 Xiaohai Zhang
Xiaohai Zhang Nancy L. Reinsmoen
Nancy L. Reinsmoen- HLA and Immunogenetics Laboratory, Comprehensive Transplant Center, Cedars-Sinai Medical Center, Los Angeles, CA, USA
The presence of donor human leukocyte antigen (HLA)-specific antibodies has been shown to be associated with graft loss and decreased patient survival, but it is not uncommon that donor-specific HLA antibodies are absent in patients with biopsy-proven antibody-mediated rejection. In this review, we focus on the latest findings on antibodies against non-HLA antigens in kidney and heart transplantation. These non-HLA antigens include myosin, vimentin, Kα1 tubulin, collagen, and angiotensin II type 1 receptor. It is suggested that the detrimental effects of HLA antibodies and non-HLA antibodies synergize together to impact graft outcome. Injury of graft by HLA antibodies can cause the exposure of neo-antigens which in turn stimulate the production of antibodies against non-HLA antigens. On the other hand, the presence of non-HLA antibodies may increase the risk for a patient to develop HLA-specific antibodies. These findings indicate it is imperative to stratify the patient’s immunologic risk by assessing both HLA and non-HLA antibodies.
Introduction
Despite the advancement of improved immunosuppression regimens and optimized patient management, antibody-mediated rejection remains a major obstacle for long-term graft survival (1). Initial emphasis has been on the identification of human leukocyte antigen (HLA)-specific antibodies directed against the donor HLA class I and/or class II antigens. With the introduction of commercially available solid phase assay reagents, a greater understanding of the specificity, strength, and function of these antibodies has been made possible. Further, the wide acceptance of these reagents in the clinical transplant testing setting, along with proficiency testing programs has allowed for increased consistency in the definition of the antibodies detected. Correlating the antibody information obtained together with the clinical graft outcome has revealed that patients with antibodies against donor HLA are at a higher risk of antibody-mediated rejection and poorer graft outcome (2, 3). However, for many patients who have allograft dysfunction and show histological characteristics of antibody-mediated rejection on biopsy, no donor HLA-specific antibodies are detected (2, 4). In heart transplant, it has been shown that 40% of patients diagnosed with biopsy-proven antibody-mediated rejection did not show donor HLA-specific antibodies in the peripheral blood (5). These studies initiated investigation into non-HLA-specific antigens, many of which are expressed by the vascular endothelium and often revealed after stress or graft injury. Even though the detection of many of these non-HLA antibodies remains elusive, antibodies to the following non-HLA antigens have been shown to be associated with graft dysfunction or rejection: major histocompatibility complex class I chain-related gene A (MICA), angiotensin II type 1 receptor (AT1R), endothelin type A receptor (ETAR), heat shock protein, phospholipid, K-α-tubulin, vimentin, and endothelial cell antigens (6). Recently, commercial reagents have become available to define antibodies to AT1R, ETAR, MICA, and endothelial cell antigens. These results have been used in the clinical testing setting to investigate their impact on graft outcome. Studies of antibodies to other non-HLA antigens have been largely single center results. The results for anti-HLA and non-HLA-specific antibodies indicate an interplay between alloimmunity and autoimmunity, impacting graft outcome. This review focuses on the contribution on antibodies against these non-HLA antigens in kidney and heart transplantation. For lung transplant outcomes, please refer to the comprehensive review by Dr. Mohanakumar in this same issue.
Antibodies to MICA
Major histocompatibility complex class I chain-related gene A gene is localized in the HLA gene cluster. The MICA protein shares similar structure with HLA class I but does not associate with β2 microglobulin at the cell surface and cannot bind peptides. MICA can activate natural killer (NK) cells via interaction with activating immunoreceptor NKG2D (7). Of the non-HLA antigens listed above, MICA is highly polymorphic with around 100 alleles identified as of July 2016. Similar to HLA molecules, the recipient and donor may carry different MICA alleles. The recipient’s immune system may develop antibodies against the donor-specific MICA allele (8). It has been reported that 5–9% of renal recipients display MICA antibodies (9). The contribution of MICA antibody to pathogenesis of antibody-mediated rejection was first demonstrated in kidney transplantation (10) and was later found to be associated with rejection in pancreas and heart transplant (11, 12). Further, the patient with antibodies against donor-specific MICA is at higher risk of antibody-mediated rejection (5). The expression of MICA is not detectable on the quiescent endothelial cells which lie at the interface of the allograft and recipient blood and are directly targeted by immune response. The expression of MICA can be induced by stress and cytokines, such as TNF-α (13). The development of MICA antibodies may indicate an underlying inflammatory status which exists in these conditions.
Studies have demonstrated that expression of MICA in tumor cells leads to the activation of NK cells via MICA/NKG2D interaction, which in turn release cytotoxic proteins and INF-γ (7). Binding of MICA antibody on endothelial cells may block interaction between MICA and NKG2D, and thereby dampen NKG2D-mediated cytotoxicity. However, NK cells may still be activated mainly through Fc receptor-dependent cytotoxicity.
