 Heather M. Wilkins
Heather M. Wilkins Ian W. Weidling
Ian W. Weidling Yan Ji
Yan Ji Russell H. Swerdlow
Russell H. Swerdlow- 1Department of Neurology, University of Kansas Medical Center, Kansas City, KS, USA
- 2University of Kansas Alzheimer’s Disease Center, Kansas City, KS, USA
- 3Department of Molecular and Integrative Physiology, University of Kansas Medical Center, Kansas City, KS, USA
- 4Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS, USA
Inflammation is increasingly implicated in neurodegenerative disease pathology. As no acquired pathogen appears to drive this inflammation, the question of what does remains. Recent advances indicate damage-associated molecular pattern (DAMP) molecules, which are released by injured and dying cells, can cause specific inflammatory cascades. Inflammation, therefore, can be endogenously induced. Mitochondrial components induce inflammatory responses in several pathological conditions. Due to evidence such as this, a number of mitochondrial components, including mitochondrial DNA, have been labeled as DAMP molecules. In this review, we consider the contributions of mitochondrial-derived DAMPs to inflammation observed in neurodegenerative diseases.
Introduction
Inflammatory pathways are activated through either pathogen-initiated or damage-initiated events. Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) activate similar inflammatory cascades and are therefore difficult to distinguish. Inflammation stimulated by DAMPs is an area of study that has recently gained notice. In particular, DAMPs derived from mitochondrial components are interesting due to the prokaryotic origin of this organelle. Furthermore, mitochondrial-derived DAMP molecules may play a role in heart disease, arthritis, liver disease, trauma, and sepsis (1–5).
Neuroinflammation and mitochondrial dysfunction are observed across numerous neurodegenerative diseases (6–8). Mitochondrial dysfunction can induce inflammation and vice versa. Mitochondrial dysfunction may modulate the release of mitochondria-derived DAMP molecules (9, 10). Here, we discuss the relationship between neuroinflammation, mitochondrial dysfunction, and mitochondria-derived DAMP molecules in the context of neurodegenerative diseases.
Neuroinflammation in Neurodegenerative Diseases
Neuroinflammation is classically defined as proliferation and activation of microglia (microgliosis), and/or astrocytes (astrogliosis). Microglia are macrophage cells of mesenchymal origin, which monitor the central nervous system (CNS) for pathogens. Astrocytes are derived from the ectoderm and have numerous functions including metabolic support for neurons, regulating synapses, brain structure, and repair. Further evidence of neuroinflammation can encompass activation of inflammatory pathways, increased expression of cytokines or chemokines, and in some cases disruption of the blood–brain barrier (BBB) accompanied by infiltration of peripheral immune cells (such as T cells). Neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) all have evidence of neuroinflammation pathology.
Alzheimer’s Disease
Initial interest in neuroinflammation as a causative factor in AD centered on the link between reduced risk for AD and long-term non-steroidal anti-inflammatory drug (NSAID) use. Early evidence from epidemiological studies suggested that long-term use of NSAIDs led to a decreased risk of developing AD (11, 12). Subsequent studies found a correlation between apolipoprotein E (APOE, a genetic risk factor for AD) genotype and the protective effects gained from NSAID use. The correlation between decreased AD risk and NSAID use is greatest in individuals who harbor an APOE ε4 allele (13, 14). Furthermore, the age of the individual taking NSAIDs and the type of NSAID administered affect the association of AD risk reduction (15).
Clinical trials investigating whether NSAIDs might benefit AD subjects, though, were disappointing. Early trials experienced high attrition rates among participants due to adverse effects and thus did not provide clear answers (16, 17). A larger trial of one NSAID, tarenflurbil, showed largely negative results (18). However, a recent publication that showed positive memory and brain inflammation outcomes for a different NSAID, fenamate, in an AD mouse model has led to renewed interest (19).
Genome-wide association studies (GWAS) have also renewed interest for neuroinflammation as a potential causative factor for AD. In particular, a rare variant of triggering receptor expressed on myeloid cells 2 (TREM2), R47H, is associated with an increased risk for late onset Alzheimer’s disease (LOAD) (20). TREM2 is a membrane protein in myeloid cells (such as microglia), which modulates inflammatory pathways by inhibiting cytokine production. TREM2 is likely responsible for determining microglial phenotypes (i.e., M1 activation versus M2 activation). Furthermore, TREM2 is also important for lipid sensing, providing a further link between neuroinflammation and bioenergetic pathways (21). The R47H TREM2 variant leads to a reduction in microglial phagocytosis (22). Soluble TREM2 levels are increased in cerebral spinal fluid (CSF) of AD subjects, a parameter that positively correlates with gray matter volume but negatively correlates with diffusivity (sometimes considered a function of cell integrity) (23). Other single-nucleotide polymorphisms (SNPs) associated with AD risk also affect inflammatory pathways. These include SNPs in complement receptor 1, clusterin (CLU), major histocompatibility complex (MHC), class II, DR beta 5, ephrin type A receptor 1, inositol polyphosphate-5-phosphatase, and or Siglec-3 (24–29). The relationship of each of these genes to inflammatory signaling has been reviewed elsewhere (30).
Microglia and reactive astrocytes are closely associated with amyloid plaques in AD brain (31, 32). The quantity of interleukin-1 (IL-1) reactive microglia is increased sixfold in AD brain (33). Furthermore, microglial IL-1 expression correlates with plaque distribution (34). Levels of macrophage colony-stimulating factor, an activator of macrophages, was found to be increased in plasma and CSF from AD subjects compared to those with mild-cognitive impairment (35). A number of cytokines and chemokines, which can be released by macrophages or other immune cells, are increased in AD, including interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor α (TNFα), interleukin-8 (IL-8), transforming growth factor β, and macrophage inflammatory protein-1α (36).
Parkinson’s Disease
The discovery of reactive microglia in the substantia nigra of PD brain tissue provided early evidence for the role of neuroinflammation in PD (37). Further evidence for microglial activation in PD brain came from positron emission tomography (PET) imaging studies. PET studies showed increased microglial activation as evidenced by increased levels of peripheral benzodiazepine sites, a selective marker of activated microglia (38). Increased levels of glial cells expressing TNFα, IL-1β, and interferon-γ (IFN-γ) have also been observed in the substantia nigra of deceased PD subjects (39). Furthermore, microglial activation is observed in animal models of PD (36, 40).
Studies on astrogliosis in the PD brain have generated conflicting results. Early studies suggested increased astrogliosis in the PD brain. More recent studies have shown minimal levels of nigral astrogliosis, although astrocytic accumulation of α-synuclein was observed. The authors speculated that α-synuclein accumulation may cause astrocytes to be less reactive (41). A subsequent report showed minimal astrogliosis and an inverse correlation between levels of α-synuclein and astrogliosis in the PD brain, suggesting α-synuclein may suppress astrogliosis (42).
Similar to AD, PD risk is reduced with regular or chronic NSAID use. Long-term aspirin use is associated with less PD risk; however, non-aspirin-based NSAID use afforded better risk reduction. Furthermore, women who used aspirin in a chronic (greater than 24 months) or regular manner had a lower risk for PD than men (43).
Genome-wide association studies have identified the gene that encodes leucine-rich repeat kinase 2 (LRRK2) as a risk factor for sporadic PD. LRRK2 mutations are also associated with autosomal dominant forms of PD (44). Similar to TREM2 in AD, LRRK2 mediates microglial function (45, 46). LRRK2 facilitates vesicle trafficking and cytoskeletal remodeling and may skew microglia toward a pro-inflammatory phenotype (45–47).
Changes in peripheral inflammation may contribute to PD. Studies of peripheral inflammation showed an increased ratio of CD8+ T cells to CD4+ T cells in the blood of PD patients (48). Peripheral inflammation may affect the brain through disruption of the BBB, an event that is observed in PD subjects (49). Furthermore, peripheral inflammatory changes correlate with PD risk. Increased plasma IL-6 levels correlated with disease risk, although with a small sample size (50). CD8+ and CD4+ T cell reactivity is increased in the substantia nigra of PD patients. Furthermore, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxin used to generate a commonly utilized PD animal model, was found to increase T cell infiltration into the substantia nigra (51).
Amyotrophic Lateral Sclerosis
Inflammatory changes have been observed in the CNS of ALS patients. Early studies in ALS spinal cord and motor cortex discovered accumulation of immunoglobulin G and complement (52). Subsequent studies found T cell infiltration and increased levels of MHC I and II antigens on macrophages and dendritic cells (53, 54). Immunohistochemical studies in postmortem brain tissue showed increased levels of phagocytic and leukocyte surface proteins on microglia, as well as evidence to support the infiltration of activated lymphocytes into the precentral gyrus (55). PET studies using PK11195, a ligand that binds microglia, have provided further evidence of microglial activation in ALS (56).
