 Gorjana Rackov
Gorjana Rackov Rahman Shokri
Rahman Shokri Melchor Álvarez De Mon
Melchor Álvarez De Mon Carlos Martínez-A.
Carlos Martínez-A. Dimitrios Balomenos
Dimitrios Balomenos- 1Department of Immunology and Oncology, Universidad Autónoma de Madrid, Centro Nacional de Biotecnología – CSIC, Madrid, Spain
- 2IMDEA Nanoscience, Universidad Autónoma de Madrid, Madrid, Spain
- 3Immune System Diseases-Rheumatology and Oncology Service, University Hospital Principe de Asturias, Alcalá de Henares, Spain
Sepsis is a complex biphasic syndrome characterized by both pro- and anti-inflammatory immune states. Whereas early sepsis mortality is caused by an acute, deleterious pro-inflammatory response, the second sepsis phase is governed by acute immunosuppression, which predisposes patients to long-term risk for life-threatening secondary infections. Despite extensive basic research and clinical trials, there is to date no specific therapy for sepsis, and mortality rates are on the rise. Although IFN-β is one of the most-studied cytokines, its diverse effects are not fully understood. Depending on the disease or type of infection, it can have beneficial or detrimental effects. As IFN-β has been used successfully to treat diverse diseases, emphasis has been placed on understanding the role of IFN-β in sepsis. Analyses of mouse models of septic shock attribute a pro-inflammatory role to IFN-β in sepsis development. As anti-inflammatory treatments in humans with antibodies to TNF-α or IL1-β resulted disappointing, cytokine modulation approaches were discouraged and neutralization of IFN-β has not been pursued for sepsis treatment. In the case of patients with delayed sepsis and immunosuppression, there is a debate as to whether the use of specific cytokines would restore the deactivated immune response. Recent reports show an association of low IFN-β levels with the hyporesponsive state of monocytes from sepsis patients and after endotoxin tolerance induction. These data, discussed here, project a role for IFN-β in restoring monocyte function and reversing immunosuppression, and suggest IFN-β-based additive immunomodulatory therapy. The dichotomy in putative therapeutic approaches, involving reduction or an increase in IFN-β levels, mirrors the contrasting nature of the early hyperinflammatory state and the delayed immunosuppression phase.
Introduction
Sepsis is a systemic inflammatory syndrome caused by massive microbial infections and is a major cause of death worldwide. Sepsis is defined as an “organ dysfunction caused by a dysregulated host response to infection,” while septic shock is associated to a greater mortality risk, caused by “underlying circulatory, cellular and metabolic abnormalities” (1, 2). Although survival of sepsis patients with overwhelming pro-inflammatory responses is greatly improved in intensive care units (ICU), most patients develop delayed sepsis with severely suppressed immune responses and succumb to secondary infections (3, 4).
IFN-β is an essential cytokine in promoting and regulating innate and adaptive immune responses; its potential as an antimicrobial agent has been studied extensively. Data from murine models have assigned IFN-β a role in septic shock development, and its neutralization is proposed as a therapeutic strategy for human sepsis (5). As recent findings show low IFN-β expression by non-responsive monocytes in delayed sepsis patients, we discuss the therapeutic value of blocking or enhancing the levels of this cytokine to modulate immunosuppression.
Sepsis Progression from Hyperinflammation and Early Death to Immunosuppression and Delayed Death
Following massive microbial infection, highly produced inflammatory cytokines, mainly TNF-α and IL-1β, drive hyperinflammation in sepsis patients (6, 7). Patients can suffer early death several days after systemic infection, due to sepsis and septic shock (8). Improvement in ICU care and compliance with the “Surviving Sepsis Campaign,” which provides clinical practice guidelines for the recognition and management of sepsis and septic shock, has reduced death incidence of sepsis patients (9–11). Early sepsis patients that survive ICU can nonetheless develop delayed sepsis and immunosuppression (12, 13). Patient death can be prolonged after initial sepsis diagnosis, due to a deactivated immune response (14, 15).
Hyporesponsiveness is considered a counteracting mechanism that regulates hyperinflammation and alleviates the deleterious effects of primary infection (16, 17). This state correlates with sepsis progression and death, as it is associated with increased risk for secondary nosocomial infections (3). In a recent review, Delano and Ward (18) present the evolution of mortality as early and late deaths and introduce a third modality of sepsis, long-term death, which can be delayed for years. As the incidence of early deaths in the ICU has diminished over the years (4), the burden of sepsis-related deaths is linked to the hyporesponsive phase of the syndrome, and late and long-term deaths are on the rise (19). The progression from initial sepsis to the prolonged syndrome is not clearly defined and the host response to sepsis might consist of concurrent inflammatory and immunosuppression processes (8, 20). Patients were recently identified that develop persistent inflammation-immunosuppression and catabolism syndrome (PICS), which leads to ongoing organ injury and death (13, 18, 21).
Mouse Models for Analysis of the Biphasic Aspects of Sepsis
Mouse models are valuable tools with which to dissect the mechanisms of human disease, and aid in discovering innovative therapeutic advances. In sepsis, there is nonetheless a disparity at the molecular level between mouse models and the human syndrome (22–25), and human and mouse inflammatory diseases show low gene correlation (26). Findings from these models must thus be evaluated critically for applicability to human sepsis.
