 Maria Carmina Castiello1
Maria Carmina Castiello1 Francesca Pala1Lucia Sereni1,2Elena Draghici1Donato Inverso3
Francesca Pala1Lucia Sereni1,2Elena Draghici1Donato Inverso3 Aisha V. Sauer1
Aisha V. Sauer1 Francesca Schena4
Francesca Schena4 Elena Fontana5
Elena Fontana5 Enrico Radaelli6Paolo Uva7Karla E. Cervantes-Luevano8Federica Benvenuti8Pietro L. Poliani5
Enrico Radaelli6Paolo Uva7Karla E. Cervantes-Luevano8Federica Benvenuti8Pietro L. Poliani5 Matteo Iannacone2,3,9Elisabetta Traggiai4
Matteo Iannacone2,3,9Elisabetta Traggiai4 Anna Villa1,10*
Anna Villa1,10* Marita Bosticardo1*
Marita Bosticardo1*
- 1San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Division of Regenerative Medicine, Stem Cells and Gene Therapy, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2Vita-Salute San Raffaele University, Milan, Italy
- 3Dynamics of Immune Responses, Division of Immunology, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 4Laboratory of Immunology and Rheumatic Disease, IGG, Genova, Italy
- 5Department of Molecular and Translational Medicine, Pathology Unit, University of Brescia, Brescia, Italy
- 6VIB11 Center for the Biology of Disease, Center for Human Genetics, KU Leuven, Leuven, Belgium
- 7CRS4, Science and Technology Park Polaris, Pula, Italy
- 8Cellular Immunology, International Centre for Genetic Engineering and Biotechnology (ICGEB), Trieste, Italy
- 9Experimental Imaging Center, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 10Milan Unit, Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche, Milan, Italy
Wiskott–Aldrich syndrome (WAS) is a primary immunodeficiency caused by mutations in the gene encoding the hematopoietic-specific WAS protein (WASp). WAS is frequently associated with autoimmunity, indicating a critical role of WASp in maintenance of tolerance. The role of B cells in the induction of autoreactive immune responses in WAS has been investigated in several settings, but the mechanisms leading to the development of autoimmune manifestations have been difficult to evaluate in the mouse models of the disease that do not spontaneously develop autoimmunity. We performed an extensive characterization of Was−/− mice that provided evidence of the potential alteration in B cell selection, because of the presence of autoantibodies against double-stranded DNA, platelets, and tissue antigens. To uncover the mechanisms leading to the activation of the potentially autoreactive B cells in Was−/− mice, we performed in vivo chronic stimulations with toll-like receptors agonists (LPS and CpG) and apoptotic cells or infection with lymphocytic choriomeningitis virus. All treatments led to increased production of autoantibodies, increased proteinuria, and kidney tissue damage in Was−/− mice. These findings demonstrate that a lower clearance of pathogens and/or self-antigens and the resulting chronic inflammatory state could cause B cell tolerance breakdown leading to autoimmunity in WAS.
Introduction
Wiskott–Aldrich syndrome (WAS) is a complex and severe X-linked disorder characterized by microthrombocytopenia, eczema, immunodeficiency, and increased risk to develop autoimmunity and lymphomas, affecting 1–10 out of a million male newborns (1). The protein encoded by the WAS gene [WAS protein (WASp)] is a hematopoietic-specific regulator of actin nucleation in response to signals arising at the cell membrane (2, 3). WAS-associated autoimmune complications are frequently observed and can occur also after hematopoietic stem cell transplantation (4). The high incidence of autoimmunity in WAS patients indicates a critical role of WASp in the maintenance of central and peripheral tolerance. Indeed, defective function and/or number of natural T regulatory cells and induced T regulatory cells have been shown in WAS patients and in the mouse model by ours and other groups (5–9). However, several recent evidences suggest a role of B cells in the development of autoimmune manifestations in WAS patients. Earlier reports identified B cell anomalies as mainly due to the defective cytoskeletal-dependent processes resulting in decreased migratory ability, adhesion, and homing (10, 11). These defects may be responsible for the inability of WAS B cells in reaching the site of infection and get properly activated. In addition, phenotypic perturbations reported in WAS patients, including marked reduction of CD21/CD35 coreceptor expression and increased representation of CD21low B cell subset (12–14), could explain abnormalities in antigen capture and presentation resulting in a defective maintenance of B cell tolerance. Immune B cell dysregulation has indeed been confirmed by the presence of circulating autoantibodies in both WAS patients (14–16) and Was−/− mice (6, 17). Moreover, in mouse models in which only B cells lacked WASp, the contribution of intrinsic B cell defects in the pathogenesis of autoimmunity has been demonstrated (18, 19).
However, the break of tolerance leading to autoimmunity onset may not be sufficient to induce disease since the contribution of environmental factors seems to be required, as in the case of systemic lupus erythematous (SLE) patients (20). The contribution of external factors to the development of autoimmunity could indeed be critical for WAS patients and could explain the high variability in the frequency of these manifestations, ranging from 25 to 72%, regardless of WASp expression (15, 21, 22). Incomplete clearance of apoptotic cells, bacterial and viral pathogens, due to the impaired functionality of WASp-deficient macrophages (23), could lead to an increased load of antigens. These antigens could be sensed by immune cells through Toll-like receptors (TLRs), whose activation could result in induction of survival and pro-inflammatory signals (24), and may favor the expansion of autoreactive cells in immune-compromised hosts, ultimately leading to autoimmune manifestations in WAS patients.
In this study, we tested the in vivo effect of several chronic stimulations (TLR agonist administrations, apoptotic cell injection, and viral infection) in the Was−/− mouse model, to define whether incomplete clearance of apoptotic cells and/or pathogen antigens could contribute to the induction of autoimmune manifestations in WAS.
Materials and Methods
Mice
C57BL/6 Was−/− mice were kindly provided by Siminovitch (25). Wild-type (wt) and Rag2−/−/γc−/− mice were purchased from Charles River Laboratories Inc. (Calco, Italy). Mice were housed under specific pathogen-free conditions. We used mice ranging from 8 to 12 weeks old, unless otherwise stated.
In Vivo Challenge with TLR Agonists and Apoptotic Cells
Wt and Was−/− mice were treated at 6 weeks of age by intravenous (i.v.) injections of LPS (from Escherichia coli, Serotype O55:B5, Alexis) at a dose of 1 µg/g once a week for 4 weeks or by intraperitoneal (i.p.) injections of oligodeoxynucleotide CpG (ODN1826, CpG-DNA TCCATGACGTTCCTGACGTT, TIB MOLBIOL s.r.l., Genova, Italy) 2 µg/g twice a week for 4 weeks. In parallel, wt and Was−/− control groups received PBS. All animals were sacrificed 1 week after the last injection. For the in vivo challenge with apoptotic cells, syngeneic thymocytes were isolated from thymus of age- and sex-matched wt and Was−/− mice. Cells were cultured overnight in RPMI 1640 supplemented with 2 mM glutamine, 100 IU/mL penicillin, 100 µg/mL streptomycin (Invitrogen), and 100 µM dexamethasone (Sigma-Aldrich) at a concentration of 2 × 107 cells/mL. The apoptotic phenotype was evaluated with Annexin V-PE and 7-AAD staining (Becton Dickinson) by flow cytometry before injections. Mice were treated once a week for 4 weeks with 107 apoptotic cells in sterile PBS injected i.v. in the tail vein. Wt and Was−/− control groups were injected with 107 live cells. Blood and serum were taken from the tail vein before treatment and 7 weeks after the last injection. Proteinuria was determined at the time of sacrifice.