Astrogliosis is observed within the spinal cord (ventral and dorsal horns) and brain (cortical gray matter and subcortical white matter) in postmortem ALS subjects (57). Microgliosis is evident in the spinal cord ventral horn, the corticospinal tract, and the motor cortex (55). The role of microglia and astrocytes in ALS disease progression is further supported in transgenic mutant superoxide dismutase 1 (SOD1) ALS mouse models. Selective deletion of mutant SOD1 from microglia (which also removes expression from peripheral myeloid cells) slowed disease progression (58). Similar results were observed when mutant SOD1 expression was deleted from astrocytes (59). Conversely, deleting mutant SOD1 expression from motor neurons had no effect on disease progression or survival, but did delay disease onset (58). These studies emphasize the potential role of neuroinflammation in ALS disease progression.
Genome-wide association studies in ALS have linked inflammatory genes with disease outcome. A SNP in CX3C chemokine receptor 1 (CXCR31, fractalkine receptor) is associated with reduced survival in sporadic ALS subjects; however, this SNP fails to associate with ALS risk (60). CXCR31 is important for the migration of leukocytes and may play a role in microglial migration (61). RNA seq and ingenuity pathway analysis of postmortem ALS spinal cord samples found upregulation of inflammatory pathways, with TNFα being a predicted upstream regulator of inflammation in these ALS samples (62).
Neuroinflammation is evident in AD, PD, and ALS. However, whether or not neuroinflammation contributes to disease onset, progression, and risk requires further study. To this end, a growing appreciation of a relationship between mitochondrial dysfunction and inflammatory pathways may provide insight into this important question. Below, we discuss evidence for mitochondrial dysfunction in AD, PD, and ALS.
Primary Mitochondrial Diseases
Primary mitochondrial diseases are caused by mutations in mitochondrial DNA (mtDNA) or nuclear DNA genes that encode mitochondrial proteins. Diseases caused by mtDNA mutations include Leber’s hereditary optic neuropathy, myoclonic epilepsy and ragged red fiber, neuropathy ataxia and retinitis pigmentosa, Kearns–Sayre syndrome, and Leigh’s syndrome. Diseases caused by a nuclear DNA mutation of which affects a mitochondrial localized protein include Friedreich’s ataxia, Wilson’s disease, and Mohr–Tranebjaerg syndrome. Commonly, these diseases manifest in neurological symptoms. A few of these diseases are associated with neuroinflammation pathology.
Friedreich’s ataxia is caused by autosomal recessive inheritance of a mutant Frataxin gene. The product of the Frataxin gene is responsible for iron homeostasis within mitochondria, and loss of this gene in Schwann cells leads to reduced mitochondrial respiration, inflammation, increased mitochondrial iron concentrations, and cell death (63–65). COX2 expression is elevated in both animal models and Friedreich’s ataxia patient lymphocytes, an indicator of increased inflammation (66).
For subjects with Leigh’s syndrome, mtDNA mutations occur in several genes including ATPase 6, ND 1–6 (NADH dehydrogenase), and CO3 [cytochrome oxidase (COX)] (67, 68). These subjects often have deficient ETC enzyme activities (67). In a mouse model of Leigh’s syndrome, evidence of neuroinflammation is abundant (69). However, inflammatory markers have not been measured from human subject tissues.
Wilson’s disease is caused by a mutation in the ATP7B (ATPase copper transporting β polypeptide) gene and is characterized by liver disease, ataxia, parkinsonism, seizures, and reduced cognition (70, 71). This gene encodes a copper transporting ATPase that localizes to mitochondria and affects mitochondrial copper levels (70, 72). Subjects with this mutation have reduced ETC function (73, 74). Pentraxin 3, a marker of inflammation, is elevated in the serum of Wilson’s disease subjects (75).
Despite the association of mitochondrial dysfunction and neuroinflammation or inflammation (discussed below), these processes have not been extensively studied in primary mitochondrial diseases. Future research endeavors into this area would likely benefit our understanding of these diseases.
Mitochondrial Dysfunction in Neurodegenerative Diseases
The Krebs cycle and oxidative phosphorylation occur in the matrix and inner mitochondrial membrane, respectively. Oxidative phosphorylation requires the mitochondrial respiratory chain. These bioenergetic pathways generate the high energy compound adenosine triphosphate (ATP) (76). Mitochondria and bioenergetic intermediates generated within mitochondria regulate cell signaling pathways (including pro-inflammatory responses, as discussed below).
The brain comprises approximately 2% of the body’s weight yet consumes about 20% of its oxygen uptake. The brain requires high amounts of energy for numerous processes, including neurotransmitter production and synaptic activity. Therefore, the brain is highly susceptible to mitochondrial dysfunction, which has been observed in several neurodegenerative diseases including (but not limited to) AD, ALS, and PD (77–80). Furthermore, mitochondrial dysfunction declines with age, and age is the greatest risk factor for these neurodegenerative diseases (78, 80). Mitochondrial dysfunction can lead to increased reactive oxygen species (ROS) production, decreased ATP production, alterations in mitochondrial membrane potential, damage to mtDNA, and activation of cell death pathways (81).
Alzheimer’s Disease
In postmortem AD brains, decreased COX function, reduced intact mitochondrial number, and increased mitochondrial autophagy have been reported (78, 82–86). Mitochondrial dysfunction appears to be systemic in AD, as deficits in COX activity are apparent in AD patient fibroblasts and platelets (83, 87–89). Changes in mtDNA may drive cell signaling changes, bioenergetic pathway deficits, and histopathological hallmarks of AD. Cytoplasmic hybrid (cybrid) studies in which mtDNA from human AD subjects is transferred into a donor cell line that lacks its own mtDNA provides a system in which mtDNA-derived biochemical and molecular consequences can be assessed. The cybrid model system controls for nuclear DNA alterations, as patient mtDNA is transferred into the context of a consistent nuclear DNA background (90). Cybrid cells generated using AD patient mtDNA have reduced COX activity, increased ROS production, and increased Aβ deposition (90, 91).
Evidence of mtDNA mutations, deletions, and oxidative modifications are present in AD subjects (92–97). mtDNA is inherited from the mother, and interestingly a maternal inheritance pattern for AD has been noted. This maternal inheritance pattern is associated with early changes in brain atrophy and mitochondrial biomarkers (98–103). Finally, mitochondrial haplotypes are associated with increased AD risk (104–106). These studies suggest changes in mitochondrial function, possibly at the level of mDNA maintenance and inheritance, are important in AD pathology.
Parkinson’s Disease
The most studied respiratory chain aberration in PD is a deficit in complex I activity. Initial insight into this deficit stems from cases of recreational drug users exposed to MPTP. After MPTP exposure, individuals developed parkinsonian symptoms and at autopsy were found to have degeneration in the substantia nigra, similar to that observed with PD. This degeneration occurred in the absence of Lewy bodies (or aggregated α-synuclein). Following its accidental discovery, MPTP was adapted to produce monkey and rodent models of PD (107–109). MPTP is oxidized to MPP+, which accumulates in neurons and is a potent complex I inhibitor (107, 110).
Complex I deficits are observed in postmortem brain, platelets, and fibroblasts from PD subjects (111, 112). The observed deficits in complex I activity could be driven by oxidative damage to its catalytic subunits or altered mtDNA (90, 113, 114). Cybrid cell lines generated from PD subject mtDNA recapitulate the complex I deficit. Sporadic PD cybrid cell lines also show reduced mtDNA copy number, reduced ATP, cell death pathway activation, and a relatively depolarized mitochondrial membrane potential (113–115).
Parkinson’s disease risk is associated with mtDNA haplotype, similar to AD (116–118). Changes to mtDNA are also observed in PD, such as mutations and deletions (119–121). Polymorphisms of mtDNA polymerase γ influences PD risk as well (122). Overall, mitochondrial function and mtDNA inheritance and maintenance are important to the pathology of PD.
Amyotrophic Lateral Sclerosis
ALS subjects have evidence of mitochondrial dysfunction within the CNS and periphery. CNS mitochondrial abnormalities include altered mitochondrial morphology, mitochondrial inclusions/aggregates, reductions in COX activity, and lower mtDNA levels with increased levels of mtDNA point mutations and deletions (123–128). COX deficits, lowered mitochondrial number, altered mitochondrial calcium levels, and mtDNA deletions, are also observed in ALS subject muscle (129–131). Lymphocytes from ALS subjects have reduced mitochondrial maximum respiration, liver mitochondria appear swollen, and platelet mitochondria manifest a depolarized mitochondrial membrane potential with increased apoptosis (132–135). Cybrid cells generated from ALS patient mtDNA have alterations in antioxidant enzyme activity and reduced complex I activity (136). The association of ALS with distinct mtDNA haplotypes is under investigation (137).
Mitochondrial Dysfunction and Inflammation
Mitochondria are important modulators of innate immunity pathways. Mitochondria-derived ROS, calcium, and ATP are signaling molecules that activate inflammatory responses. Under conditions in which mitochondria are damaged, such as accumulated mtDNA mutations or mitochondrial dysfunction (possibly through aberrant ROS production), a sustained inflammatory response and downstream pathological inflammation could ensue. A relationship between mitochondrial dysfunction and inflammatory signaling is discussed below.
