 Amy Hughes1,2,3
Amy Hughes1,2,3 Agnes S. M. Yong1,2,3*
Agnes S. M. Yong1,2,3*
- 1Department of Haematology, SA Pathology, Adelaide, SA, Australia
- 2Cancer Theme, South Australia Health and Medical Research Institute (SAHMRI), Adelaide, SA, Australia
- 3School of Medicine, The University of Adelaide, Adelaide, SA, Australia
Chronic myeloid leukemia (CML) is a hematological cancer, characterized by a reciprocal chromosomal translocation between chromosomes 9 and 22 [t(9;22)], producing the Bcr-Abl oncogene. Tyrosine kinase inhibitors (TKIs) represent the standard of care for CML patients and exert a dual mode of action: direct oncokinase inhibition and restoration of effector-mediated immune surveillance, which is rendered dysfunctional in CML patients at diagnosis, prior to TKI therapy. TKIs such as imatinib, and more potent second-generation nilotinib and dasatinib induce a high rate of deep molecular response (DMR, BCR-ABL1 ≤ 0.01%) in CML patients. As a result, the more recent goal of therapy in CML treatment is to induce a durable DMR as a prelude to successful treatment-free remission (TFR), which occurs in approximately half of all CML patients who cease TKI therapy. The lack of overt relapse in such patients has been attributed to immunological control of CML. In this review, we discuss an immunological timeline to successful TFR, focusing on the immunology of CML during TKI treatment; an initial period of immune suppression, limiting antitumor immune effector responses in newly diagnosed CML patients, linked to an expansion of immature myeloid-derived suppressor cells and regulatory T cells and aberrant expression of immune checkpoint signaling pathways, including programmed death-1/programmed death ligand-1. Commencement of TKI treatment is associated with immune system re-activation and restoration of effector-mediated [natural killer (NK) cell and T cell] immune surveillance in CML patients, albeit with differing frequencies in concert with differing levels of molecular response achieved on TKI. DMR is associated with maximal restoration of immune recovery in CML patients on TKI. Current data suggest a net balance between both the effector and suppressor arms of the immune system, at a minimum involving mature, cytotoxic CD56dim NK cells may be important in mediating TFR success. However, a major goal remains in CML to identify the most effective pathways to target to maximize an advantageous immune response and promote TFR success.
Introduction
Chronic myeloid leukemia (CML) is a hematological cancer characterized by the presence of the BCR-ABL1 oncogene, which is the product of a reciprocal translocation between chromosomes 9 and 22 [t(9;22)], in a hematopoietic stem cell. The resultant constitutively active tyrosine kinase, Bcr-Abl, mediates phosphorylation and activation of downstream signaling pathways, causing altered cell adhesion, inhibition of apoptosis, differentiation arrest, and proteasomal degradation of key proteins, which lead to the phenotype of the disease (1). CML is considered to be one of the cancers most sensitive to immunological manipulation (2). Tyrosine kinase inhibitors (TKIs) imatinib, nilotinib, and dasatinib are used as first-line treatment in CML. These small molecule inhibitors block the adenosine triphosphate-binding site of the Bcr-Abl tyrosine kinase and prevent phosphorylation of downstream effector proteins. Clinical response to treatment is assessed initially by monitoring the reduction of the peripheral white blood cell count, and subsequently by measurement of BCR-ABL1 transcript levels against a control gene (3). An optimal response following initiation of TKI treatment is a major goal, as this confers improved patient survival. Clinical guidelines on optimal molecular responses refer to achievement of target BCR-ABL1 levels [e.g., ≤0.1%, major molecular response (MMR)] at specific timepoints (4). The more recent goal in CML treatment is to induce a durable deep molecular response (DMR; BCR-ABL1 ≤ 0.01%) as a prelude to successful treatment-free remission (TFR), which occurs in approximately half of all CML patients who cease TKI therapy (5, 6). The lack of overt relapse in such patients has been attributed to immunological control of CML (7), although its precise mechanisms are as yet unclear.
Suppression of the innate and adaptive immune system, linked to an accumulation of immature myeloid cells (myeloid-derived suppressor cells, MDSCs), predominates during pathological conditions such as cancer, and leads to inhibition of the host–antitumor immunity (8). MDSC expansion in the blood and bone marrow at the time of diagnosis has been shown in patients with multiple myeloma (9), chronic lymphocytic leukemia (10), and acute myeloid leukemia (11). In CML, increased levels of MDSC, which originate from the malignant BCR-ABL1 clone, are also observed, and these MDSC subsequently reduce following highly efficacious TKI therapy (12, 13). MDSCs promote the recruitment and expansion of other suppressor cells (regulatory T cells, Treg), leading to impaired innate effector natural killer (NK) cells and inhibition of T cell proliferation and activation, further downregulating antitumor immune surveillance that subsequently influence leukemia development and progression (14). In support, quantitative and functional defects of NK cells and diminished cytotoxic T lymphocyte (CTL) function have also been described in chronic phase (CP) CML patients at diagnosis (12, 15–17). Thus, the changing ratio between resident immune effector and immune suppressor cells in untreated CML and other hematological cancers, limits the patient’s immune status such that a predominantly immune inhibitory leukemic milieu is present, accounting for a diminished anti-leukemic effector immune response to control leukemia progression and/or relapse. Very recently, an increased proportion of mature, adaptive-like CD56dim NK cells have been observed in CML patients who successfully discontinued imatinib (18). Other immunologic mediators such as plasmacytoid dendritic cells (pDCs), which may serve as promising prognostic factors for successful TFR, are also currently under investigation (19). TKIs also exert significant off-target multikinase inhibitory effects, albeit with differing potencies. Cumulative data suggest that TKIs exhibit a dual mode of action; direct oncokinase inhibition interspersed with concomitant immunomodulatory effects, particularly against key suppressor MDSC and Treg populations, conferring immune system re-activation and restoring effector-mediated immune surveillance (2, 13, 20–24). In this review, we discuss an immunological timeline to successful TFR in CML; an initial period of immune dysfunction in newly diagnosed CML patients, followed by restoration of immune effector responses and release of immune suppressors, albeit with differing frequencies in concert with differing levels of molecular response achieved on TKI. Optimum restoration of endogenous immune surveillance mechanisms may promote sustained TFR following TKI discontinuation attempt.
Immune Dysfunction in Newly Diagnosed CML Patients
The majority (~90%) of CML patients are diagnosed while in CP, characterized by an expansion of circulating myeloid cells, which are mainly mature, and maintained by a small subset of disease initiating leukemic stem cells (LSCs) (25). Persistent immune dysfunction in CML patients at the time of diagnosis, prior to the start of any therapy is well documented, precluding the development of adequate anti-leukemia immune responses and promoting disease progression in the absence of highly efficacious TKI therapy. An essential role of the immune system, in particular that of innate and adaptive immune cells (i.e., NK cells, CD8+/CD4+ T cells), effector molecules, and endogenous signaling pathways, is to confer host protection against cancer (26). However, many tumors facilitate their self preservation and progression by the recruitment of immunosuppressive cells, release of inhibitory factors including immunosuppressive and inflammatory cytokines and upregulation of immune checkpoint pathways, in particular cytotoxic T-lymphocyte-associated protein 4 and programmed death-1 (PD-1) pathways (27, 28). The ligand for PD-1, programmed death ligand-1 (PD-L1), induces a coinhibitory signal in activated T cells and promotes T cell apoptosis, anergy, and functional exhaustion (29). Further research into better understanding this altered immune balance in CML patients at diagnosis is essential for the development of new therapeutic methods, aiming to augment antitumor immune activity and enhance TFR success rates following TKI cessation.
Effector Cells of the Immune System in CML Patients at Diagnosis
The main antitumor effector cells of the immune system, NK cells, dendritic cells (DCs), and CTLs (30), play a direct role in host control of hematological malignancies, including CML. Antibody-secreting effector B cells, also called plasma cells also defend the body in an immune response, with distinct B cell subsets mediating different types of antibody responses (31). de Lavallade et al. (32) have previously reported loss of memory B cell subsets in CML patients at diagnosis; however, the clinical impact remains unclear, with no patients showing recurrent infections despite the B cell deficiency. In support, we have performed extensive characterization of major B cell subsets including transitional, naïve, non-switched memory, class-switched memory, plasmablasts, and plasma cells and observed non-switched memory B cell expression was decreased in CML patients at diagnosis compared to normal healthy donor samples (12).