Injection of lipopolysaccharide (LPS, also termed endotoxin), a constituent of Gram-negative bacteria, leads to septic shock in mice. Infection models or cecal ligation and puncture (CLP) (27) also lead to septic shock. Compared to the widely used LPS model, the CLP model is a more clear approximation of polymicrobial sepsis as it leads to bacteremia, a feature shared with human sepsis (27).
Mouse systems that emulate hyporesponsiveness and delayed sepsis in humans are limited due to the inherent complexity of sepsis and to its heterogeneity. Non-lethal CLP models adapted to restrict death of treated mice can also be used to study delayed sepsis [see review by Dejager et al. (27)]. Mice subjected to mild CLP induction or CLP models treated with antibiotics survive early hyperinflammation and show long-term immune dysfunction. Exposure of these mice to secondary bacterial infection establishes the “two-hit” model that allows the study of compromised responses (28).
Endotoxin tolerance is a convenient model for analysis of macrophage hyporesponsiveness; it is induced after exposure of mice to a non-lethal LPS dose that induces hyperinflammation (29, 30). In a few hours, macrophages from treated mice undergo functional reprogramming from activated M1 status to an M2 hyporesponsive phenotype, and epigenetic modifications could explain this polarization (31–34). Endotoxin-tolerant macrophages are hyporesponsive to subsequent LPS challenge, and produce low amounts of TNF-α, IFN-β, and inducible nitric oxide synthase (iNOS) (34). This system deviates from the CLP model and from basic human sepsis features, as it is limited to the effects of LPS and not bacterial infection, and the initial LPS treatment does not lead to trauma and death.
As in vitro LPS stimulation induces endotoxin tolerance in human monocytes (31, 35) and their refractory state shows a certain analogy to monocyte hyporesponsiveness in sepsis patients (6, 15, 20), data from this model may be useful, but are considered preliminary (16, 24) and should be verified in CPL models and in human sepsis.
Treatment Strategies for Sepsis: Past and Present
To date, intense research in the field has provided effective approaches for early sepsis treatment that have increased survival in the ICU (3, 21); there have nonetheless been no therapeutic advances for long-term sepsis-related immunosuppression.
To minimize the pro-inflammatory condition of sepsis patients, it seemed logical to antagonize hyperinflammation and to treat sepsis by neutralizing hyperinflammation through anti-TNF-α or -IL-1β specific antibodies (7). In mice, anti-TNF-α delivery protected from septic shock when delivered before or simultaneously with LPS, although patients treated with anti-TNF-α or -IL-1β antibodies failed to show sepsis improvement (7, 36). Perhaps, therapeutics directed to the early physio-pathological conditions that derive from this initial pro-inflammatory response would be more efficient in preventing early death in sepsis patients. One such condition is the activation of procoagulant pathways (27).
The ideal treatment would be based on an approach that could remedy both phases of sepsis (37). Because of the contrasting nature of early and delayed sepsis, distinct therapeutic approaches are currently considered to control hyperinflammation or immunosuppression. Before treatment, the state of each patient should thus be taken into strict account and tested, for example, by measuring HLA-DR expression in myeloid cells and evaluating overall immune cell status (3, 18).
As early sepsis survivors eventually develop immunosuppression, there is particular interest in establishing interventions for the late sepsis phase and a debate as to whether treatment for such patients should focus on boosting the pro-inflammatory response (7, 37). Macrophages are directly associated with sepsis development since Gram-negative bacteria, major constituents of infection, promote their activation through the TLR4 receptor; macrophages can then acquire a deactivated status (16). Other immune components participate in immunosuppression development in delayed sepsis. For example, T cell numbers decline due to apoptosis and attain an exhaustion state or impaired function, whereas T regulatory cells (Treg) are associated with mortality of delayed sepsis patients. NK cells and neutrophils have altered signaling functions. Moreover, a population of immature myeloid-derived suppressor cells (MDSC) arises and promotes immunosuppression (8, 18, 21). Dendritic cells undergo apoptosis in sepsis (38–41) and subsequently reemerge, but their activity is compromised due to the production of anti-inflammatory IL-10 (42–45). Because of these diverse immunosuppression features, it was suggested that intervention should not be limited to targeting a single affected immune component, but rather implement combination approaches to improve critical immune defects of sepsis-affected individuals (18). Such methods would imply delivery of immune modulators such as G-CSF, GM-CSF, IFN-γ, or IL-15 (7, 18), to reconstitute specific immune deficiencies that depend on sepsis stage and the patient’s needs.
The Role of IFN-β in Immune Disease and in Sepsis
IFN-β in Infection and Disease: Beneficial and Harmful Effects
The interferons are cytokines that modulate the immune response and antimicrobial infection; they are classified as types I, II, and III. IFN-γ is the only type-II cytokine, and IFN-α and -β (IFNα/β) of the broad IFN I family are the most studied. After microbial infection, endothelial, epithelial, and immune cells detect through their pattern-recognition receptors (PRRs), which include TLR, pathogen-associated molecular patterns (PAMPs). This interaction promotes IFN-β, which is produced by most nucleated cells (46, 47). All type-I IFNs bind to the same cell surface IFN-α/β receptor (IFNAR). Signaling through IFNAR initiates a cascade of events, which activates innate cells, and elicits chemokine/cytokine production and activation of adaptive immunity (47). IFNα/β was initially found to have antiviral activity, as their defective signaling increases viral susceptibility (48–53). Accordingly, IFN-α is effective for treatment of viral infections such as hepatitis C (54), and IFN-β is also used in cancer treatment (55). IFNα/β can nonetheless have detrimental immunosuppressive effects during chronic infection with certain viruses (56, 57). These contrasting roles of IFNα/β in controlling or exacerbating disease are a central feature of these pleiotropic cytokines (58). Similarly, IFNα/β might contribute to the development of autoimmune diseases such as lupus or psoriasis (59–61), whereas IFN-β can be beneficial in a large proportion of multiple sclerosis patients (62). In another setting, although IFNα/β are critical in the defense against bacteria (63–67), they could also impede antibacterial immunity by inducing apoptosis of immune cells, by suppressing inflammatory cytokine release, by responsiveness to IFN-γ, or by promoting IL-10 production (68–71).