Infection with Lymphocytic Choriomeningitis Virus (LCMV)
wt and Was−/− mice were infected i.v. with 1 × 106 p.f.u. non-cytopathic LCMV (Armstrong strain). Mice were followed for 1 week and then sacrificed to evaluate tissue and serum viral titers, renal damage (proteinuria), specific CD8+ T cell response, and generation of autoantibodies anti-double-stranded DNA (dsDNA). Infectious LCMV in serum and tissues of infected animals was quantified by plaque assay on MC57 cells, as previously described (26). Intracellular IFNγ staining was performed in the presence or absence of GP33 stimulation as described (26). Briefly, 5 × 105 splenocytes were cultured in RPMI medium supplemented with 50 U/mL of IL2 and 1 µL/mL of brefeldin A for 5 h in the presence or absence of the specific LCMV peptide (GP33-41). The cells were stained with monoclonal anti-mouse CD8α antibody (clone 53-6.7; Pharmingen, San Diego, CA, USA) and anti-mouse IFNγ antibody (clone XMG 1.2; Pharmingen) and its isotype control antibody [rat immunoglobulin (Ig) G1]. Samples were acquired on a FACS Canto II system (BD) and analyzed with FlowJo software (Tree Star Inc.).
ELISA
Levels of IgG isotypes, IgM, IgA, and IgE were measured in cell culture supernatants by multiplex assay [Beadlyte Mouse Immunoglobulin Isotyping kit; Millipore] on a Bio-Plex instrument (BioRad, Richmond, VA, USA). Anti-dsDNA antibodies were evaluated by ELISA assay (27). Briefly, polystyrene plates were coated with poly-l-lysine (Sigma-Aldrich, St. Louis, MO) and DNA from calf thymus (Sigma-Aldrich); after coating with 50 µg/mL polyglutamic acid for 45 min and blocking with PBS 3% BSA, serial dilutions of serum starting from 1:20 were incubated overnight. Anti-dsDNA antibodies were detected with alkaline phosphatase (AP)-conjugated goat anti-mouse IgG antibody (Southern Biotech). The score of positivity was assigned to sera that were positive for dilutions of 1:40 or higher. Mouse sera were analyzed to evaluate the presence of antiplatelet antibodies (28). Briefly, wt platelets were isolated from blood collected with the anticoagulant citrate phosphate dextrose and diluted with the same volume of tyrode buffer 1× (5 mM HEPES, 137 mM NaCl, 2.7 mM KCl, 0.4 mM NaH2PO4, 2.8 mM dextrose, pH 7.4). The platelet-rich plasma was centrifuged, and total proteins were extracted from the pellet. Ten micrograms per milliliter of protein extract was coated on 96-well plates and incubated overnight at 4°C. Diluted serum (1:100 and 1:1,000 in blocking buffer) was added and incubated for 2 h. Positivity was revealed by staining with secondary goat anti-mouse IgG-AP antibody (1:2,000 in blocking buffer) followed by pNPP conversion by AP (Sigma-Aldrich). The assay was read at 405 nm on a 680 microplate reader (BioRad).
ELISpot Assay
Immunoglobulin-secreting cells were analyzed by ELISpot assays performed in plates with nitrocellulose membrane coated with goat α-IgG, IgA, IgM (Southern Biotech), or DNA from calf thymus (Sigma-Aldrich). After blocking with PBS 1% BSA, serial dilutions of total B cells (from 2.5 × 105 to 0.3 × 105) were added and incubated o.n. at 37°C. Plates were then incubated with isotype-specific secondary antibodies, followed by streptavidin-HRP and finally developed with 3-amino-9-ethylcarbazole (Sigma-Aldrich) as a chromogenic substrate. Plates were scanned and counted using the Automated ELISA-Spot Assay Video Analysis System (AELVIS, Germany) to determine the number of spots/well.
Immunohistochemistry
Indirect immunohistochemistry was performed on tissue sections from Rag2−/−/γc−/− mice incubated with serum from wt and Was−/− mice (27). For each serum, we analyzed the reactivity to four tissues (thyroid, stomach, small intestine, and salivary gland and pancreas). Autoantibodies were revealed by using a goat anti-mouse IgG-HRP antibody (Molecular Probes; Invitrogen, Carlsbad, CA, USA) and diaminobenzidine (DBA, DAKO) followed by hematoxylin counterstaining (Sigma-Aldrich). Slides were examined on a Zeiss Axioplan2 microscope (Carl Zeiss Microimaging, Thornwood, NY, USA).
Proliferation Assay
Mononuclear cells isolated from spleens of wt and Was−/− mice in FACS buffer (PBS 0.3% BSA 0.1% NaN3) were stained with the following antibodies: anti-CD19 (1D3), anti-CD45R/B220 (RA3-6B2), anti-CD21/35 (7G6), and anti-CD23 (B3B4), all from BD Pharmingen (San Diego, CA, USA). Follicular (FO; CD21loCD23+) and marginal zone (MZ; CD21hiCD23lo) B cells were cell sorted at FACS Aria BD instrument with a purity >95% and a viability ≥85%. Sorted MZ and FO B cells were labeled with 1.5 µM CFSE (Invitrogen) for 8 min at RT. After quenching the labeling reaction by adding FCS, cells were washed twice. Labeled cells (105 cells/well) were cultured in 96-well plates with the following stimuli: 1 µg/mL CpG (ODN1826, InvivoGen, San Diego, CA, USA) or 1 µg/mL LPS (Sigma-Aldrich). Cell proliferation was analyzed at day 3 and 6 of culture by flow cytometry.
Proteinuria and Histological Analysis of Renal Damage
Proteinuria was evaluated using Albustix sticks (Bayer). Proteinuria index was scored as follows: 0 < 30 mg/dL, 1 = 30 mg/dL, 2 = 100 mg/dL, 3 = 300 mg/dL, and 4 ≥ 2,000 mg/dL. Four-micrometer thick sections of formalin-fixed, paraffin-embedded kidneys were cut on a microtome and subjected to routine hematoxylin and eosin staining. Microscopic lesions observed during histopathological examination were classified according to the INHAND system (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice), and the scores were assigned as previously described (29). In particular, histopathologic findings included tubular mineralization, tubular degeneration/regeneration, inflammatory cell foci, cystic lesions, hyaline tubular casts, glomerulopathy, and interstitial fibrosis. For each finding, we scored presence and severity as follows: 0, absent; 1, minimal; 2, mild; 3, moderate; 4, severe. The sum of all scores assigned for each histopathologic finding defines the final nephropathy score.
Autoantibody Array
Screening for a broad panel of IgM and IgG autoantibodies was performed using autoantibody arrays (UT Southwestern Medical Center, Genomic and Microarray Core Facility) as previously described (30). Statistical differences between TLR-induced autoantibody profiles were assessed by Global test (31).
Cytokine Production
Cytokine production was evaluated on serum samples from wt and Was−/− mice before and after administration of LPS, CpG, apoptotic cells, or infection with LCMV, by using the Bio-Plex Pro Mouse Cytokine Th17 Panel A 6-plex (IL6, IL17a, IFNg, IL1b, IL10, TNFa) (Biorad) and following the manufacturer’s instruction. The plate was acquired using the Bio-Plex MAGPIX Multiplex Reader (Biorad).
Statistical Analyses
All results are expressed as median and SD if not stated otherwise. Statistical significance was assessed using the two-tailed Mann–Whitney test. One-way or two-way analysis of variance tests were applied when specified. P values <0.05 were considered significant.
Results
Autoantibody Production by B Cells of Was−/− Mice
To address whether the absence of WASp affects the humoral immune response and results in an excessive reactivity against self-antigens, we tested the presence of autoreactive B cells in different settings. Significantly higher concentrations of all classes of total Igs in the sera (Figure 1A) and increased frequency of IgM- and IgG-secreting B cells were found in Was−/− mice, compared to wt animals (Figure 1B). We then evaluated the presence of circulating autoantibodies by immunohistochemistry incubating sera of wt and Was−/− mice on tissue sections of thyroid, stomach, salivary gland, small intestine, and pancreas derived from Rag2−/−/γc−/− mice, to avoid interference of endogenous Igs. We detected a strong immunoreaction in all tissues incubated with sera of Was−/− mice as opposed to wt animals (Figure 1C), suggesting the presence of autoreactive Igs targeting different tissues in the sera of mutant mice. Because of the reduced platelet counts in Was−/− mice, we evaluated whether the animals could present antiplatelet antibodies in the peripheral blood. As shown in Figure 1D, antiplatelet antibodies were detectable at levels significantly higher in Was−/− mice than in age-matched wt mice (8–10 weeks old). The presence of antibodies against dsDNA was also evaluated in wt and Was−/− mice by ELISA and ELISpot assays. Seventy-five percent of Was−/− mice resulted positive for the presence of anti-dsDNA antibodies, which were completely absent in wt mice (Figure 1E). The increased frequency of B cells producing both IgM or IgG directed against dsDNA in Was−/− compared to wt mice was confirmed in ELISpot assays performed on total splenic B cells (Figure 1F).