Natural killer cells are lymphocytes and a critical component of the innate immune system, providing a front line defense against tumor cells. NK cells exert potent cellular cytotoxicity against malignant cells (CD56dim NK cell subset) and produce immunoregulatory cytokines and chemokines, such as interferon-γ (IFN-γ) and tumor necrosis factor α (TNF-α), supporting the development of adaptive immunity (CD56bright NK cell subset). In contrast to T cells that require interaction with professional antigen-presenting cells, such as mature DCs for activation, NK cells utilize a diverse array of inhibitory and activating receptors for target cell recognition and lysis with no requirement for prior antigen stimulation or clonal expansion (33). NK cells are dysfunctional in CP CML patients at diagnosis and NK cell numbers among lymphocytes are reduced, worsening with disease progression to advanced and blast crisis phase CML (15, 16, 34). The activating C-type lectin receptor NKG2D stimulates the cytotoxicity of NK cells following recognition of the stress-induced ligand major histocompatibility complex class I chain-related A (MICA). BCR-ABL1 promotes DC-mediated NK cell activation by increasing the expression of NKG2D ligands including MICA (35). MICA releases soluble proteins produced on the surface of tumor cells, inducing negative modulation of NKG2D and facilitating tumor cell escape from NK cell killing (36–38). Reduced NKG2D-activating receptor expression has been previously identified in CML patients at diagnosis, promoting leukemic cell survival (36). Very recently, we identified reduced expression of the NKG2 family of C-type lectin receptors (CD94/NKG2A, CD94/NKG2C, and NKG2D) in CML patients at diagnosis (12). Similarly, the natural cytotoxicity receptors NKp30 and NKp46, the latter characterized as the major triggering receptor involved in NK cell cytotoxicity (39) and the killer immunoglobulin-like receptors (KIRs) KIR2DL2/DL3/DS2 were downregulated in CML patients at diagnosis compared to healthy donors. Downregulation of NKp30 and NKp46 has been reported previously in acute myeloid leukemia and chronic lymphocytic leukemia and shown to correlate with decreased NK cell cytotoxicity (40–42).
Dendritic cells are the most potent antigen-presenting cells in promoting activation of naïve T cells and play a critical role in the initiation and regulation of the immune response (43). DCs take up, process, and present antigens on major histocompatibility (MHC) class I and II molecules on the cell surface, to T cells, thus initiating antigen-specific immune responses and/or immunological tolerance (44). In normal peripheral blood, myeloid and pDC subsets are typically identified in the immature state in low numbers, in contrast, quantitative and functional defects including inefficient antigen presentation have been reported in DC subsets from CML patients (45–48).
Leukemia-associated antigen (LAA)-specific CTLs have been detected in the peripheral blood of CP CML patients, including CTLs specific for Bcr-abl and selectively expressed or overexpressed LAAs such as proteinase-3 (PR3) and Wilms’ tumor antigen 1 (WT1), and may be involved in the immunological control of CML (49–51). However, studies have shown T cells from untreated patients with CML are functionally impaired, displaying decreased TCRζ-chain expression, limited cytotoxic activity, and they do not produce immunoregulatory cytokines IFN-γ or TNF-α (52–54). TCRζ-chain expression is critical for normal T cell function, including proliferation and IFN-γ production (55). Molldrem et al. (56) suggest a novel escape mechanism from tumor immunity by leukemia-induced selective deletion of high avidity effector CTLs that have the greatest potency against CML.
CD62L downregulation may impair effector CTL immune responses in CML patients at diagnosis and abrogate anti-leukemic immune control, as CD62L is critical in controlling the traffic of T cells to secondary lymphoid tissues and priming by antigen-presenting cells. Sopper et al. (57) have reported decreased CD62L surface expression on T cells in CML patients at diagnosis. Another mechanism of impaired immune response in CML may involve aberrant PD-1/PD-L1 signaling on effector cells, and immunosuppressive Treg, which also express PD-1. PD-1 is temporarily expressed on activated immune effector cells under normal physiological conditions; however, constitutive expression results in diminished effector T cell function, and expansion of Treg with enhanced suppressor function, the latter playing a critical role in maintenance of the immunosuppressive tumor milieu (17, 58, 59). Mumprecht et al. (60) have previously reported PD-1 is upregulated on CML-specific CTLs and also on CTLs specific for unrelated antigens. Higher PD-1 expression in CTLs is related to inhibition of the effector phase of T cell responses and reduced T cell-mediated antitumor immunity (28). In addition, we have shown increased PD-1 expression on CD4+ and CD8+ T cells in CML patients at diagnosis, suggesting the presence of a broadly compromised immune system in CML patients at diagnosis (12).
Suppressor Cells of the Immune System in CML Patients at Diagnosis
Myeloid-derived suppressor cells are a heterogeneous population of immature granulocytic and monocytic cells with potent immune suppressive functions and therefore represent one of the main suppressor cell populations within the immune system (61). MDSCs expand during cancer, inflammation, and infection and are characterized by the ability to suppress the cytotoxic function of T cells, including leukemia-specific CTLs, and NK cells (62). Cancer-derived inflammation drives the expansion and suppressive activity of MDSC, this is facilitated by multiple pro-inflammatory factors including GM-CSF and VEGF, produced within the immunosuppressive tumor microenvironment (63, 64). MDSC suppressive activity is mediated via a number of mechanisms, including increased production of reactive oxygen and nitrogen species and upregulation of arginase-1, the latter culminating in local depletion of arginine, an essential amino acid for T cell function (62, 65, 66) and depletion of cysteine, required by antigen-presenting cells including DCs, for effective T cell activation (67). Arginase-1 has also been shown to inhibit NK cell proliferation and secretion of IFN-γ (68) and MDSC induce anergy of NK cells through membrane-bound TGF-β1 (69). CML patients express high levels of monocytic and granulocytic MDSC at diagnosis compared to healthy donors (12, 13, 21), and these MDSC express BCR-ABL1 and are part of the leukemia clone (13). In addition, CML serum leads to anergy of T cells, probably by increased arginase-1 expression (13) and coincubation of isolated monocytic and granulocytic MDSC of CML patients at diagnosis and autologous CFSE-labeled T cells has been shown to inhibit T cell proliferation, whereas control healthy donor MDSC displayed no suppressive activity (70).
Myeloid-derived suppressor cells also mediate the recruitment and expansion of immunosuppressive Treg, a specialized type of CD4+ T cell expressing the transcription factor forkhead box P3 (FoxP3), that can compromise the function of antitumor effector CD4+/CD8+ T cells and antigen-presenting cell activity to facilitate tumor cell immune evasion (62, 71–77). Zahran et al. (78) have reported the percentages of Tregs are significantly increased in newly diagnosed CML patients compared to controls, with lower Treg numbers in CP CML patients compared to accelerated and blast phases. In support, Bachy et al. (79) have shown that Treg are significantly increased in CML patients with intermediate or high-risk Sokal scores compared to low-risk patients. Thus, in a setting of high leukemic cell load in CP CML patients at diagnosis, CML cells can evade host immune surveillance by activation of the immune checkpoint receptor PD-1 and via PD-L1 upregulation, this signaling pathway influences immune suppression and disease progression, further supported by the recruitment of immunosuppressive MDSC and Treg cell populations (28).
Immunogenicity of CML
The constitutively active Bcr-Abl kinase, while only weakly immunogenic in itself, leads to the upregulation of multiple genes, which may result in the expression of LAAs critical for priming of a protective antitumor CTL response against CML cells (80). The cure of CML following allogeneic hematopoietic stem cell transplantation (SCT) is attributed to the immunological graft-versus-leukemia effect, which is mediated by donor-derived T cells and NK cells, targeting alloantigens, primarily minor histocompatibility antigens (mHAgs) (81) and likely also LAAs (50, 82–84) and considered to play a critical role in disease eradication. Donor lymphocyte infusion after SCT provided the first direct evidence of the graft-versus-leukemia effect, in which, CTLs with mHAg specificity could salvage disease relapse (85, 86). The presence of LAA-specific CTLs directed against PR3 was associated with clinical responses to SCT (50, 84). In earlier functional studies, LAA-specific CTL responses to WT1 and PR3 were identified in CML patients and healthy donors, albeit at a lower frequency in the latter, and shown to expand in the recipient after transplantation, contributing to remission (83). Direct evidence of high in vivo antigen-specific immunogenicity against the LAA BMI-1 has also been found in the setting of SCT in CML (82).
