 Katalin Lumniczky
Katalin Lumniczky Tünde Szatmári
Tünde Szatmári Géza Sáfrány
Géza Sáfrány- Division of Radiation Medicine, National Public Health Centre, National Research Directorate for Radiobiology and Radiohygiene, Budapest, Hungary
Radiation-induced late brain injury consisting of vascular abnormalities, demyelination, white matter necrosis, and cognitive impairment has been described in patients subjected to cranial radiotherapy for brain tumors. Accumulating evidence suggests that various degrees of cognitive deficit can develop after much lower doses of ionizing radiation, as well. The pathophysiological mechanisms underlying these alterations are not elucidated so far. A permanent deficit in neurogenesis, chronic microvascular alterations, and blood–brain barrier dysfunctionality are considered among the main causative factors. Chronic neuroinflammation and altered immune reactions in the brain, which are inherent complications of brain irradiation, have also been directly implicated in the development of cognitive decline after radiation. This review aims to give a comprehensive overview on radiation-induced immune alterations and inflammatory reactions in the brain and summarizes how these processes can influence cognitive performance. The available data on the risk of low-dose radiation exposure in the development of cognitive impairment and the underlying mechanisms are also discussed.
Introduction
Cellular and molecular mechanisms leading to radiation-induced brain injury are far from being understood. Currently, the concomitant involvement of multiple processes is thought to contribute to the development of several pathologies. Such processes are damage at the level of microvessels leading to blood–brain barrier (BBB) leakage, increased neuronal stem, and progenitor cell death as a consequence of direct cytotoxic effect of radiation, perturbations in the energy production due to mitochondrial damage, as well as direct (activation of microglia cells) and consequential (increased infiltration of immune and inflammatory cells through the damaged BBB) inflammatory and immune reactions. Although these processes are often discussed separately for didactic purposes, they are tightly interrelated where inflammation constitutes a major link. This review will focus on the role of inflammatory and immune reactions in the development of radiation-induced cognitive deficits (Figure 1).
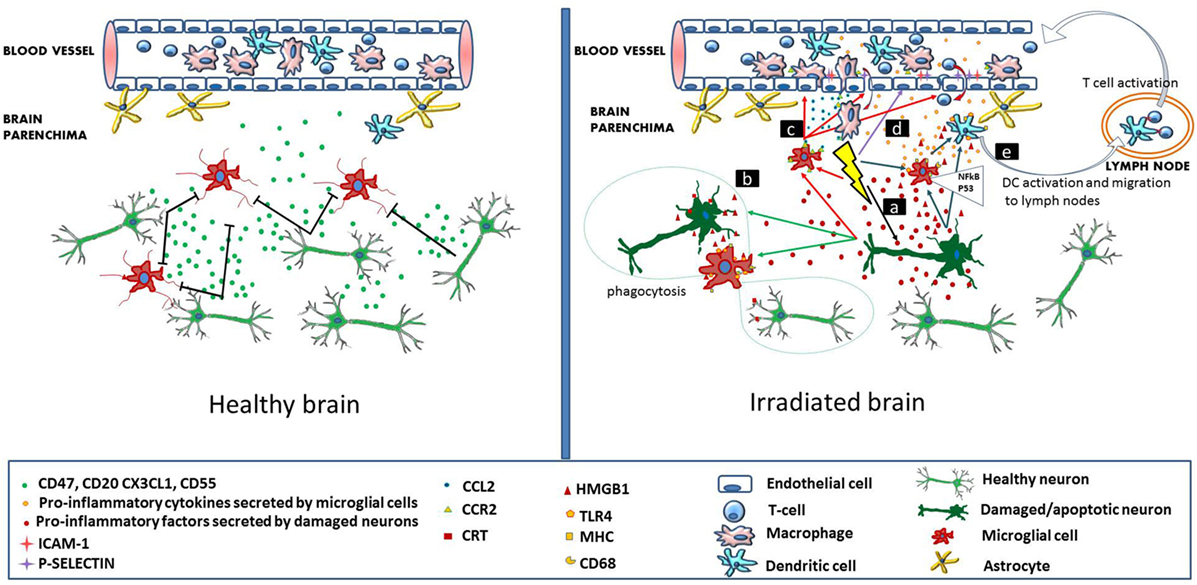
Figure 1. Immune signaling in the healthy and irradiated brain. In the healthy brain (left panel), intact neurons express and secrete molecules (CD47, CD55, CD20, and CX3CL1), which maintain adjacent microglial cells in a resting state. Brain microvascular endothelial cells, also in a resting state allow the continuous flow of blood lymphocytes and myeloid cells. In the irradiated brain (right panel), radiation-induced direct cellular damage affects neurons and microglia. Neuronal damage leads to the secretion of pro-inflammatory cytokines by the neurons, which activate microglia (mechanism a). In microglia, radiation-induced DNA damage through the NFκB pathway leads to microglia activation (MHC, CD68 upregulation) and secretion of pro-inflammatory cytokines (mechanism a). Damaged neurons secrete high-mobility group protein 1 (HMGB1) in the extracellular environment, which is a ligand for TLR4 on the activated microglia. Damaged neurons also express calreticulin on their surface, which is sensed by activated microglia and induces phagocytosis of both damaged and healthy neurons (mechanism b). Irradiation increases the secretion of CCL2 by activated microglia and also upregulates CCR2 expression. CCL2 signaling is a chemoattractant for CCR2-expressing peripheral macrophages, which penetrate the blood–brain barrier (mechanism c). Radiation induces upregulation of adhesion markers [intercellular adhesion molecule 1 (ICAM-1), P-selectin] on brain microvascular endothelial cells. Peripheral lymphocytes and monocytes adhere to activated endothelial cells and transmigrate through the microvessel wall (mechanism d). Pro-inflammatory signals and HMGB1 emitted by damaged neurons and activated microglia activate brain-residing dendritic cells, which migrate to regional lymph nodes and induce immune activation (mechanism e).