Mitochondria can directly activate inflammasome signaling. Mitochondria-derived ROS activate the NLR family pyrin domain containing 3 (NLRP3) inflammasome pathway. NLRP3 is normally associated with the endoplasmic reticulum membrane, but upon activation is redistributed to nuclear and mitochondrial membranes, where it oligomerizes with apoptosis-associated speck-like protein containing a CARD (ACS) and pro-caspase 1 to form the NLRP3 inflammasome (138–140). Mitochondria also mediate inflammatory pathway activation through redox sensitive proteins (140).
Mitochondrial dysfunction initiates inflammation across various models. In vitro and in vivo experiments demonstrate complex II inhibition using 3-nitropropionic acid (3NP) induces microglial activation and reduces the ability of microglia to undergo alternate activation (141). Human microglial cells treated with 3NP became activated and showed increased ROS production and cell death rates. Similar results were observed following intrastriatal 3NP injection in adult rats, where microglial activation, ROS production, and cell death were increased (142). Chronic subcutaneous injections of rotenone in rats (a complex I inhibitor that is used to model PD in rodents) increases IL-1β within the hypothalamus. These rats also displayed a decreased number of tyrosine hydroxylase positive neurons in the substantia nigra, perturbed locomotion, and sleep abnormalities (143).
Conversely, pro-inflammatory cytokines also modulate mitochondrial function. For example, TNFα can decrease complex I activity, reduce ATP production, depolarize the mitochondrial membrane potential, increase ROS, and lower activities of complexes II and IV (depending on the cell type). In hepatocytes, TNFα uncouples mitochondria and increases ROS production. This ROS production is generated from complex I and III of the respiratory chain and initiates NFĸB activation (144, 145). Similar effects have been reported in L929 cells (a mouse fibroblast cell line), a leukemia cell line, and 3T3-L1 adipocytes (146–148). In a mouse hippocampal cell line (HT22), TNFα induces mitochondrial respiration deficits and a loss of mitochondrial membrane potential (149). IL-1β also yields similar effects on mitochondrial function (150). Nitric oxide (NO), another inflammatory signaling molecule, disrupts mitochondrial membrane potential and inhibits COX activity. In human retinal pigment cells, TNFα, IL-1β, and IFN-γ increase the production of both mitochondrial- and NADPH oxidase-derived ROS (151).
In vivo studies also suggest pro-inflammatory molecules influence mitochondrial function. Intraperitoneal injection of lipopolysaccharide in rodents increases brain cytokine and inflammatory receptor expression [including IL-1β, toll like receptor 2 and 4, microglial scavenger receptor A (SRA), and Fc receptor (FCγRII)] in a region-specific manner. The authors also reported increased microglial number and mitochondrial functional alterations, including decreased glutathione (an antioxidant) and increased complex II/III activity (152).
A clear relationship between mitochondrial dysfunction and inflammatory cascades exists. Both of these pathological hallmarks are present across multiple neurodegenerative diseases. Mitochondria-derived DAMP molecules could provide a further link between these pathologies. Below, we discuss mitochondria-derived DAMP molecules, potential routes of release, and evidence for their role in neuroinflammation.
Mitochondria-Derived DAMPs: Inducers of Neuroinflammation?
Previously identified mitochondria-derived DAMP molecules include mtDNA, cardiolipin, ATP, mitochondrial transcription factor A (TFAM), cytochrome c, and formyl-methionine-labeled peptides (30). Formyl-methionine-labeled peptides may also include mitochondrial protein-derived cryptides. Cryptides are endogenous, fragmented functional peptides. Pro-inflammatory cryptides from mtDNA and nuclear DNA-encoded mitochondrial proteins were recently described (153–155). As reviewed in Table 1, each of these molecules has been observed to initiate a pro-inflammatory phenotype under various disease and pathological states.
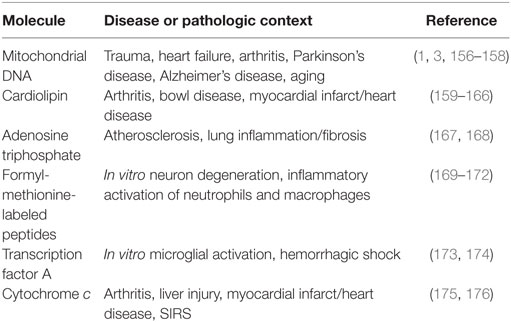
Table 1. Mitochondria-derived damage-associated molecular pattern molecules.
Damage-associated molecular pattern molecules activate inflammatory signaling in a manner similar to PAMPs. The danger molecule is recognized by a pattern-recognition receptor (PRR), and adaptor molecules initiate intracellular signaling cascades and cytokine production. Similar to PAMPs, mitochondria-derived DAMPs are recognized by various PRRs. These include TNF receptor, NLRP3, IL-1 receptor, nucleotide-binding oligomerization domain-like receptor, receptor for advanced glycation end products, formyl peptide receptors (FPR or FPRL1), and purogenic receptors. With the increasing recognition of mitochondria-derived DAMP molecules as pathogenic instigators in various diseases (Table 1), recent studies have begun to explore their contribution to neuroinflammation.
Mitochondria-Derived DAMP Molecules in Neuroinflammation
In the periphery, TFAM functions as a potent DAMP molecule (Table 1). Recently, it was found that TFAM, in combination with IFN-γ, can activate human microglial cells, human peripheral blood monocytes, and THP1 monocytic cells (173). Cotreatment of THP1 monocytic cells with IFN-γ and TFAM or TFAM alone lead to an increase in cytokine expression (including IL-1β, IL-6, and IL-8). While treatment of SH-SY5Y neuroblastoma cells with IFN-γ and TFAM was not toxic, exposure to conditioned medium from monocytic cells activated with IFN-γ and TFAM-induced SH-SY5Y cell death. Finally, mitochondrial proteins extracted from THP1 monocytic cells produced effects similar to TFAM.
Degraded and oxidized mtDNA can initiate pro-inflammatory pathways in astrocytes. Exposure of mtDNA to hydrogen peroxide can induce its degradation, and these mtDNA degradation products are found in human CSF and plasma (9). Furthermore, transfection of mouse primary astrocytes with oxidant-initiated, degraded mitochondrial polynucleotides caused a pro-inflammatory response. This inflammatory astrocyte phenotype was characterized by the upregulation of IL-6, monocyte chemotactic protein 1, TNFα, and IL-1β (9). It was also previously reported that mtDNA is degraded in response to hydrogen peroxide in HA-1 hamster ovarian cells, an effect that was not observed with nuclear DNA or cytoplasmic RNA. Degradation of the mitochondrial genome was apparent in both mtDNA and mitochondrial RNA species (10). These observations suggest a mechanism for mtDNA degradation, and also for the downstream activation of glial cell pro-inflammatory phenotypes.
Mitochondrial components induce inflammation in microglial (BV2) and neuronal (SH-SY5Y) cells (177). In the defining experiments, these cells were exposed to mitochondrial lysates prepared from SH-SY5Y cells containing mtDNA or alternatively SH-SY5Y cells lacking mtDNA. BV2 microglial cells exposed to mitochondrial lysates containing mtDNA had increased TNFα, IL-8, and matrix metalloproteinase 8 mRNA but decreased TREM2 mRNA. Furthermore, NFĸB nuclear localization was increased. These effects were not observed when BV2 microglial cells were exposed to mitochondrial lysates prepared from cells that lacked mtDNA. In SH-SY5Y neuronal cells exposed to mitochondrial lysates containing mtDNA, TNFα mRNA and NFĸB protein expression were elevated. In addition, mitochondrial lysate-exposed SH-SY5Y cells showed increased amyloid precursor protein (APP) mRNA and protein. Changes in APP expression or pro-inflammatory pathways did not occur when SH-SY5Y cells were exposed to mitochondrial lysates that lacked mtDNA.
Mitochondria-derived DAMP molecules induce neuroinflammation in vivo. We recently observed stereotactic injection of mitochondrial lysates or mtDNA into rodent hippocampi induced pro-inflammatory changes (178). Mitochondrial lysates increased hippocampal TNFα mRNA and decreased TREM2 mRNA expression. In addition, NFĸB phosphorylation was elevated in the cortex, while glial fibrillary acidic protein (GFAP) protein levels were elevated within the hippocampus. Hippocampal mtDNA injection lead to increased hippocampal TNFα mRNA but reduced hippocampal TREM2 mRNA, increased GFAP hippocampal protein expression, elevated cortical NFĸB phosphorylation, increased cortical colony-stimulating factor 1 receptor protein expression, and increased levels of phosphorylated AKT within the cortex. Beyond these inflammatory changes, whole mitochondria lysates increased protein and mRNA levels of endogenous rodent APP and Aβ1–42. These effects on APP and Aβ1–42 were not observed following injection of mtDNA. Overall, these studies provide evidence that mitochondria-derived DAMP molecules are capable of inducing neuroinflammation, as well as altering AD-related pathways.