Specific immunotherapies for CML patients targeting LAAs in combination with imatinib or other TKIs may exert a synergistic effect, inducing DMR in a high percentage of patients, while having the potential to overcome disease resistance by eliminating the quiescent CML stem cell subpopulation and enhancing specific immune responses against CML (80). In addition to BMI-1, WT1, and PR3, several other LAAs have been identified in CML patients such as RHAMM/CD186 and PRAME (82, 87–89) and thus represent candidate antigens for specific immunotherapies, with efficacy demonstrated in vaccination, including DC vaccination (90–93) and more recently, T cell receptor mimic antibodies (94, 95). Significant differences exist in the expression of LAAs relevant to CML disease progression and response, and CD34+ progenitor maturation in CML (81, 96), suggesting the importance of combining several antigens in future immunotherapeutic strategies. For effective activation of effector CTL responses, MHC molecules and costimulatory proteins are necessary, aberrant expression of these molecules can lead to immune evasion of cancer (26). Progenitor cells of CML patients (CD34+CD38+, CD34+CD38−, CD34+CD38−CD90+) actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II and its master regulator class II transactivator (CIITA), a transcription factor that functions as a molecular switch for MHC-II gene regulation, and may explain why CML stem cells persist despite lifelong TKI treatment (97). CIITA plays a central role in MHC-II antigen presentation to effector CD4+ T cells, and thus in the stimulation of the adaptive immune response. Treatment of cells with the receptor TKI ruxolitinib (and to a lesser extent IFN-γ) enhanced the expression of MHC-II on CML stem/progenitor cells and was associated with an increase in CML cell immunogenicity (enhanced CD4+ T cell proliferation) in a JAK-dependent manner (97, 98). In support, clinical studies investigating the combination regimen of ruxolitinib and TKIs imatinib, dasatinib, and nilotinib in control of CML (NCT02253277, NCT01751425, and NCT01702064) are currently ongoing. The development of immunotherapeutic strategies to enhance MHC-II expression on CML stem/progenitor cells may facilitate their elimination by host immune effectors and result in greater rates of success in TFR discontinuation studies.
The Immune System is not Permanently Compromised in CML
The introduction of TKIs has dramatically improved disease outcome in patients with CML and has replaced interferon-alpha (IFN-α) and SCT as frontline therapy (99). The therapeutic efficacy of IFN is mediated at least in part by immunological mechanisms and by the ability to cycle quiescent LSCs, responsible for disease initiation (100). The mechanism of action of TKIs, while remarkably different to that of IFN, is also accompanied by immune system re-activation, suggesting the immune system is not permanently compromised in CML (101, 102). A current limitation of immunological studies in CML is the limited investigation of immune responses longitudinally in the same patient cohort over time, for example, studies of patient samples at diagnosis, on TKI and following achievement of DMR, which may be achieved very quickly or many years later. This is further compounded by a lack of studies directly comparing the level of immune system re-activation and/or the timing of re-activation achieved with first-generation TKI imatinib compared to more potent second-generation TKIs nilotinib and dasatinib, the latter known to possess strong inhibitory activity with broader specificity against multiple other kinases including Src, Tec, and Syk family kinases, many of these being involved in innate and adaptive immune responses (103–105). Bosutinib and ponatinib are both indicated for second- or later-line treatment of CML, no studies to date appear to have explored their immunomodulatory effect on immune cell populations. We have reported maximal restoration of immune recovery in CML patients on TKI occurs only following achievement of MR4.5 (BCR-ABL1 ≤0.0032%), as demonstrated by increased effector NK cell number and function and T cell immune responses, reduced numbers of PD-1+ CD4+/CD8+ T cells and monocytic MDSC (12). This suggests there may be a staged recovery of some immune responses in CML patients on TKI, which may be linked to depth of molecular response achieved; however, the specific causal relationship between both phenomena remains to be fully elucidated.
The Immunomodulatory Effects of TKI
Peripheral blood DCs comprising plasmacytoid and myeloid subsets increase in number and function in CML patients following imatinib treatment (48, 106). In a murine model, DC treated with imatinib exhibited enhanced antigen-presenting cell function and restored the responsiveness of tolerant tumor-specific CD4+ T cells, resulting in enhanced vaccine efficacy (107). Treatment with imatinib, nilotinib, or dasatinib is associated with reduced memory B cell frequencies and significant impairment of B cell responses in CML (32). While a blunted antigen-specific immune response is observed in CML patients at diagnosis, LAA-specific CTL responses are readily detected in TKI-induced MMR and MR4.5 when the leukemic cell load is lower, suggesting effective restoration of anti-leukemic effector responses (12). Chen et al. (108) have also detected CTL responses in the majority of CML patients in remission on imatinib, confirming the immune system’s ability to respond to leukemia under certain conditions. It is possible the restoration of LAA-CTL responses in CML patients on TKI is closely linked to effective inhibition of aberrant PD-1 signaling, a potential consequence of diminishing antigenic stimulation in light of the significantly reduced leukemic cell load on TKI.
Dasatinib is unique in its ability to induce expansion of large granular lymphocytes (LGLs), consisting of mono- or oligo-clonal CD8+ T cells and NK cells, shown to correlate with better prognosis and consequently more favorable response in patients with CML (109–112). Twenty-hour pretreatment of NK cells with dasatinib followed by washout, led to dose-dependent enhancement of NK cell cytokine production, degranulation marker expression, and cytotoxicity against lymphoma and leukemia cell lines (113). Hayashi and colleagues (114) have reported dasatinib treatment markedly enhances NK cell cytotoxic function in CML patients and this is associated with an increased number of cells in the NK lineage, including CD3−CD56+ and mature CD56+CD57+ cells to levels not obtained with imatinib or nilotinib (115). CML patients in MMR and MR4.5 on TKI display a more mature, cytolytic CD57+CD62L− NK cell phenotype, consistent with restoration of NK cell activating and inhibitory receptor repertoire compared to their downregulation at diagnosis (12). Comparative analysis of NK cell and T cell receptor repertoires in patients treated with imatinib frontline or nilotinib and dasatinib as first- or second-line therapy has been performed (116). The lymphocyte count and absolute number of NK cells was not significantly different between the treatment groups. Phenotypic analysis of NK cell receptors revealed dasatinib-treated patients displayed an increased expression of KIR (KIR2DL1) receptors, while imatinib-treated patients exhibited an increased expression of activating receptors (NKp30, NKp46, NKp80, and NKG2D).
The immunostimulatory effects of dasatinib also extend to immune suppressors, as dasatinib has the potential to reduce Treg in both bone marrow and peripheral blood, skewing the balance of immune suppression toward activation and proliferation, promoting immune stimulation (115). Treg reduction has been shown to be markedly enhanced in dasatinib-treated CML patients developing LGL lymphocytosis (109, 117). Balachandran et al. (23) have found that the immune system contributes substantially to the antitumor effects of imatinib via the inhibition of indoleamine 2,3-dioxygenase (IDO), an important regulatory checkpoint influencing Treg expansion and activity (118). Imatinib activates CD8+ T cells and NK cells and leads to apoptosis of immunosuppressive Treg (23, 119). At clinically relevant doses, imatinib treatment of mice reduces Treg frequency and inhibits Treg suppressive activity and FoxP3 expression in vivo (120). The induction and persistence of Treg immunosuppressive function is dependent on FoxP3 (120). Zahran et al. (78) have reported Treg numbers are significantly lower in CML patients achieving DMR on imatinib therapy compared to diagnosis and patients not achieving DMR. Hayashi et al. (114) also revealed the frequency of Treg decreased in patients treated with imatinib, nilotinib, or dasatinib, with the percent decrease of Treg comparable among the three treatment groups (Figure 1).
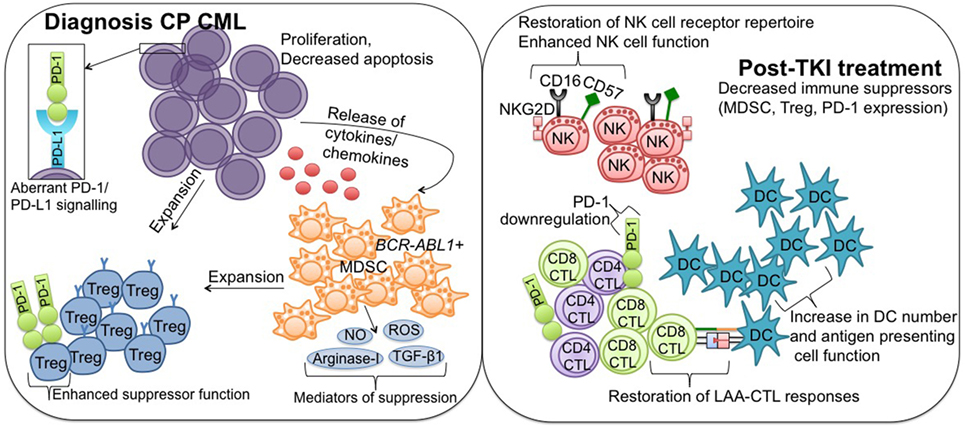
Figure 1. Immune effector recovery in CML patients achieving deep molecular response on TKI therapy. Diagnosis CP CML; suppression of the immune system in CP CML patients at diagnosis is mediated in part by hematopoietic stem cells, which acquire a proliferative/survival advantage and lose the ability to undergo apoptosis. Release of tumor-derived cytokines/chemokines drives the expansion of immune suppressor MDSC and Treg, facilitating downregulation of antitumor effector immunity. PD-L1 is upregulated on CML cells, where it interacts with the coinhibitory receptor PD-1, and contributes to protection of the malignant cells from immune destruction. MDSC originate from the malignant BCR-ABL1 clone and mediate their suppressive activity via a number of mechanisms, including increased production of reactive oxygen and nitrogen species (NO, ROS), arginase-1, and TGF-β1. MDSC can induce Treg expansion, and Treg also express PD-1 to promote enhanced suppressor function. Post-TKI treatment; TKI exert immunomodulatory effects, particularly against key suppressor MDSC and Treg populations, conferring immune system re-activation and restoring effector-mediated immune surveillance. More specifically, TKI treatment leads to restoration of NK cell receptor repertoire and enhanced NK cell function, restoration of LAA-CTL responses, including downregulation of PD-1 to normal levels, and increased DC number and antigen-presenting cell function. CML, chronic myeloid leukemia; TKI, tyrosine kinase inhibitor; CP, chronic phase; MDSCs, myeloid-derived suppressor cells; Treg, regulatory T cells; PD-L1, programmed death ligand-1; PD-1, programmed death-1; NO, nitric oxide; ROS, reactive oxygen species; TGF-β1, transforming growth factor-β1; NK, natural killer; LAA, leukemia-associated antigen; CTL, cytotoxic T lymphocyte; DC, dendritic cell.