The Immune Status of the Healthy Brain
Physiologically inflammation and subsequent immune reactions are protective mechanisms of the body by which foreign pathogens and damaged cells are eliminated and homeostasis is restored. During an inflammatory reaction, cellular and tissue damage of various extents takes place, which in the case of a tissue with a good regenerative capacity does not normally lead to functional deficit. Brain, however, is an organ with a very poor regenerative capacity. Thus, in order to minimize inflammation-induced neuronal damage, the interaction between the central nervous system (CNS) and the immune system is in several aspects different from other organs. This leads to a privileged immune status of the brain maintained by certain structural and functional features: (1) the BBB and the blood–cerebrospinal fluid barrier (BCSFB) are well-structured barrier systems that tightly control the free penetration of immune cells into the brain parenchyma. (2) Antigen presentation within the brain and at the regional lymph nodes is restricted due to (i) the absence of constitutive expression of major histocompatibility I molecules on neurons of the adult brain (1); (ii) the low number of professional antigen-presenting cells (APCs)—mainly dendritic cells (DCs)—and resident T cells in the brain parenchyma (2); and (iii) the lack of lymphatic vessels in the brain parenchyma, which would drain CNS-related antigens and APCs directly to the regional lymph nodes (3).
Microglial cells resident in the brain parenchyma are the main cellular components involved in the innate immune response. These cells possess professional antigen-presenting like characteristics, and as such show multiple similarities with DCs and macrophages. By expressing MHC molecules, microglial cells are capable of antigen presentation. Physiologically these are self-antigens and induce tolerance. Microglial cells also express danger-associated molecular pattern (DAMP) receptors able to sense various danger signals from their environment, such as infectious agents, molecular toxins, and cellular damage and by which they can trigger innate immune processes (4, 5). Microglia are inactive under normal circumstances which is partly due to a panel of anti-inflammatory factors (such as CD200, CX3CL1, CD47, and CD55) secreted by healthy neurons. However, they become activated by various chemokines, cytokines, and purine metabolites released by damaged neurons (6). The interaction between the microglia and neurons highlights the pivotal role of microglial cells in the immune surveillance of normal brain. However, microglial cells are relatively weak antigen presenters, and at present it is thought that contrary to DCs they cannot migrate to peripheral lymphoid organs to induce specific immune response (2). Thus, DCs are indispensable for a successful immune surveillance.
Conventional DCs are also present in certain well-defined brain regions in varying numbers. These are the juxtavascular spaces of the brain parenchyma, brain regions that physiologically lack an intact BBB, brain parenchyma in close contact with the cerebrospinal fluid (CSF) (along the ventricles) and the choroid plexus (CP) (7). A possible way for brain-residing DCs to become activated is through danger signals released by neuronal or other cellular damage in the brain parenchyma. Extracellular vesicles (exosomes and microvesicles) secreted by various cellular components of the brain parenchyma can also transmit inflammatory and activating signals toward DCs and other professional APCs situated around the microvessels and in the CSF (8). These signals are carried most probably by the interstitial fluid circulating in the direction of brain microvessels (9).
The presence of lymphocytes in the healthy brain is very scarce and mostly consists of CD4+ T cells and rare CD8+ T cells. A significant fraction of these lymphocytes are CD4+ memory cells. They can be found in the CSF, in the meningeal spaces, and in the stroma of the CP (the space between the blood vessel endothelium and the epithelial layer of the CP) where they continuously screen APCs presenting their cognate antigens (3). Entry of T cells at the level of the epithelial layer of the CP is facilitated by the expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) by the CP epithelial cells (10). Baruch et al. have demonstrated significant enrichment of CNS-specific T cell receptor clones within the CD4+ T cells residing in the CP stroma (11). This means that CP-residing CD4+ T cells are continuously challenged by CNS-related antigens. This phenomenon was termed as “neuroprotective autoimmunity” (12), and at present it is widely accepted that it has a fundamental role in brain regenerative processes and thus it is indispensable for the maintenance of a healthy brain homeostasis (13, 14). A tightly regulated cytokine milieu within the CP is responsible for keeping the equilibrium between protective and pathological autoimmunity. This cytokine milieu mainly consists of IFN-γ and low levels of IL-4 (11, 15, 16), indicating the presence of both Th1 and Th2 lymphocytes in the CP. Wolf et al. showed in an organotypic in vitro model using hippocampal slice cultures that both Th1 and Th2 lymphocytes could prevent neuronal damage but the neuroprotective effect of Th2 cells was superior (17). Accumulating evidence indicates that the lack of this protective autoimmunity leads to impaired hippocampal neurogenesis, cognitive deficit, and the development of neurodegenerative disorders (18, 19).
The Concept of Neuroinflammation: Neurological Pathologies with an Inflammatory Component
Neuroinflammation can be caused by exogenous (various infectious agents capable of invading the brain) and by endogenous factors (cellular damage within the brain parenchyma). Ionizing radiation, by causing various extent of cellular damage in the brain, is an important endogenous factor in inducing neuroinflammation.
