 Giada Mondanelli1†
Giada Mondanelli1† Elisa Albini1†
Elisa Albini1† Maria T. Pallotta1
Maria T. Pallotta1 Claudia Volpi1
Claudia Volpi1 Lucienne Chatenoud2
Lucienne Chatenoud2 Chantal Kuhn3
Chantal Kuhn3 Francesca Fallarino1
Francesca Fallarino1 Davide Matino1
Davide Matino1 Maria L. Belladonna1
Maria L. Belladonna1 Roberta Bianchi1
Roberta Bianchi1 Carmine Vacca1
Carmine Vacca1 Silvio Bicciato4
Silvio Bicciato4 Louis Boon5
Louis Boon5 Giovanni Ricci6
Giovanni Ricci6 Ursula Grohmann1
Ursula Grohmann1 Paolo Puccetti1
Paolo Puccetti1 Ciriana Orabona1*
Ciriana Orabona1*
- 1Section of Pharmacology, Department of Experimental Medicine, University of Perugia, Perugia, Italy
- 2INSERM U1013, Hôpital Necker-Enfants Malades, Université Paris Descartes, Paris, France
- 3VL37 Inc., Cambridge, MA, USA
- 4Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
- 5Bioceros BV, Utrecht, Netherlands
- 6Animal Facility of the University of Perugia, Perugia, Italy
Bortezomib (BTZ) is a first-in-class proteasome inhibitor approved for the therapy of multiple myeloma that also displays unique regulatory activities on immune cells. The enzyme indoleamine 2,3-dioxygenase 1 (IDO1) is a tryptophan metabolizing enzyme exerting potent immunoregulatory effects when expressed in dendritic cells (DCs), the most potent antigen-presenting cells capable of promoting either immunity or tolerance. We previously demonstrated that, in inflammatory conditions, IDO1 is subjected to proteasomal degradation in DCs, turning these cells from immunoregulatory to immunostimulatory. In non-obese diabetic (NOD) mice, an experimental model of autoimmune diabetes, we also identified an IDO1 defect such that the DCs do not develop tolerance toward pancreatic islet autoantigens. We found that BTZ rescues IDO1 protein expression in vitro in a particular subset of DCs, i.e., plasmacytoid DCs (pDCs) from NOD mice. When administered in vivo to prediabetic mice, the drug prevented diabetes onset through IDO1- and pDC-dependent mechanisms. Although the drug showed no therapeutic activity when administered alone to overtly diabetic mice, its combination with otherwise suboptimal dosages of autoimmune-preventive anti-CD3 antibody resulted in disease reversal in 70% diabetic mice, a therapeutic effect similar to that afforded by full-dosage anti-CD3. Thus, our data indicate a potential for BTZ in the immunotherapy of autoimmune diabetes and further underline the importance of IDO1-mediated immune regulation in such disease.
Introduction
Dendritic cells (DCs) have an important role as islet antigen-presenting cells in the initiation of autoimmune diabetes in the non-obese diabetic (NOD) mouse, in which defective tryptophan catabolism by immune regulatory indoleamine 2,3-dioxygenase 1 (IDO1) contributes to an overall impaired tolerogenesis. Functional flexibility represents a feature of plasmacytoid DCs (pDCs), capable of inducing either immune activation or tolerance, depending on environmental conditions (1–5). Protein expression of IDO1—and thus the functional balance between immunity and tolerance—can greatly vary in pDCs in response to transcriptional Ido1 inducers as well as post-transcriptional mechanisms. Splenic pDCs from conventional (e.g., C57BL/6 or BALB/c) mice express basal yet low levels of IDO1 protein, which can be increased by interferon (IFN)-γ, the standard IDO1 transcriptional inducer. As a result, unstimulated splenic pDCs are immunogenic, whereas IFN-γ-stimulated pDCs are tolerogenic (6, 7). The post-transcriptional mechanisms include, instead, proteasomal degradation of the enzyme, which is driven by inflammatory IL-6 and results in sustained inflammation and impaired tolerogenesis (4, 6–8).
In NOD mice, IDO1 expression and activity in pDCs are almost negligible in response to IFN-γ (9, 10), due to an impaired IFN-γ signaling activity (9) and abnormal regulatory proteolysis of the enzyme sustained by high levels of IL-6 (10). In agreement with this, systemic administration of IL-6-neutralizing antibodies results in marked suppression of the diabetic condition in NOD animals (11). Moreover, inflammatory cytokines, such as IFN-γ and tumor necrosis factor-α, induce the synthesis of catalytic subunits of the immunoproteasome—LMP2/β1i, MECL1/β2i, and LMP7/β5i—which replace their constitutive proteasome counterparts in hematopoietic cells (12). Selective inhibition of the β5i subunit has been shown to control disease progression in a series of experimental autoimmune disorders and promote long-term allograft survival (13–17).
Bortezomib (BTZ) is a reversible proteasome inhibitor that possesses immune regulatory activities that span various cellular processes of T and DCs essential for the development of adaptive immune responses (18). BTZ suppresses T-cell activation and production of cytokines, inhibits T-cell mobilization, suppresses T-cell growth and proliferation, and induces T-cell apoptosis. In DCs, BTZ suppresses maturation and function, cytokine production, and induces apoptosis. The drug can also inhibit nuclear translocation of NF-κB family members in DCs, thus supporting a pleiotropic function of BTZ in regulating DC function at the interface between immunity and tolerance (19).
Here, we report that BTZ confers tolerogenic effects on pDCs from NOD mice, resulting in enhanced generation of regulatory T cells and preventing diabetes onset in mice through an IDO1-dependent mechanism. A combination of BTZ and otherwise suboptimal dosages of anti-CD3 was found to rescue normoglycemia in overtly diabetic NOD mice.
Animals and Methods
Animals
Female BALB/c and NOD/MrkTac female mice, aged 6–16 weeks, were purchased from Charles River Breeding Laboratories (Calco, Milan, Italy) and Taconic (Albany, NY, USA), respectively. IDO1-deficient (Ido1−/−) NOD female mice were bred at the animal facility of the University of Perugia.
Cell Purification and In Vitro Treatments
Cells were isolated from pancreatic lymph nodes (pLNs) and pancreata by digestion with collagenase type IV (Sigma-Aldrich, St. Louis, MO, USA); total peritoneal cells (PeCs) were isolated from the peritoneal washes by centrifugation. Unfractionated cell populations were promptly used for cytofluorimetric analysis or incubated at 37°C for 16 h for cytokines and/or kynurenines detection in culture supernatants. All purification procedures involving pDCs and conventional DCs (cDCs, CD11c+ CD8−) have previously been described (6, 7, 9). The purity of DC populations was superior to 90%. Purified pDCs were exposed at 37°C to 200 U/ml of recombinant IFN-γ (R&D Systems, Minneapolis, MN, USA) or to 10 nM of BTZ (LC Laboratories, MA, USA) in the presence or absence of 4 μM 1-methyl-DL-tryptophan (1-MT; Sigma-Aldrich, St. Louis, MO, USA), a standard IDO1 inhibitor. In selected experiments, pDCs pretreated as described above, were co-cultured (2:1 cell ratio) with CD4+CD25− T lymphocytes purified from pLNs, and maintained at 37°C for 96 h, before harvesting the supernatants for the cytokine analysis and the cells for cytofluorimetric analysis.
Western Blot, ELISA, and Cytofluorimetric Analyses
Indoleamine 2,3-dioxygenase 1 protein expression was investigated in pDCs by immunoblot with a rabbit polyclonal anti-mouse IDO1 antibody raised in our laboratory (20). Anti-β-tubulin antibody (Sigma-Aldrich) was used as a normalizer. Mouse IL-6, IL-17A, IFN-γ, IL-10, and TGF-β1 were measured in culture supernatants by ELISA with specific kits (eBioscience, Inc., San Diego, CA, USA; Promega Italia, S.r.l., Milano, Italy). An ELISA–based TransAM Flexi NF-κB Family Kit (Active Motif, Rixensart, Belgium) was used to monitor activity of NF-κB family members, as described (6, 7, 10). Cytofluorimetric assays were conducted using the specific fluorochrome-labeled antibodies for murine CD19, CD5, CD1d, CD4, Foxp3, and RORγt (Biolegend, BD Pharmingen, eBiocience Inc., San Diego, CA, USA; Miltenyi Biotech, Germany), as described (6). For intracellular staining of Foxp3 and RORγt, cells were previously permeabilized by the Fix/Perm Buffer set (Biolegend, San Diego, CA, USA).