Antibodies against G Protein-Coupled Receptors (GPCRs)
AT1R and ETAR belong to the family of GPCRs which have seven transmembrane domains. Antibodies to the GPCRs, AT1R, and ETAR, may be relevant due to their endothelial cell surface expression and extracellular regions accessible to antibodies. Some of these antibodies, such as those to AT1R, have been shown to be involved in the pathophysiology of pregnancy preeclampsia and autoimmune diseases, including systemic sclerosis (14–16). There are several possible mechanisms relevant to explain how patients without an autoimmune disease may develop these autoantibodies. One plausible reason is the immune suppression or an underlying inflammatory process may break the self-tolerance. Also, a shearing process induced by mechanical circulatory systems or dialysis may cause proteins, such as von Willebrand factor, cleaved into smaller peptides (17). It is possible that the extracellular loop of AT1R may be clipped off the cell surface by shear stress and thereby exposing a neoantigen. The cell surface density of these GPCRs is influenced by polymorphisms and different mRNA processing mechanisms. The severity of injury by AT1R antibodies may also be influenced by the expression level of these different AT1R isotypes on the allograft. These antibodies may not only target the allograft but may also have a global effect. The impact of anti-AT1R antibodies in a clinical setting was first identified in a group of kidney-transplant recipients with malignant hypertension (18), suggesting that binding of AT1R antibodies, similar to the ligation of AT1R with angiotensin II, can also promote vasoconstriction, water intake, and sodium retention and increase blood pressure (19). Similar to HLA antibodies, AT1R antibodies may have detrimental effect on the graft survival. The presence of AT1R antibodies is associated with antibody-mediated rejection, but not cellular-mediated rejection in kidney transplant (20). In heart transplant, increased levels of AT1R have been shown to be associated with antibody-mediated rejection, cellular-mediated rejection, and early onset of microvasculopathy at 1 year posttransplant (21). In the same study, high levels of antibodies against another GPCR, ETAR, also has been reported to be associated with antibody or cell-mediated rejection. The associations observed with either antibody-mediated rejection or cellular rejection are dependent on the current pathological definition of these types of rejection in each organ group.
AT1R antibodies can synergize with HLA antibodies to predispose the graft to rejection. The presence of both strong binding AT1R antibody and HLA class II donor-specific antibodies has been found to be associated with accelerated rejection, hypertensive encephalopathy, and worse graft survival in kidney transplant (20, 22, 23). In the absence of donor-specific HLA or MICA antibodies, strong binding AT1R antibodies have been detected in patients with antibody-mediated rejection. Furthermore, transplant candidates with strong AT1R antibodies pretransplant are at a higher risk of early antibody-mediated rejection and long-term graft loss.
Activation of AT1R by its native ligand angiotensin II stimulates phospholipase C, production of cyclic adenosine monophosphate, and activation of mitogen-activated protein kinase, which promotes vasoconstriction and hypertension (24). Association between AT1R antibody and rejection was first reported in patients with malignant hypertension by Dragun and her colleagues. They found that AT1R antibodies bind to the extracellular loop 2 of AT1R and stimulate phosphorylation of extracellular signal-regulated kinase (ERK) (18). However, studies indicate that the angiotensin II binding pocket of AT1R is composed of all seven transmembrane helices and both extracellular loops 1 and 2 (25, 26), which is distinct from the antibody-binding site. It is not clear how AT1R antibody stimulates similar signals as native ligand angiotensin II. Several other studies have shown the association of AT1R antibodies with rejection, but none of these studies reported hypertension in their cohort or if these antibodies could activate signaling to stimulate hypertension (20, 21, 27–30). AT1R antibodies have been found to be the IgG1 and IgG3 subclasses which have the capacity to activate the complement cascade (18). Interestingly, detrimental effect of AT1R antibodies on the graft does not need complement activation. Reinsmoen et al. observed C4d-positive biopsy in only one out of six patients who had strong AT1R antibodies and were diagnosed as antibody-mediated rejection (20). In consistent with this observation, Fuss et al. reported that AT1R antibodies, but not donor-specific HLA antibodies, were detected in 11 cases of biopsy-proven, C4d-negative acute antibody-mediated rejection according to Banff 2013 (29). Nevertheless, AT1R antibodies may still be detrimental to the allograft through antibody-dependent cell-mediated cytotoxicity.
AT1R also plays an important role in glucose metabolism (31). It is suggested that higher expression of AT1R is associated with increased risk for diabetic nephropathy, and blockade of AT1R signaling is effective in the treatment of diabetic nephropathy (31, 32). AT1R antibody can also activate AT1R signaling. The presence of AT1R antibody in renal transplant candidate might predispose development of diabetic nephropathy. Further study on the role of AT1R antibody in diabetic nephropathy is warranted.
