomes including insulin resistance and T2D (58). In obesity, the polarization of a Th17 phenotype, especially in secondary lymphoid organs (e.g., spleen and lymph nodes), contributes to the development of other chronic inflammatory diseases, including experimental autoimmune encephalomyelitis (EAE) and asthma (58). Similarly, the severity of trinitrobenzenesulfonic acid-induced colitis in obese mice was found to be associated with Th17 expansion (58). Despite their pathogenic role in the initiation and maintenance of chronic inflammation, Th17 cells have also been implicated as critical players in the control of adipogenesis and glucose homeostasis in the context of obesity (19, 59). In a recent study, intestinal RORγt+ IL-17+ CD4+ T-cells were shown to participate in energy metabolism in mice. Indeed, a reduction in the numbers of RORγt+ and IL-17-producing CD4+ T-cells contributed to the development of insulin resistance (1). This observation supported the role of the intestinal Th17 lineage in the control of insulin sensitivity, an effect that differed from peripheral Th17 cells. Obesity also affects the numbers of Tregs and function in the regulation of whole-body metabolism. Several studies have demonstrated that visceral adipose tissues (VATs) of lean mice are highly enriched with a unique population of Foxp3+ CD4+ Tregs that possess a distinct T-cell receptor repertoire, pattern of chemokine and chemokine receptor expression, and gene expression profile compared to other tissues (4–7). Under obese conditions, the abundance of these VAT-resident Tregs is markedly reduced. Several studies have suggested that this depletion of Tregs plays a role in the inflammatory process that may contribute to obesity-associated diseases such as T2D (4–7). Foxp3+ VAT-resident Tregs express unusually high levels of the peroxisome proliferator-activator receptor (PPAR)-γ (6), a key nuclear receptor controlling adipogenesis that is also a target of the insulin-sensitizing drug thiazolidinediones (60). The functional importance of PPAR-γ expression for VAT-resident Tregs was further reinforced by the observation that Treg-specific deletion of PPAR-γ leads to a significant reduction in the number of Tregs in VAT, but not in other tissues (6). Collectively, these data highlight the role of PPAR-γ-expressing Foxp3+ Tregs in the control of inflammation induced by obesity and in metabolic diseases such as T2D (6).
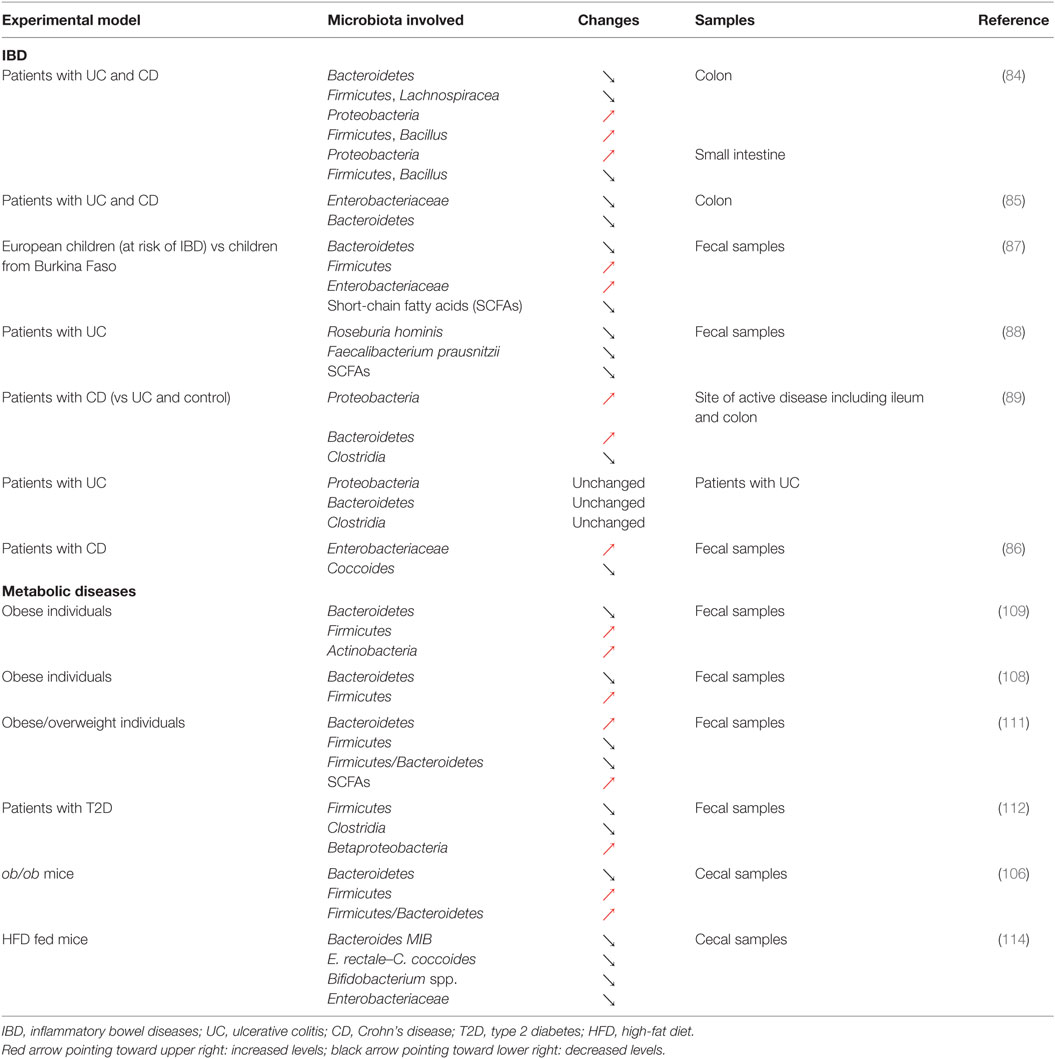
Table 2. Changes in gut microbiota in IBD and metabolic diseases.
Epigenetic Regulation of the Th17/Treg Balance
The differentiation of Th17 cells and Tregs from naïve CD4+ T-cells is not only controlled by a combination of their cytokine milieu and transcriptional activities but is also subjected to epigenetic control through various mechanisms including DNA methylation (10, 61–63), changes in microRNA (miRNA) expression (64–69), and histone modification (22, 70–74), illustrating the complexity of CD4+ T-cell lineage decisions (Figure 1).