In general terms, IFNα/β boosts pro-inflammatory cytokine/chemokine production and activates adaptive immunity, but also has diverse roles in immunity and, depending on context, might also suppress immune responses. IFN-α and IFN-β share a common receptor with apparently redundant functions. The two cytokines are used differently for treatment, and IFN-α is highly produced by plasmacytoid dendritic cells (47). The differences between IFN-α and IFN-β probably derive from the weak IFN-α binding to their common receptor (72).
IFN-β Neutralization in Treating Hyperinflammation in Acute Sepsis
Several studies in IFN-β- and IFNAR-deficient mouse models of LPS- or TNF-α-induced septic shock suggest a pro-inflammatory role for IFN-β in septic shock (Table 1) (73–75). These observations support the idea of IFN-β or IFNAR neutralization as an adjunctive immunomodulatory therapy for sepsis (5). This view was corroborated by a subsequent report showing that IFNAR is needed for CLP-triggered sepsis development (76). An anti-IFNAR antibody also reduced sepsis symptoms and was functional even when injected after CLP induction, thus reinforcing the potential of IFN-β signaling inactivation as a therapeutic approach for sepsis (76). Sepsis improvement by the anti-IFNAR antibody precludes doubts about the relevance of data from mice in which IFN signaling is genetically abolished, based on the supposition that knockout mice might not reproduce the exact function of the eliminated gene. Studies in which septic shock was reduced by IFN-α (77) or IFN-β delivery (78) that suggest an anti-inflammatory effect for these cytokines need to be reinterpreted. Perhaps, the injected cytokine dose in these two studies elicits non-physiological effects that increase survival, as there is a striking difference between physiological IFN-β levels and those used for treatment (79). The role of IFN-β appears to lie more in propagating the inflammatory response than in initiating it, as its effect differs from that of TNF-α, a potent pro-inflammatory agent that causes septic shock in mice (75). In accordance with this view, TNF-α induces IFN-β production (58).
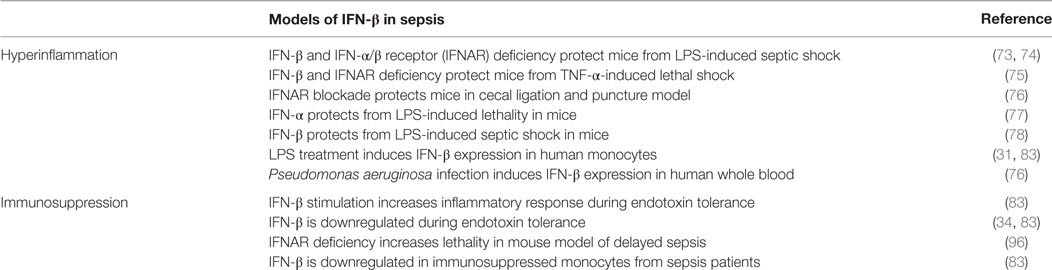
Table 1. Role of IFN-β in modulating hyperinflammatory and immunosuppressive responses in mouse models and humans.
TLR4 stimulation elicits hyperinflammation and IFN-β signaling in monocytes and macrophages as illustrated in Figure 1 (left). TLR4 triggering recruits MyD88 (myeloid differentiation primary-response protein 88) in order to activate both NF-κB and protein kinases (MAPK), which drives nuclear translocation of p65/p50 NF-κB and phospho-AP-1 and transcription of inflammatory cytokines. TLR4 stimulation also results in phosphorylation of IFN-regulatory factor 3 (IRF3), which in conjunction with NF-κB, induces expression of IFN-β (80, 81). Secretion of IFN-β activates the IFNAR complex in an autocrine manner. Subsequent STAT1 phosphorylation induces IFN-responsive elements such as iNOS, and chemokines such as CXCL9, CXCL10, and CXCL11 (Figure 1), which are potent white blood cell (WBC) attractors that further potentiate the immune response (31, 32, 82).