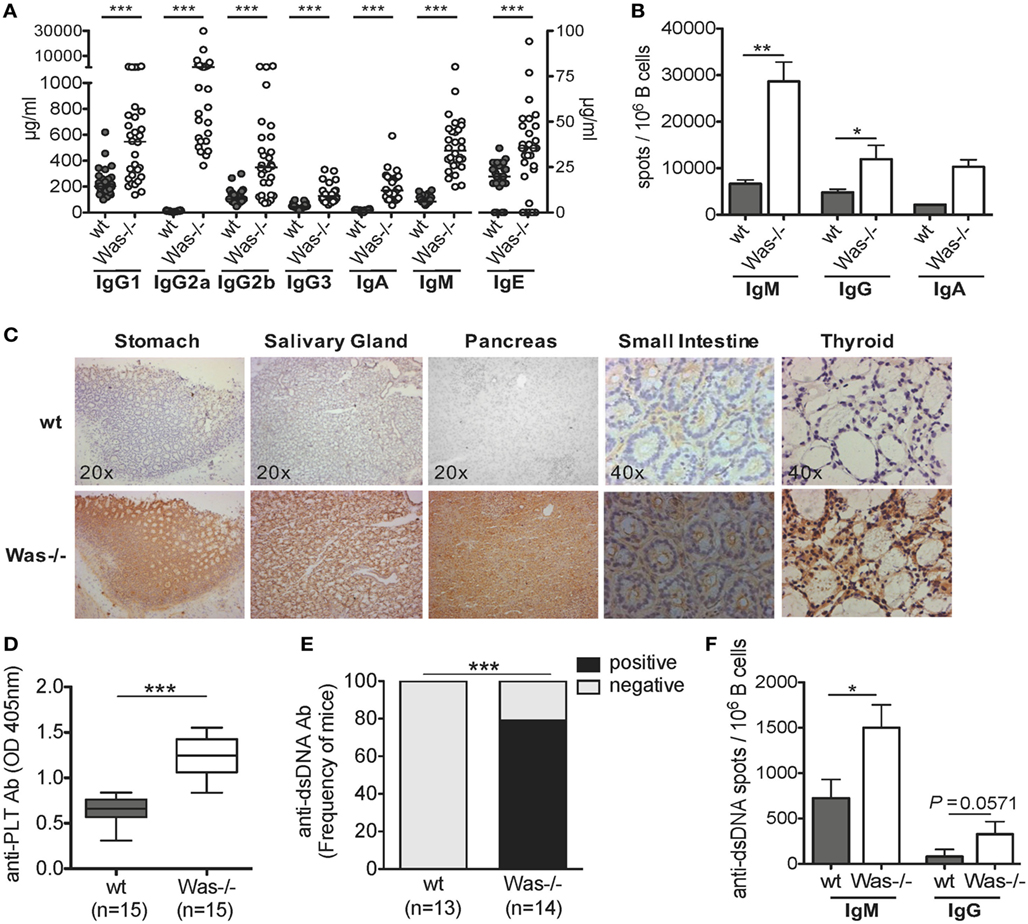
Figure 1. The presence of autoreactive B cells in Was−/− mice. (A) Serum levels of immunoglobulins (Igs) subclasses from wild-type wt (n = 28) and Was−/− (n = 30) mice are summarized in the graph. The scale on the right Y axis is specific for IgE levels. Median value is indicated as bar. (B) IgM-, IgG-, or IgA-secreting cells were evaluated by ELISpot assay performed on B cells isolated from the spleens of wt and Was−/− mice (IgM and IgG: wt n = 6, Was−/− n = 7; IgA: n = 3). The mean and SEM are shown. (C) Tissue-specific autoantibodies were detected by indirect immunohistochemistry performed on sections derived from different tissues from Rag2−/−/γc−/− mice incubated with sera of wt (n = 6) and Was−/− mice (n = 6). (D) The levels of autoantibodies against platelets present in the sera of wt and Was−/− mice were assessed by ELISA and expressed as optical density. Box and whisker plot shows the maximum and minimum value obtained. (E) Percentage of mice positive or negative for circulating autoantibodies against double-stranded DNA (dsDNA) evaluated in the sera of wt and Was−/− mice and assessed by ELISA. (F) Frequency of Ig-secreting cells against dsDNA was assessed by ELISpot assay performed with total splenic wt and Was−/− B cells. (A,B,D,F) Statistical significant differences were analyzed using the Mann–Whitney test (***P < 0.0001, **P < 0.005, and *P < 0.05). (E) Difference was statistically calculated by χ2 test (***P < 0.0005).
In conclusion, these data show that Was−/− B cells have a higher level of self-reactivity than wt B cells, highlighting a potential breakdown of B cell tolerance in WAS.
In Vivo TLR Ligand Administration Induces Production of Autoantibodies and Tissue Damage in Was−/− Mice
Given the increased production of total Igs and autoantibodies, we questioned whether the response of Was−/− B cells to in vitro stimuli could also be altered. We thus evaluated whether the response to TLRs and their ligands, important regulators of B cell functions (32), was dysfunctional in Was−/− B cells. To this aim, we tested the proliferative response of mature splenic B cells to LPS (TLR4 ligand) and CpG-containing DNAs (TLR9 ligand). To avoid biases in the proliferation assays, due to the well-known perturbation of the mature B cell subsets in the spleen of Was−/− mice with a marked reduction of MZ B cells (11, 27), the experiments were performed on sorted MZ and FO B cells. As expected, both wt and Was−/− FO B cells were unresponsive to low doses of LPS stimulation (33, 34), while stimulation with CpG resulted in a higher proliferation of FO B cells from Was−/− mice compared to wt mice (Figure 2A). Was−/− MZ B cells showed a significantly higher proliferation than wt MZ B cells to both TLR4 and TLR9 stimulations. We did not find differences in the expression of TLR4 and TLR9 in Was−/− FO and MZ B cells compared to wt B cells (data not shown). These results indicate that Was−/− B cells are hyperproliferating to TLR-dependent stimuli when compared to wt B cells.