Kynurenine Assay
The functional activity of IDO1 was measured in vitro in terms of the ability to metabolize tryptophan (Trp) to kynurenine (Kyn), whose concentration was measured by high-performance liquid chromatography (HPLC) in culture supernatants after the addition of 100 µM Trp (20). Systemic Kyn/Trp ratio was determined in deproteinated sera of mice by measuring Kyn and Trp concentrations by HPLC.
In Vivo Treatment and Histopathology in NOD Mice
Bortezomib was administered i.p. at the dose of 0.10 or 0.25 mg/kg, a dose well below the drug’s maximum tolerated dose, i.e., 0.5 mg/kg (21), every other day for 2 weeks in prediabetic or in overtly diabetic NOD mice. In selected experiments, overtly diabetic mice were coadministered with BTZ and anti-CD3 (145-2C11) F(ab′)2 fragments (22) (provided by Lucienne Chatenoud, Université Paris Descartes, Paris) i.v. at a suboptimal dose of 10 µg/day, for five consecutive days. Selected groups of mice received the standard IDO1 inhibitor 1-MT in their drinking water at the concentration of 2 mg/ml. In vivo depletion of pDCs occurred 1 week after commencing BTZ treatment, by injecting prediabetic NOD mice with 250 μg/mouse of 120G8 (IgG2a isotype) antibody i.p., as described (23). Animals were either monitored for glycemia or sacrificed for ex vivo analyses at different times of drug treatment(s). For histopathology, 3–4 µm of paraffin-embedded sections of pancreata (5/organ) were stained with hematoxylin and eosin and analyzed by light microscopy. Insulitis scoring was done according to standard criteria as described (24).
Skin Test Assay
A skin test assay was used for measuring delayed-type hypersensitivity (DTH) in response to challenge in the footpad with the diabetogenic H-2Kd-restricted IGRP peptide (VYLKTNVFL), as described (7), using prediabetic NOD mice as recipients. For in vivo immunization, 3 × 105 peptide-loaded NOD cDCs, combined with a minority fraction (5%) of peptide-loaded NOD pDCs, and pre-incubated in vitro as indicated in the relevant figure legends, were injected subcutaneously into recipient mice. Two weeks later, a DTH response was measured to intrafootpad challenge with the eliciting peptide, and results were expressed as the change in footpad weight of peptide-injected footpads over that of vehicle-injected (internal control) counterparts (4, 6, 9).
Statistical Analysis
In vitro studies comparing more than two experimental conditions were analyzed by ANOVA test followed by Dunnett’s or Bonferroni test, while Student’s t-test was used for the analysis of in vitro results comparing two conditions. In the in vivo skin test assay, a paired Student’s t-test was used for statistical analysis by comparing the weight change of the experimental footpads with vehicle-injected counterpart (9). Diabetes incidence was plotted by using the Kaplan–Meier method, i.e., a non-parametric cumulative survival plot. The statistical comparison between the curves was performed by using the log-rank (Mantel–Cox) test.
Results
BTZ Confers IDO1-Dependent Tolerogenic Effects on pDCs from NOD Mice
We initially examined whether proteasome inhibition by BTZ would upregulate IDO1 expression and function in pDCs from NOD mice. In vitro, splenic pDCs, purified from prediabetic NOD mice, were incubated with 10 nM BTZ or 200 U/ml IFN-γ and, at different times, cell lysates and supernatants were analyzed for IDO1 protein and Kyn production, respectively. As expected, IFN-γ failed to upregulate IDO1 protein (Figure 1A) and activity (Figure 1B) in NOD pDCs. By contrast, pDCs conditioned with BTZ showed higher IDO1 expression (Figure 1A) and produced significantly higher levels of Kyn (Figure 1B). Although the increase in IDO1 protein expression was rather short-lived, treatment of NOD pDCs with BTZ induced the appearance of high molecular-weight IDO1 proteins (Figure 1A), corresponding to poliubiquitinated forms of the protein. We also found significantly higher expression of Socs3—the IL-6-induced molecular driver of IDO1 proteasomal degradation—and of the immunoproteasome subunits in NOD, compared to BALB/c pancreata (Figure S1A in Supplementary Material) and in prediabetic vs. diabetic pDCs, purified from NOD pLNs (Figure S1B in Supplementary Material). Interestingly, prediabetic NOD pDCs were also featured by a higher expression of the gene set involved in the pro-inflammatory NF-κB pathway (Figure S1C in Supplementary Material), suggesting a more inflammatory-prone phenotype of pDCs in the early phase of the disease.
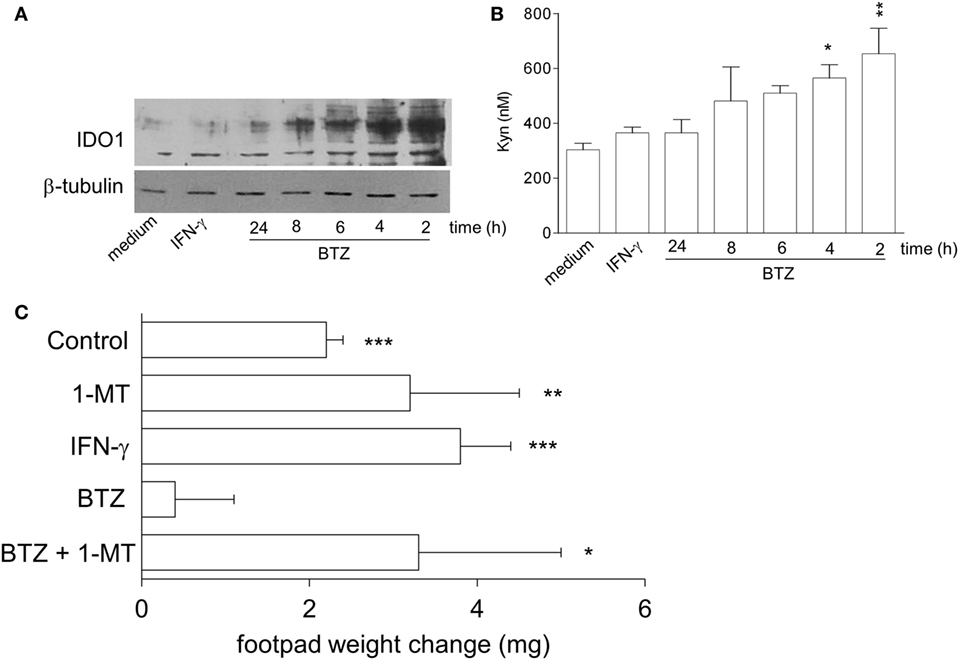
Figure 1. Bortezomib (BTZ) restores the tolerogenic potential of prediabetic non-obese diabetic (NOD) plasmacytoid DCs (pDCs) in an indoleamine 2,3-dioxygenase 1 (IDO1)-dependent fashion. (A) IDO1 immunoblot analysis of cell lysates from splenic pDCs purified from prediabetic NOD mice incubated with BTZ for the indicated time, interferon (IFN)-γ or medium alone for 24 h. One representative experiment out of three is shown. (B) IDO1 activity measured in terms of kynurenine production in cell supernatant of pDCs incubated as in panel (A). *p < 0.05; **p < 0.01 (one-way ANOVA and Dunnett’s multiple comparison test). Data are presented as means ± SD from triplicate samples. (C) IGRP-pulsed prediabetic NOD conventional DCs in combination with a minority fraction (5%) of NOD pDCs with no conditioning (control) or in vitro conditioned with 1-MT, IFN-γ, BTZ alone, or in combination with 1-MT, were transferred into NOD recipient mice (n = 6/group) to be assayed for skin reactivity to the eliciting peptide. Analysis of skin test reactivity is presented as change in footpad weight. *p < 0.05; **p < 0.01; ***p < 0.001 (paired Student’s t-test; weight change of experimental vs. control footpad).