A commercially available test reagent provides detection of AT1R antibody in a relatively easier and standardized manner compared to other non-HLA antibodies. However, this assay is enzyme-linked immunosorbent assay based and is sensitive to interference by intravenous immunoglobulin and rituximab treatment. The linear range of results obtained from this assay is also rather narrow. Improvement of the AT1R-testing reagent will promote more transplant programs to adopt the AT1R antibody test.
Antibodies to Vimentin
Vimentin is a subunit of an intermediate filament. As a cytoskeletal element, vimentin is important for stabilizing the architecture of the cytoplasm. However, vimentin is also found to be secreted by macrophages, endothelial cells, vascular smooth muscle cells, activated platelets, apoptotic T cells, and neutrophils (33). Secretion is increased by the pro-inflammatory cytokine TNF-α, but inhibited by the anti-inflammatory cytokine IL10, suggesting that vimentin may be involved in the immune response (34). Antibodies against citrullinated vimentin have been detected in sera of patients with rheumatoid arthritis (35). Vimentin antibodies also exist in pretransplant sera of patients with end-stage kidney disease (36). In this study, titers of IgM antibodies against vimentin increased every year posttransplant compared with pretransplant titers, but no difference was found in patients with interstitial fibrosis and tubular atrophy compared with a kidney recipient control group. By contrast, patients diagnosed with interstitial fibrosis and tubular atrophy have higher levels of IgG antibodies against vimentin. These results suggest that IgG, but not IgM, antibodies against vimentin may play a role in the pathology of interstitial fibrosis and tubular atrophy (37). Similarly, in heart transplant, patients with vimentin antibodies within the first 2 years of transplantation are at higher risk for cardiac allograft vasculopathy at 5 years posttransplant (38). The presence of vimentin antibodies is rather common in heart transplant recipients. Young et al. have shown 34% of patients display vimentin antibodies, but no difference in the rates of early rejection and graft survival was observed (39).
Anti-Myosin Antibodies
Myosins are a large family of proteins which bind actin cytoskeleton and move cargo proteins through ATP hydrolysis. The thymus of mice and humans does not express myosin heavy chain proteins, thus CD4+ T cells are not negatively selected for myosin in the maturation process (40). The autoantibodies frequently associated with autoimmune myocarditis may be the result of this mechanism (40, 41). The presence of myosin antibodies has been associated with antibody-mediated rejection and development of chronic allograft vasculopathy in heart transplantation (42, 43). Three hundred single-nucleotide polymorphisms have been identified in the myosin motor domain of cardiac myosin (44), but it is not yet known whether myosin antibodies detected in the patients are donor specific. The impact of cardiac myosin antibodies on heart transplantation was confirmed by a swine minor antigen-mismatch model, in which animals with strongly binding myosin antibodies after immunization of myosin pretransplant rejected grafts acutely, while animals with low and transient binding myosin antibodies had long-term allograft survival (45). The presence of donor HLA-specific antibodies in patients with antibody-mediated rejection and cardiac allograft vasculopathy precedes the detection of myosin and vimentin antibodies (43), thus again suggesting the interplay of allo- and autoimmune responses.
Anti-Perlecan Antibodies
Perlecan is a critical component of the endothelial basement membrane and serves as a barrier between the circulating blood and the surrounding tissue (46). Perlecan is a large, extracellular matrix proteoglycan with many functions. Perlecan can act as a coreceptor for fibroblast growth factor 2 to stimulate cell proliferation as demonstrated in a rat transplant model (47). Injection of LG3, a C-terminal fragment of perlecan, increases cell migration and accumulation of recipient-derived α smooth muscle actin-positive cells in neointima in a MHC-mismatched allogeneic aortic segment transplant mouse model (48). Elevated levels of LG3 have been found in the sera of kidney-transplant recipients with acute vascular rejection (49). Studies have shown that vascular injury prompts release of apoptotic exosome-like vesicles which trigger the production of antibodies to LG3 (50). Pre or posttransplant, higher levels of LG3 antibodies have been found to be associated with acute vascular rejection in kidney-transplant recipients (51). Anti-perlecan antibodies have been detected in the sera of transplant patients with glomerulopathy (52). Pretransplant LG3 antibodies have also been shown to be associated with an increased risk of delayed graft function. Passive transfer of LG3 antibodies can cause microvascular injury in a kidney ischemia-reperfusion injury mouse model (53).