While some studies suggest Aβ is pro-inflammatory and acts as a DAMP molecule, a recent study interestingly suggests Aβ has antimicrobial properties (179). In models that overproduce Aβ, including rodent, cell culture, and worm models, the severity of fungal and microbial infections was reduced. Injection of the bacterium S. typhimurium into the brain of an AD mouse model (5XFAD model) increased the propensity of Aβ to form plaques, and Aβ colocalized with the bacteria. In addition, the 5XFAD mice injected with S. typhimurium showed increased survival and reduced meningitis. This study further found that oligomerization of Aβ was required for this antimicrobial property to manifest. Earlier in vitro studies also showed Aβ initiated antimicrobial activity against several bacterial and fungal microorganisms. Aβ inhibited bacterial growth, and brain homogenates from AD subjects (containing Aβ) were also capable of inhibiting microbial growth (180). These studies suggest a possible connection between mitochondria-derived DAMP molecules, neuroinflammation, APP metabolism, and Aβ.
Mitochondria-Derived DAMP Molecules As Biomarkers of Brain Integrity
Cell-free circulating mitochondrial components (DAMPs) such as mtDNA are altered in trauma. For example, in children with traumatic brain injury (TBI), CSF mtDNA levels are elevated. CSF mtDNA levels also correlate with TBI outcome. In children who survived a TBI, CSF mtDNA levels were in the lower range, while in children whose outcome included either death or severe disability CSF mtDNA levels were in the upper range. CSF levels of another DAMP molecule, high mobility group box 1, correlated with mtDNA levels in these subjects. Overall, mtDNA appears to represent a potential CSF mitochondrial DAMP biomarker for TBI and to have the potential to correlate with patient outcomes (3).
Circulating mtDNA is also associated with aging. In a study of 831 Caucasian subjects spanning ages 1–104 years, plasma mtDNA levels began to increase after the fifth decade of life. Plasma mtDNA levels further correlated with cytokine levels, specifically TNFα, IL-6, and regulated on activation normal T cell expressed and secreted (RANTES). Subjects with the highest levels of plasma mtDNA also had the highest levels of these cytokines (TNFα, IL-6, and RANTES), while subjects with the lowest plasma mtDNA levels had the lowest amounts of measured cytokines (156).
Circulating mtDNA may also potentially have the ability to serve as an AD or PD biomarker. Cell-free CSF mtDNA levels are reduced in clinically asymptomatic subjects with a genetically defined increased AD risk and clinically symptomatic AD subjects (157). However, no differences were observed for subjects with frontotemporal lobar degeneration. In a separate study, PD subjects were found to have lower levels of cell-free CSF mtDNA (158). While some may argue lower cell-free CSF mtDNA negates the possibility of a mitochondria-derived DAMP-induced neuroinflammation, other evidence contradicts this idea. It is important to note levels of another AD biomarker, Aβ, are elevated in the brain but significantly reduced in CSF from AD subjects (181, 182). Therefore, it is difficult to draw conclusions regarding CSF biomarker data and causation without sufficient knowledge of the mechanisms that underlie those biomarker changes.
Specific Release of Mitochondria-Derived DAMP Molecules within the CNS
An important question relevant to the issue of mitochondria-derived DAMPs is how might these intracellular molecules access the extracellular space? While cellular components are released during necrotic cell death, more specific process through which mitochondria are released to the brain’s extracellular compartment have recently been described. One of these processes has been termed transcellular mitophagy. Neurons are large cells and can span long distances. Mitochondria continuously move between the cell body and dendrites, and between the cell body and axons. When peripheral mitochondria cease to function properly, they were believed to return to the cell body for disposal through the process of mitochondrial autophagy or mitophagy. Data now indicate neurons can also export mitochondria to surrounding glial cells, where they then undergo mitophagy. This process was first described to occur within the optic nerve and the cortex (183). In a separate study, it was found that astrocytes also transfer mitochondria to neurons (184). The transfer of mitochondria from astrocytes to neurons enhanced neuronal survival following ischemia reperfusion injury and required signaling from cluster of differentiation 38, cyclic ADP ribose, and calcium.
Conclusion
In specific neurodegenerative diseases, if mitochondrial dysfunction overwhelmed the ability of neurons and astrocytes to adequately perform mitophagy, then mitochondria-derived DAMP molecules could predictably facilitate neuroinflammation (Figure 1). Clearly, a rational case can be made that mitochondria-derived DAMP molecules may contribute to neurodegenerative disease-associated neuroinflammation. However, important questions remain about how mitochondrial DAMPs contribute to neurodegenerative diseases and neurodegenerative disease-related pathologies. Future directions should focus on if and how specific mitochondrial-derived molecules initiate neuroinflammation. More specifically, how does mitochondrial dysfunction contribute to the release of DAMP molecules and is this process upstream or downstream of other disease pathologies? As this line of investigation moves forward, studies to address issues of cause versus consequence should help resolve these important knowledge gaps.
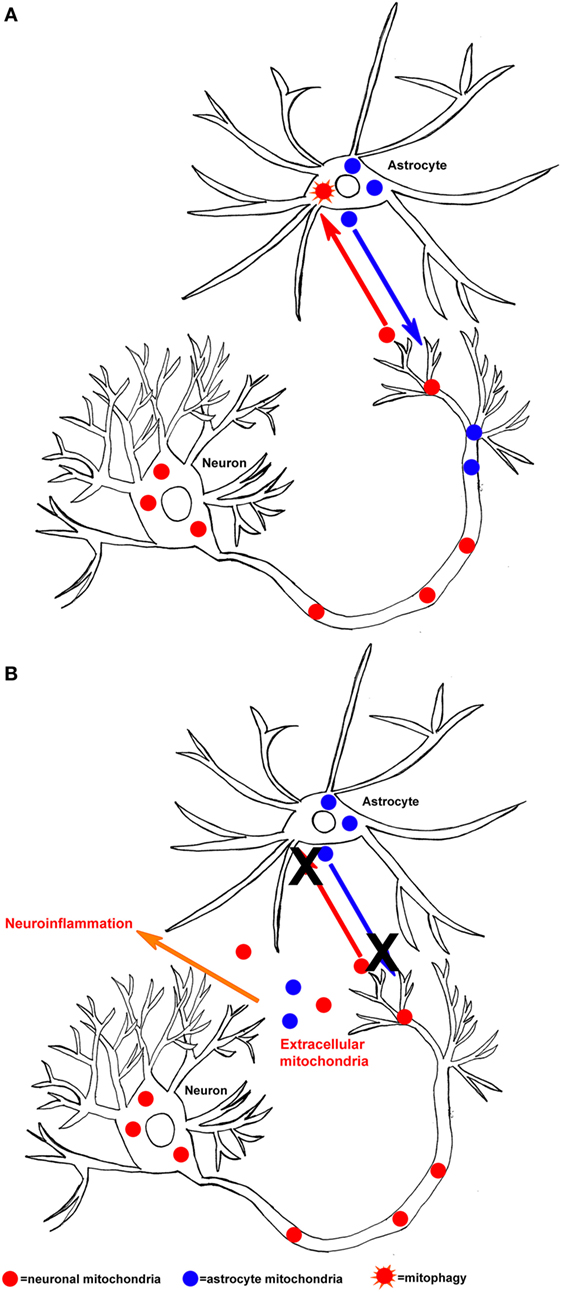
Figure 1. Hypothesized mechanism of mitochondrial-derived damage-associated molecular pattern molecule released in the central nervous system. (A) Neurons can release mitochondria (neuronal mitochondria are shown in red) to astrocytes, where they then undergo mitophagy (orange star shape). In addition, astrocytes can release mitochondria (astrocyte mitochondria are shown in blue) to neurons, under conditions of bioenergetic stress. (B) If the process of mitochondrial exchange between neurons and astrocytes malfunctions (X), then mitochondria and their components could be released into the extracellular space and initiate neuroinflammation.
Author Contributions
HW was responsible for writing the article. IW and YJ contributed to sections of the article. RS edited and provided direction for the article.
Conflict of Interest Statement
The authors declare this review article was completed in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Funding
This project was supported by the University of Kansas Alzheimer’s Disease Center (P30 AG035982), the Frank and Evangeline Thompson Alzheimer’s Treatment Program fund, the Kansas IDeA Network for Biomedical Research Excellence (KINBRE, P20GM103418), the University of Kansas Medical Center Biomedical Research Training Program, and a Mabel Woodyard Fellowship award.