It has been proposed that the induction, maintenance, and function of Foxp3 are regulated by epigenetic mechanisms (9, 10, 75). A seminal study revealed the complete demethylation in a region within the evolutionally conserved non-coding sequence elements of the Foxp3 gene and termed this region as the Treg-specific demethylation region (TSDR) (61). It was reported that demethylated TSDR is strongly associated with the stable expression of Foxp3 in Tregs (61) and germ-line deletion of the TSDR resulted in the loss of Foxp3 expression (10). However, there is evidence suggesting that TSDR demethylation is not an on or off switch but instead determines the stability of Foxp3 expression (61–63). Interestingly, demethylation of the TSDR with DNA demethylating compounds is associated with enhanced immunosuppressive function of Foxp3+ Tregs and prevents the occurrence of diabetes in mice (75). Collectively, these data established that the demethylated TSDR contributes to the stable expression of Foxp3 in Tregs.
microRNAs are emerging as critical regulators of Th17 and Treg cell differentiation and function and consequently are also implicated in a number of diseases including IBD, diabetes, neurological diseases, and cardiovascular diseases (64, 65). Several studies have demonstrated that miR-155 is a prominent regulator of both Th17 and Treg differentiation and function in vitro and in vivo (64–69). For instance, miR155 is required for Treg development as miR-155-deficient (miR-155−/−) mice show a marked decrease in the frequency and absolute numbers of Tregs in both the thymus and secondary lymphoid organs (68). Despite a reduced number of Tregs, miR-155 is dispensable for Treg immunosuppressive function. This was confirmed by the finding that miR-155−/− mice did not spontaneously develop GI diseases, as seen in patients with IBD. Moreover, Tregs from miR-155−/− mice retained the capacity to prevent colitis induced by the adoptive transfer of CD4+ CD45RBhigh T-cells into lymphopenic hosts (68). The above findings were not only recapitulated, but miR-155 was also found to be crucial in directing Th17 differentiation and enhancing IL-17A secretion by Th17 cells by directly inhibiting the SOCS1 (suppressor of cytokines signaling) signaling pathway (66). It also positively regulates Th17 cell function, but not that of Tregs (66). Together, these studies revealed that miR-155 is a critical driver of both Th17 and Treg development. Other miRNAs have also been reported to control Th17 and Treg development and function. For example, miR-182, miR-10a, and miR-17-92 are key regulators of Treg specification, stability, and their suppressor function in infectious and autoimmune diseases (76–78). miR-21 promotes Th17 differentiation and mediates the development of EAE as shown by the association between the defect in Th17 differentiation and the strong resistance to EAE in miR-21-deficient mice (79). In addition, anti-miR-21 treatments drastically reduced Th17 cell numbers as well as the severity of EAE (79). Likewise, miR-326 also drives the differentiation as well as migration of Th17 cells to inflammatory sites and the production of IL-17A which all potentiate the development of EAE (80).
Evidence from genome-wide epigenetic analysis of Th17 cells uncovered the enrichment of active histone modifications such as histone H3 acetylation and trimethyl-histone H3 lysine 4 (H3K4me3) in the promoters of cytokine and lineage-specific genes such as IL17a, IL17f, IL21, IL23R, and RORγt (22, 70–72). This suggests histone modifications such as acetylation or methylation play a role in Th17 cell lineage commitment (22). Similarly, promoters of Treg-related genes, such as Foxp3, are also marked with the permissive H3K4me3 modification, which correlates strongly with gene expression (72). Furthermore, DNA demethylation of the TSDR was reported to correlate with increased levels of H3K4me3, suggesting that specific DNA demethylation and histone modifications have similar roles in the determination and commitment of the Treg lineage (73, 74). More recently, two histone acetyltransferases, CREB-binding protein (CBP) and p300, have been shown to control Treg suppressive function and stability (81). A combined Treg-specific deletion of CBP and p300 led to rapid development of fatal autoimmunity in mice within 3–4 weeks of birth. This was accompanied by decreased expression of genes characteristic of the Treg lineage, presence of autoantibodies, and increased infiltration of mononuclear cells in multiple organs (81), which resembles the clinical phenotype of scurfy or other Foxp3 mutant mice (82). Supporting the observation that Tregs are prone to lose Foxp3 expression in inflammatory diseases, CBP, and p300 act on the TSDR to control Treg stability and function during inflammation (81). These findings not only provide insight into the importance of histone acetyltransferases in the regulation of Treg development and function but also delineate distinct roles for CBP and p300 in the control of Tregs (81).
Dysbiosis and the Th17/Treg Balance in IBD and Metabolic Diseases
As illustrated above, the Th17/Treg balance plays a critical role in control of the inflammatory process in IBD (38, 39). Although the exact etiology of IBD remains elusive, it is well accepted that this group of diseases preferentially affects genetically susceptible individuals and could result from a disproportionate activation of the immune system in response to changes affecting the gut microbiota [Tables 1 and 2; (44)]. Indeed, IBD is characterized by significant disturbances in the gut microbiota (83) and alterations in the balance between the dominant bacterial groups, depletion of particular bacteria, as well as a reduction in the diversity of the gut microbiota (84–86). A seminal study in the field recently identified that European children, considered to be at increased risk of IBD, were characterized by a reduced diversity in their gut microbiota and a total depletion of bacteria responsible for the digestion of dietary fibers when compared to their African counterparts (87). In this particular study, striking differences were observed in the diet these two groups of children were exposed to, highlighting the importance of nutritional habits in the maintenance of a healthy gut microbiota and the dramatic consequences of unfavorable dietary habits. Consistent with this observation, several studies have reported dramatic alterations in the proportion of different groups of bacteria in the context of IBD. A relative depletion of members of Bacteroidetes and Lachnospiraceae (84) or a decrease in Roseburia hominis and Faecalibacterium prausnitzii (88) has been observed in patients diagnosed with CD or UC. However, contradictory results have also been observed, with patients with IBD showing an increase in both Proteobacteria and Bacteroidetes (89) or in Enterobacteriaceae (86). This discrepancy could be the result of the variability in the type of biological samples being investigated, ranging from biopsy samples to fecal samples, or even the sites where such biopsies are taken from in the GI tract [Table 2; (90)]. Despite the inherent challenges of identifying key groups of bacteria that contribute to the development of IBD, dysbiosis is thought to be heavily involved in the pathophysiology of the disease. In particular, the ensuing immune activation and the establishment of chronic inflammation in the GI tract represent key steps in the development of IBD. Clear associations have now been established between the microbiota and T-cell differentiation or polarization and could thus link dysbiosis to changes affecting the Th17/Treg balance in IBD (91). The contribution of both dysbiosis and immune disturbance to IBD is further supported by the efficacy of anti-inflammatory, immunosuppressant, and immunomodulatory drugs, as well as microbiome modulators in the therapeutic arsenal for IBD (92, 93). In recent years, there has been a growing interest in the use of probiotics and prebiotics to treat or manage IBD, through the correction of intestinal dysbiosis (93). However, conflicting results have been reported regarding the use of probiotics or prebiotics to treat IBD, and both their efficacy and safety still remain to be established. For instance, promising results have been reported regarding the use of the probiotic, Escherichia coli Nissle 1917, to maintain remission in UC (94, 95). However, evidences supporting its use in the context of CD are still lacking (96). Some studies even reported that probiotics could cause harm (93). Similarly, if the beneficial effects of prebiotics in CD and UC remain elusive, some randomized studies using placebo as control have shown an improvement in GI symptoms and side effects following prebiotics supplementation in CD (97, 98). Another approach that has received much attention is the therapeutic manipulation of the intestinal microbiota via fecal microbial transplantation (FMT) to restore symbiosis in IBD patients (99). If some studies have reported results that could suggest some potential benefits of FMT in UC (100, 101), significant concerns remain regarding safety. In particular, screening systems to identify the nature of the stool that is likely to be safe for donation and might provide some benefits are still lacking. Indeed, adverse effects of transplanting diseased fecal microbiota have been demonstrated (102). Impaired intestinal function, including increased intestinal barrier dysfunction, faster GI transit, and innate immune activation, as well as exhibition of anxiety-like behavior, has been observed in mice that received fecal microbiota from patients with irritable bowel syndrome (102). These results suggest that the use of FMT as a management approach in IBD requires careful consideration and further research is warranted to establish its safety.