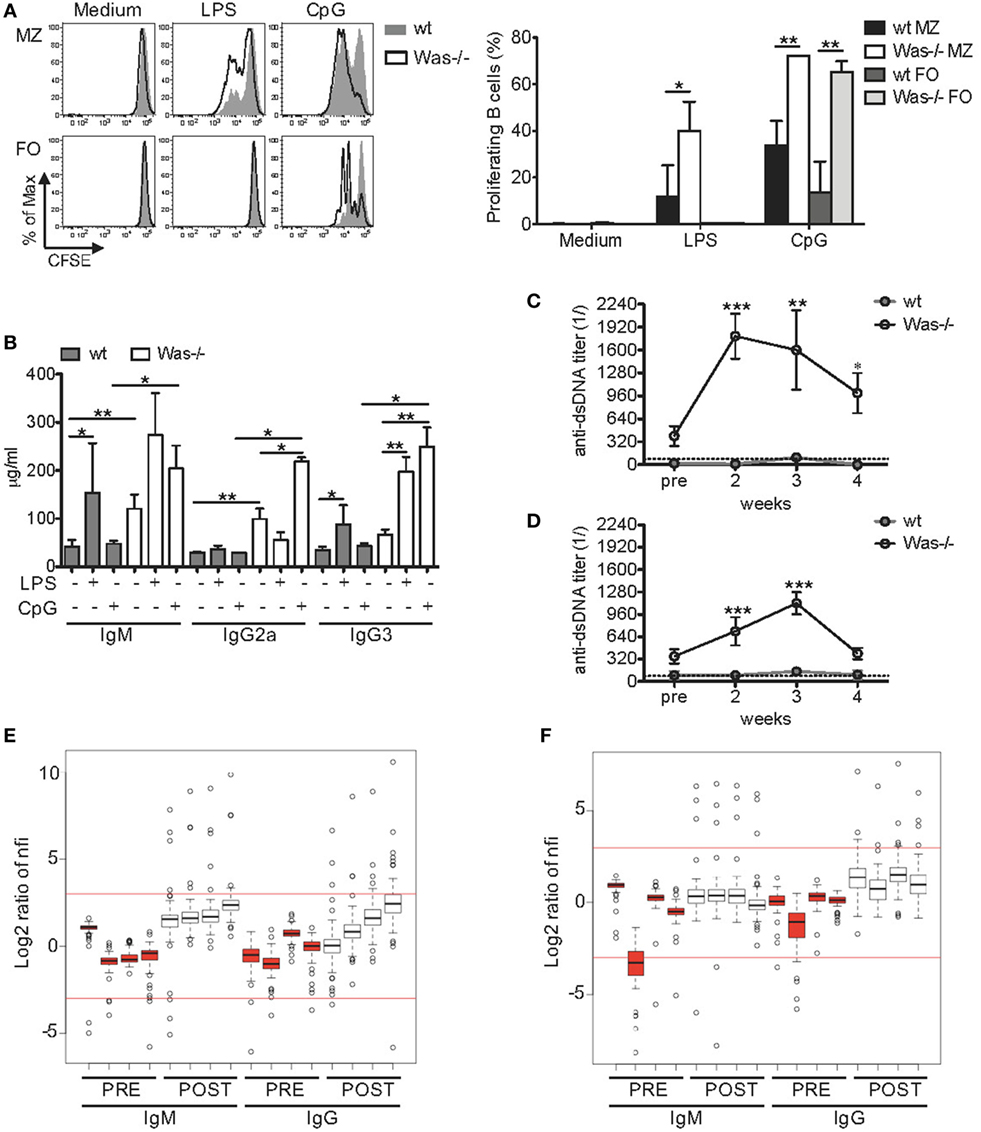
Figure 2. Increased in vitro and in vivo response of Wiskott–Aldrich syndrome protein-deficient B cells to Toll-like receptor agonists. (A) Proliferation capacity was evaluated by CFSE dilution assay in sorted marginal zone (MZ) and follicular (FO) B cells isolated from spleen of wild-type (wt) and Was−/− mice and stimulated for 72 h with LPS (1 µg/mL), CpG (1 µg/mL), or medium alone. Representative histograms are shown in the left panel, and mean values ±SD are indicated in the bar graphs (three independent experiments). Significant differences are calculated with two-way analysis of variance (ANOVA) (**P < 0.01, and *P < 0.05). (B) Total immunoglobulin (Ig) M, IgG2a, and IgG3 levels were measured in the sera of wt and Was−/− mice receiving repeated injections of LPS, CpG, or PBS. Statistical differences were analyzed with the Mann–Whitney test (**P < 0.005 and *P < 0.05). (C,D) Kinetics of serum titers of anti-double-stranded DNA (dsDNA) antibodies circulating in wt and Was−/− mice after in vivo administration of LPS (C) or CpG (D) were evaluated by ELISA. Dotted lines indicate the serum titer considered negative for anti-dsDNA antibodies. Statistical differences were evaluated with two-way ANOVA (***P < 0.001, **P < 0.01, and *P < 0.05). (E,F) The positivity of serum IgM or IgG antibodies to 74 autoantigens was analyzed in the sera of Was−/− mice (n = 4) by an autoantibody array after in vivo administration of LPS (E) or CpG (F). The signal intensity of the autoantibodies before (PRE, red) and after (POST, white) the treatments was normalized for the background fluorescence, and the normalized fluorescence intensities (nfis) are shown as log2 ratio as respect to the average nfi of PRE Was−/− mice.
Improper activation of TLRs could favor the expansion of autoreactive cells (35). To assess the role played by pattern recognition receptors in WAS autoimmunity, we evaluated the in vivo response of Was−/− mice to repeated TLR stimulations, a condition mimicking chronic inflammatory state. In fact, in WAS, it has been postulated that a chronic inflammatory state, due to an incomplete pathogen clearance, could disrupt immunological tolerance. We treated wt and Was−/− mice by i.v. injections of LPS once a week or by i.p. injections of CpG twice a week for 4 weeks. In parallel, control groups for wt and Was−/− mice received PBS. All animals were sacrificed 1 week after the last dose (scheme of treatment is summarized in Figure S1A in Supplementary Material). At the time of sacrifice, both LPS and CpG treatments caused a mild change in the amount of circulating Igs in wt mice, with only an increase in IgM and IgG3 in response to LPS (Figure 2B). On the contrary, LPS and CpG treatments significantly affected the amount of circulating Igs in Was−/− mice (Figure 2B). In particular, IgG3 was increased in response to LPS and IgG2a and IgG3 in response to CpG. Moreover, the levels of IgM, IgG2a, and IgG3 were all significantly higher in Was−/− mice after CpG treatment, compared to wt mice (Figure 2B). To evaluate the type of Igs circulating in treated mice, we monitored the kinetics of autoantibody production by measuring the levels of antibodies anti-dsDNA every week starting at 2 weeks after the first dose. In wt mice, the titers of anti-dsDNA antibodies were under or just above the threshold of positivity at all time points analyzed (Figures 2C,D). On the contrary, Was−/− mice showed high autoantibody titers, starting at 2 weeks from the first dose, which remained high until the end of treatment with both LPS and CpG (Figures 2C,D). Interestingly, LPS administration induced a more rapid production of autoantibodies, reaching titers higher than those achieved upon CpG administration. On the basis of these results, we analyzed the profile of circulating autoantibodies by performing an autoantibody array on the sera of Was−/− mice before and after the in vivo administrations of LPS and CpG to screen the positivity of IgM or IgG antibodies to 74 autoantigens (30). We noticed that Was−/− mice, injected with LPS or CpG, showed high normalized fluorescence intensity (nfi) values for many autoantigens tested with several outliers above the cutoff of +3 (which corresponds to a fold increase of 8 compared to the nfi before treatment) (Figures 2E,F). The specificity and the signal intensity of autoantibodies detected in both IgM and IgG subclasses are represented in the heat maps of LPS- and CpG-treated groups (Figures S2A,B in Supplementary Material, respectively). LPS injections in Was−/− mice led to an increased production of autoantibodies belonging to both IgM and IgG isotypes, even though the latter did not reach statistical significance (P = 0.0571) (Figure S2A in Supplementary Material). Instead, both IgM and, even more, IgG autoantibodies were significantly increased after in vivo CpG administration in Was−/− mice (Figure S2B in Supplementary Material). The specificity of autoantibodies produced by Was−/− mice challenged with LPS or CpG covered a broad panel of autoantigens that included antinuclear antibodies (with anti-dsDNA, anti-histone H2a or H3, anti-KU, antiribonucleoprotein complex, and antitopoisomerase Scl70), antineutrophil cytoplasmic antibodies [anti-bactericidal permeability-increasing protein (BPI), antiglomerular basement membrane, antilaminin, antimatrigel], anticollagen antibodies, and organ-specific autoantibodies such as antithyroglobulin protein (found in thyroid cells) or the liver-specific antiliver cytosol. In particular, three autoantibodies, of both IgM and IgG subclasses, were produced in Was−/− mice independently of the treatment: BPI (antimicrobial protein that binds and neutralizes LPS of Gram-negative bacteria) (36), anti-Ku antibody (a rare type of antinuclear antibody typical of myositis) (37), and anti-SRP54 (signal recognition particle involved in targeting secretory proteins to the rough endoplasmic reticulum membrane) (38).
To evaluate the presence of tissue damage caused by the chronic treatment with LPS and CpG, we analyzed the proteinuria of mice and found an increased proteinuria index in Was−/− mice receiving LPS compared to wt mice (Figure 3A). This difference was not present after CpG administration (Figure 3A). To validate these data, we performed a histopathological examination of kidney sections. Microscopic lesions observed were classified according to the INHAND system, and a nephropathy score was assigned as described in Section “Material and Methods.” As shown in Figure 3B, LPS administration caused a significant increase in frequency and/or severity of renal lesions as compared to wt mice. Also the Was−/− group treated with CpG showed a high nephropathy score, although it was not statistically different from wt (P = 0.0545). We could observe a glomerulopathy in Was−/− mice challenged with both LPS and CpG, while the renal parenchyma was unaffected and presented normal renal corpuscles in treated wt mice (Figure 3C).