Other Non-HLA Antibodies
The endothelium lines the interface between the graft and recipient tissue and antigens expressed by these cells are the first line targets of the recipient’s immune system. Antibodies against four non-HLA endothelial antigens: endoglin, Fms-like tyrosine kinase-3 ligand (FLT3), EGF-like repeats and discoidin I-like domains 3 (EDIL3), and intercellular adhesion molecule 4 (ICAM4) have been identified in the sera of renal transplant patients. Endoglin is a membrane glycoprotein primarily expressed on vascular endothelium. It regulates angiogenesis and revascularization (54). FLT3 is a receptor tyrosine kinase regulating cell differentiation, survival, and proliferation (55). It is suggested that activation of FLT3 signaling promotes angiogenesis of multiple myeloma (56). EDIL3 is secreted by endothelial cells and associates with extracellular matrix. The expression of EIL3 inhibits leukocyte–endothelial adhesion (57). ICAM4, also known as the Landsteiner–Wiener blood group antigen, is a single-spanning transmembrane protein. ICAM4 mediates binding of leukocytes via its interaction with integrin (58). The presence of these antibodies is associated with the production of posttransplant donor-specific HLA antibodies, antibody-mediated rejection, and early transplant glomerulopathay (59). In heart transplantation, the presence of anti-endothelial cell antibodies is also associated with increased risk of early acute rejection. Nevertheless, long-term outcome and patient survival are not affected by these antibodies (60).
Renal disease can also result from autoimmune injury, such as systemic lupus erythematosus. Anti DNA/histone antibodies found in the kidney with lupus nephritis can promote pathogenesis by stimulating cytokine production and inflammation (61, 62). Since DNA/histone are only exposed after cell breakdown as happened in cell apoptosis or necrosis, these antibodies are unlikely to be the initiating autoantibodies in lupus nephritis. However, the presence of pretransplant antibodies to apoptotic cells has been shown to correlate with allograft loss in kidney transplantation (63). Mycophenolate mofetil plus corticosteroids which is commonly used in organ transplantation is the standard-of-care treatment for lupus nephritis (64).
Conclusion
Vigorous efforts to investigate non-HLA antibodies, their impact on graft outcome, and influence on the alloimmune and autoimmune processes are still ongoing. With the availability of commercial reagents, the study of several of these antibodies to non-HLA antigens has moved to the clinical setting. As more studies populate the literature, there are common themes that appear consistently. The antibodies to these non-HLA-specific antigens do appear to be part of the overall graft dysfunction response (Table 1). These non-HLA antibodies are often checked when antibody-mediated rejection is suspected, but no donor-specific HLA antibody is detected. The presence of non-HLA antibodies may also be tested for patients who are deemed to have a high risk of antibody-mediated rejection. Commercial available Luminex bead-based reagents, which can provide detection of non-HLA antibodies in a standardized manner, may promote more transplant programs to evaluate non-HLA antibodies. Some antigens targeted by non-HLA antibodies are not polymorphic at the amino acid sequence. These antibodies may also bind the recipient’s tissue and have a global effect in the recipient as happened in patients with AT1R antibodies. It is unclear if other non-HLA antibodies also have a broader effect or if the effect is limited to the allograft because the graft expresses these antigens at the higher level. The presence of non-HLA antibodies may predispose patients to an increased risk to develop donor-specific HLA antibodies and rejection. Whether the antibodies to non-HLA antigens appear before the donor HLA-specific antibodies or the reverse is unknown at this time. The presence of these non-HLA antibodies may indicate underlying immune status. Lupus patients who usually display autoantibodies have higher level pro-inflammatory cytokines, which may promote immune response to allo-antigens. Autoantibodies may also cause allograft injury through complement-dependent cytotoxicity or antibody-dependent cell-mediated cytotoxicity. Donor HLA antigens released from injured cells may be captured by the recipient dendritic cells and trigger donor-specific HLA antibody production. Alternately, tissue injury caused by donor-specific HLA antibodies may expose neoantigens which lead to auto-antibody production. Certainly each scenario may be part of different overall immune responses in different organs, with different immunosuppressive regimes, and in different genetically disparate settings. The identity of some non-HLA antigens targeted by these antibodies remains elusive. Nonetheless, a continuing theme of these studies is that both alloimmune and autoimmune mechanisms impact graft outcome and may be synergistic to each other.