If chronic inflammation and alterations of the Th17/Treg balance are an integral component of IBD, a wide range of diseases increasingly recognized as comprising chronic inflammation, including obesity and T2D, may also be associated with such alterations. Reports supporting similarities in the physiopathology of IBD and metabolic diseases have recently started to emerge and have identified dysbiosis as a common trait shared between these conditions (83, 103). Indeed, changes in the microbiota that accompany alterations in nutritional habits and the development of obesity have been identified as critical contributors to the development of metabolic complications including insulin resistance and T2D and could even precede the establishment of local and systemic inflammation (83, 103, 104). Similar to what has been reported in IBD, both a decline in diversity and a remodeling of the bacterial component of the microbiota have been observed in the early stages of the development of obesity and insulin resistance (105). In mice, obesity has been linked to a decrease in Bacteroidetes and an increase in Firmicutes, as well as an increase in the Firmicutes/Bacteroidetes ratio (106). Similar changes have also been observed in mice fed a Western diet and these alterations could even be transmitted to germ-free mice by colonization using fecal microbiota obtained from obese mice (107). These alterations in the gut microbiota in response to obesity have since been confirmed in obese individuals or patients with T2D (108–110), although conflicting results have also been reported (111, 112). It is noteworthy that changes affecting the microbial flora associated with obesity and insulin resistance have also been shown to be reversible in response to treatment using prebiotics or probiotics (113, 114). Indeed, mice fed a high-fat diet (HFD) and supplemented with the prebiotic oligofructose showed increased Bifidobacterium spp., improved glucose metabolism, and reduced plasma and adipose tissue pro-inflammatory cytokines (114). Similarly, supplementing mice fed a HFD with the probiotic bacteria Lactobacillus paracasei ssp. paracasei F19 significantly reduced fat accumulation (113). Such beneficial effects were further confirmed in a clinical trial where adults with obese tendencies, who received the probiotic Lactobacillus gasseri SBT2055, were characterized by improved body composition and in particular decreased visceral and subcutaneous fat (115). These changes in the adipose tissue were associated with a decreased expression of pro-inflammatory genes in obese mice (116). These alteration were mediated by the regulation of macrophage infiltration and their regulatory rather than inflammatory polarization in the adipose tissue (117), confirming the effect of the gut microbiota on inflammation in metabolic diseases.
Although it is clear that a link exists between the gut microbiota and the immune system, this association is complex. While the gut microbiota can contribute to shaping or even to activating the immune system, the local immune environment can also remodel the gut microbiota (91, 118). In lean and healthy individuals, the gut microbiota and the intestinal immune system are in homeostasis, preventing bacteria and bacterial products leaking from the gut lumen to the underlying mucosa. This is partly achieved through the secretion of immunoglobulin A and the production of mucin by goblet cells. In this context, an anti-inflammatory environment is maintained through the secretion of TGF-β, as well as IL-25 and IL-33 by intestinal epithelial cells (IECs), favoring Tregs (119). In the face of dysbiosis, this fragile equilibrium is disrupted, which leads to immune activation. Both a reduction in microbiota diversity and an altered composition of the microbiota are associated with decreased mucin production and increased intestinal permeability. Increased translocation and exposure of the underlying mucosa to pathogenic bacteria and bacterial products trigger the activation of the inflammasome, and increases toll-like receptor (TLR) and NOD-like receptor signaling, resulting in the production of IL-1β, IL-6, IL-12, IL-18, and IL-23 (119). This pro-inflammatory milieu can lead to the activation of the adaptive immune system and shift the Th17/Treg balance. Although both the gut microbiota and the immune system are involved in the pathogenesis of IBD, it remains unclear which may be the initial disease trigger. As described above, dysbiosis can lead to both intestinal and systemic inflammation, but the immune system may also shape the gut microbiota (118). In the context of IBD where genetic susceptibility (e.g., genetic deficiency in TLR1) and/or environmental factors (pathogens) are present, immune activation and the resulting inflammatory process could contribute to altering IECs function and affect their secretory pattern, resulting in a remodeling of the gut microbiota (120).
The relationship between the gut microbiota and the immune system, and in particular the Th17/Treg balance, is now well established. Although it remains unclear as to which may be driving inflammatory diseases characterized by dysbiosis, their tight interaction contributes to the vicious circle of chronic intestinal inflammation.
Microbiota-Derived Metabolites and the Regulation of Th17/Treg Cells
It has been clearly established that the commensal microbial community can influence the Th17/Treg balance, as supported by the remodeling of that balance in germ-free mice or in mice treated with antibiotics (121–124). Although the exact mechanisms are still unclear, factors and metabolites derived from the gut microbiota have received significant attention. Bacteroides fragilis has been reported to protect against experimental colitis through the release of polysaccharide A [PSA; (125)]. This anti-inflammatory effect was mediated by a decreased production of IL-17 by immune cells in the intestine and through the promotion of CD4+ T-cells differentiation to IL-10-producing cells and Foxp3+ Tregs (126). Similarly, Bacteroides thetaiotaomicron has been reported to exert anti-inflammatory effects by regulating the nuclear-cytoplasmic shuttling of PPAR-γ in both epithelial and intestinal cell lines (127). Although the effect on T-cells was not tested in that study, considering the critical role played by PPAR-γ in the differentiation of Treg, these findings confirm that the gut microbiota has a strong potential to influence the T-cell landscape in the GI tract. Metabolites produced by commensal microbes, and in particular short-chain fatty acids (SCFAs) including acetate, propionate, and butyrate, also play a critical role in mediating the effect of the gut microbiota on Treg induction both in the intestine and the colon (128–130). While SCFAs levels have been correlated with the number of colonic Tregs, butyrate in particular can support Treg differentiation both in vitro and in vivo (129). Such effects are not confined to the GI tract, and both butyrate and propionate have been reported to influence peripheral Treg development (128).
Several mechanisms potentially involved in SCFAs regulation of T-cell differentiation have been suggested, including the control of cellular metabolism or G-protein-coupled receptor (GPCR) signaling (131). Indeed, through their integration in the Krebs cycle as Acetyl-CoA, SCFAs can directly affect the cellular energy status and the ATP/ADP levels. This in turn leads to the activation of the mammalian target of rapamycin (mTOR), a critical kinase involved in T-cell differentiation, and can facilitate T effector or Treg differentiation (121, 132). In addition, both butyrate and propionate have been reported to inhibit de novo fatty acid synthesis though the deactivation of acetyl-CoA carboxylase 1 (133), a process known to limit Th17 cell differentiation and promote Treg development (134). GPCRs, some of which act as receptors for various SCFAs including acetate, butyrate, propionate, or for niacin, have been linked to the regulation of inflammation in various diseases (135), and GPCR signaling plays a critical role in the regulation of Th17 cell and Treg development (131). Indeed, propionate has been reported to increase colonic Treg numbers, an effect thought to be GPR43 dependent (130). Proprionate also increases GPR15 levels, a homing receptor on colonic Tregs known to contribute to limiting inflammation (130, 136). Both butyrate and niacin have been shown to exert their anti-inflammatory effect by regulating colonic macrophages and dendritic cells in a GPR109A-dependent manner (135). In this report, colonic macrophages and dendritic cells from Niacr1−/− (the gene encoding for GPR109A) mice failed to promote the differentiation of naïve CD4+ T-cells into Tregs and IL-10-producing T-cells. However, the effect of SCFAs on T-cell differentiation has been recently reported to be GPR41 and GPR43 independent, suggesting that additional mechanisms might play a role in that process (137).