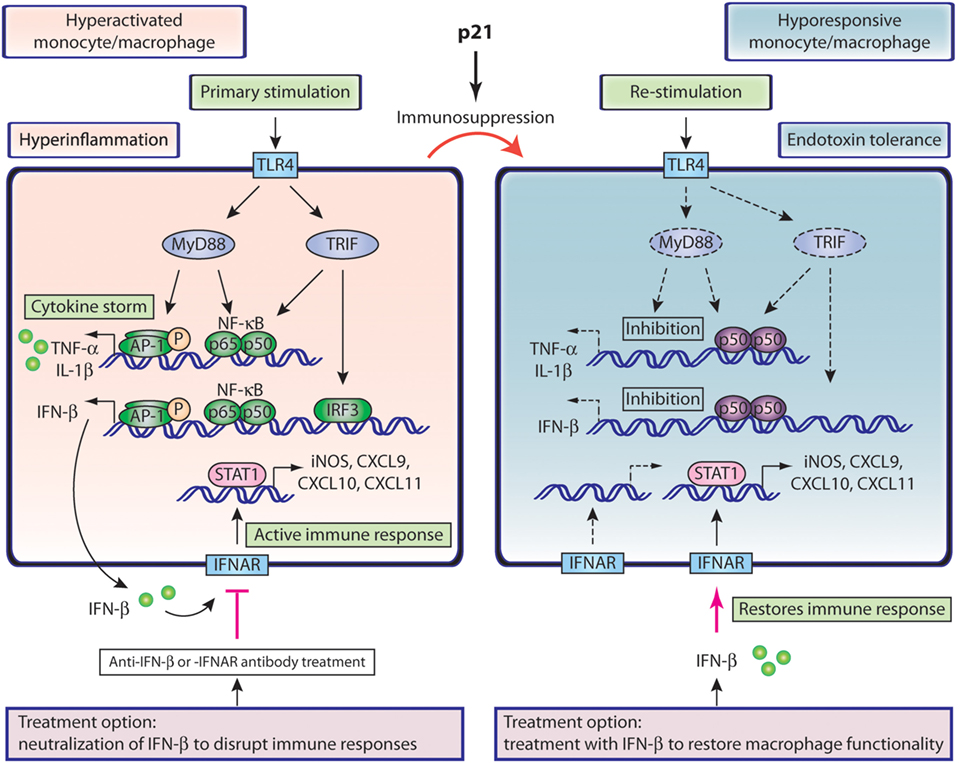
Figure 1. Role of IFN-β in hyperactivated and hyporesponsive monocytes/macrophages. Left, hyperactivated macrophages or monocytes present hyperinflammatory status and elevated IFN-β production. Secreted IFN-β interacts with its receptor and propagates the immune responses through iNOS and chemokine production. Neutralization of the IFN-β pathway interrupts these responses and could be beneficial in sepsis treatment. Right, hyporesponsive macrophages or monocytes associated with delayed sepsis or endotoxin tolerance arise from their hyperactivated counterparts, as a result of immunosuppression, driven by p21. TLR4 restimulation of such cells shows compromised activation pathways and inflammatory cytokine production, including IFN-β. IFN-β treatment could restore compromised monocyte functions and benefit immunosuppressed delayed sepsis patients.
We recently showed that neutralization of IFNAR reduces iNOS and NO production as well as WBC-attracting chemokines in a mouse model of increased response to LPS (83). Neutralization of IFN-β signaling could thus reduce the propagation of inflammation and harmful physiological effects that depend on persistent NO production (83). These findings corroborate the idea that IFN-β does not act as an immediate hyperinflammatory factor, and its neutralization could be an attractive option that allows a greater margin for intervention, as the interval of hyperinflammation induction might be too short for effective treatment.
These data reinforce the idea that neutralizing IFN-β signaling could be a therapeutic option for acute sepsis patients. As suggested by Mahiou et al. (5), such an approach might be applied to relieve acute hyperinflammation, and caution should be taken to exclude patients with delayed sepsis to avoid aggravated immunosuppression.
IFN-β in Restoring Functions of Compromised Immune Components
Boosting monocytes and other immune components to recover function is a prospective therapeutic approach for immunocompromised late sepsis patients. This idea is based on data showing that immunosuppressed monocytes from late sepsis patients recover HLA-DR levels and inflammation modulators after IFN-γ treatment (84). Although IFN-γ delivery to trauma or sepsis patients in clinical studies had some positive effects, it does not cure sepsis (7, 18). It is therefore suggested that a combination of IFN-γ treatment with GM-CSF, another monocyte booster, might prove more effective in treating immunosuppression in sepsis (7). The prevailing idea is that key cytokines or other pharmacological agents could revert immunosuppression of monocytes and other immune cells (37). Here we evaluate whether IFN-β could be included in the chart of promising factors to alleviate immunosuppression (7, 18).
IFN-β is essential for human monocyte inflammation (31), but is downmodulated in endotoxin-tolerized monocytes (34, 83). Importantly, IFN-β is also downregulated in immunosuppressed monocytes from sepsis patients (Table 1) (83), which implies that IFN-β downregulation could be critical for immunosuppression of monocytes in human sepsis, and that IFN-β treatment could reverse monocyte deactivation.
p21 was initially identified as a cell cycle and cyclin-dependent kinase 2 inhibitor (CDK2) (85). Other functions have been attributed to p21 (86, 87), and several studies designate it as an immune response modulator. p21 inhibits development of autoreactive T cells and autoimmunity (88–91). Moreover, it controls macrophage activation in septic shock and rheumatoid arthritis (92–94) and inhibits LPS-induced NF-κB activation, as well as endotoxin hyporesponsiveness of macrophages and monocytes (83). p21 regulates IFN-β levels in human monocytes, and monocytes from sepsis patients show high p21 levels, which correlate with low IFN-β expression.
These data suggest a model in which monocyte immunosuppression is controlled by p21, which promotes inhibitory p50–p50 over active p65–p50 NF-κB products (Figure 1, right). This p21 effect compromises production of inflammatory cytokines and IFN-β, and reduces iNOS induction and chemokine upregulation, which impairs WBC attraction and activation of innate and adaptive immunity (34, 47, 55, 83, 95). These IFNAR-dependent effects could theoretically be reestablished by an exogenous supply of IFN-β and thus restore monocyte functions and counteract immunosuppression (Figure 1, right). This model is further reinforced by our recent work showing that IFN-β treatment of endotoxin hyporesponsive macrophages increases expression of iNOS and CXCL11 (83). IFN-β can thus reestablish critical functional properties in immunosuppressed monocytes.