In vivo, NOD female mice were sensitized with the diabetogenic IGRP peptide loaded on highly immunogenic NOD cDCs. The cDCs were administered alone or in combination with a minority fraction of NOD pDCs, treated with control media, IFN-γ, or BTZ (the latter alone or in combination with the IDO1 inhibitor 1-MT). Two weeks later, in vivo IGRP-specific immune reactivity was assayed. As expected, the priming ability of immunostimulatory cDCs was not affected by the presence of untreated or IFN-γ-stimulated NOD pDCs. However, co-administration of BTZ-treated pDCs ablated the IGRP-specific reactivity elicited by cDCs. The suppressive effect of BTZ-conditioned pDCs was, however, lost when 1-MT treatment was applied only in concurrence with BTZ exposure (Figure 1C). These data clearly show that BTZ—unlike IFN-γ—regulates the quantitative and qualitative expression of IDO1 protein in prediabetic NOD pDCs, restoring their tolerogenic potential in vivo. In addition to the signaling defect previously described (9), post-transcriptional anomalies were found to contribute to the failure of IFN-γ to enhance IDO1 expression in NOD mice.
BTZ-Conditioned NOD pDCs Favor the Differentiation of Regulatory Rather Than Effector T Cells In Vitro
Next, we investigated the ability of BTZ-conditioned NOD pDCs to affect T-cell differentiation and/or function when cocultured with CD4+ T cells. In the pDC-CD4+ T-cell cocultures, BTZ-pretreated pDCs significantly decreased IL-6, IFN-γ (as T helper 1 marker), and IL-17 (as T helper 17 marker) in the coculture supernatant, and IL-10 but not TGF-β was instead increased (Figure 2A). An analogous cytokine profile was observed after BTZ conditioning of the pDCs alone (Figure S2 in Supplementary Material). Interestingly, the co-presence of 1-MT at the time of pDCs exposure to BTZ selectively negated the drug effect on the pro-inflammatory cytokine (i.e., IL-6 and IFN-γ) secretion profile (Figure 2A), suggesting that IDO1 upregulation by BTZ could play a role in controlling those cytokines. Cytofluorimetric analysis conducted at the end of the coculture showed that BTZ-pretreated pDCs induced a twofold increase in the percentage of CD4+Foxp3+ T cells, an effect negated by 1–MT, the IDO1 inhibitor (Figures 2B,C).
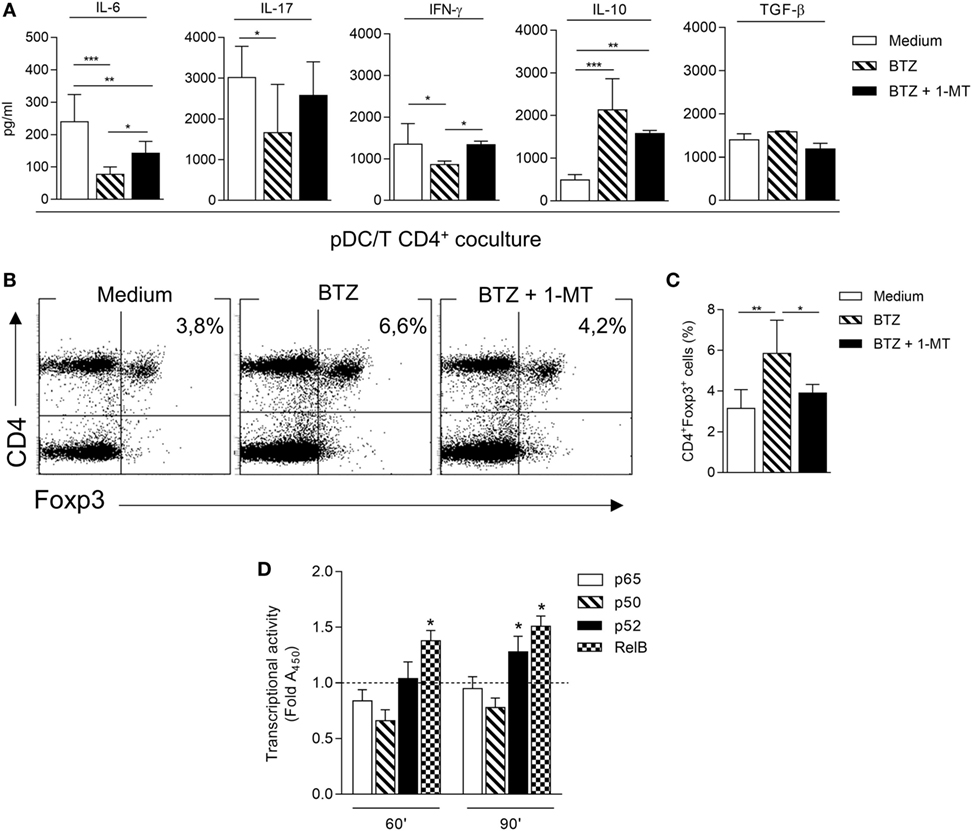
Figure 2. Bortezomib (BTZ) conditions inflammatory-prone non-obese diabetic plasmacytoid DCs (pDCs) toward immunoregulatory signals. (A) Secretion of IL-6, IL-17, interferon (IFN)-γ, IL-10, and TGF-β in supernatant of pDC-CD4+ T cell cocultures, where pDCs were pretreated with BTZ alone or in combination with 1-MT, or left untreated (medium) for 24 h. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA and Dunnett’s multiple comparison test). Results in panel (A) are representative of three different experiments (mean ± SD). (B) Representative flow cytometric dot plots illustrating the frequency of CD4+Foxp3+ cells among CD4+CD25− T cells cultured for 4 days together with pDCs treated as in panel (A). (C) Percentage of CD4+Foxp3+ cells in a CD4+CD25− T cells population cultured for 4 days together with pDCs treated as in panel (A). *p < 0.05; **p < 0.01 (one-way ANOVA followed by Bonferroni multiple comparison test). Results are representative of three different experiments (mean ± SD). (D) ELISA detection of p65, p50, p52, and RelB subunits of NF-κB in nuclear extracts of pDCs untreated (dotted line) or treated with BTZ for 60 and 90 min. Results are presented as fold change of the absorbance at 450 nm (Fold A450). *p < 0.05 (paired Student’s t-test; treated vs. untreated samples). Data are presented as means ± SD from triplicate samples.
In immune cells, BTZ has been shown to downregulate the production of several pro-inflammatory cytokines, presumably via inhibition of NF-κB activation (25, 26). Moreover, prediabetic pDCs showed a higher expression of the pro-inflammatory NF-κB pathway compared to diabetic pDCs (Figure S1C in Supplementary Material). To evaluate any role for impaired NF-κB activation by BTZ in our setting, pDCs purified from NOD mice were incubated with 10 nM BTZ or medium alone and the activation of NF-κB was evaluated in nuclear cell lysates after 60 and 90 min. A significant reduced nuclear translocation of p50 and p65 canonical subunits could be observed after 60 min in BTZ-treated as compared to untreated NOD pDCs; at a later time (90 min) a significant nuclear translocation of the non-canonical subunits p52 and RelB could be observed (Figure 2D). The presence of BTZ in the cell culture significantly modified the transcriptional activity of NF-κB, shifting the balance toward the non-canonical pathway, which we have previously shown as being involved in the transcription of Ido1 (6, 7).
Overall, these data suggested that the BTZ exposure of inflammation-prone pDCs—such as those from NOD mice (27, 28)—would tip the balance between inflammatory and immunoregulatory signals in favor of the latter, via upregulation of the IDO1 mechanism and also via correction of a dysfunctional NF-κB activity.