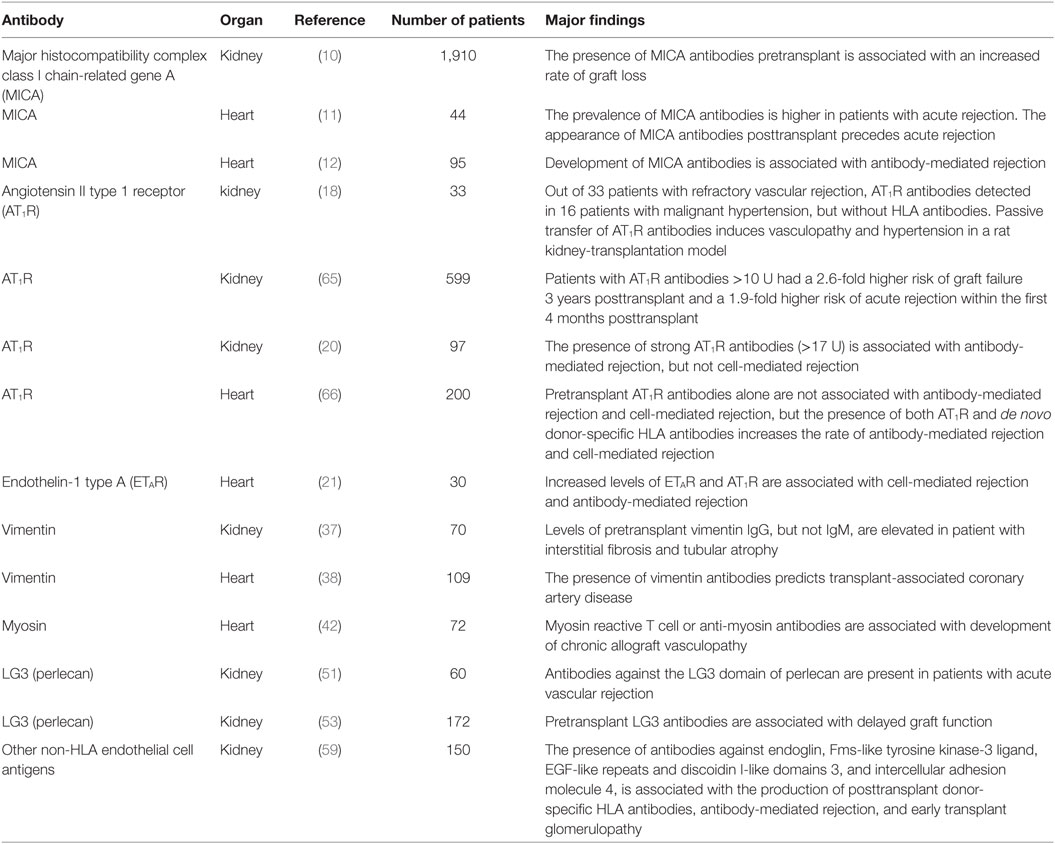
Table 1. Non-human leukocyte antigen (HLA) antibodies in solid organ transplantation.
Author Contributions
XZ and NLR participated in literature review and writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, DW, and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Abbreviations
HLA, human leukocyte antigen; MICA, major histocompatibility complex class I chain-related gene A; AT1R, angiotensin II type 1 receptor; ETAR, endothelin type A receptor; NK, natural killer; GPCR, G protein-coupled receptor; SNPs, single-nucleotide polymorphisms; PLC, phospholipase C; cAMP, cyclic adenosine monophosphate; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; FLT3, Fms-like tyrosine kinase-3 ligand; EDIL3, EGF-215-like repeats and discoidin I-like domains 3; ICAM4, intercellular adhesion molecule 4.
References
1. Singh N, Pirsch J, Samaniego M. Antibody-mediated rejection: treatment alternatives and outcomes. Transplant Rev (Orlando) (2009) 23(1):34–46. doi: 10.1016/j.trre.2008.08.004
2. Clerkin KJ, Farr MA, Restaino SW, Zorn E, Latif F, Vasilescu ER, et al. Donor-specific anti-HLA antibodies with antibody-mediated rejection and long-term outcomes following heart transplantation. J Heart Lung Transplant (2016). doi:10.1016/j.healun.2016.10.016
3. Kauke T, Oberhauser C, Lin V, Coenen M, Fischereder M, Dick A, et al. De novo donorspecific anti-HLA antibodies after kidney transplantation are associated with impaired graft outcome independently of their C1q-binding ability. Transpl Int (2016) 30(4):360–70. doi:10.1111/tri.12887
4. Michielsen LA, van Zuilen AD, Krebber MM, Verhaar MC, Otten HG. Clinical value of non-HLA antibodies in kidney transplantation: still an enigma? Transplant Rev (Orlando) (2016) 30(4):195–202. doi:10.1016/j.trre.2016.06.001
5. Zhang Q, Cecka JM, Gjertson DW, Ge P, Rose ML, Patel JK, et al. HLA and MICA: targets of antibody-mediated rejection in heart transplantation. Transplantation (2011) 91(10):1153–8. doi:10.1097/TP.0b013e3182157d60
6. Zhang X, Reed EF. Effect of antibodies on endothelium. Am J Transplant (2009) 9(11):2459–65. doi:10.1111/j.1600-6143.2009.02819.x
7. Carapito R, Bahram S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol Rev (2015) 267(1):88–116. doi:10.1111/imr.12328
8. Tonnerre P, Gerard N, Chatelais M, Poli C, Allard S, Cury S, et al. MICA variant promotes allosensitization after kidney transplantation. J Am Soc Nephrol (2013) 24(6):954–66. doi:10.1681/ASN.2012080814
9. Terasaki PI, Ozawa M, Castro R. Four-year follow-up of a prospective trial of HLA and MICA antibodies on kidney graft survival. Am J Transplant (2007) 7(2):408–15. doi:10.1111/j.1600-6143.2006.01644.x
10. Zou Y, Stastny P, Susal C, Dohler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med (2007) 357(13):1293–300. doi:10.1056/NEJMoa067160
11. Suarez-Alvarez B, Lopez-Vazquez A, Gonzalez MZ, Fdez-Morera JL, Diaz-Molina B, Blanco-Gelaz MA, et al. The relationship of anti-MICA antibodies and MICA expression with heart allograft rejection. Am J Transplant (2007) 7(7):1842–8. doi:10.1111/j.1600-6143.2007.01838.x
12. Nath DS, Angaswamy N, Basha HI, Phelan D, Moazami N, Ewald GA, et al. Donor-specific antibodies to human leukocyte antigens are associated with and precede antibodies to major histocompatibility complex class I-related chain A in antibody-mediated rejection and cardiac allograft vasculopathy after human cardiac transplantation. Hum Immunol (2010) 71(12):1191–6. doi:10.1016/j.humimm.2010.09.012
13. Lin D, Lavender H, Soilleux EJ, O’Callaghan CA. NF-kappaB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J Biol Chem (2012) 287(6):4299–310. doi:10.1074/jbc.M111.282152
14. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest (1999) 103(7):945–52. doi:10.1172/JCI4106