Epigenetic Regulation of the Th17/Treg Balance by the Gut Microbiota
The inhibitory action of SCFAs on histone deacetylase (HDAC) activity suggests that the gut microbiota could contribute to shaping the host epigenome. This is supported by the recently emerging evidence linking dysbiosis to epigenetic changes in the GI tract, the adipose tissue, or the liver. Such changes have even been suggested to play a role in the development of pathological conditions including obesity and GI cancer (8, 138). The effect of SCFAs on T-cell differentiation could thus be mediated by reshaping the T-cell epigenome (Figure 2). In agreement with this, the effect of butyrate on Treg differentiation is thought to be linked to increased histone H3 acetylation in the Foxp3 locus (129). Indeed, increased peripheral Treg differentiation in response to butyrate is mediated through increased acetylation of histone 3 at lysine 27 in the Foxp3 locus (128). In the same study, propionate potentiated peripheral Treg generation through the same HDAC inhibitory effect, while acetate failed to produce such action. The effect of propionate on HDAC inhibition has since been shown to specifically affect HDAC6 and HDAC9 and to be GPR43 dependent (130). Propionate increased not only the frequency and number of colonic Tregs but also their suppressive capacity, potentially through the increased expression of Foxp3 and Il10, and reduced intestinal inflammation. However, it must be noted that both GPR41 and GPR43 have been recently suggested to be dispensable for the inhibition of HDACs by SCFAs in T-cells (137). In this particular study, acetate, propionate, and butyrate were shown to present similar HDAC inhibitory effects and to regulate the mTOR pathway, affecting T-cell differentiation in a cytokine milieu-dependent manner. Although the exact role of GPCRs in SCFA-induced HDAC inhibition still remains to be clarified, there is no doubt that SCFAs play a key role in T-cell differentiation by altering the T-cell epigenome. Several inflammatory diseases have been linked to epigenetic changes that could affect the Th17/Treg balance. In a murine model of colitis, the methyltransferase G9A has been identified as playing a key role in T-cell differentiation, in particular by facilitating dimethylation of histone H3 at lysine 9, supporting Th17 cell and Treg differentiation both in vivo and in vitro (18). A significant hypomethylation on MIR21 and increased expression of MIR21 mRNA has recently been reported in circulating leukocytes from patients diagnosed with CD (139). This could have important consequences on the T-cell profile in these patients as miR-21 has been linked to Th17 cell differentiation (64, 79). The gut microbiota has been shown to downregulate miR-10a in the intestine and dendritic cells in specific pathogen-free mice (140). In this model, miR-10a downregulation was associated with increased expression of IL-12/IL23p40 in dendritic cells. These changes could affect the regulation of intestinal inflammation, as IL-12/IL23p40 play a key role in Th1/Th17 cell differentiation (141). One of the potential mechanisms by which commensal bacteria could influence miRNA expression is through SCFAs. Butyrate has been reported to affect the expression of 44 different miRNAs in a human colon cancer cell line, and in particular members of the miR-106b family (142). These miRNAs included miR-17, a member of the miR-17-92 cluster which is known to promote Th17-mediated inflammation (143). The same miR cluster has further been reported to be critical for the differentiation of Treg, with the loss of miR-17-92 significantly affecting the accumulation of activated Tregs and their differentiation into IL-10-producing cells (76).
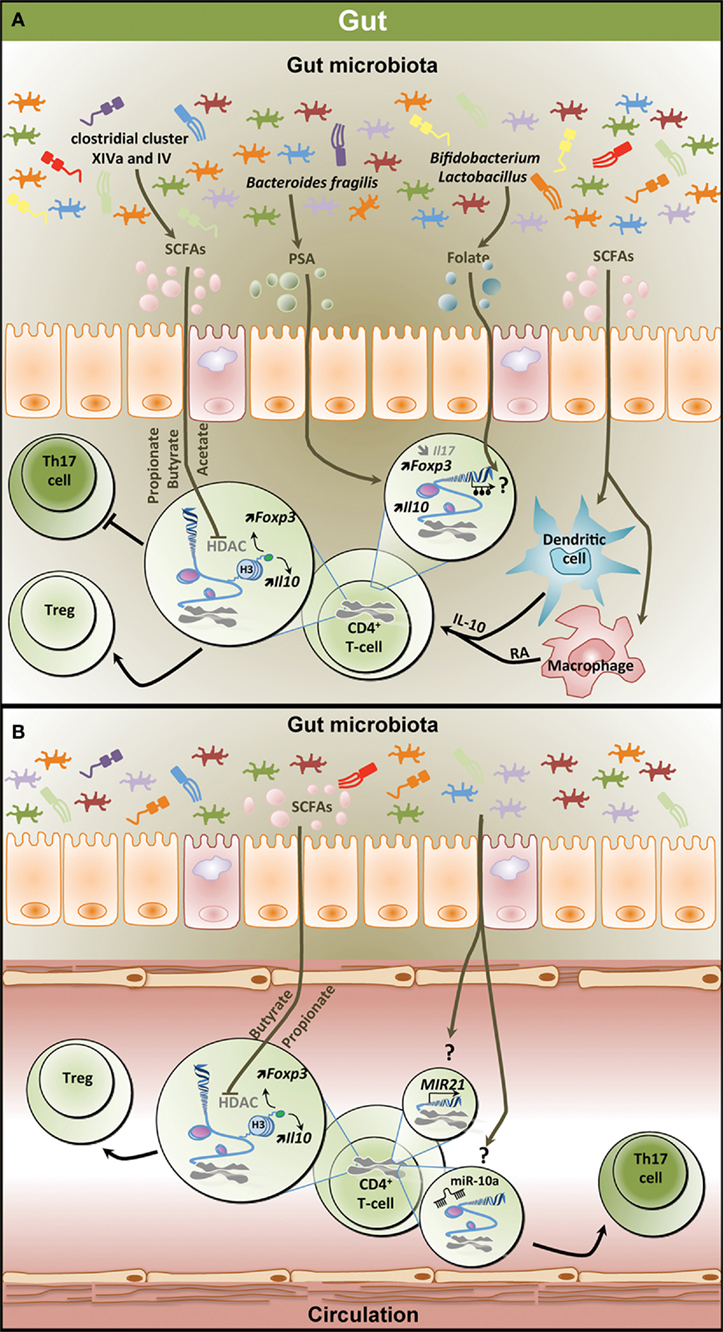
Figure 2. Epigenetic regulation of the T helper 17/regulatory T cell (Th17/Treg) balance by the gut microbiota. (A) Metabolites derived from the gut microbiota affect the T-cell epigenome influencing the Th17/Treg balance in the gastrointestinal (GI) tract. SCFAs including propionate, butyrate, and acetate which are mostly produced by clostridial clusters VIV and IV exert HDAC inhibitory activity, thereby increasing Foxp3 and Il10 expression and promoting Treg differentiation and function. SCFAs can also affect dendritic cells and macrophages, thereby inducing IL-10 and RA production and promoting Treg differentiation. PSA derived from Bacteroides fragilis contributes to increased expression of Il10 and Foxp3 in T-cell and reduces Il17 expression. Both Bifidobacterium and Lactobacillus contribute to the production of the methyl donor folate potentially influencing the methylome in T-cells. (B) Effect of microbiota-derived metabolites on the epigenome of peripheral T-cells. Similar to the GI tract, SCFAs increase Foxp3 and Il10 expression in peripheral T-cells through HDAC inhibition favoring Treg differentiation. Through unknown mechanisms, the gut microbiota increases the expression of MIR21 and the levels of miR-10a, potentially promoting Th17 differentiation. SCFAs, short-chain fatty acids; PSA, polysaccharide A; HDAC, histone deacetylase; H3, histone 3; Foxp3, forkhead box P3; Il10, interleukin 10; RA, retinoic acid; Treg, regulatory T cell.