Abbreviations
3NP, 3-nitropropionic acid; ACS, apoptosis-associated speck-like protein containing a CARD; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; APOE, apolipoprotein E; APP, amyloid precursor protein; ATP, adenosine triphosphate; BBB, blood–brain barrier; CD33, Siglec-3; CD38, cluster of differentiation 38; CLU, clusterin; CNS, central nervous system; COX, cytochrome oxidase; CR1, complement rector 1; CSF, cerebral spinal fluid; CSF1R, colony-stimulating factor 1 receptor; CXCR31, CX3C chemokine receptor 1 or fractalkine receptor; cybrid, cytoplasmic hybrid; DAMP, damage-associated molecular pattern; EPHA1, ephrin type A receptor 1; ER, endoplasmic reticulum; FCγRII, Fc receptor or CD32; FPR or FPRL1, formyl peptide receptors; FTLD, frontotemporal lobar degeneration; GFAP, glial fibrillary acidic protein; GWAS, genome-wide association study; HLA-DRB5/DRB1, major histocompatibility complex, class II, DR beta 5; HMGB1, high mobility group box 1; IFN-γ, interferon-γ; IgG, immunoglobulin G; IL-1, interleukin-1; IL-1β, interleukin-1β; IL-6, interleukin-6; IL-8, interleukin-8; INPPD5, inositol polyphosphate-5-phosphatase; IP, intraperitoneal; LPS, lipopolysaccharide; LRRK2, leucine-rich repeat kinase 2; MCI, mild-cognitive impairment; MCP-1, monocyte chemotactic protein 1; M-CSF, macrophage colony-stimulating factor; MHC, major histocompatibility complex; MIP-1α, macrophage inflammatory protein-1α; MMP-8, matrix metalloproteinase 8; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; mtDNA, mitochondrial DNA; NLRP3, NLR family pyrin domain containing 3; NO, nitric oxide; NOD, nucleotide-binding oligomerization domain-like receptors; NSAID, non-steroidal anti-inflammatory drug; PAMP, pathogen-associated molecular pattern; PD, Parkinson’s disease; PET, positron emission tomography; PRR, pattern-recognition receptor; RAGE, receptor for advanced glycation end products; RANTES, regulated on activation, normal T cell expressed and secreted; ROS, reactive oxygen species; SNP, single-nucleotide polymorphism; SOD1, superoxide dismutase 1; SRA, microglial scavenger receptor A; TBI, traumatic brain injury; TFAM, transcription factor A; TGF-β, transforming growth factor β; TNFα, tumor necrosis factor α; TREM2, triggering receptor expressed on myeloid cells 2.
References
1. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature (2012) 485(7397):251–5. doi: 10.1038/nature10992
2. Lam NY, Rainer TH, Chiu RW, Joynt GM, Lo YM. Plasma mitochondrial DNA concentrations after trauma. Clin Chem (2004) 50(1):213–6. doi:10.1373/clinchem.2003.025783
3. Walko TD III, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, et al. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock (2014) 41(6):499–503. doi:10.1097/SHK.0000000000000160
4. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
5. Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol (2004) 75(6):995–1000. doi:10.1189/jlb.0703328
6. Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis (2010) 20(Suppl 2):S369–79. doi:10.3233/JAD-2010-100543
7. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell (2010) 140(6):918–34. doi:10.1016/j.cell.2010.02.016
8. Hensley K. Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis (2010) 21(1):1–14. doi:10.3233/JAD-2010-1414
9. Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, et al. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis (2012) 30(3):617–27. doi:10.3233/JAD-2012-120145
10. Abramova NE, Davies KJ, Crawford DR. Polynucleotide degradation during early stage response to oxidative stress is specific to mitochondria. Free Radic Biol Med (2000) 28(2):281–8. doi:10.1016/S0891-5849(99)00239-7
11. in ’t Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med (2001) 345(21):1515–21. doi:10.1056/NEJMoa010178
12. Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology (1997) 48(3):626–32. doi:10.1212/WNL.48.3.626
13. Szekely CA, Breitner JC, Fitzpatrick AL, Rea TD, Psaty BM, Kuller LH, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology (2008) 70(1):17–24. doi:10.1212/01.wnl.0000284596.95156.48
14. Cornelius C, Fastbom J, Winblad B, Viitanen M. Aspirin, NSAIDs, risk of dementia, and influence of the apolipoprotein E epsilon 4 allele in an elderly population. Neuroepidemiology (2004) 23(3):135–43. doi:10.1159/000075957
15. Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology (2008) 70(19):1672–7. doi:10.1212/01.wnl.0000311269.57716.63
16. Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology (1993) 43(8):1609–11. doi:10.1212/WNL.43.8.1609
17. Scharf S, Mander A, Ugoni A, Vajda F, Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology (1999) 53(1):197–201. doi:10.1212/WNL.53.1.197
18. Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA, et al. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: a randomised phase II trial. Lancet Neurol (2008) 7(6):483–93. doi:10.1016/s1474-4422(08)70090-5
19. Daniels MJD, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun (2016) 7:12504. doi:10.1038/ncomms12504
20. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med (2012) 368(2):117–27. doi:10.1056/NEJMoa1211851
21. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell (2015) 160(6):1061–71. doi:10.1016/j.cell.2015.01.049
22. Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med (2014) 6(243):243ra86. doi:10.1126/scitranslmed.3009093
23. Gispert JD, Suarez-Calvet M, Monte GC, Tucholka A, Falcon C, Rojas S, et al. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement (2016) 12(12):1259–72. doi:10.1016/j.jalz.2016.06.005
24. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet (2009) 41(10):1088–93. doi:10.1038/ng.440
25. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet (2009) 41(10):1094–9. doi:10.1038/ng.439
26. McGeer PL, McGeer EG. Polymorphisms in inflammatory genes and the risk of Alzheimer disease. Arch Neurol (2001) 58(11):1790–2. doi:10.1001/archneur.58.11.1790
27. Hazrati LN, Van Cauwenberghe C, Brooks PL, Brouwers N, Ghani M, Sato C, et al. Genetic association of CR1 with Alzheimer’s disease: a tentative disease mechanism. Neurobiol Aging (2012) 33(12): 2949.e5–12. doi:10.1016/j.neurobiolaging.2012.07.001
28. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet (2011) 43(5):429–35. doi:10.1038/ng.803
29. Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet (2013) 45(12):1452–8. doi:10.1038/ng.2802
30. Wilkins HM, Carl SM, Greenlief AC, Festoff BW, Swerdlow RH. Bioenergetic dysfunction and inflammation in Alzheimer’s disease: a possible connection. Front Aging Neurosci (2014) 6:311. doi:10.3389/fnagi.2014.00311
31. Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci Lett (1990) 119(1):32–6. doi:10.1016/0304-3940(90)90748-X
32. Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol (2013) 239:133–8. doi:10.1016/j.expneurol.2012.10.007
33. Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A (1989) 86(19):7611–5. doi:10.1073/pnas.86.19.7611
34. Sheng JG, Mrak RE, Griffin WST. Microglial inter leukin-1α expression in brain regions in Alzheimer’s disease: correlation with neuritic plaque distribution. Neuropathol Appl Neurobiol (1995) 21(4):290–301. doi:10.1111/j.1365-2990.1995.tb01063.x
35. Laske C, Stransky E, Hoffmann N, Maetzler W, Straten G, Eschweiler GW, et al. Macrophage colony-stimulating factor (M-CSF) in plasma and CSF of patients with mild cognitive impairment and Alzheimer’s disease. Curr Alzheimer Res (2010) 7(5):409–14. doi:10.2174/156720510791383813
36. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging (2000) 21(3):383–421. doi:10.1016/S0197-4580(00)00124-X
37. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology (1988) 38(8):1285–91. doi:10.1212/WNL.38.8.1285
38. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis (2006) 21(2):404–12. doi:10.1016/j.nbd.2005.08.002
39. Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, et al. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci (1999) 19(9):3440–7.
40. Barcia C, Ros CM, Ros-Bernal F, Gómez A, Annese V, Carrillo-de Sauvage MA, et al. Persistent phagocytic characteristics of microglia in the substantia nigra of long-term parkinsonian macaques. J Neuroimmunol (2013) 261(1–2):60–6. doi:10.1016/j.jneuroim.2013.05.001
41. Song YJC, Halliday GM, Holton JL, Lashley T, Sullivan SS, McCann H, et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol (2009) 68(10):1073. doi:10.1097/NEN.0b013e3181b66f1b
42. Tong J, Ang L-C, Williams B, Furukawa Y, Fitzmaurice P, Guttman M, et al. Low levels of astroglial markers in Parkinson’s disease: relationship to α-synuclein accumulation. Neurobiol Dis (2015) 82:243–53. doi:10.1016/j.nbd.2015.06.010
43. Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology (2007) 69(19):1836–42. doi:10.1212/01.wnl.0000279519.99344.ad
44. Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med (2012) 2(1):a008888. doi:10.1101/cshperspect.a008888
45. Ma B, Xu L, Pan X, Sun L, Ding J, Xie C, et al. LRRK2 modulates microglial activity through regulation of chemokine (C-X3-C) receptor 1-mediated signalling pathways. Hum Mol Genet (2016) 25(16):3515–23. doi:10.1093/hmg/ddw194
46. Russo I, Bubacco L, Greggio E. LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation (2014) 11:52. doi:10.1186/1742-2094-11-52
47. Choi I, Kim B, Byun JW, Baik SH, Huh YH, Kim JH, et al. LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nat Commun (2015) 6:8255. doi:10.1038/ncomms9255
48. Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord (2005) 11(8):493–8. doi:10.1016/j.parkreldis.2005.07.005
49. Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol (2005) 57(2):176–9. doi:10.1002/ana.20369
50. Chen H, O’Reilly EJ, Schwarzschild MA, Ascherio A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am J Epidemiol (2008) 167(1):90–5. doi:10.1093/aje/kwm260
51. Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest (2009) 119(1):182–92. doi:10.1172/jci36470
52. Donnenfeld H, Kascsak RJ, Bartfeld H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol (1984) 6(1):51–7. doi:10.1016/0165-5728(84)90042-0
53. Lampson LA, Kushner PD, Sobel RA. Major histocompatibility complex antigen expression in the affected tissues in amyotrophic lateral sclerosis. Ann Neurol (1990) 28(3):365–72. doi:10.1002/ana.410280311
54. Troost D, Van den Oord JJ, Vianney de Jong JM. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol (1990) 16(5):401–10. doi:10.1111/j.1365-2990.1990.tb01276.x
55. Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol (1992) 140(3):691–707.