15. Kill A, Tabeling C, Undeutsch R, Kuhl AA, Gunther J, Radic M, et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res Ther (2014) 16(1):R29. doi:10.1186/ar4457
16. Gunther J, Kill A, Becker MO, Heidecke H, Rademacher J, Siegert E, et al. Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res Ther (2014) 16(2):R65. doi:10.1186/ar4503
17. Nascimbene A, Neelamegham S, Frazier OH, Moake JL, Dong JF. Acquired von Willebrand syndrome associated with left ventricular assist device. Blood (2016) 127(25):3133–41. doi:10.1182/blood-2015-10-636480
18. Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med (2005) 352(6):558–69. doi:10.1056/NEJMoa035717
19. Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol (2016) 310(2):H137–52. doi:10.1152/ajpheart.00618.2015
20. Reinsmoen NL, Lai CH, Heidecke H, Haas M, Cao K, Ong G, et al. Anti-angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation (2010) 90(12):1473–7. doi:10.1097/TP.0b013e3181fd97f1
21. Hiemann NE, Meyer R, Wellnhofer E, Schoenemann C, Heidecke H, Lachmann N, et al. Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplantation (2012) 94(9):919–24. doi:10.1097/TP.0b013e3182692ad2
22. Kelsch R, Everding AS, Kuwertz-Broking E, Brand E, Spriewald BM, Sibrowski W, et al. Accelerated kidney transplant rejection and hypertensive encephalopathy in a pediatric patient associated with antibodies against angiotensin type 1 receptor and HLA class II. Transplantation (2011) 92(10):e57–9. doi:10.1097/TP.0b013e318234b337
23. Amico P, Honger G, Bielmann D, Lutz D, Garzoni D, Steiger J, et al. Incidence and prediction of early antibody-mediated rejection due to non-human leukocyte antigen-antibodies. Transplantation (2008) 85(11):1557–63. doi:10.1097/TP.0b013e31816f612a
24. Vinturache AE, Smith FG. Angiotensin type 1 and type 2 receptors during ontogeny: cardiovascular and renal effects. Vascul Pharmacol (2014) 63(3):145–54. doi:10.1016/j.vph.2014.11.001
25. Zhang H, Unal H, Desnoyer R, Han GW, Patel N, Katritch V, et al. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J Biol Chem (2015) 290(49):29127–39. doi:10.1074/jbc.M115.689000
26. Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, et al. Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell (2015) 161(4):833–44. doi:10.1016/j.cell.2015.04.011
27. Guzzo I, Morolli F, Camassei FD, Piazza A, Poggi E, Dello Strologo L. Acute kidney transplant rejection mediated by angiotensin II type 1 receptor antibodies in a pediatric hyperimmune patient. Pediatr Nephrol (2017) 32(1):185–8. doi:10.1007/s00467-016-3500-8
28. Lee J, Huh KH, Park Y, Park BG, Yang J, Jeong JC, et al. The clinicopathological relevance of pretransplant anti-angiotensin II type 1 receptor antibodies in renal transplantation. Nephrol Dial Transplant (2015). doi:10.1093/ndt/gfv375
29. Fuss A, Hope CM, Deayton S, Bennett GD, Holdsworth R, Carroll RP, et al. C4d-negative antibody-mediated rejection with high anti-angiotensin II type I receptor antibodies in absence of donor-specific antibodies. Nephrology (Carlton) (2015) 20(7):467–73. doi:10.1111/nep.12441
30. Philogene MC, Bagnasco S, Kraus ES, Montgomery RA, Dragun D, Leffell MS, et al. Anti-angiotensin II type 1 receptor and anti-endothelial cell antibodies: a cross-sectional analysis of pathological findings in allograft biopsies. Transplantation (2017) 101(3):608–15. doi:10.1097/TP.0000000000001231
31. Rahimi Z. The role of renin angiotensin aldosterone system genes in diabetic nephropathy. Can J Diabetes (2016) 40(2):178–83. doi:10.1016/j.jcjd.2015.08.016
32. Bichu P, Nistala R, Khan A, Sowers JR, Whaley-Connell A. Angiotensin receptor blockers for the reduction of proteinuria in diabetic patients with overt nephropathy: results from the AMADEO study. Vasc Health Risk Manag (2009) 5(1):129–40. doi:10.2147/VHRM.S3121
33. Rose ML. Role of anti-vimentin antibodies in allograft rejection. Hum Immunol (2013) 74(11):1459–62. doi:10.1016/j.humimm.2013.06.006
34. Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol (2003) 5(1):59–63. doi:10.1038/ncb898
35. Van Steendam K, Tilleman K, Deforce D. The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) (2011) 50(5):830–7. doi:10.1093/rheumatology/keq419
36. Liebscher F, Arnold T, Liang YY, Reiter T, Bohmig G, Oehler R. Vimentin cleavage in end-stage renal disease is not related to apoptosis. Cent Eur J Med (2013) 8(3):297–301. doi:10.2478/s11536-012-0131-8
37. Besarani D, Cerundolo L, Smith JD, Procter J, Barnardo MCN, Roberts ISD, et al. Role of anti-vimentin antibodies in renal transplantation. Transplantation (2014) 98(1):72–8. doi:10.1097/01.TP.0000443224.66960.37
38. Jurcevic S, Ainsworth ME, Pomerance A, Smith JD, Robinson DR, Dunn MJ, et al. Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation (2001) 71(7):886–92. doi:10.1097/00007890-200104150-00011
39. Young RK, Dale B, Russell SD, Zachary AA, Tedford RJ. Incidence and early outcomes associated with pre-transplant antivimentin antibodies in the cardiac transplantation population. Clin Transplant (2015) 29(8):685–8. doi:10.1111/ctr.12567
40. Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest (2011) 121(4):1561–73. doi:10.1172/JCI44583
41. Lichtman AH. The heart of the matter: protection of the myocardium from T cells. J Autoimmun (2013) 45:90–6. doi:10.1016/j.jaut.2013.05.004
42. Kalache S, Dinavahi R, Pinney S, Mehrotra A, Cunningham MW, Heeger PS. Anticardiac myosin immunity and chronic allograft vasculopathy in heart transplant recipients. J Immunol (2011) 187(2):1023–30. doi:10.4049/jimmunol.1004195
43. Nath DS, Ilias Basha H, Tiriveedhi V, Alur C, Phelan D, Ewald GA, et al. Characterization of immune responses to cardiac self-antigens myosin and vimentin in human cardiac allograft recipients with antibody-mediated rejection and cardiac allograft vasculopathy. J Heart Lung Transplant (2010) 29(11):1277–85. doi:10.1016/j.healun.2010.05.025
44. Colegrave M, Peckham M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken) (2014) 297(9):1670–80. doi:10.1002/ar.22973
45. Veillette GR, Sahara H, Meltzer AJ, Weiss MJ, Iwamoto Y, Kim KM, et al. Autoimmune sensitization to cardiac myosin leads to acute rejection of cardiac allografts in miniature swine. Transplantation (2011) 91(11):1187–91. doi:10.1097/TP.0b013e318218415d
46. Farach-Carson MC, Warren CR, Harrington DA, Carson DD. Border patrol: insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol (2014) 34:64–79. doi:10.1016/j.matbio.2013.08.004
47. Katta K, Boersema M, Adepu S, Rienstra H, Celie JW, Mencke R, et al. Renal heparan sulfate proteoglycans modulate fibroblast growth factor 2 signaling in experimental chronic transplant dysfunction. Am J Pathol (2013) 183(5):1571–84. doi:10.1016/j.ajpath.2013.07.030
48. Pilon EA, Dieude M, Qi S, Hamelin K, Pomerleau L, Beillevaire D, et al. The perlecan fragment LG3 regulates homing of mesenchymal stem cells and neointima formation during vascular rejection. Am J Transplant (2015) 15(5):1205–18. doi:10.1111/ajt.13119
49. Soulez M, Pilon EA, Dieude M, Cardinal H, Brassard N, Qi S, et al. The perlecan fragment LG3 is a novel regulator of obliterative remodeling associated with allograft vascular rejection. Circ Res (2012) 110(1):94–104. doi:10.1161/CIRCRESAHA.111.250431
50. Dieude M, Bell C, Turgeon J, Beillevaire D, Pomerleau L, Yang B, et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Transl Med (2015) 7(318):318ra200. doi:10.1126/scitranslmed.aac9816
51. Cardinal H, Dieude M, Brassard N, Qi S, Patey N, Soulez M, et al. Antiperlecan antibodies are novel accelerators of immune-mediated vascular injury. Am J Transplant (2013) 13(4):861–74. doi:10.1111/ajt.12168