Future Perspectives and Conclusion
Changes in the gut microbiota are intimately linked to significant alterations in Th17/Treg balance, potentially mediated by epigenetic mechanisms, and contributing to the development and establishment of IBD, obesity, and T2D, and possibly other chronic inflammatory conditions. Targeting such epigenetic changes through the use of HDAC inhibitors and miRNA-based therapies has recently started to emerge as a novel therapeutic option in the context of inflammatory diseases (144). This raises the question whether manipulating the gut microbiota could provide a novel approach to reshape the T-cell epigenome and influence the Th17/Treg balance in inflammatory diseases (Figure 2B). Several dietary interventions targeting the gut microbiome have reported positive outcomes in both animal models and human cohorts in IBD, obesity, or T2D (114, 116, 117, 122, 145). Perhaps, some of the most promising dietary interventions include the use of prebiotics and probiotics. Prebiotics are defined as fermented ingredients capable of stimulating bacterial growth and activity in the GI microbiota providing health benefits to the host and include oligosaccharides that are non-digestible, poorly digested carbohydrates and dietary fibers. Supplementation with prebiotics can increase the representation of butyrogenic strains in the gut microbiota, thus supporting an increased production of SCFAs. Increased levels of SCFAs could remodel the T-cell epigenome, favoring Treg differentiation (128–130), in line with the anti-inflammatory effect of SCFAs (129, 130). Even direct oral administration of SCFAs, and in particular butyrate, successfully remodeled the T-cell profile and improved inflammation in mice models of colitis (129, 146). Probiotics are live organisms which, when administered to the host, provide beneficial health effects. They represent an alternative option to prebiotics in modulating the gut microbiota and leading to positive immune adaptations in inflammatory diseases (116, 117). Several microorganisms presenting beneficial immunomodulatory properties that could help protect the host and provide novel therapeutic options for chronic inflammatory disorders have been identified (125, 147). B. fragilis, through the expression of PSA, is critical for the suppression of IL-17 production and the protection against inflammation (125). Bifidobacterium alone or in combination with prebiotics favors the production of the methyl donor folate (148), which may contribute to Treg survival through epigenetic mechanisms (149), in addition to its well-documented role in health and diseases prevention (150, 151). Similarly, probiotics (Bifidobacterium and Lactobacillus species) contained in maternal milk have even been suggested to mediate some of the effects of breastfeeding on Treg differentiation (152). These observations highlight the importance of the specific components of the gut microbiota in the establishment and the maintenance of immune homeostasis. Alternatively, untested combinations of bacterial species may produce maximized responses.
Although there is still limited information on the direct effect of microbiota-based therapies on epigenetic regulation of the Th17/Treg balance, the continuously growing literature focusing on such interventions should soon provide the much needed evidence to guide future therapeutic strategies against the inflammatory component of IBD, obesity, or T2D. The systematic investigation of the effect of prebiotics or probiotics on the T-cell epigenome could provide invaluable information on their potential as immunomodulatory therapeutics and support the refining of such interventions. Modulation of the epigenome at gene specific loci, for example, using methyltransferases or acetyltransferases fused to a nuclease-null CRISPR-Cas9 could be used to improve the specificity of such therapies. In the face of the ever-growing pandemic of inflammatory diseases, we are indeed in urgent need of innovative strategies that could specifically target the immune system while also affecting additional contributors to altered immune homeostasis. Challenges that still remain to be overcome include the identification of prebiotics, probiotics, or related metabolites with the highest potential to promote a balanced immune system and the best modalities of administration, as well as the translation of such findings in the context of human diseases. Achieving these outcomes will undoubtedly place us in a better position to tackle inflammatory diseases.
Author Contributions
AL, SL, RB, LH, MG, and DS contributed to the writing, review, and approval of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation.
References
1. Pomié C, Garidou L, Burcelin R. Intestinal RORrt-generated Th17 cells control type 2 diabetes: a first antidiabetic target identified from the host to microbiota crosstalk. Inflamm Cell Signal (2016) 3(1):1–7. doi: 10.14800/ics.1074
2. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52(1):65–70. doi:10.1136/gut.52.1.65
3. Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, et al. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol (2010) 160(3):386–93. doi:10.1111/j.1365-2249.2010.04093.x
4. Cipolletta D. Adipose tissue-resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology (2014) 142(4):517–25. doi:10.1111/imm.12262
5. Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D. Appearance and disappearance of the mRNA signature characteristic of T(reg) cells in visceral adipose tissue: age, diet, and PPARγ effects. Proc Natl Acad Sci U S A (2015) 112(2):482–7. doi:10.1073/pnas.1423486112
6. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPARγ is a major driver of the accumulation and phenotype of adipose-tissue T(reg) cells. Nature (2012) 486(7404):549–53. doi:10.1038/nature11132
7. Cipolletta D, Kolodin D, Benoist C, Mathis D. Tissular Tregs: a unique population of adipose-tissue-resident Foxp3+CD4+ T cells that impacts organismal metabolism. Semin Immunol (2011) 23(6):431–7. doi:10.1016/j.smim.2011.06.002
8. Kuroiwa-Trzmielina J, Hesson LB. Epigenetic effects of gut microbiota on obesity and gastrointestinal cancers. In: Berger NA, editor. Epigenetics, Energy Balance, and Cancer. (Chap. 11), Switzerland: Springer International Publishing (2016). p. 167–89.
9. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol (2009) 9(2):83–9. doi:10.1038/nri2474
10. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463(7282):808–12. doi:10.1038/nature08750
11. Adams JB, Johansen LJ, Powell LD, Quig D, Rubin RA. Gastrointestinal flora and gastrointestinal status in children with autism – comparisons to typical children and correlation with autism severity. BMC Gastroenterol (2011) 11(1):22. doi:10.1186/1471-230X-11-22
12. Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2016) 22(6):598–605. doi:10.1038/nm.4102
13. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22(6):586–97. doi:10.1038/nm.4106
14. Tilg H, Cani PD, Mayer EA. Gut microbiome and liver diseases. Gut (2016) 65(12):2035–44. doi:10.1136/gutjnl-2016-312729
15. Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med (2016) 22(10):1079–89. doi:10.1038/nm.4185
16. Becattini S, Taur Y, Pamer EG. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol Med (2016) 22(6):458–78. doi:10.1016/j.molmed.2016.04.003
17. Kamada N, Seo S-U, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13(5):321–35. doi:10.1038/nri3430
18. Antignano F, Burrows K, Hughes MR, Han JM, Kron KJ, Penrod NM, et al. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J Clin Invest (2014) 124(5):1945–55. doi:10.1172/JCI69592
19. Zúñiga LA, Shen W-J, Joyce-Shaikh B, Pyatnova EA, Richards AG, Thom C, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol (2010) 185(11):6947–59. doi:10.4049/jimmunol.1001269
20. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi:10.1038/nm.2002
21. Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol (2007) 19(6):409–17. doi:10.1016/j.smim.2007.10.011
22. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev (2010) 238(1):247–62. doi:10.1111/j.1600-065X.2010.00951.x
23. Chen Z, Lin F, Gao Y, Li Z, Zhang J, Xing Y, et al. FOXP3 and RORγt: transcriptional regulation of Treg and Th17. Int Immunopharmacol (2011) 11(5):536–42. doi:10.1016/j.intimp.2010.11.008
24. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27(1):485–517. doi:10.1146/annurev.immunol.021908.132710
25. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs and skin. Annu Rev Pathol (2013) 8:477–512. doi:10.1146/annurev-pathol-011110-130318
26. van Wijk F, Cheroutre H. Mucosal T cells in gut homeostasis and inflammation. Expert Rev Clin Immunol (2010) 6(4):559–66. doi:10.1586/eci.10.34
27. Cheroutre H. Starting at the beginning: new perspectives on the biology of mucosal T cells. Annu Rev Immunol (2004) 22:217–46. doi:10.1146/annurev.immunol.22.012703.104522
28. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
29. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
30. Roncarolo MG, Gregori S. Is FOXP3 a bona fide marker for human regulatory T cells? Eur J Immunol (2008) 38(4):925–7. doi:10.1002/eji.200838168
31. Zahorsky-Reeves JL, Wilkinson JE. The murine mutation scurfy (sf) results in an antigen-dependent lymphoproliferative disease with altered T cell sensitivity. Eur J Immunol (2001) 31(1):196–204. doi:10.1002/1521-4141(200101)31:1<196:AID-IMMU196>3.0.CO;2-9
32. Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol (2003) 15(4):430–5. doi:10.1097/00002281-200307000-00010
33. Le Bras S, Geha RS. IPEX and the role of Foxp3 in the development and function of human Tregs. J Clin Invest (2006) 116(6):1473–5. doi:10.1172/JCI28880
34. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest (2006) 116(6):1713–22. doi:10.1172/JCI25112
35. Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol (2009) 21(10):1105–11. doi:10.1093/intimm/dxp095
36. Jonuleit H, Schmitt E. The regulatory T cell family: distinct subsets and their interrelations. J Immunol (2003) 171(12):6323–7. doi:10.4049/jimmunol.171.12.6323
37. Ziegler SF, Buckner JH. FOXP3 and the regulation of Treg/Th17 differentiation. Microbes Infect (2009) 11(5):594–8. doi:10.1016/j.micinf.2009.04.002
38. Raza A, Yousaf W, Giannella R, Shata MT. Th17 cells: interactions with predisposing factors in the immunopathogenesis of inflammatory bowel disease. Expert Rev Clin Immunol (2012) 8(2):161–8. doi:10.1586/eci.11.96
39. Galvez J. Role of Th17 cells in the pathogenesis of human IBD. ISRN Inflamm (2014) 2014:928461. doi:10.1155/2014/928461
40. Lee S-Y, Lee SH, Yang E-J, Kim E-K, Kim J-K, Shin D-Y, et al. Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS One (2015) 10(9):e0135858. doi:10.1371/journal.pone.0135858
41. Xu XR, Liu CQ, Feng BS, Liu ZJ. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J Gastroenterol (2014) 20(12):3255–64. doi:10.3748/wjg.v20.i12.3255
42. Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res (2007) 149(4):173–86. doi:10.1016/j.trsl.2006.11.009
43. Arseneau KO, Tamagawa H, Pizarro TT, Cominelli F. Innate and adaptive immune responses related to IBD pathogenesis. Curr Gastroenterol Rep (2007) 9(6):508–12. doi:10.1007/s11894-007-0067-3
44. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448(7152):427–34. doi:10.1038/nature06005
45. Keubler LM, Buettner M, Hager C, Bleich A. A multihit model: colitis lessons from the interleukin-10-deficient mouse. Inflamm Bowel Dis (2015) 21(8):1967–75. doi:10.1097/MIB.0000000000000468
46. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61:1693–700. doi:10.1136/gutjnl-2011-301668
47. Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte J-M, et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol (2006) 290(4):G827. doi:10.1152/ajpgi.00513.2005
48. Wolk K, Witte E, Hoffmann U, Doecke W-D, Endesfelder S, Asadullah K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn’s disease. J Immunol (2007) 178(9):5973–81. doi:10.4049/jimmunol.178.9.5973
49. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T cell function in the intestine. Nature (2014) 513(7519):564–8. doi:10.1038/nature13577
50. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev (2014) 13(1):3–10. doi:10.1016/j.autrev.2013.06.004
51. Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol (2009) 86(4):959–69. doi:10.1189/jlb.0109006
52. Sakaguchi S, Wing K, Yamaguchi T. Dynamics of peripheral tolerance and immune regulation mediated by Treg. Eur J Immunol (2009) 39(9):2331–6. doi:10.1002/eji.200939688
53. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity (2011) 34(2):237–46. doi:10.1016/j.immuni.2011.01.016
54. Veenbergen S, Samsom JN. Maintenance of small intestinal and colonic tolerance by IL-10-producing regulatory T cell subsets. Curr Opin Immunol (2012) 24(3):269–76. doi:10.1016/j.coi.2012.03.004
55. Ip B, Cilfone N, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M, et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity (2016) 24(1):102–12. doi:10.1002/oby.21243
56. Endo Y, Asou Hikari K, Matsugae N, Hirahara K, Shinoda K, Tumes Damon J, et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep (2015) 12(6):1042–55. doi:10.1016/j.celrep.2015.07.014
57. Chen X, Wu Y, Wang L. Fat-resident Tregs: an emerging guard protecting from obesity-associated metabolic disorders. Obes Rev (2013) 14(7):568–78. doi:10.1111/obr.12033
58. Endo Y, Yokote K, Nakayama T. The obesity-related pathology and Th17 cells. Cell Mol Life Sci (2017) 74(7):1231–45. doi:10.1007/s00018-016-2399-3
59. Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem Pharmacol (2009) 77(12):1835–44. doi:10.1016/j.bcp.2009.03.008
60. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem (2008) 77(1):289–312. doi:10.1146/annurev.biochem.77.061307.091829
61. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5(2):e38. doi:10.1371/journal.pbio.0050038
62. Nagar M, Vernitsky H, Cohen Y, Dominissini D, Berkun Y, Rechavi G, et al. Epigenetic inheritance of DNA methylation limits activation-induced expression of FOXP3 in conventional human CD25-CD4+ T cells. Int Immunol (2008) 20(8):1041–55. doi:10.1093/intimm/dxn062
63. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38(6):1654–63. doi:10.1002/eji.200838105
64. Kalla R, Ventham NT, Kennedy NA, Quintana JF, Nimmo ER, Buck AH, et al. microRNAs: new players in IBD. Gut (2015) 64(3):504–13. doi:10.1136/gutjnl-2014-307891
65. Yang TT, Song SJ, Xue HB, Shi DF, Liu CM, Liu H. Regulatory T cells in the pathogenesis of type 2 diabetes mellitus retinopathy by miR-155. Eur Rev Med Pharmacol Sci (2015) 19(11):2010–5.