The first step in mounting an acute inflammatory reaction within the brain consists of microglia and astrocyte activation, which sense neuronal damage in their environment. It has been already mentioned that neurons express soluble factors that inhibit microglia activation (Figure 1). It is most likely that a CNS insult leading to neuronal damage and/or death reduces/eliminates this suppression. Microglial cells remove cellular debris through phagocytosis, upregulate their MHC molecules (enhancing antigen presentation), and together with the astrocytes secrete a panel of pro-inflammatory cytokines (among others: TNFα, IL-1β, and IL-6), chemokines (CX3CL1 or fractalkine, CCL3 or macrophage inhibitory factor 1), reactive oxygen, and nitrogen species (ROS and RNS), which activate the brain-resident APCs (20, 21). It should be mentioned that while there is a certain level of immune cell trafficking through the BCSFB under physiological conditions (as detailed above), the BBB is physiologically impermeable to immune cells. However, these same cytokines can lead to endothelial cell activation and a subsequent increase in BBB permeability, as well as an increased penetrability of the BCSFB (22).
Once activated, brain-residing DCs migrate to the regional lymph nodes by the lymphatic drainage of the CSF (23), where they interact with T cells and immune activation takes place. Activated T cells reach the CNS and penetrate into the brain parenchyma via the altered barrier systems. A certain level of lymphocyte infiltration within the brain parenchyma during the acute phase of a neuroinflammation is needed for a quick resolution of the inflammatory process and for rapid neuroregeneration. The immunological profile of the immune cells penetrating the brain parenchyma is an immune suppressive one, consisting of Th2 lymphocytes, regulatory T cells, and M2 macrophages, which produce IL-10 and TGFβ (24, 25). Their main role is microglia suppression.
A persistent activation of the microglia and astrocytes is the hallmark of a chronic neuroinflammation. This is believed to develop when the rate of leukocyte infiltration during an acute inflammatory process is not sufficiently abundant to halt the process. A prolonged activation of microglia leads to a vicious circle, where secretion of pro-inflammatory cytokines and other neurotoxic agents (ROS and RNS) leads to further neuronal damage and cell death, which maintains microglial cells in their activated status. Interestingly, systemic immune suppressive strategies for resolving neuroinflammation are often counterproductive because they inhibit CD4+ T cell activation, which is needed for these cells to enter brain parenchyma and resolve the inflammatory process as described above (14).
Chronic neuroinflammation has been shown in the aging brain. In many aspects, this process is driven by and resembles systemic immune senescence, which is also an accompanying process of aging. During aging, the equilibrium between systemic immune-stimulating and -suppressive mechanisms is shifted toward immune suppression with an increase in the systemic ratio of regulatory T cells and CD4+ cells with a Th2 phenotype and elevated T cell anergy (26). These systemic changes impede an efficient resolution of neuroinflammation. A similar process resembling systemic immune senescence takes place within the CP as well. The level of IFNγ production by the CP epithelial cells and residing immune cells decreases and is replaced by IFNI production, while IL-4 secretion increases. This drives an increase in the production of the CCL11 chemokine by the epithelial cells, which in turn negatively regulates neurogenesis and induces cognitive decline (27).
An inflammatory component has long been known in the pathophysiology of several neurodegenerative diseases (28) and increasing evidence suggests that inflammation is involved in the pathophysiology of neurovascular and certain psychiatric disorders, as well (29, 30). The inflammation-related mechanistic link between these different diseases has been recently reviewed by several research groups. They show that DAMP-associated activation of inflammasomes in various cell types within the brain (mainly microglia, astrocytes, neurons, and endothelial cells) constitute a common mechanism in the development of different types of neurological and psychiatric disorders (31, 32). In Alzheimer’s disease, for example, where extracellular deposition of the β-amyloid (Aβ) peptide, forming the typical neuritic plaque is a major hallmark of the disease, it seems that Aβ represents a DAMP for microglial cells and causes their continuous activation through their toll-like receptors 2, 4, 9 (TLR-2, -4, -9) (33). Increased ROS levels, as activators of inflammasomes have been recognized in certain cerebrovascular diseases, whereas animal data and limited human studies indicate TLR-triggered activation of the inflammasomes in depression, bipolar disorders, and other psychological diseases (34–36). It has not been yet clarified whether inflammatory reactions are the cause or the consequence of these diseases. However, several research groups showed that in the case of Alzheimer’s disease inflammatory reactions were present already at an early stage of the disease before the appearance of the neurofibrillary pathology, suggesting a causative effect for inflammation (37, 38).
Radiation-Induced Late Brain Injuries
Therapeutic and diagnostic medical interventions represent the main source of radiation exposure for the brain. Radiotherapy constitutes a first-line treatment option for various primary or metastatic brain tumors, as well as head and neck cancers, where a high dose (on average 50–60 Gy) is delivered in multiple fractions of approximately 1.8–2 Gy either to the whole brain or to restricted brain regions. Despite the fact that treatment schedules are planned in a way to avoid or minimize toxic side effects in healthy tissues, they still occur in a certain number of sensitive patients.
Classically, radiation-induced brain damage can be divided into acute, early delayed, and late injury based on the time of onset and includes both morphologic and functional deficits (39). Acute damage manifests itself as headache and drowsiness within hours to days after radiotherapy and is caused by brain edema. It is a fully reversible condition, and it appears rarely with modern radiation techniques. Early delayed injury is characterized by somnolence, short-term memory loss, and attention deficits and morphologically by transient demyelination. These are transient symptoms, which resolve in approximately 3 weeks without leading to long-lasting cognitive disturbances. The so-called radiation somnolence syndrome has been described mainly in children receiving whole brain radiotherapy for brain tumor treatment or prophylactic irradiation for acute lymphoid leukemia. Although it has been attributed to a transient demyelination process, recent evidence supports the inflammatory nature of this condition, where various pro-inflammatory cytokines (most notably IL-1β) play an important role. This is further supported by the fact that steroid administration can improve the symptoms (40, 41).