The role of IFN-β in controlling infection in long-term sepsis is supported by a mild CLP sepsis model. In such settings, WT mice survive the initial inflammatory shock, control bacteremia, and elude delayed death, whereas IFN-β-deficient mice, also unaffected by early inflammation, succumb to infection, and undergo late death (96). The data from this model, which in a way resembles delayed sepsis, show association of IFN-β expression with production of CXCL10, a chemokine that promotes homing of immune cells such as neutrophils (95, 96). In this mild CLP model, immunosuppression is possibly partial and IFN-β is produced. The results, however, support our view (Figure 1, right) that IFN-β, which must be supplied exogenously in severe immunosuppression, is essential for chemokine-mediated WBC attraction and antimicrobial action.
The model in Figure 1 (right) shows the possible effect of exogenous IFN-β in reversing monocyte hyporesponsiveness. This IFN-β effect can be extended to other immunosuppressed immune cells, as it can increase effector T cells, antibody responses in B cells, and activate innate immunity and antigen presentation (47, 95). As IFN-β can induce dendritic cell maturation (95), this cytokine could enhance the generation of dendritic cells, which are reduced by apoptosis in sepsis patients. Similarly to monocytes, exogenous IFN-β could reverse immunosuppressive aspects of dendritic cells, including IL-10 production, which inhibits IL-12 synthesis and T cell stimulation (45, 97).
Apart from its activating effect in monocytes and consistent with its pleiotropic antimicrobial responses, IFN-β could thus abrogate a wide range of sepsis-associated immunosuppressive responses. As little is known about its positive immunomodulatory effects in sepsis, establishing a role for IFN-β in antagonizing immunosuppression requires experimental evidence. Testing the effect of exogenous IFN-β in the “two hit” mouse model (28) could impart some early answers on its suitability for treatment of sepsis, which should be evaluated in human sepsis.
IFN-β has adverse immune effects that hinder microbial clearance in some systems (47, 98), such as proapoptotic effects on innate cells and T cells, inhibition of the IFN-γ pathway in macrophages, as well as generation of IL-10-producing Treg cells (58). The idea that IFN-β delivery might benefit immunosuppressed patients with delayed sepsis thus needs to be assessed meticulously. As the negative immune impact of IFN-β in infection is manifested in the context of certain but not all microbial infections, sepsis treatment might not be affected by these discordant IFN-β effects.
Much experimentation remains in order to elucidate the potential immunomodulatory effect of IFN-β in sepsis therapeutics and to estimate whether, in addition to IFN-β, any other stimulus could aid in efficient immune response reactivation in patients with delayed sepsis.
Concluding Remarks
The failure of anti-TNF-α- or IL-1β-based therapies to decrease the death toll in sepsis patients has generated doubts as to whether cytokine-based treatments can be effective. Recent research has given new impetus to the field, and appropriate cytokine combinations are being considered for restoring suppressed immune functions in delayed sepsis patients. IFN-β-based therapeutic approaches such as neutralization could be used during the hyperinflammation phase of sepsis, but also during the immunosuppression phase, with IFN-β delivery to boost suppressed immunity. A thorough analysis of individual sepsis patients is needed before applying such radically opposed treatments for hyperinflammation or hyporesponsiveness. Patients with established immunosuppression might thus be a clearer target for IFN-β treatment to refurbish immune responses. Research is needed in mouse models and in humans to determine the precise mechanistic aspects and effectiveness of IFN-β-based treatments.
Author Contributions
GR composed the figures and contributed in writing the manuscript; RS composed the final versions of the figures and organised the argument and the manuscript; MM and CM-A contributed to the intellectual content of the manuscript. DB conceived and wrote the manuscript; all authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank C. Mark for editorial assistance.
Funding
GR was funded by a predoctoral fellowship from the “La Caixa” program. This work was supported by grants from the Ministry of Economy and Competitivity (MINECO/FEDER PI11/00950 and SAF2016-80803-R to DB, and SAF2013-42289-R and SAF2016-75456-R to CM-A), from the Fundación Alfonso Martin Escudero to CM-A, and from the Community of Madrid (MITIC S2011/BMD2502 to DB, MM, and CM-A).