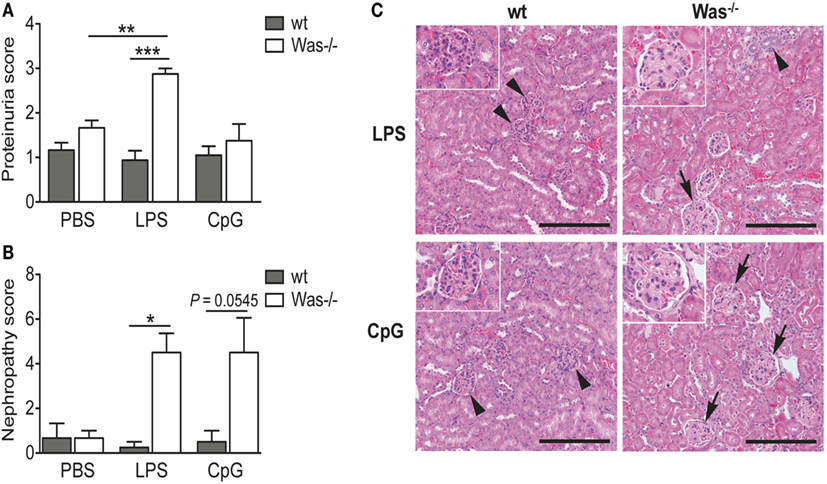
Figure 3. Toll-like receptor-induced kidney damage in Was−/− mice. (A) Proteinuria was determined at the time of sacrifice of mice treated with PBS, LPS, or CpG (n = 5 per group), and the score was assigned starting from 0 (absence or trace of proteins, <30 mg/dL) to 4 (heavy proteinuria, >2,000 mg/dL). (B) The nephropathy score was based on the frequency and the severity of different renal lesions observed in wild-type (wt) and Was−/− mice (n = 5 per group) and ranged from 0, with no lesions, to 4, with severe histopathological changes. Mean values and SEM are reported in the graphs. The Mann–Whitney test was applied to assess whether differences between wt and Was−/− were significant (***P < 0.0001, **P < 0.005, and *P < 0.05). (C) Hematoxylin and eosin staining of kidney sections of wt and Was−/− mice after LPS or CpG treatment. Unaffected renal parenchyma with normal corpuscles is shown in wt mice (arrowheads). Glomerulopathy (arrows) characterized by segmental to global hyaline thickening of glomerular basement membrane with hypertrophic cells populating the affected mesangiocapillary areas was present in Was−/− mice. Scale bar = 200 µm (main panel) and 100 µm (inset).
Altogether, these data demonstrate that in vivo TLR4 and TLR9 stimulations trigger activation of autoreactive B cells leading to increased production of autoantibodies and renal damage in Was−/− mice.
In Vivo Response to Challenge with Apoptotic Cells
An antigen overload in immunodeficient conditions could trigger development of autoimmunity. To test the effect of an overload of apoptotic cells on the development of autoimmunity in Was−/− mice, we i.v. injected syngeneic apoptotic cells in wt and Was−/− mice, once a week for 4 weeks, to induce an overload of self-antigens. Live cells were injected in control groups. Seven weeks after the last administration of apoptotic cells, we sacrificed the mice and examined the response to apoptotic cell injections in terms of levels of autoantibodies against dsDNA and proteinuria (schematic representation of experimental protocol is shown in Figure S1B in Supplementary Material). We observed that Was−/− mice injected with apoptotic cells produced significantly higher levels of anti-dsDNA antibodies compared with wt mice treated with apoptotic cells or with the same group before treatment (Figure 4A). The reaction was specific to apoptotic cells, since in mice injected with live cells, the autoantibody levels were higher than in wt mice, but similar to those of Was−/− mice before treatment. In addition, the increased response to apoptotic cells was associated with an increased proteinuria, suggestive of kidney tissue damage, only in Was−/− mice injected with apoptotic cells (Figure 4B). Immunohistochemical analysis of splenic sections stained with B220 and peanut agglutinin showed a reduced frequency of germinal centers on the total number of foci in Was−/− mice injected with apoptotic cells compared to wt mice challenged with apoptotic cells or Was−/− mice injected with live cells, likely indicating different kinetics of germinal center formation between wt and Was−/− mice (Figures 4C,D).
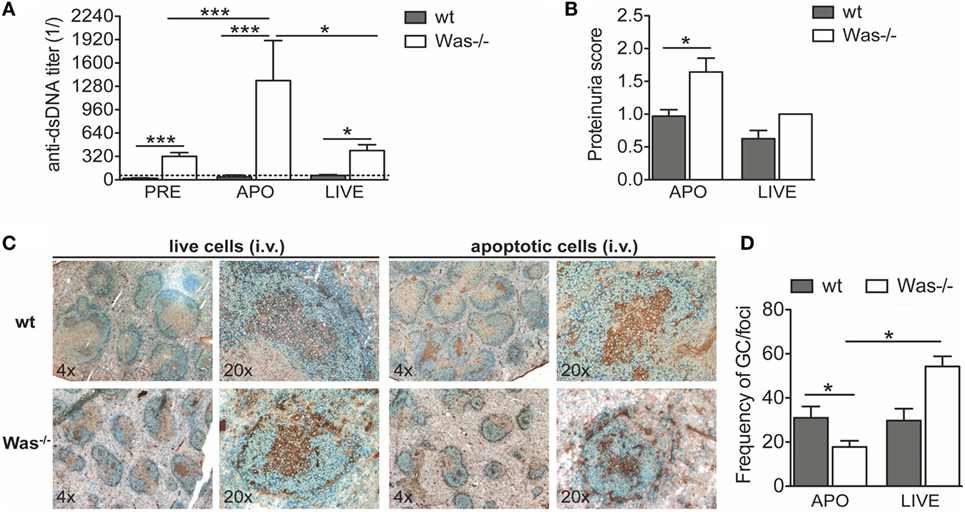
Figure 4. In vivo challenge with apoptotic cells triggered autoreactive B cells and kidney damage in Was−/− mice. (A) Serum titers of anti-double-stranded DNA (dsDNA) circulating antibodies in wild-type (wt) and Was−/− mice evaluated by ELISA before (PRE, n = 16 per group) and 8 weeks after the first injection with apoptotic (APO, n = 7–8 per group) or live cells (LIVE, n = 4 per group). (B) Proteinuria scores were determined at the time of the sacrifice. (C) Splenic sections of wt and Was−/− mice treated with live or apoptotic cells were stained with peanut agglutinin (PNA) (brown) and B220 Ab (blue). (D) The frequency of germinal centers on the total number of foci was evaluated by immunohistochemistry analysis of splenic sections stained with PNA and B220 Ab. Mean values ±SD are reported in the graphs. Mann–Whitney test was applied to assess the significant differences between wt and Was−/− groups (***P < 0.0001 and *P < 0.05).
These results demonstrate that Was−/− B cells have a lower activation threshold than wt B cells and that an overload of self-antigens is sufficient to overcome tolerance mechanisms.
In Vivo Response to Viral Infection
To test whether also incomplete pathogen clearance following viral infection could disrupt immunological tolerance and trigger development of autoimmunity, we performed acute LCMV infection in Was−/− mice. wt and Was−/− mice were infected i.v. with 1 × 106 p.f.u. LCMV (Armstrong strain). Mice were monitored for 7 days and then sacrificed. We observed a 30% mortality in Was−/− mice (data not shown), as they were unable to completely clear the infection. In fact, higher viral titers were detected in the serum of mutant mice at day 4 in the serum and at day 7 in the liver (Figure 5A). Consistently, in vitro stimulation of CD8+ T cells obtained from the spleens of infected mice with GP33-specific LCMV peptide revealed a decreased CD8-mediated specific response to the virus, as shown by the reduced production of IFNγ in Was−/− mice (Figure 5B). We then evaluated the effect of acute LCMV infection on the generation of autoantibodies. Serum from LCMV-infected Was−/− mice showed a significantly increased positivity for anti-dsDNA antibodies compared to wt animals (Figure 5C). Finally, we evaluated renal tissue damage by dosing proteinuria, and we detected a significant increase of proteinuria score at day 7 after infection, only in Was−/− mice (Figure 5D).