Radiation-induced late brain injuries develop more than 6 months after irradiation and are mostly irreversible changes. Morphological damage consists of vascular abnormalities, demyelination, gliosis, and in extreme cases white matter necrosis. Functionally, it is associated with two main alterations: endocrinopathy and cognitive impairment. Endocrinopathy develops mainly after higher radiation doses delivered to the hypothalamic–pituitary axis. Its most frequent manifestations are hypothyroidism (due to direct radiation damage on the thyroid or to decreased production of the thyroid-stimulating hormone or TSH as a consequence of radiation damage to the hypophysis), growth retardation (due to growth hormone deficiency), and gonadal dysfunction (due to gonadotropin deficiency). Several comprehensive reviews have been published in this topic (42–45). Since it is out of the scope of this review to detail radiation-induced endocrine dysfunctions, we recommend all interested readers to consult these. Cognitive deficit manifests itself in various degrees of memory impairment, learning difficulties, declined flexibility in thinking and IQ performance, and, in extreme cases, full dementia. Radiation-induced cognitive impairment is the most debilitating late sequel of brain irradiation, and it has a great impact on the quality of life of the individuals. Importantly, it often develops even in the absence of detectable morphological abnormalities (46).
Certain patient cohorts can be used to study radiation-induced cognitive impairment. Long-term survivors of glioma constitute an important group; however, in this case, a number of confounding factors (such as short follow-up period due to limited survival rates, neuropsychological symptoms attributable to the malignant disease, the impact of chemotherapy) make the correct evaluation of radiotherapy effects on the cognitive performance more difficult. Within this group, low-grade glioma patients’ follow-up is of particular interest due to their much better prognosis in terms of overall survival. Most studies agree that radiotherapy poses a significant risk of late cognitive impairment in adult patients with low-grade gliomas (47, 48), but conclusions are contradictory whether focal radiotherapy with fractional doses less than 2 Gy is associated with an increased risk of cognitive deficit (49, 50). In long-term survivors of childhood brain tumors, on the other hand, there is an agreement in the literature that the most important risk factor for impaired intellectual outcome is radiotherapy, especially in children irradiated before the age of 15 (51–53), indicating the higher vulnerability of pediatric patients to brain irradiation.
The brain can be exposed to substantial doses of irradiation during the radiotherapy of various head and neck tumors as well. However, in these cases, radiation exposure is restricted to certain brain regions only. Various trials demonstrated an increased risk of cognitive impairment in these patients (54). Meyers et al. studied the cognitive performance of patients who received paranasal sinus irradiation, where the mean delivered dose was 60 Gy in fractions of 1.8–2 Gy. They found memory impairment in 80%, learning difficulties in more than 50%, difficulty with visual–motor speed, frontal lobe executive functions, and fine motor coordination in more than 30% of the patients. Cognitive performance could be correlated with total dose delivered to the brain but not with the volume of the irradiated brain or chemotherapy treatment (55). Severe cognitive deficit was reported also in children treated with radiotherapy for head and neck rhabdomyosarcoma with symptoms manifesting within 10 years after radiotherapy (56).
Lung cancer patients receiving prophylactic brain irradiation to reduce the rate of brain metastasis are also at risk for developing late cognitive alterations (57, 58). Children with acute lymphoid leukemia constitute another important study group who, for prophylactic reasons, received cranial irradiation. In a study conducted at the Children’s Hospital in Philadelphia in the early 80s, an average total dose of 24 Gy cranial irradiation, combined with intrathecal methotrexate were applied to these children. The authors demonstrated significant reduction in the overall IQ score for the majority of children, younger patients being more affected. Notably, even in those patients who did not have any IQ decline, learning deficit was still present. However, cognitive deficits were absent in children treated with intrathecal chemotherapy only (59), indicating that chemotherapy per se was not a toxic agent for cognitive outcome. Waber et al. reported slightly different findings in a study conducted 15 years later at the Dana Farber Cancer Institute. In this study, cranial irradiation could not be directly linked with cognitive damage, most probably because the applied average total dose was much lower (18 Gy) (60).
It is very important to note that all of these cohorts were treated with conventional X-ray or gamma ray techniques. While major technical improvements were done to reduce irradiation of healthy tissues (such as the development of different intensity-modulated radiotherapy techniques), due to the energy deposition characteristics of these radiation types it is impossible to completely spare non-tumorous tissues. Proton radiation therapy has emerged as a novel therapeutic modality that is beginning to be largely applied for the treatment of various brain tumors. Protons are charged particles, which deposit their energy over a narrow range, and have little lateral scatters in the tissues. Due to these properties, the proton beam focuses on the tumor and doses delivered to surrounding normal tissues are much lower than in the case of X-ray-based techniques. While proton beam therapy (PBT) is a relatively new technology, and there are no large patient cohorts yet which allow a thorough evaluation of the developing side effects in the brain, the already available data indicate its suitability to reduce late toxicities. This is especially important in children whose brain is very sensitive to irradiation (as discussed above). An essential dose reduction by using PBT compared to conformal radiotherapy was shown particularly in contralaterally located critical neuronal structures (61). Different clinical studies measured superior quality of life, physical, and IQ scores in children with brain tumors receiving PBT compared to those treated with X-rays (62–65). However, all of these studies agree that additional long-term data and larger cohorts are needed to correctly evaluate the impact of PBT on neurocognitive performance and to determine whether PBT is associated with a clinically relevant cognitive sparing compared to X-ray protocols.