BTZ Prevents Diabetes Development in NOD Mice
Because IDO1 has been shown to play a major role in the protection from the experimental autoimmune diabetes (10, 24, 29, 30), we performed in vivo experiments to ascertain whether BTZ could exert protective effects in NOD mice. Groups of female NOD mice in prediabetes were thus administered BTZ i.p. at 0.10 or 0.25 mg/kg every other day for 2 weeks, a well-tolerated regimen, and glycemia was regularly measured. Over time, approximately 80% of control mice, and mice given the lower BTZ dose, developed overt diabetes, with glycemia values exceeding 250 mg/dl. By contrast, only 20% of mice administered BTZ at 0.25 mg/kg became hyperglycemic (Figure 3A), and glycemia values of protected mice remained in the range of 120–170 mg/dl over time (Figure 3B). In mice administered 0.25 mg/kg BTZ, histology of the pancreata revealed the presence of preserved islets, without invasive insulitis or with minimum or no peri-insulitis. The great majority of islets of the control and mice receiving the lower BTZ dose was characterized by immune cell infiltration (Figures 3C,D).
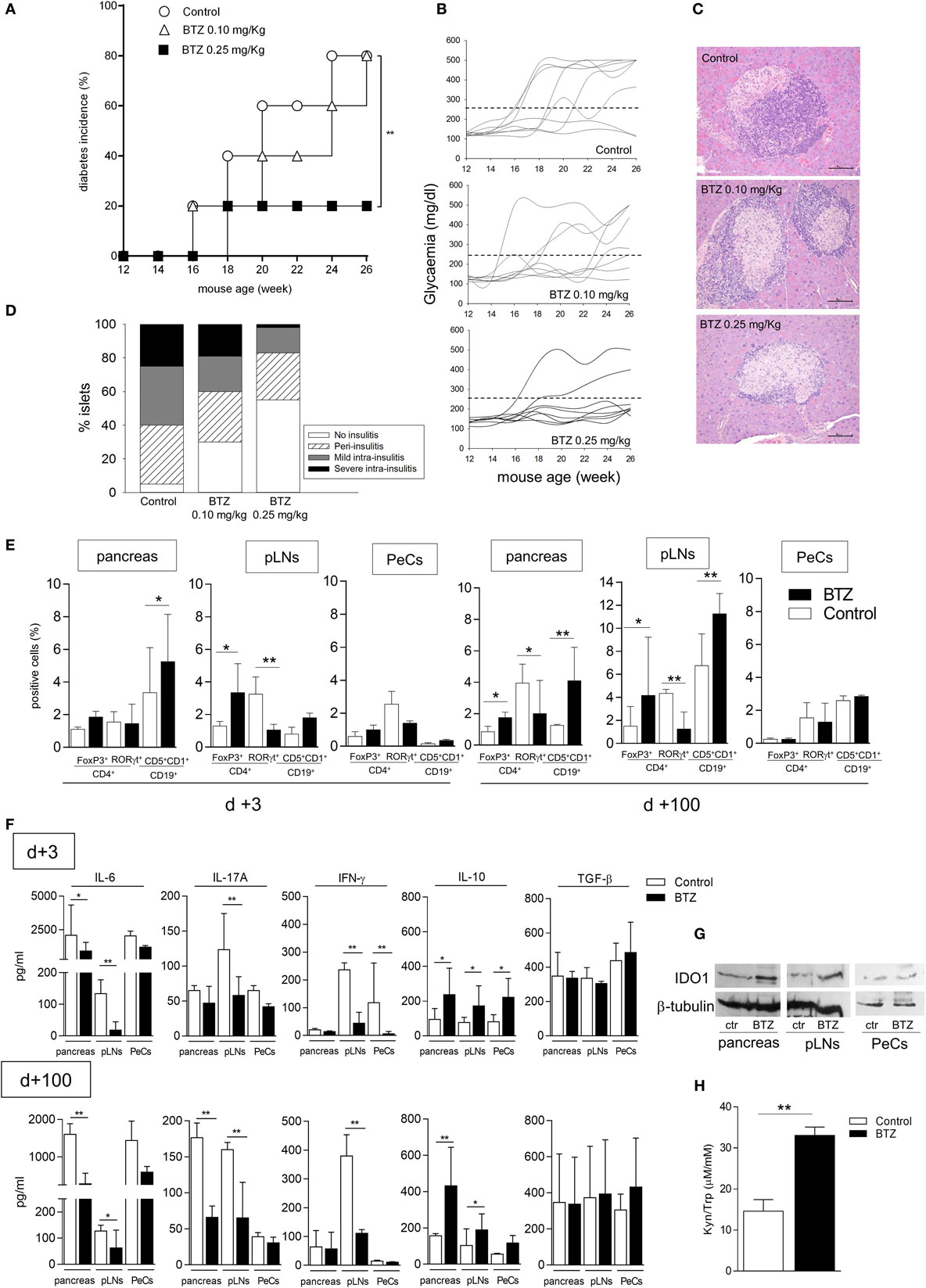
Figure 3. Administration of bortezomib (BTZ) prevents the development of autoimmune diabetes in non-obese diabetic (NOD) mice. (A) Prediabetic female NOD mice (n = 8) were intraperitoneally administered with vehicle alone (control; white circles), BTZ at 0.10 mg/kg (white triangles) or 0.25 mg/kg (black squares), and blood glucose was monitored over time. Diabetes was diagnosed in mice with blood glucose level ≥250 mg/dl and results are reported as diabetes incidence. **p < 0.01 (log-rank Mantel–Cox test). (B) Single blood glucose concentrations of NOD mice treated as in panel (A). (C) Pancreatic histology of mice treated as in panel (A) (scale bar, 100 µm; representative islet area for each group are shown). (D) Islets were scored to record percentages of islets of a given score over total number of islets (20–30/pancreas). One experiment is depicted of three with similar results. (E) Cytofluorimetric analysis of the frequency of Foxp3+ and RORγt+ cells among CD4+ T cell population and CD19+CD5+CD1d+ cells in pancreas, pancreatic lymph nodes (pLNs) and peritoneal cells (PeCs) from prediabetic female NOD mice treated with BTZ at 0.25 mg/kg or vehicle alone (control) and sacrificed at day +3 or day +100 after the last drug treatment. *p < 0.05 and **p < 0.01 (unpaired Student’s t-test, BTZ-treated vs. control mice). (F) Detection of IL-6, IL-17A, IFN-γ, IL-10, and TGF-β in supernatant of unfractionated cells from pancreas, pLNs, and PeCs of mice treated as in panel (E) and cultured for 24 h. *p < 0.05 and **p < 0.01 (unpaired Student’s t-test, BTZ-treated vs. control mice for each cell type). (G) Indoleamine 2,3-dioxygenase 1 immunoblot analysis of cell lysates from pancreas, pLNs, and PeCs of mice treated as in panel (E). One representative experiment out of three is shown. (H) Kynurenine to tryptophan ratio (Kyn/Trp) measured in sera from mice treated as in panel (E). **p < 0.01 (unpaired Student’s t-test, BTZ-treated vs. control mice). Data are presented as means ± SD for each group (n = 8 per group).