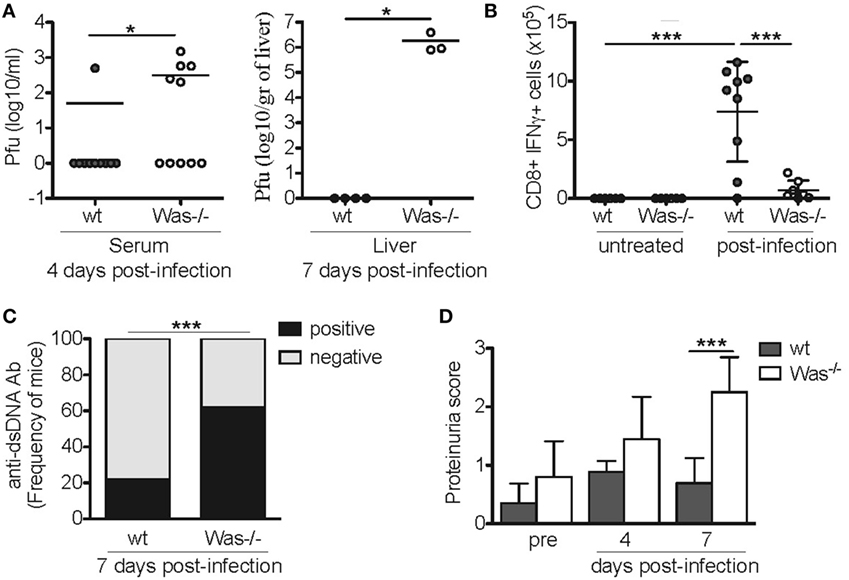
Figure 5. Lymphocytic choriomeningitis virus (LCMV) infection in Was−/− mice. (A) Quantification by plaque assay of the viral titers in the serum (left panel) and liver (right panel) of wild-type (wt) and Was−/− mice at the indicated time points after LCMV arm infection (106 p.f.u./mouse). Mann–Whitney test was applied to assess the significant differences between wt and Was−/− groups (*P < 0.05). (B) Assessment by intracellular IFNγ staining of the absolute number of GP33-specific CD8+ T cells recovered from the spleens of infected animals at the time of sacrifice. Mean values ±SD are reported in the graphs, and significant differences between wt and Was−/− groups were evaluated by one-way ANOVA test (***P < 0.0001). (C) Graph shows the percentage of mice positive or negative for circulating autoantibodies against double-stranded DNA (dsDNA) after LCMV arm infection, evaluated in the sera by ELISA (n = 10 mice per group). Significant differences were statistically calculated by χ2 test (***P < 0.0005). (D) Proteinuria scores were assessed before (pre) and 4 and 7 days after LCMV arm infection. Two-way ANOVA test was applied to assess the significant differences between wt and Was−/− groups (***P < 0.001, n = 10 mice per group).
Taken together, our results indicate that incomplete clearance of viral infections can lead to a break of tolerance and trigger an autoimmune-like response in Was−/− mice.
Increased Production of Pro-inflammatory Cytokines in Was−/− Mice after In Vivo Chronic Stimulations
We performed in vivo chronic stimulation in Was−/− mice by means of administration of TLR agonists (LPS and CpG), injection of apoptotic cells, and LCMV infection. In all cases, we observed the increase of autoantibody production and the development of signs of autoimmunity, which could indicate a common mechanism of peripheral break of tolerance induction in Was−/− mice. To this end, we tested whether chronic stimulations might induce an inflammatory state that could trigger the activation of potentially autoreactive B cells in Was−/− mice by evaluating the production of pro-inflammatory cytokines (IL6, IL17a, IFNg, IL1b, IL10, and TNFa) in the serum of animals, before and after in vivo chronic stimulations. As shown in Figure 6, production of both IL6 and IL17a was strongly increased in Was−/− mice, compared to the wt counterpart, after all different chronic stimulations (LPS, CpG, apoptotic cells, and LCMV infection). Upon LPS administration, also IFNg production was increased in Was−/− mice (Figure S3 in Supplementary Material). No difference in the production of the other cytokines evaluated (IL1b, IL10, and TNFa) was found between wt and Was−/− mice (data not shown).
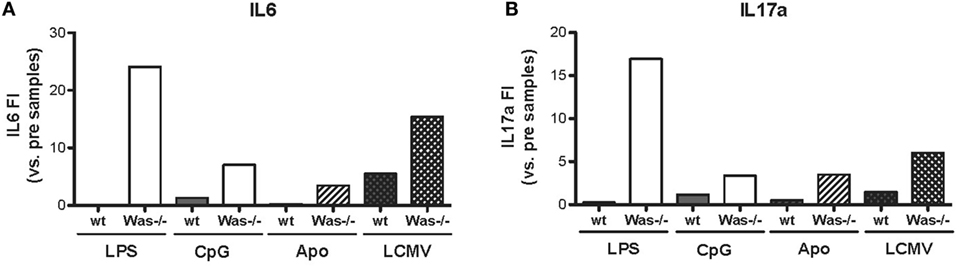
Figure 6. Cytokine production in Was−/− mice after in vivo chronic stimulations. The fold increase of IL6 (A) and IL17a (B) concentrations was calculated as the ratio between the mean value of cytokine production after the indicated in vivo treatment over the mean value before the treatment for each group of mice [LPS/CpG, n = 4 mice; Apo, n = 7–8 mice; lymphocytic choriomeningitis virus (LCMV), n = 5 mice].
Discussion
Many cellular defects contributing to the immunopathology of WAS have been identified over the years; however, the mechanisms leading to the development of autoimmunity still remain not completely understood. In particular, although several groups have demonstrated that the B cell intrinsic defect contributes to WAS-related autoimmunity (12, 18, 19, 39, 40), the pathogenic mechanisms are still matter of study. In this article, we took advantage of the Was−/− mouse model to gain new insights in the mechanisms responsible for the humoral autoimmunity observed in the absence of WASp.
In naïve Was−/− mice, we found high levels of circulating Igs and increased frequency of Ig-secreting cells, plasma cells, and GC B cells (data not shown), suggesting a critical role for WASp in the differentiation toward Ig-secreting cells, both in vitro and ex vivo, in line with results previously published (19, 41, 42). Was−/− mice are also characterized by the presence of autoreactive B cells, which produce autoantibodies against dsDNA, tissue antigens, and platelets. Antinuclear and anti-dsDNA antibodies were initially described in WAS by Humblet-Baron et al. (6) and later observed by other groups in Was−/− mice with different gene targeting strategies and/or genetic mutations/backgrounds (17–19, 39). Thus, the presence of autoantibodies is a consistent finding of Was−/− mice, often without any overt autoimmune manifestations. In fact, only the 129Sv Was−/− strain develops spontaneous, radiation-induced colitis, which resembles autoimmune inflammatory bowel disease (43), and IgA nephropathy-like glomerulonephritis (44, 45).
We showed a hyperactivation of WASp-negative B cells to TLR-dependent stimuli both in vitro and in vivo. Alterations in the B cell cytoskeleton of Was−/− mice could influence their activation threshold, and, in this respect, it is known that membrane lipid rafts participate in many of the cell surface events involved in B cell activation, including signaling by the BCR, endocytosis of antigen bound to the BCR, and TLR activation (46, 47). We know that WASp is required for the dynamics of lipid rafts, which are necessary for immunological synapse assembly and amplification of downstream signals during T-cell activation (48, 49). Thus, it is likely that an alteration of the movements of lipid rafts in WASp-negative B cells could sustain the abnormal responses to TLR engagement.