56. Turner MR, Cagnin A, Turkheimer FE, Miller CCJ, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis (2004) 15(3):601–9. doi:10.1016/j.nbd.2003.12.012
57. Schiffer D, Cordera S, Cavalla P, Migheli A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci (1996) 139(Suppl):27–33. doi:10.1016/0022-510X(96)00073-1
58. Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science (2006) 312(5778):1389–92. doi:10.1126/science.1123511
59. Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci (2008) 11(3):251–3. doi:10.1038/nn2047
60. Lopez-Lopez A, Gamez J, Syriani E, Morales M, Salvado M, Rodriguez MJ, et al. CX3CR1 is a modifying gene of survival and progression in amyotrophic lateral sclerosis. PLoS One (2014) 9(5):e96528. doi:10.1371/journal.pone.0096528
61. Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci (2013) 7:26. doi:10.3389/fncel.2013.00026
62. Brohawn DG, O’Brien LC, Bennett JP Jr. RNAseq analyses identify tumor necrosis factor-mediated inflammation as a major abnormality in ALS spinal cord. PLoS One (2016) 11(8):e0160520. doi:10.1371/journal.pone.0160520
63. Lu C, Cortopassi G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch Biochem Biophys (2007) 457(1):111–22. doi:10.1016/j.abb.2006.09.010
64. Hayashi G, Cortopassi G. Oxidative stress in inherited mitochondrial diseases. Free Radic Biol Med (2015) 88(Pt A):10–7. doi:10.1016/j.freeradbiomed.2015.05.039
65. Kaplan J. Friedreich’s ataxia is a mitochondrial disorder. Proc Natl Acad Sci U S A (1999) 96(20):10948–9. doi:10.1073/pnas.96.20.10948
66. Hayashi G, Shen Y, Pedersen TL, Newman JW, Pook M, Cortopassi G. Frataxin deficiency increases cyclooxygenase 2 and prostaglandins in cell and animal models of Friedreich’s ataxia. Hum Mol Genet (2014) 23(25):6838–47. doi:10.1093/hmg/ddu407
67. Makino M, Horai S, Goto Y, Nonaka I. Mitochondrial DNA mutations in Leigh syndrome and their phylogenetic implications. J Hum Genet (2000) 45(2):69–75. doi:10.1007/s100380050014
68. Thorburn DR, Rahman S. Mitochondrial DNA-associated Leigh syndrome and NARP. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle, WA (1993).
69. Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science (2013) 342(6165):1524–8. doi:10.1126/science.1244360
70. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet (1993) 5(4):327–37. doi:10.1038/ng1293-327
71. Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol (2006) 2(9):482–93. doi:10.1038/ncpneuro0291
72. Lutsenko S, Cooper MJ. Localization of the Wilson’s disease protein product to mitochondria. Proc Natl Acad Sci U S A (1998) 95(11):6004–9. doi:10.1073/pnas.95.11.6004
73. Gu M, Cooper JM, Butler P, Walker AP, Mistry PK, Dooley JS, et al. Oxidative-phosphorylation defects in liver of patients with Wilson’s disease. Lancet (2000) 356(9228):469–74. doi:10.1016/S0140-6736(00)02556-3
74. Rossi L, Lombardo MF, Ciriolo MR, Rotilio G. Mitochondrial dysfunction in neurodegenerative diseases associated with copper imbalance. Neurochem Res (2004) 29(3):493–504. doi:10.1023/B:NERE.0000014820.99232.8a
75. Wang H, Cheng N, Dong J, Wang X, Han Y, Yang R, et al. Serum pentraxin 3 is elevated in patients with neurological Wilson’s disease. Clin Chim Acta (2016) 462:178–82. doi:10.1016/j.cca.2016.08.010
76. Scheffler IE. A century of mitochondrial research: achievements and perspectives. Mitochondrion (2001) 1(1):3–31. doi:10.1016/S1567-7249(00)00002-7
77. Swerdlow RH. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals (Basel) (2009) 2(3):150–67. doi:10.3390/ph2030150
78. Swerdlow RH. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal (2012) 16(12):1434–55. doi:10.1089/ars.2011.4149
79. Lezi E, Swerdlow RH. Mitochondria in neurodegeneration. Adv Exp Med Biol (2012) 942:269–86. doi:10.1007/978-94-007-2869-1_12
80. Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol (2005) 58(4):495–505. doi:10.1002/ana.20624
81. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature (2006) 443(7113):787–95. doi:10.1038/nature05292
82. Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci (2001) 21(9):3017–23.
83. Parker WD Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology (1990) 40(8):1302–3. doi:10.1212/WNL.40.8.1302
84. Parker WD Jr, Parks JK. Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterization. Neurology (1995) 45(3 Pt 1):482–6. doi:10.1212/WNL.45.3.482
85. Kish SJ. Brain energy metabolizing enzymes in Alzheimer’s disease: alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann N Y Acad Sci (1997) 826:218–28. doi:10.1111/j.1749-6632.1997.tb48473.x
86. Mutisya EM, Bowling AC, Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J Neurochem (1994) 63(6):2179–84. doi:10.1046/j.1471-4159.1994.63062179.x
87. Cardoso SM, Proenca MT, Santos S, Santana I, Oliveira CR. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol Aging (2004) 25(1):105–10. doi:10.1016/S0197-4580(03)00033-2
88. Curti D, Rognoni F, Gasparini L, Cattaneo A, Paolillo M, Racchi M, et al. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer’s disease (AD) patients. Neurosci Lett (1997) 236(1):13–6. doi:10.1016/S0304-3940(97)00741-6
89. Mancuso M, Filosto M, Bosetti F, Ceravolo R, Rocchi A, Tognoni G, et al. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp Neurol (2003) 182(2):421–6. doi:10.1016/S0014-4886(03)00092-X
90. Wilkins HM, Carl SM, Swerdlow RH. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol (2014) 2:619–31. doi:10.1016/j.redox.2014.03.006
91. Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, et al. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol (2000) 48(2):148–55. doi:10.1002/1531-8249(200008)48:2<148::AID-ANA3>3.0.CO;2-7
92. Chang SW, Zhang D, Chung HD, Zassenhaus HP. The frequency of point mutations in mitochondrial DNA is elevated in the Alzheimer’s brain. Biochem Biophys Res Commun (2000) 273(1):203–8. doi:10.1006/bbrc.2000.2885
93. Hamblet NS, Castora FJ. Elevated levels of the Kearns-Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer’s patients. Mutat Res (1997) 379(2):253–62. doi:10.1016/S0027-5107(97)00158-9
94. Krishnan KJ, Ratnaike TE, De Gruyter HL, Jaros E, Turnbull DM. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol Aging (2012) 33(9):2210–4. doi:10.1016/j.neurobiolaging.2011.08.009
95. Lovell MA, Markesbery WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res (2007) 35(22):7497–504. doi:10.1093/nar/gkm821
96. Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol (1994) 36(5):747–51. doi:10.1002/ana.410360510
97. Phillips NR, Simpkins JW, Roby RK. Mitochondrial DNA deletions in Alzheimer’s brains: a review. Alzheimers Dement (2014) 10(3):393–400. doi:10.1016/j.jalz.2013.04.508
98. Mosconi L, Berti V, Swerdlow RH, Pupi A, Duara R, de Leon M. Maternal transmission of Alzheimer’s disease: prodromal metabolic phenotype and the search for genes. Hum Genomics (2010) 4(3):170–93. doi:10.1186/1479-7364-4-3-170
99. Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, et al. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A (2007) 104(48):19067–72. doi:10.1073/pnas.0705036104
100. Mosconi L, de Leon M, Murray J, Lezi E, Lu J, Javier E, et al. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J Alzheimers Dis (2011) 27(3):483–90. doi:10.3233/JAD-2011-110866
101. Honea RA, Vidoni ED, Swerdlow RH, Burns JM; Alzheimer’s Disease Neuroimaging Initiative. Maternal family history is associated with Alzheimer’s disease biomarkers. J Alzheimers Dis (2012) 31(3):659–68. doi:10.3233/JAD-2012-120676
102. Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology (1996) 47(1):254–6. doi:10.1212/WNL.47.1.254
103. Andrawis JP, Hwang KS, Green AE, Kotlerman J, Elashoff D, Morra JH, et al. Effects of ApoE4 and maternal history of dementia on hippocampal atrophy. Neurobiol Aging (2012) 33(5):856–66. doi:10.1016/j.neurobiolaging.2010.07.020