In order to investigate the in vivo effects of BTZ on immune cells, additional groups of mice, receiving 0.25 mg/kg BTZ or vehicle treatment, were sacrificed at different times (day +3 and day +100 after the last drug administration) and used as source of pancreas, pLNs, and PeCs—peritoneum being the site of BTZ injection—to be analyzed for T and B cell populations, cytokine production, and IDO1 expression. Cytofluorimetric analyses showed that, at 3 and more so 100 days, the drug significantly increased percentages of CD4+ Foxp3+ (Treg) and CD19+ CD5+ CD1d+ (Breg) cells in pancreas and pLNs but not in PeCs. In parallel, a decrease in CD4+ RORγt+ T cells (i.e., Th17 subset) was found in pLNs and later on (day +100) in pancreata (Figure 3E). Cytokine analysis of the same sources cultured for 24 h revealed most evident changes in IL-6 production, which started to decrease as soon as on day +3 in the pancreas of BTZ-treated mice relative to controls and in pLNs as well, though later in time (day +100; Figure 3F). An early (day +3) IL–17A decrease was also found in cultured pLNs and it persisted longer (day +100), whereas, in the pancreas, IL-17A decreased only later in time (day +100). IFN-γ decreased early in pLNs and PeCs and the effect persisted in pLNs up to day +100. Release of IL-10, but not of TGF-β, was increased on day +3 in all supernatants from whichever source and it remained increased in the pancreas and pLNs on day +100 (Figure 3F). IDO1 protein expression appeared to be upregulated in pancreas and pLNs, but not in PeCs from mice administered BTZ (Figure 3G). Moreover, the Kyn/Trp ratio increased in sera from mice treated with BTZ (Figure 3H).
Thus, our in vivo data indicated that proteasomal inhibition is effective in preventing development of autoimmune diabetes in a significant percentage of mice and that the protective effect is accompanied by a decrease in pro-inflammatory effector (Th17) cells and cytokines (IL-6, IL-17A, and IFN-γ), as well as by an increase in regulatory (Treg and Breg) cells and anti-inflammatory IL-10. Moreover, the in vivo treatment with BTZ upregulated IDO1 protein in the pancreas and pLNs and increased the systemic Kyn/Trp ratio. Importantly, changes were most evident in the pancreas, in immune cells draining (pLNs), and in proximity of the pancreas (namely, peritoneum, which was also the injection site).
Prevention of Diabetes by BTZ Requires IDO1 and pDCs
Based on the ability of BTZ to rectify IDO1 expression and activity in pDCs in vitro, we evaluated whether the protective effect of the drug in vivo could be mediated by IDO1 and pDCs. To this purpose, mice were administered BTZ at 0.25 mg/kg as in Figure 3A, with or without 1-MT, the inhibitor of IDO1. Mice receiving vehicle alone were used as a control. IDO1 blockade completely abrogated the protective effect of BTZ (Figure 4A). Similarly, BTZ protection was lost in Ido1−/− NOD mice (Figure 4B). In vivo depletion of pDCs (23) occurring at the time of BTZ administration negated the protective effect of the proteasomal inhibitor against diabetes development (Figure 4C), whereas depletion of pDCs occurring before BTZ treatment would not ablate its anti-diabetic effect (data not shown). Of interest, administration of anti-PDCA1 in the absence of BTZ also significantly reduced the percentage of animals that developed diabetes over time (Figure 4D). Although difficult to explain at this time, the fact that pDC depletion per se exerted protective effects in untreated prediabetic NOD mice may confirm previous results indicating that pDCs can be pathogenic in NOD mice (31), and BTZ may help restoring an anti-inflammatory phenotype in NOD pDCs, so that depletion of pDCs during the treatment turns out to be detrimental as it abrogates the protective effect of BTZ.
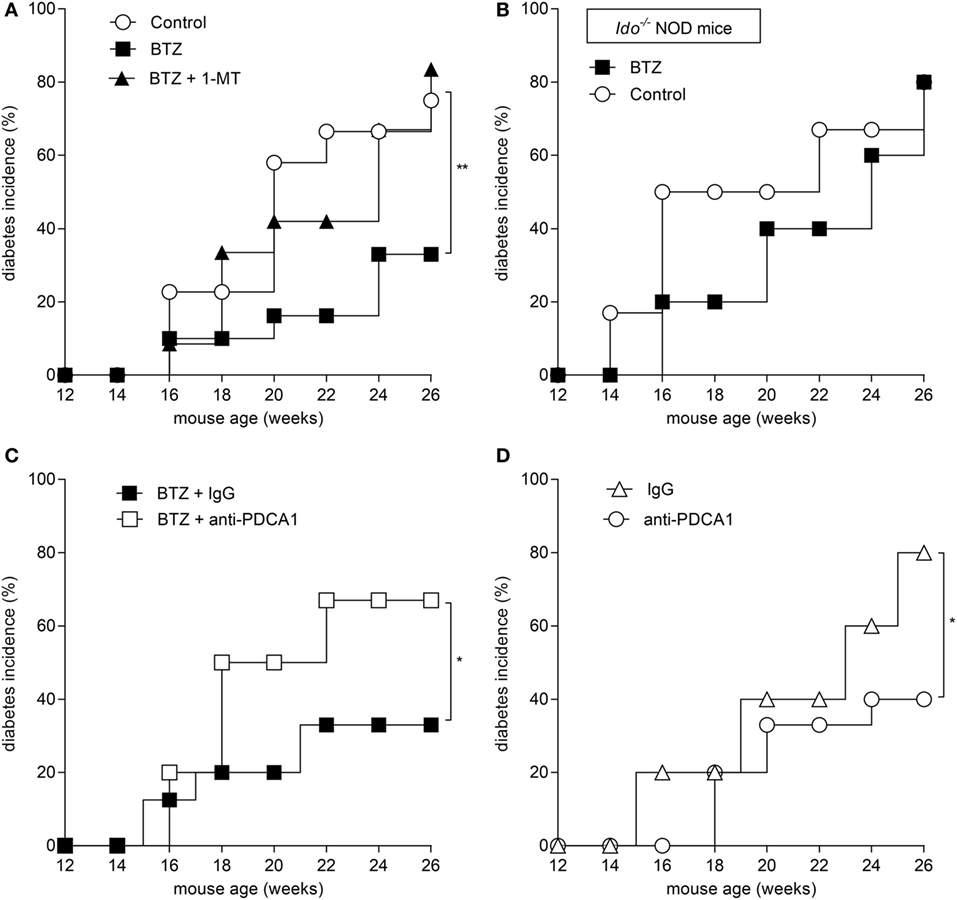
Figure 4. Prevention of autoimmune diabetes by bortezomib (BTZ) requires plasmacytoid DCs and indoleamine 2,3-dioxygenase 1 (IDO1)-mediated tryptophan catabolism. Diabetes was diagnosed in mice with blood glucose level ≥250 mg/dl and results are reported as diabetes incidence. (A) Prediabetic female non-obese diabetic (NOD) mice (n = 8) were intraperitoneally administered with vehicle (control; white circles), BTZ at 0.25 mg/kg alone (black squares) or in combination with 1-MT (black triangles), and blood glucose was monitored over time. (B) Prediabetic female Ido1−/− NOD mice (n = 8) were intraperitoneally administered with vehicle alone (control; white circles) or BTZ at 0.25 mg/kg (black squares), and blood glucose was monitored over time. (C) Prediabetic female NOD mice (n = 8) were intraperitoneally administered with BTZ at 0.25 mg/kg in combination with anti-PDCA1 depleting antibody (250 μg/mouse; white squares) or with the isotype control (rat IgG; black squares), and blood glucose was monitored over time. (D) Diabetic incidence of prediabetic NOD mice treated with anti-PDCA1 depleting antibody (white circles) or isotype rat IgG (white triangles). *p < 0.05 and **p < 0.01 (log-rank Mantel–Cox test).
Overall, our data indicated that proteasome inhibition is effective in the prevention of experimental autoimmune diabetes and that the effect requires pDCs and IDO1-mediated tryptophan catabolism.
A Combination of BTZ and Suboptimal Anti-CD3 Antibody Rescues Normoglycemia in Overtly Diabetic NOD Mice
Prompted by BTZ efficacy in preventing diabetes, we interrogated whether the drug would revert an ongoing disease. NOD female mice with hyperglycemia were i.p. administered BTZ at 0.1 or 0.25 mg/kg every other day for 2 weeks. Apart from a transient and not significant reduction in glycemia at 1 week post-initiation of drug treatment, no therapeutic effect was afforded by BTZ (Figure 5A).