All the abovementioned clinical studies demonstrate that cognitive impairment is a relatively frequent consequence of high-dose therapeutic brain irradiation. While the severity of the damage is influenced by multiple factors, the most important ones are the young age at irradiation and the irradiated brain region. The exquisite sensitivity of the hippocampus to irradiation, where the neuronal stem cells are located has been shown by numerous animal experiments (66–69) and clinical studies (67, 70, 71), and it is evidenced also by the fact that the most common neurological alterations are hippocampal-related memory deficits. On the other hand, as stated by Greene-Schloesser et al. in a recent review (39), hippocampal sparing radiotherapy might not be sufficient to avoid cognitive impairment since brain regions other than hippocampus are also involved in cognitive processes. Furthermore, neuronal stem cell death is only one component in the mechanism of radiation-induced brain injury.
Inflammation-Mediated Mechanisms in Radiation-Induced Brain Injury
Radiation-Induced Activation of the Microglia
It is well established that ionizing radiation induces inflammatory reactions in the brain mainly via microglia and endothelial cell activation (72) (Figure 1). A possible mechanism on how microglia are activated is by IR-induced double-strand breaks, which trigger the NFκB pathway-mediated production of inflammatory proteins (73). Microglial cells in their activated state secrete a panel of pro-inflammatory cytokines, which inhibit neurogenesis in the hippocampus by disrupting neurogenic signaling pathways. It was shown that neuroinflammation induced a long-term disruption of hippocampal network activity and had a significant impact on the recruitment of adult-born neurons into hippocampal networks encoding spatial information. Increased levels of cyclooxygenase-2, IL-1β, IL-6, IL-18, TNFα, and interferon-gamma-inducible protein-10, as well as several chemokines such as monocyte chemoattractant protein (MCP-1/CCL2) and macrophage inflammatory protein 2 (MIP-2/CXCL2) were measured in microglial cells after radiation doses higher than 7 Gy both in vitro and in vivo (72, 74–77). Microglia activation was detected even months after irradiation indicating the persistence of the neuroinflammatory process (78). Selective inhibition of microglia-mediated neuroinflammation was able to ameliorate radiation-induced late cognitive impairment (79). Schindler et al. investigated radiation-induced neuronal loss and microglial activation in young, adult, and aged rats. They found that in younger animals 10 Gy whole brain irradiation induced a more pronounced and persistent reduction in the number of immature neurons than in aged rats. On the other hand, microglial activation was more prevalent in older animals, where 10 weeks after irradiation the proportion of activated/resting microglial cells was 60%, compared to a rate of 20% found in young animals (80). Furthermore, irradiation induced an RNA expression profile resembling to the transcriptome of the aging microglia (81). These findings are very important in our opinion since they highlight that the mechanisms responsible for radiation-induced cognitive impairment might be different in young and aged individuals. While at young age radiation-induced direct alteration in neurogenesis is the major factor, at older ages the preponderant mechanism for the development of radiation-induced cognitive deficit is neuroinflammation, which in turn impacts neurogenesis. These findings are in concordance with other reports indicating that radiation induces a premature aging process in the brain and accelerates and/or aggravates the onset of chronic degenerative disorders characteristic for elderly (82, 83).
Chemokine receptors, due to their central role in attracting immune cells to the site of inflammation, are considered as key components in mediating neuroinflammation. A panel of chemokine receptors and their ligands such as CCL7, CCL8, CCL12, CXCL4, CCR1, and CCR2 were shown to be upregulated as a result of brain irradiation (84). Among these, CCR2 has a prominent role in enhancing macrophage infiltration at the sites of injury in the brain (85) and in modulating several neurodegenerative disorders (86). It was postulated that irradiation influenced neurogenesis and cognitive functions by altering CCR2 signaling pathways in the brain. Recently, Belarbi et al. proved the direct involvement of CCR2 expression in the development of radiation-induced cognitive alterations. Using CCR2 knockout mice, they showed that CCR2 deficiency prevented cranial irradiation-induced neuronal damage and cognitive impairment (84). The protective effect of CCR2 deficiency against radiation-induced neuronal damage was identified after low-dose irradiation, as well (doses below 2 Gy) (87).
Since the phenotype of activated microglia is difficult to discern from brain-infiltrating activated macrophages (88), it is possible that the main inflammatory cells within the brain parenchyma are originating from blood-derived macrophages penetrating into the brain parenchyma, which becomes permissive for them in an inflamed state. Several lines of evidence support this hypothesis. Burrell et al. demonstrated that bone marrow-derived cells were recruited specifically to the site of cranial irradiation in a dose-dependent manner and differentiated predominantly into inflammatory cells and microglia (89). Mildner et al. conducted a very elegant experiment in which they proved the role of cranial irradiation in the engraftment of blood-derived macrophages into the brain parenchyma. They identified a specific monocyte subpopulation (Ly-6ChiGr-1 + CCR2 + CX3CR1lo cells), as the precursor of adult murine microglia in the peripheral blood and showed that microglia engraftment during postnatal life was enhanced by various degenerative brain disorders. However, these monocytes were preferentially recruited to the brain and differentiated into microglia only if the brain was “preconditioned” by irradiation. The authors explained this enhanced cell engraftment primarily by a radiation-induced production of CCL2 in the brain, which attracted blood-derived CCR2-expressing monocytes and by an inactivation of the repository signals and to a lesser extent by a radiation-induced damage in BBB integrity, although they admitted that subtle BBB alterations might have been present (90). Similar findings were reported by Lampron et al., who induced myeloablation either by chemotherapy or by total body irradiation and followed the repopulation of the hematopoietic niche, as well as the entry of bone marrow-derived cells into the brain. While repopulation was equally efficient after both chemo- and radioablation, brain penetration of bone marrow cells was only observed after irradiation (91). Morganti et al. showed that a single dose of cranial irradiation with 10 Gy induced a significant decrease in brain-residing microglia, while significantly increasing the penetration of blood-derived CCR2+ macrophages. They also proved that penetrating macrophages adopted a microglia-like phenotype. Similar to Mildner et al., they also did not detect BBB damage, which could be responsible for the increased penetration of monocytes, but demonstrated a radiation-induced increase in the secretion of a panel of chemoattractant molecules implicated in the recruitment, adhesion, and migration of monocytes (92). On the other hand, it seems that repopulation of brain parenchyma with peripheral microglia progenitors does not necessarily happen under physiological conditions, since these bone marrow-residing progenitors do not mobilize spontaneously to the peripheral blood and can only reach the CNS if artificially delivered into the circulation (93).