104. Fesahat F, Houshmand M, Panahi MS, Gharagozli K, Mirzajani F. Do haplogroups H and U act to increase the penetrance of Alzheimer’s disease? Cell Mol Neurobiol (2007) 27(3):329–34. doi:10.1007/s10571-006-9126-9
105. Lakatos A, Derbeneva O, Younes D, Keator D, Bakken T, Lvova M, et al. Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol Aging (2010) 31(8):1355–63. doi:10.1016/j.neurobiolaging.2010.04.031
106. Maruszak A, Canter JA, Styczynska M, Zekanowski C, Barcikowska M. Mitochondrial haplogroup H and Alzheimer’s disease – is there a connection? Neurobiol Aging (2009) 30(11):1749–55. doi:10.1016/j.neurobiolaging.2008.01.004
107. Sian J, Youdim MBH, Riederer P, Gerlach M. MPTP-induced parkinsonian syndrome. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th ed. Philadelphia: Lippincott-Raven (1999). Available from: https://www.ncbi.nlm.nih.gov/books/NBK27974/
108. Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson’s disease: an update. J Parkinsons Dis (2011) 1(1):19–33. doi:10.3233/JPD-2011-11023
109. Porras G, Li Q, Bezard E. Modeling Parkinson’s disease in primates: the MPTP model. Cold Spring Harb Perspect Med (2012) 2(3):a009308. doi:10.1101/cshperspect.a009308
110. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem (1990) 54(3):823–7. doi:10.1111/j.1471-4159.1990.tb02325.x
111. Yoshino H, Nakagawa-Hattori Y, Kondo T, Mizuno Y. Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson’s disease. J Neural Transm Park Dis Dement Sect (1992) 4(1):27–34. doi:10.1007/BF02257619
112. Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, et al. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann Neurol (1995) 37(6):714–22. doi:10.1002/ana.410370604
113. Keeney PM, Xie J, Capaldi RA, Bennett JP Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci (2006) 26(19):5256–64. doi:10.1523/JNEUROSCI.0984-06.2006
114. Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, et al. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol (1996) 40(4):663–71. doi:10.1002/ana.410400417
115. Esteves AR, Domingues AF, Ferreira IL, Januario C, Swerdlow RH, Oliveira CR, et al. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion (2008) 8(3):219–28. doi:10.1016/j.mito.2008.03.004
116. Autere J, Moilanen JS, Finnila S, Soininen H, Mannermaa A, Hartikainen P, et al. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum Genet (2004) 115(1):29–35. doi:10.1007/s00439-004-1123-9
117. Ghezzi D, Marelli C, Achilli A, Goldwurm S, Pezzoli G, Barone P, et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson’s disease in Italians. Eur J Hum Genet (2005) 13(6):748–52. doi:10.1038/sj.ejhg.5201425
118. Pyle A, Foltynie T, Tiangyou W, Lambert C, Keers SM, Allcock LM, et al. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol (2005) 57(4):564–7. doi:10.1002/ana.20417
119. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet (2006) 38(5):515–7. doi:10.1038/ng1769
120. Lin MT, Cantuti-Castelvetri I, Zheng K, Jackson KE, Tan YB, Arzberger T, et al. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol (2012) 71(6):850–4. doi:10.1002/ana.23568
121. Parker WD Jr, Parks JK. Mitochondrial ND5 mutations in idiopathic Parkinson’s disease. Biochem Biophys Res Commun (2005) 326(3):667–9. doi:10.1016/j.bbrc.2004.11.093
122. Luoma PT, Eerola J, Ahola S, Hakonen AH, Hellstrom O, Kivisto KT, et al. Mitochondrial DNA polymerase gamma variants in idiopathic sporadic Parkinson disease. Neurology (2007) 69(11):1152–9. doi:10.1212/01.wnl.0000276955.23735.eb
123. Dhaliwal GK, Grewal RP. Mitochondrial DNA deletion mutation levels are elevated in ALS brains. Neuroreport (2000) 11(11):2507–9. doi:10.1097/00001756-200008030-00032
124. Sasaki S, Horie Y, Iwata M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol (2007) 114(6):633–9. doi:10.1007/s00401-007-0299-1
125. Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol (2007) 66(1):10–6. doi:10.1097/nen.0b013e31802c396b
126. Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol (1999) 46(5):787–90. doi:10.1002/1531-8249(199911)46:5<787::AID-ANA17>3.0.CO;2-8
127. Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem (2002) 80(4):616–25. doi:10.1046/j.0022-3042.2001.00731.x
128. Mawrin C, Kirches E, Krause G, Wiedemann FR, Vorwerk CK, Bogerts B, et al. Single-cell analysis of mtDNA deletion levels in sporadic amyotrophic lateral sclerosis. Neuroreport (2004) 15(6):939–43. doi:10.1097/00001756-200404290-00002
129. Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol (2010) 67(7):849–54. doi:10.1001/archneurol.2010.128
130. Siklos L, Engelhardt J, Harati Y, Smith RG, Joo F, Appel SH. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann Neurol (1996) 39(2):203–16. doi:10.1002/ana.410390210
131. Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, et al. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain (2000) 123(Pt 7):1339–48. doi:10.1093/brain/123.7.1339
132. Curti D, Malaspina A, Facchetti G, Camana C, Mazzini L, Tosca P, et al. Amyotrophic lateral sclerosis: oxidative energy metabolism and calcium homeostasis in peripheral blood lymphocytes. Neurology (1996) 47(4):1060–4. doi:10.1212/WNL.47.4.1060
133. Nakano Y, Hirayama K, Terao K. Hepatic ultrastructural changes and liver dysfunction in amyotrophic lateral sclerosis. Arch Neurol (1987) 44(1):103–6. doi:10.1001/archneur.1987.00520130079022
134. Shrivastava M, Das TK, Behari M, Pati U, Vivekanandhan S. Ultrastructural variations in platelets and platelet mitochondria: a novel feature in amyotrophic lateral sclerosis. Ultrastruct Pathol (2011) 35(2):52–9. doi:10.3109/01913123.2010.541985
135. Shrivastava M, Vivekanandhan S, Pati U, Behari M, Das TK. Mitochondrial perturbance and execution of apoptosis in platelet mitochondria of patients with amyotrophic lateral sclerosis. Int J Neurosci (2011) 121(3):149–58. doi:10.3109/00207454.2010.537416
136. Swerdlow RH, Parks JK, Cassarino DS, Trimmer PA, Miller SW, Maguire DJ, et al. Mitochondria in sporadic amyotrophic lateral sclerosis. Exp Neurol (1998) 153(1):135–42. doi:10.1006/exnr.1998.6866
137. Mancuso M, Conforti FL, Rocchi A, Tessitore A, Muglia M, Tedeschi G, et al. Could mitochondrial haplogroups play a role in sporadic amyotrophic lateral sclerosis? Neurosci Lett (2004) 371(2–3):158–62. doi:10.1016/j.neulet.2004.08.060
138. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med (2015) 21(3):193–201. doi:10.1016/j.molmed.2014.11.008
139. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol (2013) 191(10):5230–8. doi:10.4049/jimmunol.1301490
140. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi:10.1038/nature09663
141. Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, Ludolph AC, et al. Effects of mitochondrial dysfunction on the immunological properties of microglia. J Neuroinflammation (2010) 7:45. doi:10.1186/1742-2094-7-45
142. Ryu JK, Nagai A, Kim J, Lee MC, McLarnon JG, Kim SU. Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: in vitro and in vivo studies. Neurobiol Dis (2003) 12(2):121–32. doi:10.1016/S0969-9961(03)00002-0
143. Yi PL, Tsai CH, Lu MK, Liu HJ, Chen YC, Chang FC. Interleukin-1beta mediates sleep alteration in rats with rotenone-induced parkinsonism. Sleep (2007) 30(4):413–25. doi:10.1093/sleep/30.4.413
144. Kastl L, Sauer S, Beissbarth T, Becker M, Krammer P, Gulow K. TNF-a stimulation enhances ROS-dependent cell migration via NF-κB activation in liver cells. Free Radic Biol Med (2014) 75(Suppl 1):S32. doi:10.1016/j.freeradbiomed.2014.10.765
145. Kastl L, Sauer SW, Ruppert T, Beissbarth T, Becker MS, Suss D, et al. TNF-alpha mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-kappaB activation in liver cells. FEBS Lett (2014) 588(1):175–83. doi:10.1016/j.febslet.2013.11.033
146. Goossens V, Stange G, Moens K, Pipeleers D, Grooten J. Regulation of tumor necrosis factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron flux through the electron transport chain complex I. Antioxid Redox Signal (1999) 1(3):285–95. doi:10.1089/ars.1999.1.3-285