Giallongo et al. (13) have previously shown MDSC decrease to normal levels in CML patients following imatinib therapy, and more recently, both imatinib and dasatinib have been shown to modulate the immunosuppressive CML tumor milieu, leading to decreased numbers of MDSC and associated inhibitory molecule arginase-1 expression (121). The TKI sunitinib reduces the frequency of MDSC and reverses T cell immune suppression in the peripheral blood of metastatic renal cell carcinoma patients (22).
Contributing Immunological Factors in TFR
Several clinical trials including the landmark STIM (122, 123) and CML 8 (TWISTER) (5) trials have demonstrated that ~40% of CML patients in stable DMR successfully maintain TFR after stopping imatinib therapy. Importantly, all patients in molecular relapse (MolR) remained responsive to imatinib re-treatment, and most patients who relapsed did so within 6 months of imatinib cessation. Many other TFR stopping trials are currently ongoing, some with less stringent criteria for relapse than earlier studies, such as loss of MMR for re-starting TKI therapy and others evaluating discontinuation of second-generation TKIs (124–130). Rea and colleagues (130) provided the first report of cessation of second-generation TKI dasatinib or nilotinib (STOP 2G-TKI) in CML patients, TFR rates at 12 and 48 months were 63% (CI: 51–76%) and 55% (CI: 40–67%), respectively. Prior suboptimal TKI response or TKI resistance was the only baseline patient characteristic associated with a significantly higher incidence of MolR following cessation. The largest discontinuation study to date, the European stop TKI (EURO-SKI) trial has recently reported a molecular recurrence-free survival of 52% (CI: 48–56%) in 750 CP CML patients treated with imatinib, nilotinib, or dasatinib at 24 months, using loss of MMR as the criteria for relapse (126).
The lack of overt relapse in such patients, despite the presence of very low levels of residual disease has been attributed to immunological control of CML (131), challenging the long-held belief that SCT represents the only “curative” therapy for CML and indicating that complete elimination of residual CML stem cells is not necessary in all patients to successfully achieve TFR (132). DMR occurs in ~20% of imatinib treated patients in the first 2–3 years of therapy; however, using more potent TKIs dasatinib and nilotinib, the rates of DMR are faster and deeper, and it is possible that this will lead to more patients eligible to attempt TFR in a shorter time frame (133, 134). However, it is also likely that the optimum duration of TKI treatment before discontinuation attempt is heterogeneous and varies between patients (135). To this end, identifying predictive factors for successful discontinuation of TKI and thus, identification of CML patients who would benefit most from discontinuation remains a key issue (6).
Role of the Immune System for Successful TFR
To date, more than 2,000 CML patients worldwide have attempted to discontinue TKI after achieving DMR in the clinical trial setting (6). More recently, several immunological sub-studies within larger discontinuation trials have attempted to identify immunological predictive factors of TFR maintenance, typically prior to TKI cessation (baseline immune markers) (6). Results from Immunostim, performed on a subset of 51 patients enrolled in the STIM trial, identified an association between elevated peripheral blood NK cells and positive clinical outcome following imatinib discontinuation (136). Accordingly, these observations provided some of the first evidence that NK cell based immune surveillance may contribute to CML control following TKI cessation. More recent molecular data (A-STIM and EURO-SKI trials) demonstrating fluctuating BCR-ABL1 levels just below MMR without loss of MMR, and therefore without TKI resumption, has all but confirmed the importance of the role of immune surveillance for sustained TFR in CML. Immunostim did not observe any association between CD3+/CD4+/CD8+ T cells, the CD4/CD8 ratio, or Treg numbers in patients who relapsed compared to those who did not. Patient characteristics revealed 52.9% of the patients had received prior IFN therapy, shown in other studies to contribute to higher TFR rates (5, 137). Induction of a PR3-specific CTL response by IFN has been shown to contribute to this effect (138). Prior to the advent of TKI therapy, patients treated with IFN who discontinued treatment without relapse showed increased NK cell counts (139). IFN/imatinib induction treatment followed by a temporary IFN maintenance may enable a higher rate of treatment discontinuation in CML patients in at least MMR when stopping TKI (140). The TIGER study (NCT01657604) is currently investigating de-escalating maintenance therapy using low dose IFN as an inducer of immune surveillance following nilotinib discontinuation.
In keeping with the NK cell effector responses observed in Immunostim, a subgroup analysis of 45 patients in the EURO-SKI clinical trial has also shown that patients with higher NK cell counts at the time of TKI discontinuation are more likely to have successful TFR, linked to an increased frequency of more mature (CD57+) and cytotoxic (CD16+ and CD57+) NK cells (18). Prior IFN treatment showed no statistically significant correlation between relative or absolute numbers of NK cells. Similar association to successful TFR was not found with T cells or B cells or their subsets. The low affinity Fcγ receptor III (CD16) facilitates antibody-dependent cellular cytotoxicity, involved in triggering NK cell cytotoxicity (141). However, evaluation of activating NK cell surface receptors: NKG2C, NKp46, and NKG2D, which control NK cell cytotoxic responses revealed no significant difference between the patient groups. We have examined expression levels of all three natural cytotoxicity receptors NKp30, NKp44, and NKp46 and additional C-type lectin receptors NKG2A, NKG2C, NKG2D, CD161, and CD69 in Australian CML patients at time of TKI cessation and 3 and 6 months after (142). NKG2D represented the only differentially expressed receptor, decreased in MolR patients when compared to TFR at all three time points assessed. The functional degranulation response of CD3−CD56dim NK cells following target cell stimulation does not appear to be different between TFR and MolR (18, 136, 142). In further analysis by EURO-SKI, TFR patients had more NK cells that had downregulated CD16 (CD16−) upon K562 stimulation, suggesting an increased activity of these cells. In addition, the TNF-α/IFN-γ cytokine secretion by these activated CD56dimCD16− NK cells correlated with TFR success (18). A separate sub-study of 122 EURO-SKI patients has recently reported that increased CD86+ pDCs, which mediate immune tolerance are found in MolR patients at the time of TKI cessation, and thus low CD86+ pDC might be predictive of TFR (19). This study also found that higher numbers of pDCs correlated with increased PD-1-expression on PR3-specific CD8+ CTLs, suggesting immune exhaustion contributes to relapse risk.
The specific role and/or predictive ability of NK cells in successful dasatinib cessation is currently unclear. In the Japanese D-STOP trial, patients with DMR on TKI received dasatinib consolidation therapy for 2 years prior to cessation, and at the end of the consolidation period, there was a significant increase in the proportion of CD3−CD56+ NK cells in patients who relapsed (127). In contradistinction, the Japanese DADI trial (125) where patients received dasatinib consolidation for 1 year prior to cessation showed high NK cell (CD3−CD56+ and CD16+/CD56+) and NK cell LGL (CD56+CD57+) numbers, and low γδ T cells and Treg (CD25+CD127low) counts prior to stopping second line dasatinib treatment were associated with an increased likelihood of TFR success. Treg facilitate tumor cell immune evasion and may be a contributing factor responsible for relapse in these patients. By contrast, other studies have reported no difference in Treg or Treg naïve/memory subsets in TFR patients compared to those who relapsed (136, 142). This difference may reflect heterogeneity of distinct immune subsets, which are pre-conditioned by different TKI treatment modalities prior to cessation attempt. For example, DADI requires 1 year of dasatinib consolidation therapy prior to cessation, while Immunostim and our recently reported Australian data represent a predominantly imatinib treated patient cohort. Yoshida et al. (143) have suggested a critical role of Treg inhibition by dasatinib for the induction of NK cell effector differentiation and achievement of DMR and dasatinib has been previously shown to potently inhibit the proliferation and function of CD4+CD25+ Treg (24). Thus, it is possible the enhanced NK cell immune effector phenotype observed in patients who did not relapse in DADI, acting in concert with reduced immune suppressive Treg, reduces the likelihood of relapse in CML patients following TKI discontinuation. Further studies are needed to ascertain the mechanisms responsible for achievement of TFR in the absence of reduced Treg as it does not appear that reduced Treg numbers are critical in all TFR scenarios.