The way a cell is dying greatly impacts the immune and inflammatory response of the host. The characteristics of an immunogenic cell death have been initially described for cancer cells (94). One of the most important features of an immunogenic cell death is that dying cells expose so-called “eat-me” signals sensed by nearby tissue-residing phagocytes (95) and the physiologically present phagocytic barrier is lost. CD47 is considered a typical phagocytic barrier or “don’t eat me” signal, which in the context of cellular apoptosis is frequently lost and this phenomenon is paralleled with the cell surface exposure of the endoplasmatic reticulum-associated calreticulin (CRT) (96). Cell surface bound CRT is the most important “eat-me” signal for surrounding phagocytes. It seems that “eat-me” and “don’t eat me” signaling molecules are present in neurons as well, indicating that interactions between neurons and activated microglia are in multiple aspects similar to those seen outside the brain (97). Although the presence of cell surface CRT is usually characteristic for dying cells, it has been shown that neurons constitutively express it (98). Resting microglia do not react with CRT-expressing neurons. However, as shown by Fricker et al., microglia activation via ligands binding to their TLR4 receptor has led to the phagocytosis of CRT-expressing both viable and apoptotic neurons, significantly contributing to the amplification of a neurodegenerative condition (98). Irradiation can impact this process in multiple ways. Radiation induces apoptosis among neuronal stem and progenitor cells (99). Whether IR-induced apoptosis is de facto accompanied by increased cell surface CRT levels on neurons has not been reported yet, but it has been shown in carcinoma cells (100), and this phenomenon was directly linked with the induction of an immunogenic type of apoptotic cell death (101). Experiments related to CD47 changes in apoptotic cells after ionizing radiation are also lacking. However, it was shown that UV-induced apoptosis induced CD47 redistribution on the cell surface associated with a significant reduction in the binding efficiency of CD47 to its natural ligand on phagocytes. This resulted in facilitating the clearance of apoptotic cells by phagocytes (102).
We have previously discussed that IR can directly activate microglial cells. It is very probable that IR can contribute to microglia activation via their TLR4 receptor as well. The prototypic TLR4 ligand is lipopolysaccharide (LPS), which is an endotoxin released by bacterial cells during an infection. On the other hand, the endogenous LPS-like molecule high-mobility group protein 1 (HMGB1) is a danger signal (or alarmin), which is released in the extracellular medium under cellular stress. It was shown that HMGB1 by binding to the TLR4 receptor could promote microglia activation under stress conditions associated with neuronal damage such as traumatic brain injury, ischemic injury, and methamphetamine treatment (103–105). Studies investigating the direct effect of IR on HMGB1 release and TLR4 activation in the brain are not available yet. However, given the fact that IR is a strong cellular stressor, it is plausible to hypothesize that it induces similar stress-related pathways than other stressors.
Radiation Effects on Brain Endothelial Cells, BBB Integrity, and Immune Cell Infiltration in the Brain
Blood–brain barrier is a major route for the systemic supply of immune and inflammatory cells during neuroinflammation. There are not too many in vivo studies referring to the impact of acute cranial irradiation on BBB integrity. The previously mentioned studies reported no significant BBB damage after high-dose irradiation (around 10 Gy), though they did not exclude the possibility of minor BBB alterations (90, 92). On the other hand, other studies detected significant alterations in BBB damage with or without alterations in endothelial tight junctions after high-dose irradiation, albeit this damage was transient, and its severity varied in the different brain regions (106–108). In vitro models also demonstrated that alterations in BBB integrity were detected after much lower doses (4 Gy). These alterations were relatively long lasting and were accompanied by increased permeability for both low- and high-molecular weight proteins. Morphologically, a rarefaction of the endothelial layer was seen, which could lead to the opening of the endothelial tight junctions, despite the fact that no gross alterations were observed in the immunolabelling of a panel of tight junction proteins (ZO-1, claudin-5, and occludin) (109).
Endothelial cells are among the most radiosensitive cellular structures in the brain. Direct IR induces endothelial cell death by various mechanisms. Several in vitro and in vivo studies demonstrated endothelial cell apoptosis as an early event after irradiation. However, it was induced only by high doses of irradiation and was accompanied by strong inhibition of endothelial cell proliferation capacity (110, 111). The rate of apoptotic endothelial cells was estimated to be around 15% within 24 h after irradiation with high doses (112, 113). Li et al. demonstrated a direct link between radiation-induced endothelial cell apoptosis and acute increase in BBB permeability (110).