147. Chen XH, Zhao YP, Xue M, Ji CB, Gao CL, Zhu JG, et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol Cell Endocrinol (2010) 328(1–2):63–9. doi:10.1016/j.mce.2010.07.005
148. Higuchi M, Proske RJ, Yeh ET. Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene (1998) 17(19):2515–24. doi:10.1038/sj.onc.1202485
149. Doll DN, Rellick SL, Barr TL, Ren X, Simpkins JW. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J Neurochem (2015) 132(4):443–51. doi:10.1111/jnc.13008
150. Yasuhara R, Miyamoto Y, Akaike T, Akuta T, Nakamura M, Takami M, et al. Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem J (2005) 389(Pt 2):315–23. doi:10.1042/BJ20041996
151. Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res (2007) 85(4):462–72. doi:10.1016/j.exer.2007.06.013
152. Noh H, Jeon J, Seo H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int (2014) 69:35–40. doi:10.1016/j.neuint.2014.02.008
153. Hokari Y, Seki T, Nakano H, Matsuo Y, Fukamizu A, Munekata E, et al. Isolation and identification of novel neutrophil-activating cryptides hidden in mitochondrial cytochrome c. Protein Pept Lett (2012) 19(6):680–7. doi:10.2174/092986612800494048
154. Marutani T, Hattori T, Tsutsumi K, Koike Y, Harada A, Noguchi K, et al. Mitochondrial protein-derived cryptides: are endogenous N-formylated peptides including mitocryptide-2 components of mitochondrial damage-associated molecular patterns? Biopolymers (2016) 106(4):580–7. doi:10.1002/bip.22788
155. Seki T, Fukamizu A, Kiso Y, Mukai H. Mitocryptide-2, a neutrophil-activating cryptide, is a specific endogenous agonist for formyl-peptide receptor-like 1. Biochem Biophys Res Commun (2011) 404(1):482–7. doi:10.1016/j.bbrc.2010.12.007
156. Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur J Immunol (2014) 44(5):1552–62. doi:10.1002/eji.201343921
157. Podlesniy P, Figueiro-Silva J, Llado A, Antonell A, Sanchez-Valle R, Alcolea D, et al. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol (2013) 74(5):655–68. doi:10.1002/ana.23955
158. Pyle A, Brennan R, Kurzawa-Akanbi M, Yarnall A, Thouin A, Mollenhauer B, et al. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann Neurol (2015) 78(6):1000–4. doi:10.1002/ana.24515
159. Aichbichler BW, Petritsch W, Reicht GA, Wenzl HH, Eherer AJ, Hinterleitner TA, et al. Anti-cardiolipin antibodies in patients with inflammatory bowel disease. Dig Dis Sci (1999) 44(4):852–6. doi:10.1023/A:1026646816672
160. Erkkila AT, Narvanen O, Lehto S, Uusitupa MI, Yla-Herttuala S. Autoantibodies against oxidized low-density lipoprotein and cardiolipin in patients with coronary heart disease. Arterioscler Thromb Vasc Biol (2000) 20(1):204–9. doi:10.1161/01.ATV.20.1.204
161. Koutroubakis IE, Petinaki E, Anagnostopoulou E, Kritikos H, Mouzas IA, Kouroumalis EA, et al. Anti-cardiolipin and anti-beta2-glycoprotein I antibodies in patients with inflammatory bowel disease. Dig Dis Sci (1998) 43(11):2507–12. doi:10.1023/A:1026602803622
162. Nityanand S, Bergmark C, de Faire U, Swedenborg J, Holm G, Lefvert AK. Antibodies against endothelial cells and cardiolipin in young patients with peripheral atherosclerotic disease. J Intern Med (1995) 238(5):437–43. doi:10.1111/j.1365-2796.1995.tb01221.x
163. Thong BY, Chng HH, Ang CL, Ho MS. Recurrent venous thromboses, anti-cardiolipin antibodies and Crohn’s disease. QJM (2002) 95(4):253–5. doi:10.1093/qjmed/95.4.253-a
164. Wan M, Hua X, Su J, Thiagarajan D, Frostegard AG, Haeggstrom JZ, et al. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis (2014) 235(2):592–8. doi:10.1016/j.atherosclerosis.2014.05.913
165. Wolf P, Gretler J, Aglas F, Auer-Grumbach P, Rainer F. Anticardiolipin antibodies in rheumatoid arthritis: their relation to rheumatoid nodules and cutaneous vascular manifestations. Br J Dermatol (1994) 131(1):48–51. doi:10.1111/j.1365-2133.1994.tb08456.x
166. Wu R, Nityanand S, Berglund L, Lithell H, Holm G, Lefvert AK. Antibodies against cardiolipin and oxidatively modified LDL in 50-year-old men predict myocardial infarction. Arterioscler Thromb Vasc Biol (1997) 17(11):3159–63. doi:10.1161/01.ATV.17.11.3159
167. Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med (2010) 182(6):774–83. doi:10.1164/rccm.201003-0359OC
168. Stachon P, Geis S, Peikert A, Heidenreich A, Michel NA, Unal F, et al. Extracellular ATP induces vascular inflammation and atherosclerosis via purinergic receptor Y2 in mice. Arterioscler Thromb Vasc Biol (2016) 36(8):1577–86. doi:10.1161/ATVBAHA.115.307397
169. Raoof M, Zhang Q, Itagaki K, Hauser CJ. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J Trauma (2010) 68(6):1328–32, discussion 32–4. doi:10.1097/TA.0b013e3181dcd28d
170. Gao X, Hu X, Qian L, Yang S, Zhang W, Zhang D, et al. Formyl-methionyl-leucyl-phenylalanine-induced dopaminergic neurotoxicity via microglial activation: a mediator between peripheral infection and neurodegeneration? Environ Health Perspect (2008) 116(5):593–8. doi:10.1289/ehp.11031
171. Pan ZK, Chen LY, Cochrane CG, Zuraw BL. fMet-Leu-Phe stimulates proinflammatory cytokine gene expression in human peripheral blood monocytes: the role of phosphatidylinositol 3-kinase. J Immunol (2000) 164(1):404–11. doi:10.4049/jimmunol.164.1.404
172. Sodhi A, Biswas SK. fMLP-induced in vitro nitric oxide production and its regulation in murine peritoneal macrophages. J Leukoc Biol (2002) 71(2):262–70.
173. Little JP, Simtchouk S, Schindler SM, Villanueva EB, Gill NE, Walker DG, et al. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol Cell Neurosci (2014) 60:88–96. doi:10.1016/j.mcn.2014.04.003
174. Chaung WW, Wu R, Ji Y, Dong W, Wang P. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med (2012) 30(1):199–203. doi:10.3892/ijmm.2012.959
175. Pullerits R, Bokarewa M, Jonsson IM, Verdrengh M, Tarkowski A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology (2005) 44(1):32–9. doi:10.1093/rheumatology/keh406
176. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol (2011) 32(4):157–64. doi:10.1016/j.it.2011.01.005
177. Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, et al. Mitochondrial lysates induce inflammation and Alzheimer’s disease-relevant changes in microglial and neuronal cells. J Alzheimers Dis (2015) 45(1):305–18. doi:10.3233/JAD-142334
178. Wilkins HM, Koppel SJ, Weidling IW, Roy N, Ryan LN, Stanford JA, et al. Extracellular mitochondria and mitochondrial components act as damage-associated molecular pattern molecules in the mouse brain. J Neuroimmune Pharmacol (2016) 11(4):622–8. doi:10.1007/s11481-016-9704-7
179. Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med (2016) 8(340):340ra72. doi:10.1126/scitranslmed.aaf1059
180. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One (2010) 5(3):e9505. doi:10.1371/journal.pone.0009505
181. Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol (2009) 66(3):382–9. doi:10.1001/archneurol.2008.596
182. Galasko D, Chang L, Motter R, Clark CM, Kaye J, Knopman D, et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol (1998) 55(7):937–45. doi:10.1001/archneur.55.7.937
183. Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A (2014) 111(26):9633–8. doi:10.1073/pnas.1404651111
Keywords: damage-associated molecular pattern, mitochondria, neuroinflammation, neurodegeneration, sterile inflammation
Citation: Wilkins HM, Weidling IW, Ji Y and Swerdlow RH (2017) Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 8:508. doi: 10.3389/fimmu.2017.00508
Received: 26 October 2016; Accepted: 12 April 2017;
Published: 26 April 2017
Edited by:
Walter Gottlieb Land, University of Strasbourg, FranceReviewed by:
R. Clinton Webb, Georgia Health Sciences University, USAFlorence Burté, Newcastle University, UK
Copyright: © 2017 Wilkins, Weidling, Ji and Swerdlow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Russell H. Swerdlow, cnN3ZXJkbG93QGt1bWMuZWR1