66. Yao R, Ma Y-L, Liang W, Li H-H, Ma Z-J, Yu X, et al. microRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS One (2012) 7(10):e46082. doi:10.1371/journal.pone.0046082
67. Baumjohann D, Ansel KM. microRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol (2013) 13(9):666–78. doi:10.1038/nri3494
68. Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol (2009) 182(5):2578–82. doi:10.4049/jimmunol.0803162
69. Yang B-H, Floess S, Hagemann S, Deyneko IV, Groebe L, Pezoldt J, et al. Development of a unique epigenetic signature during in vivo Th17 differentiation. Nucleic Acids Res (2015) 43(3):1537–48. doi:10.1093/nar/gkv014
70. Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem (2007) 282(9):5969–72. doi:10.1074/jbc.C600322200
71. Tripathi SK, Lahesmaa R. Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol Rev (2014) 261(1):62–83. doi:10.1111/imr.12204
72. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30(1):155–67. doi:10.1016/j.immuni.2008.12.009
73. Kitagawa Y, Wing JB, Sakaguchi S. Transcriptional and epigenetic control of regulatory T cell development. Prog Mol Biol Transl Sci (2015) 136:1–33. doi:10.1016/bs.pmbts.2015.07.011
74. Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res (2009) 19(7):1165–74. doi:10.1101/gr.091470.109
75. Zheng Q, Xu Y, Liu Y, Zhang B, Li X, Guo F, et al. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J Mol Med (Berl) (2009) 87(12):1191–205. doi:10.1007/s00109-009-0530-8
76. de Kouchkovsky D, Esensten JH, Rosenthal WL, Morar MM, Bluestone JA, Jeker LT. microRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J Immunol (2013) 191(4):1594–605. doi:10.4049/jimmunol.1203567
77. Jeker LT, Zhou X, Gershberg K, de Kouchkovsky D, Morar MM, Stadthagen G, et al. microRNA 10a marks regulatory T cells. PLoS One (2012) 7(5):e36684. doi:10.1371/journal.pone.0036684.g001
78. Kelada S, Sethupathy P, Okoye IS, Kistasis E, Czieso S, White SD, et al. miR-182 and miR-10a are key regulators of Treg specialisation and stability during Schistosome and Leishmania-associated inflammation. PLoS Pathog (2013) 9(6):e1003451. doi:10.1371/journal.ppat.1003451
79. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. microRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest (2015) 125(3):1069–80. doi:10.1172/JCI74347
80. Wei B, Pei G. microRNAs: critical regulators in Th17 cells and players in diseases. Nature (2010) 7(3):175–81. doi:10.1038/cmi.2010.19
81. Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T, et al. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol Cell Biol (2014) 34(21):3993–4007. doi:10.1128/MCB.00919-14
82. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi:10.1038/83784
83. Michalak A, Mosińska P, Fichna J. Common links between metabolic syndrome and inflammatory bowel disease: current overview and future perspectives. Pharmacol Rep (2016) 68(4):837–46. doi:10.1016/j.pharep.2016.04.016
84. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A (2007) 104(34):13780–5. doi:10.1073/pnas.0706625104
85. Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut (2004) 53(5):685–93. doi:10.1136/gut.2003.025403
86. Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut (2003) 52(2):237–42. doi:10.1136/gut.52.2.237
87. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A (2010) 107(33):14691–6. doi:10.1073/pnas.1005963107/-/DCSupplemental
88. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut (2014) 63(8):1275–83. doi:10.1136/gutjnl-2013-304833
89. Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJO. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol (2006) 44(11):4136–41. doi:10.1128/JCM.01004-06
90. Miyoshi J, Chang EB. The gut microbiota and inflammatory bowel diseases. Transl Res (2017) 179:38–48. doi:10.1016/j.trsl.2016.06.002
91. Atarashi K, Umesaki Y, Honda K. Microbiotal influence on T cell subset development. Semin Immunol (2011) 23(2):146–53. doi:10.1016/j.smim.2011.01.010
92. Bernstein CN. Treatment of IBD: where we are and where we are going. Am J Gastroenterol (2015) 110(1):114–26. doi:10.1038/ajg.2014.357
93. Triantafillidis JK, Merikas E, Georgopoulos F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des Devel Ther (2011) 5:185–210. doi:10.2147/DDDT.S11290
94. Matthes H, Krummenerl T, Giensch M, Wolff C, Schulze J. Clinical trial: probiotic treatment of acute distal ulcerative colitis with rectally administered Escherichia coli Nissle 1917 (EcN). BMC Complement Altern Med (2010) 10:13. doi:10.1186/1472-6882-10-13
95. Sood A, Midha V, Makharia GK, Ahuja V, Singal D, Goswami P, et al. The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin Gastroenterol Hepatol (2009) 7(11):1202–9,9.e1. doi:10.1016/j.cgh.2009.07.016
96. Jonkers D, Penders J, Masclee A, Pierik M. Probiotics in the management of inflammatory bowel disease: a systematic review of intervention studies in adult patients. Drugs (2012) 72(6):803–23. doi:10.2165/11632710-000000000-00000
97. Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Hart AL, et al. Randomised, double-blind, placebo-controlled trial of fructo-oligosaccharides in active Crohn’s disease. Gut (2011) 60(7):923–9. doi:10.1136/gut.2010.232025
98. De Preter V, Joossens M, Ballet V, Shkedy Z, Rutgeerts P, Vermeire S, et al. Metabolic profiling of the impact of oligofructose-enriched inulin in Crohn’s disease patients: a double-blinded randomized controlled trial. Clin Transl Gastroenterol (2013) 4:e30. doi:10.1038/ctg.2012.24
99. van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med (2013) 368(5):407–15. doi:10.1056/NEJMoa1205037
100. Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, van den Bogaerde J, Samuel D, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet (2017) 389(10075):1218–28. doi:10.1016/S0140-6736(17)30182-4
101. Shi Y, Dong Y, Huang W, Zhu D, Mao H, Su P. Fecal microbiota transplantation for ulcerative colitis: a systematic review and meta-analysis. PLoS One (2016) 11(6):e0157259. doi:10.1371/journal.pone.0157259
102. De Palma G, Lynch MD, Lu J, Dang VT, Deng Y, Jury J, et al. Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice. Sci Transl Med (2017) 9(379):eaaf6397. doi:10.1126/scitranslmed.aaf6397
103. Ding S, Lund PK. Role of intestinal inflammation as an early event in obesity and insulin resistance. Curr Opin Clin Nutr Metab Care (2011) 14(4):328–33. doi:10.1097/MCO.0b013e3283478727
104. Reijnders D, Goossens GH, Hermes GDA, Neis EPJG, van der Beek CM, Most J, et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metab (2016) 24(1):63–74. doi:10.1016/j.cmet.2016.06.016
105. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature (2013) 500(7464):541–6. doi:10.1038/nature12506
106. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A (2005) 102(31):11070–5. doi:10.1073/pnas.0504978102
107. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med (2009) 1(6):6ra14. doi:10.1126/scitranslmed.3000322
108. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444(7122):1022–3. doi:10.1038/4441022a
109. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature (2009) 457(7228):480–4. doi:10.1038/nature07540
110. Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, et al. The gut microbiota and its relationship to diet and obesity. Gut Microbes (2014) 3(3):186–202. doi:10.4161/gmic.20168
111. Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (2010) 18(1):190–5. doi:10.1038/oby.2009.167
112. Larsen N, Vogensen FK, van den Berg FWJ, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One (2010) 5(2):e9085. doi:10.1371/journal.pone.0009085
113. Aronsson L, Huang Y, Parini P, Korach-André M, Håkansson J, Gustafsson J-A, et al. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PLoS One (2010) 5(9):e13087. doi:10.1371/journal.pone.0013087
114. Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia (2007) 50(11):2374–83. doi:10.1007/s00125-007-0791-0
115. Kadooka Y, Sato M, Imaizumi K, Ogawa A, Ikuyama K, Akai Y, et al. Regulation of abdominal adiposity by probiotics (Lactobacillus gasseri SBT2055) in adults with obese tendencies in a randomized controlled trial. Eur J Clin Nutr (2010) 64(6):636–43. doi:10.1038/ejcn.2010.19
116. Miyoshi M, Ogawa A, Higurashi S, Kadooka Y. Anti-obesity effect of Lactobacillus gasseri SBT2055 accompanied by inhibition of pro-inflammatory gene expression in the visceral adipose tissue in diet-induced obese mice. Eur J Nutr (2013) 53(2):599–606. doi:10.1007/s00394-013-0568-9
117. Ukibe K, Miyoshi M, Kadooka Y. Administration of Lactobacillus gasseri SBT2055 suppresses macrophage infiltration into adipose tissue in diet-induced obese mice. Br J Nutr (2015) 114(8):1180–7. doi:10.1017/S0007114515002627
118. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28(1):573–621. doi:10.1146/annurev-immunol-030409-101225
119. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489(7415):231–41. doi:10.1038/nature11551
120. Kamdar K, Khakpour S, Chen J, Leone V, Brulc J, Mangatu T, et al. Genetic and metabolic signals during acute enteric bacterial infection alter the microbiota and drive progression to chronic inflammatory disease. Cell Host Microbe (2016) 19(1):21–31. doi:10.1016/j.chom.2015.12.006
121. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria TH17 cell differentiation. Nature (2008) 455(7214):808–12. doi:10.1038/nature07240
122. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331(6015):337–41. doi:10.1126/science.1198469
123. Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity (2008) 29(4):637–49. doi:10.1016/j.immuni.2008.08.009
124. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe (2008) 4(4):337–49. doi:10.1016/j.chom.2008.09.009
125. Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature (2008) 453(7195):620–5. doi:10.1038/nature07008
126. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107(27):12204–9. doi:10.1073/pnas.0909122107/-/DCSupplemental
127. Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AGP, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-γ and RelA. Nat Immunol (2003) 5(1):104–12. doi:10.1038/ni1018
128. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi:10.1038/nature12726
129. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi:10.1038/nature12721
130. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi:10.1126/science.1241165
131. Kim CH, Park J, Kim M. Gut microbiota-derived short-chain fatty acids, T cells, and inflammation. Immune Netw (2014) 14(6):277–212. doi:10.4110/in.2014.14.6.277
132. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi:10.1016/j.immuni.2009.04.014
133. Heimann E, Nyman M, Pålbrink A-K, Lindkvist-Petersson K, Degerman E. Branched short-chain fatty acids modulate glucose and lipid metabolism in primary adipocytes. Adipocyte (2016) 5(4):359–68. doi:10.1080/21623945.2016.1252011
134. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi:10.1038/nm.3704
135. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40(1):128–39. doi:10.1016/j.immuni.2013.12.007
136. Kim SV, Xiang WV, Kwak C, Yang Y, Lin XW, Ota M, et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science (2013) 340(6139):1456–9. doi:10.1126/science.1237013
137. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol (2015) 8(1):80–93. doi:10.1038/mi.2014.44
138. Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell (2016) 24:1–11. doi:10.1016/j.molcel.2016.10.025
139. Adams AT, Kennedy NA, Hansen R, Ventham NT, O’Leary KR, Drummond HE, et al. Two-stage genome-wide methylation profiling in childhood-onset Crohn’s disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm Bowel Dis (2014) 20(10):1784–93. doi:10.1097/MIB.0000000000000179
140. Xue X, Feng T, Yao S, Wolf KJ, Liu CG, Liu X, et al. Microbiota downregulates dendritic cell expression of miR-10a, which targets IL-12/IL-23p40. J Immunol (2011) 187(11):5879–86. doi:10.4049/jimmunol.1100535
141. Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest (2006) 116(5):1218–22. doi:10.1172/JCI28508
142. Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH, et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PLoS One (2011) 6(1):e16221. doi:10.1371/journal.pone.0016221
143. Liu SQ, Jiang S, Li C, Zhang B, Li QJ. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros Family Zinc Finger 4 to promote TH17-mediated inflammation. J Biol Chem (2014) 289(18):12446–56. doi:10.1074/jbc.M114.550723
144. Raghuraman S, Donkin I, Versteyhe S, Barrès R, Simar D. The emerging role of epigenetics in inflammation and immunometabolism. Trends Endocrinol Metab (2016) 27(11):782–95. doi:10.1016/j.tem.2016.06.008
145. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature (2013) 500(7461):232–6. doi:10.1038/nature12331
146. Vieira ELM, Leonel AJ, Sad AP, Beltrão NRM, Costa TF, Ferreira TMR, et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J Nutr Biochem (2012) 23(5):430–6. doi:10.1016/j.jnutbio.2011.01.007
147. Narushima S, Sugiura Y, Oshima K, Atarashi K, Hattori M, Suematsu M, et al. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut Microbes (2014) 5(3):333–9. doi:10.4161/gmic.28572
148. Pompei A, Cordisco L, Amaretti A, Zanoni S, Raimondi S, Matteuzzi D, et al. Administration of folate-producing bifidobacteria enhances folate status in Wistar rats. J Nutr (2007) 137(12):2742–6.
149. Kunisawa J, Hashimoto E, Ishikawa I, Kiyono H. A pivotal role of vitamin B9 in the maintenance of regulatory T cells in vitro and in vivo. PLoS One (2012) 7(2):e32094. doi:10.1371/journal.pone.0032094
150. Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr (2012) 3(1):21–38. doi:10.3945/an.111.000992
151. Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab (2016) 25(1):27–42. doi:10.1016/j.cmet.2016.08.009
Keywords: gut microbiota, epigenetics, regulatory T cell, T helper 17 cell, inflammatory bowel diseases, obesity, type 2 diabetes
Citation: Luo A, Leach ST, Barres R, Hesson LB, Grimm MC and Simar D (2017) The Microbiota and Epigenetic Regulation of T Helper 17/Regulatory T Cells: In Search of a Balanced Immune System. Front. Immunol. 8:417. doi: 10.3389/fimmu.2017.00417
Received: 21 December 2016; Accepted: 23 March 2017;
Published: 10 April 2017
Edited by:
Laurence Macia, University of Sydney, AustraliaReviewed by:
Mourad Aribi, University of Tlemcen, AlgeriaMaria Buoncervello, Istituto Superiore di Sanità, Italy
Copyright: © 2017 Luo, Leach, Barres, Hesson, Grimm and Simar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Simar, ZC5zaW1hckB1bnN3LmVkdS5hdQ==