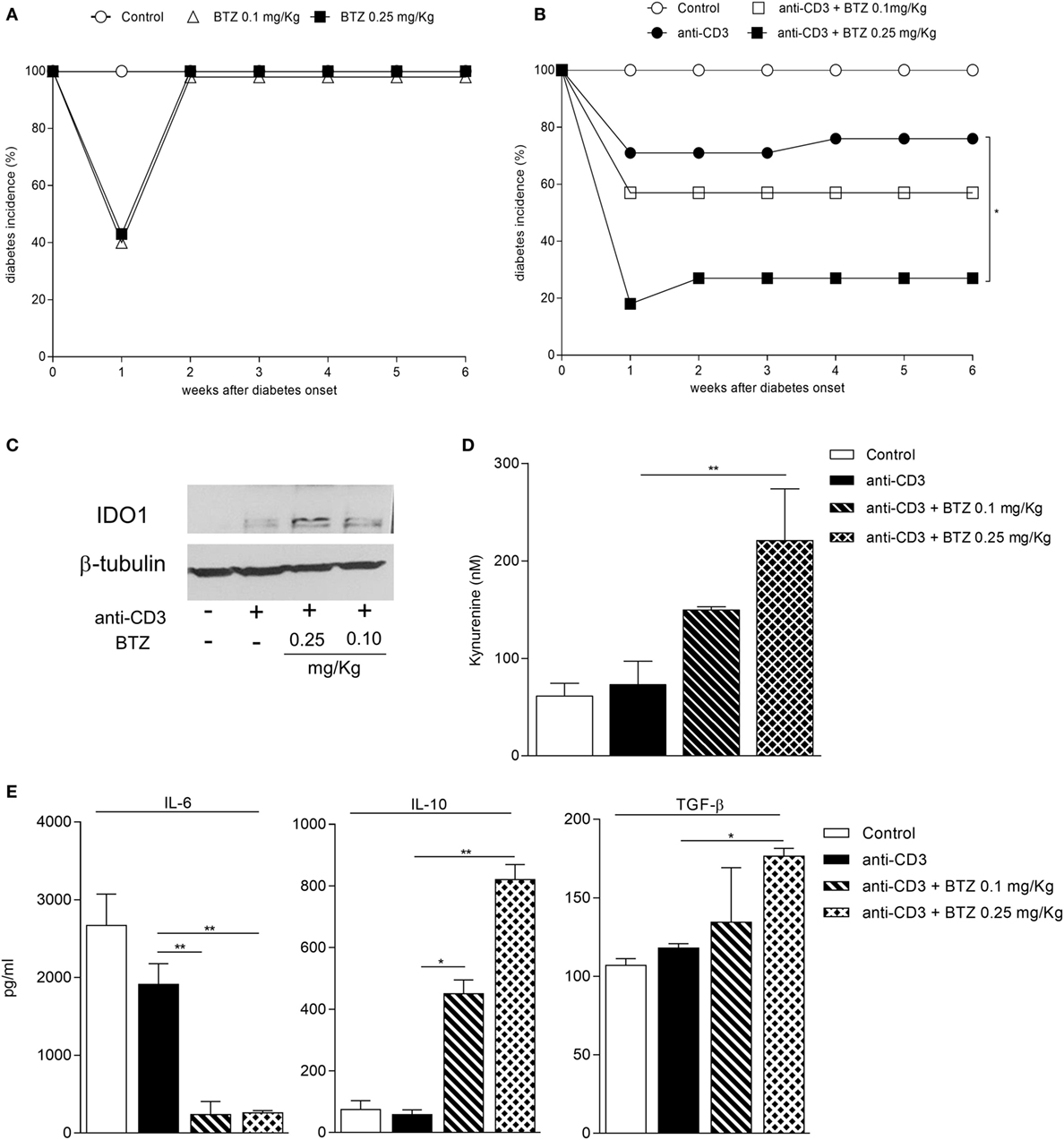
Figure 5. A combination of bortezomib (BTZ) and suboptimal anti-CD3 antibody restores normoglycemia in overtly diabetic mice. (A) Hyperglycemic female non-obese diabetic (NOD) mice (n = 8) were intraperitoneally administered with vehicle (white circles), BTZ at 0.1 mg/kg (white triangles) or 0.25 mg/kg (black squares). Blood glucose was monitored over time, and results are reported as diabetes incidence. (B) Hyperglycemic female NOD mice (n = 8) were administered with vehicle (white circles), with anti-CD3 alone (10 μg/mouse; black circles) or combined with BTZ at 0.1 mg/kg (white squares) or 0.25 mg/kg (black squares). Blood glucose was monitored over time and results are reported as diabetes incidence. *p < 0.05 (log-rank Mantel–Cox test). (C) Indoleamine 2,3-dioxygenase 1 (IDO1) immunoblot analysis of cell lysates from pancreatic lymph nodes (pLNs) of mice treated as in panel (B). One representative experiment out of three is shown. (D) IDO1 activity measured in terms of kynurenine production in supernatant of cells from pLNs of mice treated as in panel (B) and cultured for 24 h. **p < 0.01 (one-way ANOVA and Dunnett’s multiple comparison test). (E) Detection of IL-6, IL-10, and TGF-β in supernatant of cells from pLNs of mice treated as in panel (B) and cultured for 24 h. *p < 0.05 and **p < 0.01 (one-way ANOVA and Dunnett’s multiple comparison test).
Previous studies have demonstrated that administration of non-mitogenic anti-CD3 represents an effective maneuver capable of rescuing normoglycemia in approximately 80% diabetic NOD mice (22). Maximum therapeutic effects are achieved at the dose of 50 µg administered i.v. for five consecutive days, and they relied on the release of TGF-β by Treg cells (32).
We thus became interested in ascertaining whether a combination of BTZ and anti-CD3 would result in additive effects. Previous experiments (Chantal Kuhn and Lucienne Chatenoud, unpublished data) found that the suboptimal dose of 10 µg is capable of reverting hyperglycemia in a minority (less than 30%) of diabetic animals (Figure 5B). In our setting, a combination of BTZ at 0.25—but not 0.1—mg/kg and 10 µg anti-CD3 restored normoglycemia in 70% diabetic mice, a value significantly different from that of mice on suboptimal anti-CD3 alone (Figure 5B).
Groups of mice, treated as in Figure 5B, were sacrificed at the end of the experiment and used as a source of pLNs to be assayed for IDO1 expression and activity as well as for cytokine production. The combination of the two drugs greatly upregulated expression of IDO1 protein (Figure 5C) and activity (Figure 5D) in pLNs, compared to suboptimal anti-CD3 alone. Perhaps most importantly, at variance with the results in Figure 3F, the combined in vivo treatment with BTZ and anti-CD3—but not either treatment alone—resulted in a significant increase in the production of TGF-β by pLNs, and it significantly reduced IL-6 and increased IL-10 (Figure 5E).
Because we have previously demonstrated that TGF-β is a long-term inducer of IDO1 (6), our current data would further substantiate the importance of this cytokine in the effective control of a chronic autoimmune disease such as T1D, as previously reported (33). Moreover, the inhibition of IDO1 proteasomal degradation may represent a valuable therapeutic option to be considered in associative regimens with low-dose anti-CD3, so perhaps to reduce the inflammatory side effects observed in some patients on optimal-dose anti-CD3 treatment (34).
Discussion
Initially, the antiproliferative capacity of proteasome inhibitors has received considerable attention because of the success of their first prototypical representative, BTZ, in the treatment of B cell and plasma cell-related hematological malignancies. However, the emerging role of immunoproteasome involvement in autoantigen presentation as well as the ability of BTZ to inhibit the activation of NF-κB and suppress the release of pro-inflammatory cytokines, including IL-6, have increased interest in exploring the therapeutic potential of proteasome inhibitors in inflammatory and autoimmune disease settings (35). Selective inhibitors of the immunoproteasome and constitutive proteasome have recently been tested in models of autoimmune disease and allograft survival (17, 36). In experimental settings of inflammation and autoimmunity, BTZ does ameliorate disease severity, yet it has not been possible to identify a unitary mechanism of action, leading to the conclusion that the drug affects chronic inflammation and autoimmunity in a pleiotropic and/or model-specific fashion (37).