It has been demonstrated that MyD88 signaling is essential for development of systemic autoimmune disease in Was−/− chimeras (18). Accordingly, we observed that in vivo triggering of TLRs stimulates the production of anti-dsDNA autoantibodies with renal damage in Was−/− mice, demonstrating for the first time the in vivo hyperresponsiveness of WASp-negative B cells to TLR ligands. These findings suggest that, in the absence of WASp, improper activation of TLRs could favor expansion of autoreactive B cells, as also observed in SLE (50). DNA ligands for TLR9 may be provided in vivo by apoptotic bodies that are incompletely cleared and could lead to uncontrolled activation of the TLR9-MyD88 pathway promoting anti-DNA autoantibody generation (51, 52). Delayed phagocytosis of apoptotic cells has been reported in WASp deficiency (23) and proposed as another potential mechanism of loss of peripheral tolerance (53). We indeed demonstrated here that an overload of apoptotic cells perturbs the tolerance threshold resulting in high autoantibodies titers in Was−/− mice. In particular, the overload of apoptotic cells activates autoreactive B cells, which have increased access to self-antigens derived from apoptotic cells, thus inducing the increased production of autoantibodies in Was−/− mice. However, we detected smaller GC in Was−/− mice after injection of apoptotic cells, compared to wt mice, differently from what has been described in literature (41). We believe that there is no discrepancy with our results, since we evaluated GC at a later time point compared to the article by Dahlberg and colleagues. We think that the germinal centers in our mice were past their maximum expansion, and the fact that in Was−/− mice they were even smaller than in wt mice could indicate that they were more exhausted due to the extensive Ig production and proliferation typical of Was−/− mice after in vivo stimuli.
Along the same line, we tested whether the infection of Was−/− mice with viral pathogens, e.g., LCMV, could trigger the development of autoimmunity. Indeed, Was−/− mice infected with LCMV showed a defective clearance of the virus, and the resulting chronic exposure to viral antigens or the persistence of an inflammatory state led to the development of signs of autoimmunity, with an increased production of anti-dsDNA autoantibodies and the presence of renal tissue damage.
In summary, all chronic stimulations tested in Was−/− mice were able to generate similar phenotypes, characterized by an increased production of autoantibodies and signs of autoimmunity, including proteinuria and renal tissue damage. Thus, we hypothesize that a similar mechanism of break of tolerance might take place in Was−/− mice. Indeed, we saw that all treatments led to an overproduction of pro-inflammatory cytokines, such as IL6 and IL17a, only in Was−/− mice and not in wt animals. These results indicate that the chronic stimulations were inducing a persistent inflammatory state able to promote the expansion of the potentially autoreactive B cells present in Was−/− mice, allowing them escape tolerance mechanisms and become pathogenic. These mechanisms, together with defective T cell tolerance present in WAS, could add an additional layer of immune dysregulation contributing to development of autoimmunity in WAS.
Ethics Statement
This study was carried out in accordance with the recommendations of the Italian Ministry of Health. The protocol was approved by the Animal Care and Use Committee of the San Raffaele Scientific Institute (IACUC 406 and 557).
Author Contributions
MC, MI, ET, AV, and MB designed the experiments. MC, FP, ED, LS, DI, AS, FS, EF, KC-L, and ER performed the experiments. MC, PU, FB, PP, MI, ET, AV, and MB analyzed the data. MI and ET revised the manuscript. MC, AV, and MB wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank H. D. Ochs and L. D. Notarangelo for the anti-WASp antibody. The autoantibody arrays (UT Southwestern Medical Center, Genomic and Microarray Core Facility) were performed in collaboration with Q.-Z. Li (Center for Immunology, University of Texas Southwestern Medical Center at Dallas, Dallas TX, USA).
Funding
This work was supported by grants from the Fondazione Telethon Italia (TGT11A02 to AV and MB; TGT16F03 to MB), European Union (project CELL PID FP7 no. 261387 to AV), ERC grant (281648 to MI), and Italian Association for Cancer Research (AIRC grant 15350 to MI). DI was funded by a EMBO long-term fellowship program (EMBO ALTF 1018-2016). LS conducted this study as partial fulfillment of her PhD in Molecular Medicine, Program in Basic and Applied Immunology, Vita-Salute San Raffaele University, Milan, Italy.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00490/full#supplementary-material.
References
1. Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol (2006) 117(4):725–38. doi: 10.1016/j.jaci.2006.02.005
2. Stewart DM, Treiber-Held S, Kurman CC, Facchetti F, Notarangelo LD, Nelson DL. Studies of the expression of the Wiskott-Aldrich syndrome protein. J Clin Invest (1996) 97(11):2627–34. doi:10.1172/JCI118712
3. Symons M, Derry JM, Karlak B, Jiang S, Lemahieu V, McCormick F, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell (1996) 84(5):723–34. doi:10.1016/S0092-8674(00)81050-8
4. Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood (2008) 111(1):439–45. doi:10.1182/blood-2007-03-076679
5. Adriani M, Aoki J, Horai R, Thornton AM, Konno A, Kirby M, et al. Impaired in vitro regulatory T cell function associated with Wiskott-Aldrich syndrome. Clin Immunol (2007) 124(1):41–8. doi:10.1016/j.clim.2007.02.001
6. Humblet-Baron S, Sather B, Anover S, Becker-Herman S, Kasprowicz DJ, Khim S, et al. Wiskott-Aldrich syndrome protein is required for regulatory T cell homeostasis. J Clin Invest (2007) 117(2):407–18. doi:10.1172/JCI29539
7. Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, et al. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J Exp Med (2007) 204(2):381–91. doi:10.1084/jem.20061338
8. Marangoni F, Trifari S, Scaramuzza S, Panaroni C, Martino S, Notarangelo LD, et al. WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med (2007) 204(2):369–80. doi:10.1084/jem.20061334
9. Adriani M, Jones KA, Uchiyama T, Kirby MR, Silvin C, Anderson SM, et al. Defective inhibition of B-cell proliferation by Wiskott-Aldrich syndrome protein-deficient regulatory T cells. Blood (2011) 117(24):6608–11. doi:10.1182/blood-2010-12-322834
10. Westerberg L, Greicius G, Snapper SB, Aspenstrom P, Severinson E. Cdc42, Rac1, and the Wiskott-Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood (2001) 98(4):1086–94. doi:10.1182/blood.V98.4.1086
11. Westerberg L, Larsson M, Hardy SJ, Fernandez C, Thrasher AJ, Severinson E. Wiskott-Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood (2005) 105(3):1144–52. doi:10.1182/blood-2004-03-1003
12. Castiello MC, Bosticardo M, Pala F, Catucci M, Chamberlain N, van Zelm MC, et al. Wiskott-Aldrich syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans. J Autoimmun (2014) 50:42–50. doi:10.1016/j.jaut.2013.10.006
13. Simon KL, Anderson SM, Garabedian EK, Moratto D, Sokolic RA, Candotti F. Molecular and phenotypic abnormalities of B lymphocytes in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol (2014) 133(3):896–9.e4. doi:10.1016/j.jaci.2013.08.050
14. Castiello MC, Scaramuzza S, Pala F, Ferrua F, Uva P, Brigida I, et al. B-cell reconstitution after lentiviral vector-mediated gene therapy in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol (2015) 136(3):692–702.e2. doi:10.1016/j.jaci.2015.01.035
15. Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics (2003) 111(5 Pt 1):e622–7. doi:10.1542/peds.111.5.e622
16. Schurman SH, Candotti F. Autoimmunity in Wiskott-Aldrich syndrome. Curr Opin Rheumatol (2003) 15(4):446–53. doi:10.1097/00002281-200307000-00012