References
1. Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA (2016) 315:762–74. doi: 10.1001/jama.2016.0288
2. Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS, et al. Developing a new definition and assessing new clinical criteria for septic shock. JAMA (2016) 315:775. doi:10.1001/jama.2016.0289
3. Angus D, van der Poll T. Severe sepsis and septic shock. N Engl J Med (2013) 369:840–51. doi:10.1056/NEJMra1208623
4. Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity (2014) 40:463–75. doi:10.1016/j.immuni.2014.04.001
5. Mahieu T, Libert C. Should we inhibit type I interferons in sepsis? Infect Immun (2007) 75:22–9. doi:10.1128/IAI.00829-06
6. Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care (2006) 10:233. doi:10.1186/cc5055
7. Brown KA, Brown GA, Lewis SM, Beale R, Treacher DF. Targeting cytokines as a treatment for patients with sepsis: a lost cause or a strategy still worthy of pursuit? Int Immunopharmacol (2016) 36:291–9. doi:10.1016/j.intimp.2016.04.041
8. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol (2013) 13:862–74. doi:10.1038/nri3552
9. Castellanos-Ortega A, Suberviola B, Garcia-Astudillo LA, Holanda MS, Ortiz F, Llorca J, et al. Impact of the Surviving Sepsis Campaign protocols on hospital length of stay and mortality in septic shock patients: results of a three-year follow-up quasi-experimental study. Crit Care Med (2010) 38:1036–43. doi:10.1097/CCM.0b013e3181d455b6
10. Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev (2016) 274:330–53. doi:10.1111/imr.12499
11. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med (2017) 45(3):486–552. doi:10.1097/CCM.0000000000002255
12. Adib-Conquy M, Cavaillon J-M. Compensatory anti-inflammatory response syndrome. Thromb Haemost (2009) 101(1):36–47. doi:10.1160/TH08-07-0421
13. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg (2012) 72:1491–501. doi:10.1097/TA.0b013e318256e000.Persistent
14. Escoll P, del Fresno C, García L, Vallés G, Lendínez MJ, Arnalich F, et al. Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun (2003) 311:465–72. doi:10.1016/j.bbrc.2003.10.019
15. Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernández-Jiménez E, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity (2015) 42:484–98. doi:10.1016/j.immuni.2015.02.001
16. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol (2009) 30:475–87. doi:10.1016/j.it.2009.07.009
17. López-Collazo E, Del Fresno C. Pathophysiology of endotoxin tolerance: mechanisms and clinical consequences. Crit Care (2013) 17:242. doi:10.1186/cc13110
18. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest (2016) 126:23–31. doi:10.1172/JCI82224
19. Winters BD, Eberlein M, Leung J, Needham DM, Pronovost PJ, Sevransky JE. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med (2010) 38:1276–83. doi:10.1097/CCM.0b013e3181d8cc1d
20. Pena OM, Hancock DG, Lyle NH, Linder A, Russell JA, Xia J, et al. An endotoxin tolerance signature predicts sepsis and organ dysfunction at initial clinical presentation. EBioMedicine (2014) 1:64–71. doi:10.1016/j.ebiom.2014.10.003.An
21. Rosenthal MD, Moore FA. Persistent inflammation, immunosuppression, and catabolism: evolution of multiple organ dysfunction. Surg Infect (Larchmt) (2016) 17:167–72. doi:10.1089/sur.2015.184
22. Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity (2004) 21:227–39. doi:10.1016/j.immuni.2004.07.010
23. Liew FY, Xu D, Brint EK, O’Neill LAJ. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol (2005) 5:446–58. doi:10.1038/nri1630
24. Del Fresno C, García-Rio F, Gómez-Piña V, Soares-Schanoski A, Fernández-Ruíz I, Jurado T, et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol (2009) 182:6494–507. doi:10.4049/jimmunol.0803350
25. van ’t Veer C, van den Pangaart PS, van Zoelen MA, de Kruif M, Birjmohun RS, Stroes ES, et al. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol (2007) 179:7110–20. doi:10.4049/jimmunol.179.10.7110
26. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A (2013) 110:3507–12. doi:10.1073/pnas.1222878110
27. Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol (2011) 19:198–208. doi:10.1016/j.tim.2011.01.001
28. Nascimento DC, Alves-Filho JC, Sonego F, Fukada SY, Pereira MS, Benjamim C, et al. Role of regulatory T cells in long-term immune dysfunction associated with severe sepsis. Crit Care Med (2010) 38:1718–25. doi:10.1097/CCM.0b013e3181e78ad0
29. Berg DJ, Kühn R, Rajewsky K, Müller W, Menon S, Davidson N, et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest (1995) 96:2339–47. doi:10.1172/JCI118290
30. Wysocka M, Robertson S, Riemann H, Caamano J, Hunter C, Mackiewicz A, et al. IL-12 suppression during experimental endotoxin tolerance: dendritic cell loss and macrophage hyporesponsiveness. J Immunol (2001) 166:7504–13. doi:10.4049/jimmunol.166.12.7504
31. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A (2009) 106:14978–83. doi:10.1073/pnas.0809784106
32. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643DS1
33. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. doi:10.1038/nature05836
34. Piao W, Song C, Chen H, Diaz MA, Wahl LM, Fitzgerald KA, et al. Endotoxin tolerance dysregulates MyD88- and toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol (2009) 86:863–75. doi:10.1189/jlb.0309189
35. Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol (2011) 186:7243–54. doi:10.4049/jimmunol.1001952