Recently, it has been shown that senescence is another major cell death mechanism developing at a later time point in the surviving endothelial cells. Irradiation doses in the range of 2–8 Gy led to increased DNA damage and a reduced repair efficiency in rat primary cerebrovascular endothelial cells, which were accompanied by increased yields of endothelial cells showing premature senescence and acquiring a senescence-associated secretory profile. Endothelial senescence could be a consequence of pro-inflammatory cytokines secreted by activated glial cells and astrocytes such as TNFα or IL-6 (114, 115). These senescent cells acquired certain phenotypical features resembling activated endothelial cells. Senescent endothelial cells significantly contributed to the onset and progression of neuroinflammation by secreting a panel of pro-inflammatory molecules (IL-1α, IL-6, and MCP1), upregulating adhesion molecules on their surface, and increasing their ROS production (116, 117).
Changes in the activation status of microvascular brain endothelial cells can facilitate immune cell transmigration even in the absence of an overt BBB damage. Several studies reported that high doses of IR could directly activate brain microvascular endothelial cells by increasing ICAM-1, VCAM-1, and P-selectin expression (118–120). ICAM-1 induction on brain endothelial cells is a rapid but persistent process, appearing as soon as 4 h after irradiation and being detectable even 6 months later (119, 121). Since ICAM-1 expression has a major role in facilitating leukocyte trafficking into the brain parenchyma, its persistent presence contributes to the slow resolution of the neuroinflammatory process. Another important molecule regulating monocyte and leukocyte transmigration through the BBB is CD47 expressed on endothelial cells. CD47 plays an active role in immune cell diapedesis by interacting with the signal-regulatory protein alpha on monocytes, activating signaling pathways that induce cytoskeleton remodeling and cadherin redistribution. CD47 activation was shown to occur after ischemic neurovascular injury, and its overexpression on brain endothelial cells significantly enhanced monocyte transmigration and contributed to BBB injury and edema (122–124). It remains to be determined whether radiation injury to the brain induces similar CD47 changes.
Moravan et al. performed a systematic longitudinal analysis of brain-infiltrating immune cells after irradiation. According to this study, neutrophil penetration in the irradiated brain was a transient effect, which could be detected only in the first 12 h after irradiation. CD3+ T cells penetrated the brain as early as day 7 after irradiation and persisted even 12 months later. DC penetration was also seen, and similar to T cells, it was a rather late process persisting up to 6 months after irradiation. Several of the penetrating DCs acquired an activated phenotype and often colocalized with T cells suggesting a possible interaction between the two cell types. Penetration of myeloid cells in the brain was dose dependent within the range of 5–35 Gy radiation dose and was dependent on CCR2 signaling (121, 125).
Low-Dose Radiation Effects on the Brain
The vast majority of radiation exposures delivered to the brain in the population are for diagnostic purposes, where absorbed doses are in the low-dose range (below 100 mGy). Recent epidemiological data pose serious concerns regarding long-term health consequences of these low doses. It was shown by several epidemiological studies that cranial CT exposure increased the risk of brain tumors in children (126–128). Similar conclusions were drawn after interventional radiology exposures to the brain (127) as well as in hemangioma cohorts subjected to head irradiation for hemangioma treatment (129). A recent report indicated a twofold increased risk of brain cancer mortality among technologists who performed fluoroscopically guided interventional procedures (130). These observations raise the possibility that low-dose radiation might cause cognitive alterations as well. We found one report in the literature about the risk of late cognitive deficit in humans subjected to low-dose cranial irradiation. A population-based cohort study was performed in Sweden involving 3,030 boys who were treated with IR for cutaneous hemangioma before the age of 18. The study could not show any difference regarding logical, spatial, and technical test scores between IR-treated subjects and controls, but verbal test scores displayed a significant trend for decreasing scores with increasing doses to the hippocampus. The authors also concluded that hippocampal dose was a better predictor of late cognitive side effects than doses delivered to other brain regions (131). While human epidemiological data are almost absent, several animal experiments indicate cognitive damage as a potential long-term risk of low-dose cranial irradiation. Altered adult spontaneous behavior and impaired habituation capacity was found in mice exposed to low doses (500 mGy) total-body irradiation at a very young age (postnatal day 3 and 10) but not later, indicating an exquisite sensitivity of the young brain to IR. The same group showed significantly higher alterations in the behavior of these mice if they were coexposed to IR and nicotine (132, 133). Gene expression studies performed in the brain or various brain structures repeatedly report mRNA expression profiles characteristic for low-dose exposure. Low-dose exposures (100 mGy) induced genes that were not affected by high doses (2 Gy), and low-dose genes were associated with unique pathways and functions similar to those seen in the aging brain and in the brain tissue from patients with Alzheimer’s disease (134). Yin et al. also showed qualitatively different gene expression profiles after 0.1 and 2 Gy, where low-dose-regulated genes were involved in protective and reparative functions such as stress response, cell cycle control, and DNA repair as well as in neural signaling activity (135). Dose-dependent changes in gene expression profiles were seen in human neuronal progenitor cells, where very low-dose chronic irradiation (31 mGy/72 h) induced alterations in inflammatory pathways related to interferon signaling, while higher doses induced different signaling pathways (136). It was reported that low-dose chronic irradiation stimulated leptin production in mice (137, 138). Leptin is a member of the cytokine superfamily, resembling IL-6 also known as the “saturation hormone” produced mainly by adipocytes. It acts on receptors in the hypothalamus to inhibit hunger and thus has major role in maintaining a metabolic balance. It has important effects on the immune system as well, by shifting the Th1/Th2 balance in favor of Th1 cells, by regulating monocyte–macrophage activation, by inducing T cell proliferation, and by suppressing apoptosis (139). Since leptin levels were directly correlated with cognitive performance and higher leptin levels could even ameliorate cognitive deterioration seen in Alzheimer’s disease (140–142), low-dose radiation-induced increase in circulating leptins might be a favorable parameter in the risk of radiation-induced cognitive alterations.