17. Nikolov NP, Shimizu M, Cleland S, Bailey D, Aoki J, Strom T, et al. Systemic autoimmunity and defective Fas ligand secretion in the absence of the Wiskott-Aldrich syndrome protein. Blood (2010) 116(5):740–7. doi:10.1182/blood-2009-08-237560
18. Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med (2011) 208(10):2033–42. doi:10.1084/jem.20110200
19. Recher M, Burns SO, de la Fuente MA, Volpi S, Dahlberg C, Walter JE, et al. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein causes severe abnormalities of the peripheral B-cell compartment in mice. Blood (2012) 119:2819–28. doi:10.1182/blood-2011-09-379412
20. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol (2006) 6(11):823–35. doi:10.1038/nri1957
21. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr (1994) 125(6 Pt 1):876–85. doi:10.1016/S0022-3476(05)82002-5
22. Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, et al. Clinical course of patients with WASP gene mutations. Blood (2004) 103(2):456–64. doi:10.1182/blood-2003-05-1480
23. Leverrier Y, Lorenzi R, Blundell MP, Brickell P, Kinnon C, Ridley AJ, et al. Cutting edge: the Wiskott-Aldrich syndrome protein is required for efficient phagocytosis of apoptotic cells. J Immunol (2001) 166(8):4831–4. doi:10.4049/jimmunol.166.8.4831
24. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature (2007) 449(7164):819–26. doi:10.1038/nature06246
25. Zhang J, Shehabeldin A, da Cruz LA, Butler J, Somani AK, McGavin M, et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J Exp Med (1999) 190(9):1329–42. doi:10.1084/jem.190.9.1329
26. Guidotti LG, Inverso D, Sironi L, Di Lucia P, Fioravanti J, Ganzer L, et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell (2015) 161(3):486–500. doi:10.1016/j.cell.2015.03.005
27. Bosticardo M, Draghici E, Schena F, Sauer AV, Fontana E, Castiello MC, et al. Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott-Aldrich syndrome. J Allergy Clin Immunol (2011) 127(6):1376–84.e5. doi:10.1016/j.jaci.2011.03.030
28. Sauer AV, Brigida I, Carriglio N, Hernandez RJ, Scaramuzza S, Clavenna D, et al. Alterations in the adenosine metabolism and CD39/CD73 adenosinergic machinery cause loss of Treg cell function and autoimmunity in ADA-deficient SCID. Blood (2012) 119(6):1428–39. doi:10.1182/blood-2011-07-366781
29. Radaelli E, Del Piero F, Aresu L, Sciarrone F, Vicari N, Mattiello S, et al. Expression of major histocompatibility complex class II antigens in porcine leptospiral nephritis. Vet Pathol (2009) 46(5):800–9. doi:10.1354/vp.08-VP-0078-R-FL
30. Li QZ, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol (2007) 147(1):60–70. doi:10.1111/j.1365-2249.2006.03251.x
31. Goeman JJ, van de Geer SA, van Houwelingen HC. Testing against a high dimensional alternative. J R Stat Soc Series B Stat Methodol (2006) 68:477–93. doi:10.1111/J.1467-9868.2006.00551.X
32. Peng SL. Signaling in B cells via toll-like receptors. Curr Opin Immunol (2005) 17(3):230–6. doi:10.1016/j.coi.2005.03.003
33. Snapper CM, Yamada H, Smoot D, Sneed R, Lees A, Mond JJ. Comparative in vitro analysis of proliferation, Ig secretion, and Ig class switching by murine marginal zone and follicular B cells. J Immunol (1993) 150(7):2737–45.
34. Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur J Immunol (1997) 27(9):2366–74. doi:10.1002/eji.1830270935
35. Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol (2011) 23(2):106–12. doi:10.1016/j.smim.2011.01.016
36. Schinke S, Fellermann K, Herlyn K, Reichel PH, Fundke R, Stange EF, et al. Autoantibodies against the bactericidal/permeability-increasing protein from inflammatory bowel disease patients can impair the antibiotic activity of bactericidal/permeability-increasing protein. Inflamm Bowel Dis (2004) 10(6):763–70. doi:10.1097/00054725-200411000-00011
37. Belizna C, Henrion D, Beucher A, Lavigne C, Ghaali A, Levesque H. Anti-Ku antibodies: clinical, genetic and diagnostic insights. Autoimmun Rev (2010) 9(10):691–4. doi:10.1016/j.autrev.2010.05.020
38. Romisch K, Miller FW, Dobberstein B, High S. Human autoantibodies against the 54 kDa protein of the signal recognition particle block function at multiple stages. Arthritis Res Ther (2006) 8(2):R39. doi:10.1186/ar1895
39. Kolhatkar NS, Brahmandam A, Thouvenel CD, Becker-Herman S, Jacobs HM, Schwartz MA, et al. Altered BCR and TLR signals promote enhanced positive selection of autoreactive transitional B cells in Wiskott-Aldrich syndrome. J Exp Med (2015) 212(10):1663–77. doi:10.1084/jem.20150585
40. Pala F, Morbach H, Castiello MC, Schickel JN, Scaramuzza S, Chamberlain N, et al. Lentiviral-mediated gene therapy restores B cell tolerance in Wiskott-Aldrich syndrome patients. J Clin Invest (2015) 125(10):3941–51. doi:10.1172/JCI82249
41. Dahlberg CI, Torres ML, Petersen SH, Baptista MA, Keszei M, Volpi S, et al. Deletion of WASp and N-WASp in B cells cripples the germinal center response and results in production of IgM autoantibodies. J Autoimmun (2015) 62:81–92. doi:10.1016/j.jaut.2015.06.003
42. Volpi S, Santori E, Abernethy K, Mizui M, Dahlberg CI, Recher M, et al. N-WASP is required for B-cell-mediated autoimmunity in Wiskott-Aldrich syndrome. Blood (2016) 127(2):216–20. doi:10.1182/blood-2015-05-643817
43. Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, Liu CH, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity (1998) 9(1):81–91. doi:10.1016/S1074-7613(00)80590-7
44. Shimizu M, Nikolov NP, Ueno K, Ohta K, Siegel RM, Yachie A, et al. Development of IgA nephropathy-like glomerulonephritis associated with Wiskott-Aldrich syndrome protein deficiency. Clin Immunol (2012) 142(2):160–6. doi:10.1016/j.clim.2011.10.001
45. Nguyen DD, Muthupalani S, Goettel JA, Eston MA, Mobley M, Taylor NS, et al. Colitis and colon cancer in WASP-deficient mice require helicobacter species. Inflamm Bowel Dis (2013) 19(10):2041–50. doi:10.1097/Mib.0b013e318295fd8f
46. Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci (2002) 115:2603–11.
47. Gupta N, DeFranco AL. Lipid rafts and B cell signaling. Semin Cell Dev Biol (2007) 18(5):616–26. doi:10.1016/j.semcdb.2007.07.009
48. Dupre L, Aiuti A, Trifari S, Martino S, Saracco P, Bordignon C, et al. Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation. Immunity (2002) 17(2):157–66. doi:10.1016/S1074-7613(02)00360-6
49. Kumari S, Depoil D, Martinelli R, Judokusumo E, Carmona G, Gertler FB, et al. Actin foci facilitate activation of the phospholipase C-gamma in primary T lymphocytes via the WASP pathway. Elife (2015) 4:e04953. doi:10.7554/eLife.04953
50. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med (2006) 203(3):553–61. doi:10.1084/jem.20052438
51. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature (2002) 416(6881):603–7. doi:10.1038/416603a
52. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med (2005) 202(2):321–31. doi:10.1084/jem.20050338
Keywords: Wiskott–Aldrich syndrome, autoimmunity, B cells, toll-like receptors, apoptotic cells, lymphocytic choriomeningitis virus
Citation: Castiello MC, Pala F, Sereni L, Draghici E, Inverso D, Sauer AV, Schena F, Fontana E, Radaelli E, Uva P, Cervantes-Luevano KE, Benvenuti F, Poliani PL, Iannacone M, Traggiai E, Villa A and Bosticardo M (2017) In Vivo Chronic Stimulation Unveils Autoreactive Potential of Wiskott–Aldrich Syndrome Protein-Deficient B Cells. Front. Immunol. 8:490. doi: 10.3389/fimmu.2017.00490
Received: 09 February 2017; Accepted: 10 April 2017;
Published: 02 May 2017
Edited by:
Fabio Candotti, Vaudois University Hospital Centre (CHUV), SwitzerlandReviewed by:
Lisa S. Westerberg, Karolinska Institutet, SwedenKohsuke Imai, Tokyo Medical and Dental University, Japan
Copyright: © 2017 Castiello, Pala, Sereni, Draghici, Inverso, Sauer, Schena, Fontana, Radaelli, Uva, Cervantes-Luevano, Benvenuti, Poliani, Iannacone, Traggiai, Villa and Bosticardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Villa, dmlsbGEuYW5uYUBoc3IuaXQ=;
Marita Bosticardo, Ym9zdGljYXJkby5tYXJpdGFAaHNyLml0