36. Lyle NH, Pena OM, Boyd JH, Hancock RE. Barriers to the effective treatment of sepsis: antimicrobial agents, sepsis definitions, and host-directed therapies. Ann N Y Acad Sci (2014) 1323:101–14. doi:10.1111/nyas.12444
37. Cavaillon JM, Eisen D, Annane D. Is boosting the immune system in sepsis appropriate? Crit Care (2014) 18(2):216. doi:10.1186/cc13787
38. Grimaldi D, Louis S, Pene F, Sirgo G, Rousseau C, Claessens YE, et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med (2011) 37:1438–46. doi:10.1007/s00134-011-2306-1
39. Guisset O, Dilhuydy MS, Thiebaut R, Lefevre J, Camou F, Sarrat A, et al. Decrease in circulating dendritic cells predicts fatal outcome in septic shock. Intensive Care Med (2007) 33:148–52. doi:10.1007/s00134-006-0436-7
40. Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol (2002) 168:2493–500. doi:10.4049/jimmunol.168.5.2493
41. Efron PA, Martins A, Minnich D, Tinsley K, Ungaro R, Bahjat FR, et al. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol (2004) 173:3035–43. doi:10.4049/jimmunol.173.5.3035
42. Riccardi F, Della Porta MG, Rovati B, Casazza A, Radolovich D, De Amici M, et al. Flow cytometric analysis of peripheral blood dendritic cells in patients with severe sepsis. Cytometry B Clin Cytom (2011) 80:14–21. doi:10.1002/cyto.b.20540
43. Poehlmann H, Schefold JC, Zuckermann-Becker H, Volk HD, Meisel C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit Care (2009) 13:R119. doi:10.1186/cc7969
44. Pastille E, Didovic S, Brauckmann D, Rani M, Agrawal H, Schade FU, et al. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J Immunol (2011) 186:977–86. doi:10.4049/jimmunol.1001147
45. Fallarino F, Pallotta MT, Matino D, Gargaro M, Orabona C, Vacca C, et al. LPS-conditioned dendritic cells confer endotoxin tolerance contingent on tryptophan catabolism. Immunobiology (2015) 220:315–21. doi:10.1016/j.imbio.2014.09.017
46. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta (2017). doi:10.1016/j.bbadis.2017.01.013
47. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2013) 14:36–49. doi:10.1038/nri3581
48. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet (2003) 33:388–91. doi:10.1038/ng1097
49. Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity (2012) 36:515–28. doi:10.1016/j.immuni.2012.03.016
50. Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, et al. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci U S A (1995) 92:11284–8. doi:10.1073/pnas.92.24.11284
51. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7. doi:10.1038/nature03464
52. Rudd PA, Wilson J, Gardner J, Larcher T, Babarit C, Le TT, et al. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J Virol (2012) 86:9888–98. doi:10.1128/JVI.00956-12
53. Daffis S, Lazear HM, Liu WJ, Audsley M, Engle M, Khromykh AA, et al. The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol (2011) 85:5664–8. doi:10.1128/JVI.00232-11
54. Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A (2008) 105:7034–9. doi:10.1073/pnas.0707882105
55. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12:125–35. doi:10.1038/nri3133
56. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340:207–11. doi:10.1126/science.1235214
57. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340:202–7. doi:10.1126/science.1235208
58. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi:10.1084/jem.20101664
59. Ronnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Curr Opin Rheumatol (2013) 25:248–53. doi:10.1097/BOR.0b013e32835c7e32
60. Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, Theofilopoulos AN. Anti-IFN-α/β receptor antibody treatment ameliorates disease in lupus-predisposed mice. J Immunol (2012) 189:5976–84. doi:10.4049/jimmunol.1201477
61. Grine L, Dejager L, Libert C, Vandenbroucke RE. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev (2015) 26:25–33. doi:10.1016/j.cytogfr.2014.10.009
62. Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung H-P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol (2006) 2:201–11. doi:10.1038/ncpneuro0154
63. De la Maza LM, Peterson EM, Goebel JM, Fennie CW, Czarniecki CW. Interferon-induced inhibition of Chlamydia trachomatis: dissociation from antiviral and antiproliferative effects. Infect Immun (1985) 47:719–22.
64. Rothfuchs AG, Gigliotti D, Palmblad K, Andersson U, Wigzell H, Rottenberg ME. IFN-alpha beta-dependent, IFN-gamma secretion by bone marrow-derived macrophages controls an intracellular bacterial infection. J Immunol (2001) 167:6453–61. doi:10.4049/jimmunol.167.11.6453
65. Rothfuchs AG, Trumstedt C, Mattei F, Schiavoni G, Hidmark A, Wigzell H, et al. STAT1 regulates IFN-alpha beta- and IFN-gamma-dependent control of infection with Chlamydia pneumoniae by nonhemopoietic cells. J Immunol (2006) 176:6982–90. doi:10.4049/jimmunol.176.11.6982
66. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 178:3126–33. doi:10.4049/jimmunol.178.5.3126
67. Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, et al. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio (2011) 2:e00016–11. doi:10.1128/mBio.00016-11
68. Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med (2004) 200:527–33. doi:10.1084/jem.20040976
69. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med (2004) 200:535–40. doi:10.1084/jem.20040769
70. O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med (2004) 200:437–45. doi:10.1084/jem.20040712
71. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol (2014) 193:3600–12. doi:10.4049/jimmunol.1401088
72. Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, van der Heyden J, et al. Inquiring into the differential action of interferons (IFNs): an IFN-α2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-β. Mol Cell Biol (2006) 26:1888–97. doi:10.1128/MCB.26.5.1888
73. Karaghiosoff M, Steinborn R, Kovarik P, Kriegshäuser G, Baccarini M, Donabauer B, et al. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat Immunol (2003) 4:471–7. doi:10.1038/ni910
74. Mahieu T, Park JM, Revets H, Pasche B, Lengeling A, Staelens J, et al. The wild-derived inbred mouse strain SPRET/Ei is resistant to LPS and defective in IFN-beta production. Proc Natl Acad Sci U S A (2006) 103:2292–7. doi:10.1073/pnas.0510874103
75. Huys L, Van Hauwermeiren F, Dejager L, Dejonckheere E, Lienenklaus S, Weiss S, et al. Type I interferon drives tumor necrosis factor-induced lethal shock. J Exp Med (2009) 206:1873–82. doi:10.1084/jem.20090213
76. Dejager L, Vandevyver S, Ballegeer M, Van Wonterghem E, An L-L, Riggs J, et al. Pharmacological inhibition of type I interferon signaling protects mice against lethal sepsis. J Infect Dis (2013) 209:960–70. doi:10.1093/infdis/jit600
77. Tzung SP, Mahl TC, Lance P, Andersen V, Cohen SA. Interferon-alpha prevents endotoxin-induced mortality in mice. Eur J Immunol (1992) 22:3097–101. doi:10.1002/eji.1830221211
78. Yoo CH, Yeom JH, Heo JJ, Song EK, Lee SI, Han MK. Interferon β protects against lethal endotoxic and septic shock through SIRT1 upregulation. Sci Rep (2014) 4:4220. doi:10.1038/srep04220
79. Berry CM. Understanding interferon subtype therapy for viral infections: harnessing the power of the innate immune system. Cytokine Growth Factor Rev (2016) 31:83–90. doi:10.1016/j.cytogfr.2016.08.001
80. Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med (2007) 13:460–9. doi:10.1016/j.molmed.2007.09.002
81. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
82. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4:499–511. doi:10.1038/nri1391
83. Rackov G, Hernández-Jiménez E, Shokri R, Carmona-Rodríguez L, Mañes S, Álvarez-Mon M, et al. p21 mediates macrophage reprogramming through regulation of p50-p50 NF-κB and IFN-β. J Clin Invest (2016) 126:3089–103. doi:10.1172/JCI83404
84. Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med (1997) 3:678–81. doi:10.1038/nm0697-678
85. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell (1993) 75:805–16. doi:10.1016/0092-8674(93)90499-G
86. Lloberas J, Celada A. p21(waf1/CIP1), a CDK inhibitor and a negative feedback system that controls macrophage activation. Eur J Immunol (2009) 39:691–4. doi:10.1002/eji.200939262
87. Perkins ND. Not just a CDK inhibitor: regulation of transcription by p21(WAF1/CIP1/SDI1). Cell Cycle (2002) 1:35–7. doi:10.4161/cc.1.1.98
88. Zhu B, Symonds AL, Martin JE, Kioussis D, Wraith DC, Li S, et al. Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med (2008) 205:2295–307. doi:10.1084/jem.20080187
89. Chen H, Li C, Huang J, Cung T, Seiss K, Beamon J, et al. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J Clin Invest (2011) 121:1549–60. doi:10.1172/JCI44539
90. Arias CF, Ballesteros-tato A, Isabel M, Martín-caballero J, Flores JM, Martı C, et al. p21 controls proliferation of activated/memory T cells and affects homeostasis and memory T cell responses. J Immunol (2007) 178(4):2296–306. doi:10.4049/jimmunol.178.4.2296
91. Daszkiewicz L, Vázquez-Mateo C, Rackov G, Ballesteros-Tato A, Weber K, Madrigal-Avilés A, et al. Supplemental methods distinct p21 requirements for regulating normal and self-reactive T cells through IFN-γ production. Sci Rep (2015) 5:7691. doi:10.1038/srep07691
92. Trakala M, Arias CF, García MI, Moreno-Ortiz MC, Tsilingiri K, Fernández PJ, et al. Regulation of macrophage activation and septic shock susceptibility via p21(WAF1/CIP1). Eur J Immunol (2009) 39:810–9. doi:10.1002/eji.200838676
93. Scatizzi JC, Mavers M, Hutcheson J, Young B, Shi B, Pope RM, et al. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol (2009) 39:820–5. doi:10.1002/eji.200838683
94. Mavers M, Cuda CM, Misharin AV, Gierut AK, Agrawal H, Weber E, et al. Cyclin-dependent kinase inhibitor p21, via its C-terminal domain, is essential for resolution of murine inflammatory arthritis. Arthritis Rheum (2012) 64:141–52. doi:10.1002/art.33311
95. Uematsu S, Akira S. Toll-like receptors and type I interferons. J Biol Chem (2007) 282:15319–23. doi:10.1074/jbc.R700009200
96. Kelly-Scumpia KM, Scumpia PO, Delano MJ, Weinstein JS, Cuenca AG, Wynn JL, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med (2010) 207:319–26. doi:10.1084/jem.20091959
97. Fan X, Liu Z, Jin H, Yan J, Liang HP. Alterations of dendritic cells in sepsis: featured role in immunoparalysis. Biomed Res Int (2015) 2015:903720. doi:10.1155/2015/903720
Keywords: IFN, IFN-β, sepsis, macrophages, monocytes, immunosuppression, M1–M2 polarization, p21
Citation: Rackov G, Shokri R, De Mon MÁ, Martínez-A. C and Balomenos D (2017) The Role of IFN-β during the Course of Sepsis Progression and Its Therapeutic Potential. Front. Immunol. 8:493. doi: 10.3389/fimmu.2017.00493
Received: 01 December 2016; Accepted: 10 April 2017;
Published: 08 May 2017
Edited by:
Jorg Hermann Fritz, McGill University, CanadaReviewed by:
Lyle Leonard Moldawer, University of Florida College of Medicine, USAStella E. Autenrieth, University of Tübingen, Germany
Copyright: © 2017 Rackov, Shokri, De Mon, Martínez-A. and Balomenos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Balomenos, ZGJhbG9tZW5vc0BjbmIuY3NpYy5lcw==