The only difference in immune suppressors observed in our Australian patient cohort was increased monocytic MDSC in MolR patients at the time of TKI discontinuation. It is possible monocytic MDSC, which play an important role in suppression of host immune responses, including effector NK responses (144) block or dampen immune surveillance, a necessary component against relapse in CML. Therapeutic strategies aimed at targeting the number and/or function of MDSC, such as promoting their differentiation into mature myeloid cells that do not have suppressive abilities, inhibition of the signaling pathways that regulate MDSC suppressive factors, or elimination of MDSC using chemotherapeutic drugs may further enhance TFR success rates (62). We have also identified reduced KIR2DL2/DL3/DS2 positive NK cells in TFR patients at 3 months and 6 months post-TKI cessation compared to MolR patients (142). In this setting, it is possible antibodies that block KIR on NK cells may enhance TFR success rates. However, inhibition of KIR2D with the monoclonal antibody IPH2101 has been shown recently to induce contraction and hyporesponsiveness of NK cells in patients with myeloma, with reductions in NK function directly correlating with loss of free KIR2D surface molecules (145). This finding may compromise antibody-based strategies designed at augmenting NK cell tumor killing via KIR inhibition in CML.
Killer immunoglobulin-like receptor genotypes have been reported to influence the probability of achieving molecular response in CML; however, none has yet emerged as a reliable predictor of TFR after DMR has been achieved (135). Caocci et al. (146) reported KIR polymorphisms that could be significantly associated with increased likelihood of TFR success. Interestingly, one of the haplotypes has already been reported to be significantly linked to the TKI response (KIR2DL5B) (147). However, there was no difference observed in KIR genotypes in EURO-SKI patients with TFR success compared with MolR (18).
Concluding Remarks and Future Directions
Treatment-free remission attempts are safe and are feasible in most well-responding CP CML patients, and the impact of implementing stopping strategies as a routine part of clinical practice in CML would be significant, from both a patient-centered and medico-economic viewpoint. At present, available data show that MolR typically occurs within 6 months of TKI discontinuation, and most studies have identified the majority of clinical prognostic factors in patients prior to stopping TKI. Normalization of many immune cell populations early in the setting of TFR and MolR, at least by 3 months following the initial timepoint of TKI cessation, may be linked to the dynamic nature of the immune system as evidenced by its constant immune surveillance and the enhanced net effector immune responses and decreased PD-1 and immune suppressors observed in CML patients in DMR compared to diagnosis (12). Thus, longer follow-up of patients in all TFR studies is critically needed to determine how long patients will ultimately be able to maintain TFR (148). In this regard, immune monitoring may reveal an optimum threshold level of immune effector responses in which TKI cessation is more achievable and successful, which could prove especially informative for patients contemplating a second TKI discontinuation attempt following initial failure. Preliminary data suggest a net balance between both the effector and suppressor arms of the immune system may be important in mediating TFR success (142); however, a major goal remains to identify the most effective pathways to target to maximize an advantageous immune response and promote TFR success. Based on what is known regarding NK cell frequency in TFR, trials are underway using pharmacological manipulation, such as lenalidomide, which stimulates NK cells and enhances antitumor responses, in combination with TKI to further augment TFR success in CML (ACTRN12615001169538), or using IFN in combination with a second-generation TKI such as nilotinib (NCT02001818) to enhance immune-modulation prior to a TFR attempt. In conclusion, the identification of a robust immunological biomarker that accurately predicts which patient may stop TKI without relapse, and delineation of the precise mechanisms involved in TFR success should become a primary goal of future discontinuation studies in CML.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
AH declares no competing financial interests. AY has received research funding from Novartis, Bristol-Myers Squibb, and Celgene and honoraria from Novartis and Bristol-Myers Squibb.
Funding
This study was funded by Royal Adelaide Hospital Contributing Haematologists’ Committee, Sail for Cancer Research Fellowship, Leukaemia Foundation of Australia, and the Australian National Health and Medical Research Council grant (APP1059165) (AY) and Cancer Council SA Beat Cancer Project (AH and AY).
References
1. Bennour A, Saad A, Sennana H. Chronic myeloid leukemia: relevance of cytogenetic and molecular assays. Crit Rev Oncol Hematol (2016) 97:263–74. doi: 10.1016/j.critrevonc.2015.08.020
2. Vonka V, Petrackova M. Immunology of chronic myeloid leukemia: current concepts and future goals. Expert Rev Clin Immunol (2015) 11(4):511–22. doi:10.1586/1744666X.2015.1019474
3. Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood (2012) 120(7):1390–7. doi:10.1182/blood-2012-03-378919
4. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood (2013) 122(6):872–84. doi:10.1182/blood-2013-05-501569
5. Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood (2013) 122(4):515–22. doi:10.1182/blood-2013-02-483750
6. Saussele S, Richter J, Hochhaus A, Mahon FX. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia (2016) 30(8):1638–47. doi:10.1038/leu.2016.115
7. Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA, et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia (2010) 24(10):1719–24. doi:10.1038/leu.2010.185
8. De Veirman K, Van Valckenborgh E, Lahmar Q, Geeraerts X, De Bruyne E, Menu E, et al. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front Oncol (2014) 4:349. doi:10.3389/fonc.2014.00349
9. Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, et al. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR(-)/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol (2010) 72(6):540–7. doi:10.1111/j.1365-3083.2010.02463.x
10. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood (2014) 124(5):750–60. doi:10.1182/blood-2013-12-546416
11. Sun H, Li Y, Zhang ZF, Ju Y, Li L, Zhang BC, et al. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int J Hematol (2015) 102(5):579–86. doi:10.1007/s12185-015-1865-2
12. Hughes A, Clarson J, Tang C, Vidovic L, White DL, Hughes TP, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood (2017) 129(9):1166–76. doi:10.1182/blood-2016-10-745992
13. Giallongo C, Parrinello N, Tibullo D, La Cava P, Romano A, Chiarenza A, et al. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS One (2014) 9(7):e101848. doi:10.1371/journal.pone.0101848
14. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology (2013) 138(2):105–15. doi:10.1111/imm.12036
15. Mellqvist UH, Hansson M, Brune M, Dahlgren C, Hermodsson S, Hellstrand K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood (2000) 96(5):1961–8.
16. Chen CI, Koschmieder S, Kerstiens L, Schemionek M, Altvater B, Pscherer S, et al. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia (2012) 26(3):465–74. doi:10.1038/leu.2011.239
17. Ahmad SM, Svane IM, Andersen MH. The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J (2014) 4:e230. doi:10.1038/bcj.2014.50
18. Ilander M, Olsson-Strömberg U, Schlums H, Guilhot J, Brück O, Lähteenmäki H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia (2016). doi:10.1038/leu.2016.360
19. Schütz C, Inselmann S, Sausslele S, Dietz CT, Muller MC, Eigendorff E, et al. Expression of the CTLA-4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML. Leukemia (2017) 31(4):829–36. doi:10.1038/leu.2017.9
20. Zitvogel L, Rusakiewicz S, Routy B, Ayyoub M, Kroemer G. Immunological off-target effects of imatinib. Nat Rev Clin Oncol (2016) 13(7):431–46. doi:10.1038/nrclinonc.2016.41
21. Giallongo C, Parrinello N, Brundo MV, Raccuia SA, Di Rosa M, La Cava P, et al. Myeloid derived suppressor cells in chronic myeloid leukemia. Front Oncol (2015) 5:107. doi:10.3389/fonc.2015.00107
22. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15(6):2148–57. doi:10.1158/1078-0432.CCR-08-1332
23. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med (2011) 17(9):1094–100. doi:10.1038/nm.2438
24. Fei F, Yu Y, Schmitt A, Rojewski MT, Chen B, Götz M, et al. Dasatinib inhibits the proliferation and function of CD4+CD25+ regulatory T cells. Br J Haematol (2009) 144(2):195–205. doi:10.1111/j.1365-2141.2008.07433.x
25. Nievergall E, Ramshaw HS, Yong AS, Biondo M, Busfield SJ, Vairo G, et al. Monoclonal antibody targeting of IL-3 receptor alpha with CSL362 effectively depletes CML progenitor and stem cells. Blood (2014) 123(8):1218–28. doi:10.1182/blood-2012-12-475194
26. Corthay A. Does the immune system naturally protect against cancer? Front Immunol (2014) 5:197. doi:10.3389/fimmu.2014.00197
27. Stevens WB, Netea MG, Kater AP, van der Velden WJ. ‘Trained immunity’: consequences for lymphoid malignancies. Haematologica (2016) 101(12):1460–8. doi:10.3324/haematol.2016.149252
28. Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol (2013) 6(1):74. doi:10.1186/1756-8722-6-74
29. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27(1):111–22. doi:10.1016/j.immuni.2007.05.016
30. Gismondi A, Stabile H, Nisti P, Santoni A. Effector functions of natural killer cell subsets in the control of hematological malignancies. Front Immunol (2015) 6:567. doi:10.3389/fimmu.2015.00567
31. Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol (2005) 5(3):230–42. doi:10.1038/nri1572
32. de Lavallade H, Khoder A, Hart M, Sarvaria A, Sekine T, Alsuliman A, et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood (2013) 122(2):227–38. doi:10.1182/blood-2012-11-465039