52. Joosten SA, van Dixhoorn MG, Borrias MC, Benediktsson H, van Veelen PA, van Kooten C, et al. Antibody response against perlecan and collagen types IV and VI in chronic renal allograft rejection in the rat. Am J Pathol (2002) 160(4):1301–10. doi:10.1016/S0002-9440(10)62557-6
53. Yang B, Dieude M, Hamelin K, Henault-Rondeau M, Patey N, Turgeon J, et al. Anti-LG3 antibodies aggravate renal ischemia-reperfusion injury and long-term renal allograft dysfunction. Am J Transplant (2016) 16(12): 3416–29. doi:10.1111/ajt.13866
54. Nunez-Gomez E, Pericacho M, Ollauri-Ibanez C, Bernabeu C, Lopez-Novoa JM. The role of endoglin in post-ischemic revascularization. Angiogenesis (2017) 20(1):1–24. doi:10.1007/s10456-016-9535-4
55. Nguyen B, Williams AB, Young DJ, Ma H, Li L, Levis M, et al. FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget (2017) 8(7):10931–44. doi:10.18632/oncotarget.14539
56. Kokonozaki M, Tsirakis G, Devetzoglou M, Kyriakaki S, Antonakis A, Vyzoukaki R, et al. Potential role of FLT3-ligand in the angiogenic process of multiple myeloma. Leuk Res (2015) 39(12):1467–72. doi:10.1016/j.leukres.2015.10.009
57. Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science (2008) 322(5904):1101–4. doi:10.1126/science.1165218
58. Delahunty M, Zennadi R, Telen MJ. LW protein: a promiscuous integrin receptor activated by adrenergic signaling. Transfus Clin Biol (2006) 13(1–2):44–9. doi:10.1016/j.tracli.2006.02.022
59. Jackson AM, Sigdel TK, Delville M, Hsieh SC, Dai H, Bagnasco S, et al. Endothelial cell antibodies associated with novel targets and increased rejection. J Am Soc Nephrol (2015) 26(5):1161–71. doi:10.1681/ASN.2013121277
60. Lehle K, Kroher J, Kolat P, von Susskind-Schwendi M, Schmid C, Haneya A, et al. Existence of circulating anti-endothelial cell antibodies after heart transplantation is associated with post-transplant acute allograft rejection. Heart Vessels (2016) 31(5):752–7. doi:10.1007/s00380-015-0666-0
61. Pisetsky DS. Anti-DNA antibodies – quintessential biomarkers of SLE. Nat Rev Rheumatol (2016) 12(2):102–10. doi:10.1038/nrrheum.2015.151
62. Yung S, Ng CY, Au KY, Cheung KF, Zhang Q, Zhang C, et al. Binding of anti-dsDNA antibodies to proximal tubular epithelial cells contributes to renal tubulointerstitial inflammation. Clin Sci (Lond) (2017) 131(1):49–67. doi:10.1042/CS20160421
63. Gao B, Moore C, Porcheray F, Rong C, Abidoglu C, DeVito J, et al. Pretransplant IgG reactivity to apoptotic cells correlates with late kidney allograft loss. Am J Transplant (2014) 14(7):1581–91. doi:10.1111/ajt.12763
64. Henderson L, Masson P, Craig JC, Flanc RS, Roberts MA, Strippoli GF, et al. Treatment for lupus nephritis. Cochrane Database Syst Rev (2012) 12:CD002922. doi:10.1002/14651858.CD002922.pub3
65. Giral M, Foucher Y, Dufay A, Van Huyen JP, Renaudin K, Moreau A, et al. Pretransplant sensitization against angiotensin II type 1 receptor is a risk factor for acute rejection and graft loss. Am J Transplant (2013) 13(10):2567–76. doi:10.1111/ajt.12397
Keywords: human leukocyte antigen, non-human leukocyte antigen antibody, kidney transplant, heart transplant, angiotensin II type 1 receptor antibody
Citation: Zhang X and Reinsmoen NL (2017) Impact of Non-Human Leukocyte Antigen-Specific Antibodies in Kidney and Heart Transplantation. Front. Immunol. 8:434. doi: 10.3389/fimmu.2017.00434
Received: 17 October 2016; Accepted: 28 March 2017;
Published: 13 April 2017
Edited by:
Brian Duncan Tait, Australian Red Cross Blood Service, AustraliaReviewed by:
Andrew Gennery, Newcastle University, UKJames Drake, Albany Medical College, USA
Narinder K. Mehra, All India Institute of Medical Sciences, India
Duncan C. Walton, Australian Red Cross Blood Service, Australia
Copyright: © 2017 Zhang and Reinsmoen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohai Zhang, eGlhb2hhaS56aGFuZ0Bjc2hzLm9yZw==;
Nancy L. Reinsmoen, bmFuY3kucmVpbnNtb2VuQGdtYWlsLmNvbQ==