In the NOD mouse model system of autoimmune diabetes, we have previously demonstrated that impaired tryptophan catabolism contributes to defective tolerance to pancreatic antigens (9, 38). The condition results from a combination of functional anomalies, including poor signaling activity of IFN-γ and poor transcriptional activation of Ido1 (4), and increased proteasomal degradation of the IDO1 protein. The latter is largely sustained by high-level production of IL-6 (8, 39), which operates through SOCS3—a negative regulator of IDO1’s enzymatic and signaling functions (40)—which acts, in turn, to ubiquitinate and degrade IDO1. One major effect of the reduced tryptophan catabolic function by IDO1 could be the defective production of kynurenine-type ligands of the aryl hydrocarbon receptor (41), which is involved in the generation of regulatory T cells (42, 43).
Plasmacytoid DCs have an ambivalent role in the pathogenesis of autoimmune diabetes (44). The protective mechanisms are thought to involve inhibition of effector T cells, induction of regulatory T cells, and production of anti-inflammatory cytokines and IDO1. Although the exact mechanism of tolerance induction by pDCs in diabetes remains to be established, the intrinsic tolerogenic properties of pDCs provide a promising, yet perhaps underestimated, target for therapeutic intervention. Here, we found that BTZ treatment in vitro rescued IDO1 protein expression in prediabetic NOD pDCs, which were confirmed to manifest basally high transcriptional expression of Socs3, β-subunits of the immunoproteasome and the pro-inflammatory pathway of NF-κB. When administered in vivo to NOD mice, the BTZ-conditioned pDCs blocked the immunogenic presentation of a diabetogenic peptide, and BTZ itself would oppose diabetes onset in prediabetic mice. This was reflected in vitro by changes in the activation patterns of NF-κB family members and in cytokine secretion profiles—most notably, IL-6—and by generation of Foxp3+ regulatory T cells. Nevertheless, the inhibition of IDO1 by 1-MT during the in vitro conditioning of pDCs with BTZ would not consistently abrogate BTZ effects, suggesting the involvement of additional mechanisms beside the IDO1-mediated effect of BTZ. Mice protected from diabetes by BTZ in vivo also displayed increased percentages of Foxp3+CD4+ and IDO1 protein expression in both pLNs and pancreata, and an increased systemic Kyn/Trp ratio. The in vivo effect of BTZ required IDO1-mediated tryptophan catabolism and immune regulatory pDCs. Although pDC-depleted NOD mice in the absence of BTZ treatment showed a reduction in diabetes incidence, as previously reported (31), depletion of pDCs in BTZ-treated NOD mice would abrogate the protective effect of the drug. Unlike inflammatory-prone pDCs, which contribute to the pathogenesis of autoimmune diabetes, the immune regulatory properties of BTZ-conditioned pDCs are pivotal in the preventive effect of the drug in autoimmune diabetes. BTZ, however, showed no therapeutic activity when administered alone to overtly diabetic mice. Nevertheless, its combination with suboptimal dosages of autoimmune-preventive anti-CD3 antibody resulted in disease reversal in 70% diabetic mice, a therapeutic effect similar to that afforded by full-dosage anti-CD3 (22).
Clinical trials in type 1 diabetes showed that adequate dosing of anti-CD3 resulted in improved C-peptide responses and reduced exogenous insulin need (34, 45). Teplizumab, in particular, has been shown to preserve β-cell function in patients, failing, however, to represent a “cure” for patients (46). Side effects, including fever, anemia, urticarial rash, and a high incidence of anti-drug antibody development, occur frequently in patients treated with Teplizumab. For safety reasons, the dose of anti-CD3 used in the phase 3 trials was reduced as compared with that used in the pilot studies and was therefore probably insufficient (47).
Thus, effective therapies in humans that ensure efficacy while minimizing side effects remain a challenge. In addition, monotherapy with anti-CD3 also has its limits from the perspective of targeting only a single arm of the immune process. For this reason, several recent experimental studies have been exploiting synergy between anti-CD3 and various immunotherapeutic modalities (48). Our current results show that a combination of BTZ and low-dosage anti-CD3 results in a marked increase in the therapeutic efficacy of the antibody. We speculate that the synergistic effect may stem from a two-pronged approach to the mechanisms whereby NOD pDCs affect CD4+ T-cell differentiation toward a regulatory phenotype, by altering the cytokine milieu and the antigen presentation profile of the pDCs, via restoration of IDO1 functions, and by the non-lytic modulation of T cells by anti-CD3. Specifically, by reducing the proteolytic degradation of IDO1, BTZ might, at least in part, compensate for the defective Ido1 transcription by IFN-γ as well as for the IL-6/SOCS3-driven degradation of the IDO1 protein.
In conclusion, our current data point to a potential for BTZ in the management of autoimmune diabetes in humans and, at the same time, they provide mechanistic into how the drug acts to restore immune homeostasis in autoimmune settings via potentiation of the IDO1 tolerogenic mechanism.
Ethics Statement
All in vivo procedures were conducted in compliance with the Directive 2010/63/EU for animal experiments, according the protocols approved by the Animal Ethics Committee of the University of Perugia.
Author Contributions
GM, EA, MP, and MB performed in vitro experiments; CVacca and RB performed in vivo experiments; CK and LC performed in vivo experiments with anti-CD3; CVolpi, FF, and GR performed ex vivo analysis; DM performed the statistical analysis; SB performed microarray analysis; LB produced in vivo reagents; UG and PP wrote and reviewed the manuscript; CO researched data and reviewed/edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Italian Ministry of Education, Universities and Research (PRIN2012-2012S47X27 to CO), by the Foundation “Cassa di Risparmio di Perugia” (2015.0330.021 to CO), and by the European Research Council (338954-DIDO to UG).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00428/full#supplementary-material.
References
1. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol (2011) 29:163–83. doi: 10.1146/annurev-immunol-031210-101345
2. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol (2004) 5(12):1219–26. doi:10.1038/ni1141
3. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol (2008) 8(8):594–606. doi:10.1038/nri2358
4. Fallarino F, Bianchi R, Orabona C, Vacca C, Belladonna ML, Fioretti MC, et al. CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J Exp Med (2004) 200(8):1051–62. doi:10.1084/jem.20040942
5. Lippens C, Duraes FV, Dubrot J, Brighouse D, Lacroix M, Irla M, et al. IDO-orchestrated crosstalk between pDCs and Tregs inhibits autoimmunity. J Autoimmun (2016) 75:39–49. doi:10.1016/j.jaut.2016.07.004
6. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol (2011) 12(9):870–8. doi:10.1038/ni.2077
7. Pallotta MT, Orabona C, Bianchi R, Vacca C, Fallarino F, Belladonna ML, et al. Forced IDO1 expression in dendritic cells restores immunoregulatory signalling in autoimmune diabetes. J Cell Mol Med (2014) 18(10):2082–91. doi:10.1111/jcmm.12360
8. Orabona C, Pallotta MT, Volpi C, Fallarino F, Vacca C, Bianchi R, et al. SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc Natl Acad Sci U S A (2008) 105(52):20828–33. doi:10.1073/pnas.0810278105
9. Grohmann U, Fallarino F, Bianchi R, Orabona C, Vacca C, Fioretti MC, et al. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J Exp Med (2003) 198(1):153–60. doi:10.1084/jem.20030633
10. Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, et al. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J Immunol (2009) 183(10):6303–12. doi:10.4049/jimmunol.0901577
11. Campbell IL, Kay TW, Oxbrow L, Harrison LC. Essential role for interferon-gamma and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Wehi mice. J Clin Invest (1991) 87(2):739–42. doi:10.1172/JCI115055
12. Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M. The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol (2015) 33(4 Suppl 92):S74–9.
13. Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J Immunol (2010) 185(1):634–41. doi:10.4049/jimmunol.0903182
14. Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum (2012) 64(2):493–503. doi:10.1002/art.33333
15. Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med (2009) 15(7):781–7. doi:10.1038/nm.1978
16. Belogurov A Jr, Kuzina E, Kudriaeva A, Kononikhin A, Kovalchuk S, Surina Y, et al. Ubiquitin-independent proteosomal degradation of myelin basic protein contributes to development of neurodegenerative autoimmunity. FASEB J (2015) 29(5):1901–13. doi:10.1096/fj.14-259333
17. Sula Karreci E, Fan H, Uehara M, Mihali AB, Singh PK, Kurdi AT, et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc Natl Acad Sci U S A (2016) 113(52):E8425–32. doi:10.1073/pnas.1618548114
18. Fierabracci A. Proteasome inhibitors: a new perspective for treating autoimmune diseases. Curr Drug Targets (2012) 13(13):1665–75. doi:10.2174/138945012803530053
19. Wang Y, Liang Y, Zhang Y, Wu D, Liu H. Bortezomib inhibits bone marrow-derived dendritic cells. Int J Clin Exp Pathol (2015) 8(5):4857–62.
20. Albini E, Rosini V, Gargaro M, Mondanelli G, Belladonna ML, Pallotta MT, et al. Distinct roles of immunoreceptor tyrosine-based motifs in immunosuppressive indoleamine 2,3-dioxygenase 1. J Cell Mol Med (2017) 21:165–76. doi:10.1111/jcmm.12954
21. LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res (2002) 62(17):4996–5000.
22. Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol (1997) 158(6):2947–54.
23. Ioannou M, Alissafi T, Boon L, Boumpas D, Verginis P. In vivo ablation of plasmacytoid dendritic cells inhibits autoimmunity through expansion of myeloid-derived suppressor cells. J Immunol (2013) 190(6):2631–40. doi:10.4049/jimmunol.1201897
24. Fallarino F, Luca G, Calvitti M, Mancuso F, Nastruzzi C, Fioretti MC, et al. Therapy of experimental type 1 diabetes by isolated Sertoli cell xenografts alone. J Exp Med (2009) 206(11):2511–26. doi:10.1084/jem.20090134
25. Pellom ST Jr, Dudimah DF, Thounaojam MC, Sayers TJ, Shanker A. Modulatory effects of bortezomib on host immune cell functions. Immunotherapy (2015) 7(9):1011–22. doi:10.2217/imt.15.66
27. Wheat W, Kupfer R, Gutches DG, Rayat GR, Beilke J, Scheinman RI, et al. Increased NF-kappa B activity in B cells and bone marrow-derived dendritic cells from NOD mice. Eur J Immunol (2004) 34(5):1395–404. doi:10.1002/eji.200324490
28. Weaver DJ Jr, Poligone B, Bui T, Abdel-Motal UM, Baldwin AS Jr, Tisch R. Dendritic cells from nonobese diabetic mice exhibit a defect in NF-kappa B regulation due to a hyperactive I kappa B kinase. J Immunol (2001) 167(3):1461–8. doi:10.4049/jimmunol.167.3.1461
29. Alexander AM, Crawford M, Bertera S, Rudert WA, Takikawa O, Robbins PD, et al. Indoleamine 2,3-dioxygenase expression in transplanted NOD islets prolongs graft survival after adoptive transfer of diabetogenic splenocytes. Diabetes (2002) 51(2):356–65. doi:10.2337/diabetes.51.2.356
30. Ueno A, Cho S, Cheng L, Wang J, Hou S, Nakano H, et al. Transient upregulation of indoleamine 2,3-dioxygenase in dendritic cells by human chorionic gonadotropin downregulates autoimmune diabetes. Diabetes (2007) 56(6):1686–93. doi:10.2337/db06-1727
31. Allen JS, Pang K, Skowera A, Ellis R, Rackham C, Lozanoska-Ochser B, et al. Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes (2009) 58(1):138–45. doi:10.2337/db08-0964
32. Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med (2003) 9(9):1202–8. doi:10.1038/nm924
33. You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A (2007) 104(15):6335–40. doi:10.1073/pnas.0701171104
34. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352(25):2598–608. doi:10.1056/NEJMoa043980
35. Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD, Jansen G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res Ther (2015) 17:17. doi:10.1186/s13075-015-0529-1
36. Kisselev AF, Groettrup M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr Opin Chem Biol (2014) 23:16–22. doi:10.1016/j.cbpa.2014.08.012
37. Yannaki E, Papadopoulou A, Athanasiou E, Kaloyannidis P, Paraskeva A, Bougiouklis D, et al. The proteasome inhibitor bortezomib drastically affects inflammation and bone disease in adjuvant-induced arthritis in rats. Arthritis Rheum (2010) 62(11):3277–88. doi:10.1002/art.27690
38. Grohmann U, Fallarino F, Bianchi R, Vacca C, Orabona C, Belladonna ML, et al. Tryptophan catabolism in nonobese diabetic mice. Adv Exp Med Biol (2003) 527:47–54. doi:10.1007/978-1-4615-0135-0_5
39. Orabona C, Pallotta MT, Grohmann U. Different partners, opposite outcomes: a new perspective of the immunobiology of indoleamine 2,3-dioxygenase. Mol Med (2012) 18:834–42. doi:10.2119/molmed.2012.00029
40. Fallarino F, Grohmann U, Puccetti P. Indoleamine 2,3-dioxygenase: from catalyst to signaling function. Eur J Immunol (2012) 42(8):1932–7. doi:10.1002/eji.201242572
41. Grohmann U, Puccetti P. The coevolution of IDO1 and AhR in the emergence of regulatory T-cells in mammals. Front Immunol (2015) 6:58. doi:10.3389/fimmu.2015.00058
42. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature (2014) 511(7508):184–90. doi:10.1038/nature13323
43. Fallarino F, Romani L, Puccetti P. AhR: far more than an environmental sensor. Cell Cycle (2014) 13(17):2645–6. doi:10.4161/15384101.2014.954219
44. Morel PA. Dendritic cell subsets in type 1 diabetes: friend or foe? Front Immunol (2013) 4:415. doi:10.3389/fimmu.2013.00415
45. Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol (2013) 149(3):268–78. doi:10.1016/j.clim.2013.05.001
46. Vudattu NK, Herold KC. Treatment of new onset type 1 diabetes with teplizumab: successes and pitfalls in development. Expert Opin Biol Ther (2014) 14(3):377–85. doi:10.1517/14712598.2014.881797
47. Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr, et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet (2011) 378(9790):487–97. doi:10.1016/S0140-6736(11)60931-8
Keywords: tryptophan metabolism, indoleamine 2,3-dioxygenase 1 enzyme, dendritic cells, proteasome, autoimmune diabetes, immune regulation, anti-CD3 antibody
Citation: Mondanelli G, Albini E, Pallotta MT, Volpi C, Chatenoud L, Kuhn C, Fallarino F, Matino D, Belladonna ML, Bianchi R, Vacca C, Bicciato S, Boon L, Ricci G, Grohmann U, Puccetti P and Orabona C (2017) The Proteasome Inhibitor Bortezomib Controls Indoleamine 2,3-Dioxygenase 1 Breakdown and Restores Immune Regulation in Autoimmune Diabetes. Front. Immunol. 8:428. doi: 10.3389/fimmu.2017.00428
Received: 02 February 2017; Accepted: 27 March 2017;
Published: 13 April 2017
Edited by:
Duncan Howie, University of Oxford, UKReviewed by:
Penelope Anne Morel, University of Pittsburgh, USAPeter M. Van Endert, Institut national de la santé et de la recherche médicale (INSERM), France
Robert Hilbrands, Vrije Universiteit Brussel, Belgium
Maja Wallberg, University of Cambridge, UK
Copyright: © 2017 Mondanelli, Albini, Pallotta, Volpi, Chatenoud, Kuhn, Fallarino, Matino, Belladonna, Bianchi, Vacca, Bicciato, Boon, Ricci, Grohmann, Puccetti and Orabona. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ciriana Orabona, Y2lyaWFuYS5vcmFib25hQHVuaXBnLml0
†These authors have contributed equally to this work.