33. Caligiuri MA. Human natural killer cells. Blood (2008) 112(3):461–9. doi:10.1182/blood-2007-09-077438
34. Pierson BA, Miller JS. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood (1996) 88(6):2279–87.
35. Terme M, Borg C, Guilhot F, Masurier C, Flament C, Wagner EF, et al. BCR/ABL promotes dendritic cell-mediated natural killer cell activation. Cancer Res (2005) 65(14):6409–17. doi:10.1158/0008-5472.CAN-04-2675
36. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol (2006) 176(8):5108–16. doi:10.4049/jimmunol.176.8.5108
37. Sconocchia G, Lau M, Provenzano M, Rezvani K, Wongsena W, Fujiwara H, et al. The antileukemia effect of HLA-matched NK and NK-T cells in chronic myelogenous leukemia involves NKG2D-target-cell interactions. Blood (2005) 106(10):3666–72. doi:10.1182/blood-2005-02-0479
38. Davies JO, Stringaris K, Barrett AJ, Rezvani K. Opportunities and limitations of natural killer cells as adoptive therapy for malignant disease. Cytotherapy (2014) 16(11):1453–66. doi:10.1016/j.jcyt.2014.03.009
39. Sivori S, Pende D, Bottino C, Marcenaro E, Pessino A, Biassoni R, et al. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol (1999) 29(5):1656–66. doi:10.1002/(SICI)1521-4141(199905)29:05<1656::AID-IMMU1656>3.0.CO;2-1
40. Costello RT, Knoblauch B, Sanchez C, Mercier D, Le Treut T, Sebahoun G. Expression of natural killer cell activating receptors in patients with chronic lymphocytic leukaemia. Immunology (2012) 135(2):151–7. doi:10.1111/j.1365-2567.2011.03521.x
41. Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood (2002) 99(10):3661–7. doi:10.1182/blood.V99.10.3661
42. Fauriat C, Just-Landi S, Mallet F, Arnoulet C, Sainty D, Olive D, et al. Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood (2007) 109(1):323–30. doi:10.1182/blood-2005-08-027979
43. Ginhoux F, Guilliams M, Naik SH. Editorial: dendritic cell and macrophage nomenclature and classification. Front Immunol (2016) 7:168. doi:10.3389/fimmu.2016.00168
44. Poltorak MP, Schraml BU. Fate mapping of dendritic cells. Front Immunol (2015) 6:199. doi:10.3389/fimmu.2015.00199
45. Mohty M, Isnardon D, Vey N, Brière F, Blaise D, Olive D, et al. Low blood dendritic cells in chronic myeloid leukaemia patients correlates with loss of CD34+/CD38- primitive haematopoietic progenitors. Br J Haematol (2002) 119(1):115–8. doi:10.1046/j.1365-2141.2002.03831.x
46. Mohty M, Jarrossay D, Lafage-Pochitaloff M, Zandotti C, Brière F, de Lamballeri XN, et al. Circulating blood dendritic cells from myeloid leukemia patients display quantitative and cytogenetic abnormalities as well as functional impairment. Blood (2001) 98(13):3750–6. doi:10.1182/blood.V98.13.3750
47. Dong R, Cwynarski K, Entwistle A, Marelli-Berg F, Dazzi F, Simpson E, et al. Dendritic cells from CML patients have altered actin organization, reduced antigen processing, and impaired migration. Blood (2003) 101(9):3560–7. doi:10.1182/blood-2002-06-1841
48. Boissel N, Rousselot P, Raffoux E, Cayuela JM, Maarek O, Charron D, et al. Defective blood dendritic cells in chronic myeloid leukemia correlate with high plasmatic VEGF and are not normalized by imatinib mesylate. Leukemia (2004) 18(10):1656–61. doi:10.1038/sj.leu.2403474
49. Li Y, Lin C, Schmidt CA. New insights into antigen specific immunotherapy for chronic myeloid leukemia. Cancer Cell Int (2012) 12(1):52. doi:10.1186/1475-2867-12-52
50. Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med (2000) 6(9):1018–23. doi:10.1038/79526
51. Butt NM, Rojas JM, Wang L, Christmas SE, Abu-Eisha HM, Clark RE. Circulating bcr-abl-specific CD8+ T cells in chronic myeloid leukemia patients and healthy subjects. Haematologica (2005) 90(10):1315–23.
52. Chen S, Yang L, Chen S, Li Y. TCR zeta chain expression in T cells from patients with CML. Hematology (2009) 14(2):95–100. doi:10.1179/102453309X385241
53. Li Y, Geng S, Du X, Chen S, Yang L, Wu X, et al. Restricted TRBV repertoire in CD4+ and CD8+ T-cell subsets from CML patients. Hematology (2011) 16(1):43–9. doi:10.1179/102453311X12902908411634
54. Zha X, Yan X, Shen Q, Zhang Y, Wu X, Chen S, et al. Alternative expression of TCRzeta related genes in patients with chronic myeloid leukemia. J Hematol Oncol (2012) 5:74. doi:10.1186/1756-8722-5-74
55. Baniyash M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol (2004) 4(9):675–87. doi:10.1038/nri1434
56. Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, Lu S, et al. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest (2003) 111(5):639–47. doi:10.1172/JCI200316398
57. Sopper S, Mustjoki S, White D, Hughes T, Valent P, Burchert A, et al. Reduced CD62L expression on T cells and increased soluble CD62L levels predict molecular response to tyrosine kinase inhibitor therapy in early chronic-phase chronic myelogenous leukemia. J Clin Oncol (2017) 35(2):175–84. doi:10.1200/JCO.2016.67.0893
58. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi:10.1111/j.1600-065X.2010.00923.x
59. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi:10.1084/jem.20090847
60. Mumprecht S, Schurch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood (2009) 114(8):1528–36. doi:10.1182/blood-2008-09-179697
61. Gantt S, Gervassi A, Jaspan H, Horton H. The role of myeloid-derived suppressor cells in immune ontogeny. Front Immunol (2014) 5:387. doi:10.3389/fimmu.2014.00387
62. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
63. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182(8):4499–506. doi:10.4049/jimmunol.0802740
64. Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol (2010) 40(1):22–35. doi:10.1002/eji.200939903
65. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
66. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182(9):5693–701. doi:10.4049/jimmunol.0900092
67. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi:10.1158/0008-5472.CAN-09-2587
68. Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Müller I, et al. Regulation of NK cell function by human granulocyte arginase. J Immunol (2009) 182(9):5259–67. doi:10.4049/jimmunol.0803523
69. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182(1):240–9. doi:10.4049/jimmunol.182.1.240
70. Giallongo C, Parrinello N, Tibullo D, Cava P, Romano A, Chiarenza A, et al. Monocytic myeloid derived suppressor cells (M-MDSC) as prognostic factor in chronic myeloid leukemia patients treated with dasatinib. Blood (2015) 126(23):2767.
71. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi:10.1158/0008-5472.CAN-05-1299
72. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68(13):5439–49. doi:10.1158/0008-5472.CAN-07-6621
73. Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol (2005) 174(5):2591–601. doi:10.4049/jimmunol.174.5.2591
74. Larmonier N, Marron M, Zeng Y, Cantrell J, Romanoski A, Sepassi M, et al. Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol Immunother (2007) 56(1):48–59. doi:10.1007/s00262-006-0160-8
75. Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res (2009) 15(10):3325–32. doi:10.1158/1078-0432.CCR-08-3010
76. Idris SZ, Hassan N, Lee LJ, Md Noor S, Osman R, Abdul-Jalil M, et al. Increased regulatory T cells in acute lymphoblastic leukemia patients. Hematology (2015) 20(9):523–9. doi:10.1179/1607845415Y.0000000025
77. D’Arena G, Laurenti L, Minervini MM, Deaglio S, Bonello L, De Martino L, et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res (2011) 35(3):363–8. doi:10.1016/j.leukres.2010.08.010
78. Zahran AM, Badrawy H, Ibrahim A. Prognostic value of regulatory T cells in newly diagnosed chronic myeloid leukemia patients. Int J Clin Oncol (2014) 19(4):753–60. doi:10.1007/s10147-013-0615-9
79. Bachy E, Bernaud J, Roy P, Rigal D, Nicolini FE. Quantitative and functional analyses of CD4(+) CD25(+) FoxP3(+) regulatory T cells in chronic phase chronic myeloid leukaemia patients at diagnosis and on imatinib mesylate. Br J Haematol (2011) 153(1):139–43. doi:10.1111/j.1365-2141.2010.08453.x
80. Scheich F, Duyster J, Peschel C, Bernhard H. The immunogenicity of Bcr-Abl expressing dendritic cells is dependent on the Bcr-Abl kinase activity and dominated by Bcr-Abl regulated antigens. Blood (2007) 110(7):2556–60. doi:10.1182/blood-2007-01-071001
81. Yong AS, Keyvanfar K, Eniafe R, Savani BN, Rezvani K, Sloand EM, et al. Hematopoietic stem cells and progenitors of chronic myeloid leukemia express leukemia-associated antigens: implications for the graft-versus-leukemia effect and peptide vaccine-based immunotherapy. Leukemia (2008) 22(9):1721–7. doi:10.1038/leu.2008.161
82. Yong AS, Stephens N, Weber G, Li Y, Savani BN, Eniafe R, et al. Improved outcome following allogeneic stem cell transplantation in chronic myeloid leukemia is associated with higher expression of BMI-1 and immune responses to BMI-1 protein. Leukemia (2011) 25(4):629–37. doi:10.1038/leu.2010.325
83. Rezvani K, Grube M, Brenchley JM, Sconocchia G, Fujiwara H, Price DA, et al. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood (2003) 102(8):2892–900. doi:10.1182/blood-2003-01-0150
84. Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S, et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia (2017) 31(3):697–704. doi:10.1038/leu.2016.254
85. Falkenburg JH, Wafelman AR, Joosten P, Smit WM, van Bergen CA, Bongaerts R, et al. Complete remission of accelerated phase chronic myeloid leukemia by treatment with leukemia-reactive cytotoxic T lymphocytes. Blood (1999) 94(4):1201–8.
86. Marijt WA, Heemskerk MH, Kloosterboer FM, Goulmy E, Kester MG, van der Hoorn MA, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci U S A (2003) 100(5):2742–7. doi:10.1073/pnas.0530192100
87. Rezvani K, Yong AS, Tawab A, Jafarpour B, Eniafe R, Mielke S, et al. Ex vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood (2009) 113(10):2245–55. doi:10.1182/blood-2008-03-144071
88. Rezvani K, Brenchley JM, Price DA, Kilical Y, Gostick E, Sewell AK, et al. T-cell responses directed against multiple HLA-A*0201-restricted epitopes derived from Wilms’ tumor 1 protein in patients with leukemia and healthy donors: identification, quantification, and characterization. Clin Cancer Res (2005) 11(24 Pt 1):8799–807. doi:10.1158/1078-0432.CCR-05-1314
89. Greiner J, Ringhoffer M, Taniguchi M, Schmitt A, Kirchner D, Krähn G, et al. Receptor for hyaluronan acid-mediated motility (RHAMM) is a new immunogenic leukemia-associated antigen in acute and chronic myeloid leukemia. Exp Hematol (2002) 30(9):1029–35. doi:10.1016/S0301-472X(02)00874-3
90. Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood (2008) 111(1):236–42. doi:10.1182/blood-2007-08-108241
91. Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, et al. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood (2009) 113(26):6541–8. doi:10.1182/blood-2009-02-202598
92. Maslak PG, Dao T, Krug LM, Chanel S, Korontsvit T, Zakhaleva V, et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood (2010) 116(2):171–9. doi:10.1182/blood-2009-10-250993
93. Smahel M. Antigens in chronic myeloid leukemia: implications for vaccine development. Cancer Immunol Immunother (2011) 60(12):1655–68. doi:10.1007/s00262-011-1126-z
94. Dubrovsky L, Pankov D, Brea EJ, Dao T, Scott A, Yan S, et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood (2014) 123(21):3296–304. doi:10.1182/blood-2014-01-549022
95. Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood (2011) 117(16):4262–72. doi:10.1182/blood-2010-07-299248
96. Radich JP, Dai H, Mao M, Oehler V, Schelter J, Druker B, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A (2006) 103(8):2794–9. doi:10.1073/pnas.0510423103
97. Tarafdar A, Hopcroft LE, Gallipoli P, Pellicano F, Cassels J, Hair A, et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood (2017) 129(2):199–208. doi:10.1182/blood-2016-09-742049
98. Nicolini FE. CML stem cells: evasion for better invasion. Blood (2017) 129(2):141–2. doi:10.1182/blood-2016-11-750554
99. Goldman JM. Chronic myeloid leukemia: a historical perspective. Semin Hematol (2010) 47(4):302–11. doi:10.1053/j.seminhematol.2010.07.001
100. de Castro FA, Palma PV, Morais FR, Simões BP, Carvalho PV, Ismael SJ, et al. Immunological effects of interferon-alpha on chronic myelogenous leukemia. Leuk Lymphoma (2003) 44(12):2061–7. doi:10.1080/1042819031000110973
101. Seggewiss R, Price DA, Purbhoo MA. Immunomodulatory effects of imatinib and second-generation tyrosine kinase inhibitors on T cells and dendritic cells: an update. Cytotherapy (2008) 10(6):633–41. doi:10.1080/14653240802317639
102. Graham SM, Jørgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood (2002) 99(1):319–25. doi:10.1182/blood.V99.1.319
103. Rix U, Hantschel O, Dürnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood (2007) 110(12):4055–63. doi:10.1182/blood-2007-07-102061
104. Lee KC, Ouwehand I, Giannini AL, Thomas NS, Dibb NJ, Bijlmakers MJ. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia (2010) 24(4):896–900. doi:10.1038/leu.2010.11
105. Rohon P, Porkka K, Mustjoki S. Immunoprofiling of patients with chronic myeloid leukemia at diagnosis and during tyrosine kinase inhibitor therapy. Eur J Haematol (2010) 85(5):387–98. doi:10.1111/j.1600-0609.2010.01501.x
106. Mohty M, Jourdan E, Mami NB, Vey N, Damaj G, Blaise D, et al. Imatinib and plasmacytoid dendritic cell function in patients with chronic myeloid leukemia. Blood (2004) 103(12):4666–8. doi:10.1182/blood-2003-09-3220
107. Wang H, Cheng F, Cuenca A, Horna P, Zheng Z, Bhalla K, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood (2005) 105(3):1135–43. doi:10.1182/blood-2004-01-0027
108. Chen CI, Maecker HT, Lee PP. Development and dynamics of robust T-cell responses to CML under imatinib treatment. Blood (2008) 111(11):5342–9. doi:10.1182/blood-2007-12-128397
109. Mustjoki S, Ekblom M, Arstila TP, Dybedal I, Epling-Burnette PK, Guilhot F, et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia (2009) 23(8):1398–405. doi:10.1038/leu.2009.46
110. Nagata Y, Ohashi K, Fukuda S, Kamata N, Akiyama H, Sakamaki H. Clinical features of dasatinib-induced large granular lymphocytosis and pleural effusion. Int J Hematol (2010) 91(5):799–807. doi:10.1007/s12185-010-0565-1
111. Kim DH, Kamel-Reid S, Chang H, Sutherland R, Jung CW, Kim HJ, et al. Natural killer or natural killer/T cell lineage large granular lymphocytosis associated with dasatinib therapy for Philadelphia chromosome positive leukemia. Haematologica (2009) 94(1):135–9. doi:10.3324/haematol.13151
112. Mizoguchi I, Yoshimoto T, Katagiri S, Furusawa J, Chiba Y, Mizuguchi J, et al. Immunological control of chronic myeloid leukemia leading to treatment-free remission. J Hematol Blood Transfus (2014) 2(3):1024.
113. Hassold N, Seystahl K, Kempf K, Urlaub D, Zekl M, Einsele H, et al. Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int J Cancer (2012) 131(6):E916–27. doi:10.1002/ijc.27537
114. Hayashi Y, Nakamae H, Katayama T, Nakane T, Koh H, Nakamae M, et al. Different immunoprofiles in patients with chronic myeloid leukemia treated with imatinib, nilotinib or dasatinib. Leuk Lymphoma (2012) 53(6):1084–9. doi:10.3109/10428194.2011.647017
115. El Missiry M, Adnan Awad S, Rajala HL, Al-Samadi A, Ekblom M, Markevän B, et al. Assessment of bone marrow lymphocytic status during tyrosine kinase inhibitor therapy and its relation to therapy response in chronic myeloid leukaemia. J Cancer Res Clin Oncol (2016) 142(5):1041–50. doi:10.1007/s00432-015-2101-4
116. Binotto G, Frison L, Boscaro E, Zambello R, Lessi F, Parolo A, et al. Comparative analysis of NK receptor and T-cell receptor repertoires in patients with chronic myeloid leukemia treated with different tyrosine kinase inhibitors. Blood (2014) 124(21):5508.
117. Qiu ZY, Xu W, Li JY. Large granular lymphocytosis during dasatinib therapy. Cancer Biol Ther (2014) 15(3):247–55. doi:10.4161/cbt.27310
118. Munn DH. Indoleamine 2,3-dioxygenase, Tregs and cancer. Curr Med Chem (2011) 18(15):2240–6. doi:10.2174/092986711795656045
119. Borg C, Terme M, Taïeb J, Ménard C, Flament C, Robert C, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest (2004) 114(3):379–88. doi:10.1172/JCI21102
120. Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol (2008) 181(10):6955–63. doi:10.4049/jimmunol.181.10.6955
121. Christiansson L, Söderlund S, Mangsbo S, Hjorth-Hansen H, Höglund M, Markevärn B, et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol Cancer Ther (2015) 14(5):1181–91. doi:10.1158/1535-7163.MCT-14-0849
122. Mahon FX, Réa D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre stop imatinib (STIM) trial. Lancet Oncol (2010) 11(11):1029–35. doi:10.1016/S1470-2045(10)70233-3
123. Etienne G, Guilhot J, Rea D, Rigal-Huguet F, Nicolini F, Charbonnier A, et al. Long-term follow-up of the French stop imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol (2017) 35(3):298–305. doi:10.1200/JCO.2016.68.2914
124. Mori S, Vagge E, le Coutre P, Abruzzese E, Martino B, Pungolino E, et al. Age and dPCR can predict relapse in CML patients who discontinued imatinib: the ISAV study. Am J Hematol (2015) 90(10):910–4. doi:10.1002/ajh.24120
125. Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol (2015) 2(12):e528–35. doi:10.1016/S2352-3026(15)00196-9
126. Mahon FX, Richter J, Guilhot J, Hjorth-Hansen H, Almeida A, Janssen J, et al. Cessation of tyrosine kinase inhibitors treatment in chronic myeloid leukemia patients with deep molecular response: results of the Euro-Ski Trial. Blood (2016) 128(22):787.
127. Kumagai T, Nakaseko C, Nishiwaki K, Yoshida C, Ohashi K, Takezako N, et al. Discontinuation of dasatinib after deep molecular response for over 2 years in patients with chronic myelogenous leukemia and the unique profiles of lymphocyte subsets for successful discontinuation: a prospective, multicenter Japanese Trial (D-STOP Trial). Blood (2016) 128(22):791.
128. Clark RE, Polydoros F, Apperley JF, Pocock C, Smith G, Byrne JL, et al. Chronic myeloid leukaemia patients with stable molecular responses (at least MR3) may safely decrease the dose of their tyrosine kinase inhibitor: data from the British Destiny Study. Blood (2016) 128(22):938.
129. Lee SE, Choi SY, Song HY, Kim SH, Choi MY, Park JS, et al. Imatinib withdrawal syndrome and longer duration of imatinib have a close association with a lower molecular relapse after treatment discontinuation: the KID study. Haematologica (2016) 101(6):717–23. doi:10.3324/haematol.2015.139899
130. Rea D, Nicolini FE, Tulliez M, Guilhot F, Guilhot J, Guerci-Bresler A, et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: interim analysis of the STOP 2G-TKI study. Blood (2016) 129(7):846–54. doi:10.1182/blood-2016-09-742205
131. Kimura S. Current status of ABL tyrosine kinase inhibitors stop studies for chronic myeloid leukemia. Stem Cell Investig (2016) 3:36. doi:10.21037/sci.2016.07.08
132. Radich J. When to consider allogeneic transplantation in CML. Clin Lymphoma Myeloma Leuk (2016) 16(Suppl):S93–5. doi:10.1016/j.clml.2016.02.008
133. Hughes TP, Saglio G, Kantarjian HM, Guilhot F, Niederwieser D, Rosti G, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood (2014) 123(9):1353–60. doi:10.1182/blood-2013-06-510396
134. Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boque C, et al. Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-naive chronic myeloid leukemia patients trial. J Clin Oncol (2016) 34(20):2333–40. doi:10.1200/JCO.2015.64.8899
135. Hughes TP, Ross DM. Moving treatment-free remission into mainstream clinical practice in CML. Blood (2016) 128(1):17–23. doi:10.1182/blood-2016-01-694265
136. Rea D, Dulphy N, Henry G, Guilhot J, Guilhot F, Nicolini FE, et al. Low natural killer (NK) cell counts and functionality are associated with molecular relapse after imatinib discontinuation in patients (pts) with chronic phase (CP)-chronic myeloid leukemia (CML) with undetectable BCR-ABL transcripts for at least 2 years: preliminary results from immunostim, on behalf of STIM investigators. Blood (2013) 122(21):856.
137. Takahashi N, Kyo T, Maeda Y, Sugihara T, Usuki K, Kawaguchi T, et al. Discontinuation of imatinib in Japanese patients with chronic myeloid leukemia. Haematologica (2012) 97(6):903–6. doi:10.3324/haematol.2011.056853
138. Burchert A, Müller MC, Kostrewa P, Erben P, Bostel T, Liebler S, et al. Sustained molecular response with interferon alfa maintenance after induction therapy with imatinib plus interferon alfa in patients with chronic myeloid leukemia. J Clin Oncol (2010) 28(8):1429–35. doi:10.1200/JCO.2009.25.5075
139. Kreutzman A, Rohon P, Faber E, Indrak K, Juvonen V, Kairisto V, et al. Chronic myeloid leukemia patients in prolonged remission following interferon-alpha monotherapy have distinct cytokine and oligoclonal lymphocyte profile. PLoS One (2011) 6(8):e23022. doi:10.1371/journal.pone.0023022
140. Burchert A, Saussele S, Eigendorff E, Müller MC, Sohlbach K, Inselmann S, et al. Interferon alpha 2 maintenance therapy may enable high rates of treatment discontinuation in chronic myeloid leukemia. Leukemia (2015) 29(6):1331–5. doi:10.1038/leu.2015.45
141. Tu MM, Mahmoud AB, Makrigiannis AP. Licensed and unlicensed NK cells: differential roles in cancer and viral control. Front Immunol (2016) 7:166. doi:10.3389/fimmu.2016.00166
142. Hughes A, Clarson J, White DL, Ross DM, Hughes TP, Yong AS. Enhanced natural killer and cytotoxic T lymphocyte responses, with decreased monocytic myeloid derived suppressor cells may promote treatment free remission in chronic myeloid leukaemia patients following tyrosine kinase inhibitor cessation. Blood (2016) 128(22):1122.
143. Yoshida C, Iriyama N, Najima Y, Fujisawa S, Wakita H, Chiba S, et al. Association of peripheral regulatory T cells with achievement of deep molecular response in newly diagnosed chronic phase chronic myeloid leukemia treated with dasatinib – the final results of D-First Study. Blood (2016) 128(22):1916.
144. Wesolowski R, Markowitz J, Carson W. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer (2013) 1:10. doi:10.1186/2051-1426-1-10
145. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res (2016) 22(21):5211–22. doi:10.1158/1078-0432.CCR-16-1108
146. Caocci G, Martino B, Greco M, Abruzzese E, Trawinska MM, Lai S, et al. Killer immunoglobulin-like receptors can predict TKI treatment-free remission in chronic myeloid leukemia patients. Exp Hematol (2015) 43(12):1015–8.e1011. doi:10.1016/j.exphem.2015.08.004
147. Yeung DT, Tang C, Vidovic L, White DL, Branford S, Hughes TP, et al. KIR2DL5B genotype predicts outcomes in CML patients treated with response-directed sequential imatinib/nilotinib strategy. Blood (2015) 126(25):2720–3. doi:10.1182/blood-2015-07-655589
Keywords: immunology, chronic myeloid leukemia, treatment-free remission, immune surveillance, deep molecular response
Citation: Hughes A and Yong ASM (2017) Immune Effector Recovery in Chronic Myeloid Leukemia and Treatment-Free Remission. Front. Immunol. 8:469. doi: 10.3389/fimmu.2017.00469
Received: 20 January 2017; Accepted: 05 April 2017;
Published: 24 April 2017
Edited by:
Masoud H. Manjili, Virginia Commonwealth University Massey Cancer Center, USAReviewed by:
Francesco Dazzi, King’s College London School of Medicine, UKShahram Kordasti, King’s College London, UK
Copyright: © 2017 Hughes and Yong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnes S. M. Yong, YWduZXMueW9uZ0BzYS5nb3YuYXU=