Very interesting data start to emerge regarding the impact of low-dose or low-dose rate irradiation on endothelial cell integrity. A premature senescence was observed in human umbilical vein endothelial cells exposed to low-dose rate irradiation delivered by 2.4 or 4.1 mGy/h dose rates. Transcriptomic and proteomic studies revealed the activation of signaling pathways related to cell–cell communication, adhesion, and inflammation in these cells with a special involvement of the insulin-like growth factor-binding protein 5 in this process (143, 144). Endothelial damage in the brain was reflected in a rarefaction of capillary density after low-dose (0.1 Gy) whole brain irradiation (66). These data indicate that doses well below those considered damaging for various brain structures lead to microvascular disturbances and endothelial dysfunction promoting the onset of a neuroinflammatory process.
Exposition of astronauts to cosmic rays during deep space flights represents another source of low-dose irradiation to the brain. Cosmic rays are mainly composed of high-atomic number and energy charged particles (high-energy protons and fully ionized atomic nuclei). These are densely ionizing radiations, which differ from main terrestrial radiation types (X and γ-rays) in terms of biological damage. The density of ionizing events deposited in tissues by charged particles produces a track of biological damage (mostly complex DNA double-strand breaks), which is very difficult to be repaired through the cellular repair processes. Exposure to heavy ion irradiation as low as 0.5 Gy was supposed to induce impaired neurogenesis with a very poor or no recovery (145). A long-lasting functional damage induced by low-dose heavy particles was shown in the hippocampus, leading to cell type-specific alterations in both the excitatory and inhibitory synaptic microcircuits (146). Significant dose-dependent and long-lasting reductions in dendritic complexity, spine density, and morphology (147) as well as altered neurogenesis (148) were observed in hippocampal neurons after low-dose total-body proton irradiation. At molecular level, long-term changes in DNA methylation patterns (149), distinctive miRNA signatures (150) were described in the brain following proton irradiation. Similar to γ-rays, heavy ion exposure also increased circulating leptin levels (151). It was reported by Baluchamy et al. that high-energy protons induced a dose-dependent increase in reactive oxygen species and lipid peroxidation as well as a reduction in antioxidant levels in the brain, mainly in the neural stem cells, followed by apoptotic cell death (152–155).
Very few studies investigated the effect of low doses of proton and heavy ion irradiation on inflammatory and immune parameters in the brain. Vlkolinsky et al. showed that LPS treatment of mice in the absence of (56)Fe-particle irradiation induced a reduction in the hippocampal long-term potentiation capacity, while this inhibition was abolished and a reversal effect was registered after irradiation of the brain with (56)Fe ions. This phenomenon persisted for months, indicating that heavy ion irradiation stably altered hippocampal reactivity to immunological stressors (156). Regarding the direct effect of protons or heavy ions on brain inflammation existing reports are contradictory. Raber et al. demonstrated microglia activation in the hippocampus of mice exposed to low-dose proton, heavy ion, or combined irradiation, which correlated well with deterioration in novel object recognition, suggesting a role for neuroinflammation in the development of cognitive impairment (157). On the other hand, Sweet et al., investigating low-dose effects of high-energy proton particles on inflammatory reactions in the hippocampus, could not detect significant astrocyte and microglia activation indicating lack of neuroinflammation. They also found significantly reduced ICAM-1 levels selectively in the hippocampus, pointing to a lack of endothelial activation and/or to a capillary rarefaction and endothelial cell loss (148).
Conclusion
In this review, we presented data proving a direct link between ionizing radiation-induced neuroinflammation and the development of late neurodegenerative disorders and cognitive deficit. It has been shown that the most common radiation-related alterations after brain irradiation are various forms of cognitive deficit. Some of the most representative epidemiological cohorts presenting an elevated risk for late cognitive sequela have been reviewed highlighting the increased sensitivity of the developing brain (and thus children) for radiation damage. The second part of the review focused on the description of the mechanisms on how IR can induce inflammatory reactions and can perturb brain immune homeostasis. IR-induced neuroinflammation develops as a result of a complex signaling between various cellular components residing in the brain (neurons, microglia, astrocytes, and endothelial cells) as well as the peripheral immune system. These data clearly prove that immune reactions in the brain are in many aspects similar to systemic immune reactions. Finally, we have discussed the long-term risk of low-dose radiation on the brain and presented the already available epidemiological and experimental data supporting this increased risk. These findings showed that molecular and cellular mechanisms within the low-dose range are often different from those elicited by high-dose irradiation. The relevance of these data is huge, since this means that even doses in the range used for diagnostic purposes might have long-lasting consequences and might contribute to the development of radiation-induced late cognitive impairment.
Although much progress has been made in the field, the mechanisms that govern IR-induced inflammatory and immune reactions in the brain, their relationship with IR-related functional deficit and consequently the optimal therapeutic countermeasures are far from being elucidated. While formerly research work focused almost exclusively on therapeutic radiation doses, new and accumulating data regarding the risk of low-dose radiation highlight the importance of studies within this dose range as well.
Author Contributions
KL designed the review. KL, TS, and GS wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by the following grants: DoReMi FP7 project (grant agreement number: 249689), CEREBRAD FP7 project (grant agreement number: 295552), and Hungarian research grant funded by the National Research, Development and Innovation Office (grant agreement number: VKSZ_14-1-2015-0021).
References
1. Zhang A, Yu H, He Y, Shen Y, Pan N, Liu J, et al. The spatio-temporal expression of MHC class I molecules during human hippocampal formation development. Brain Res (2013) 1529:26–38. doi: 10.1016/j.brainres.2013.07.001
2. Ludewig P, Gallizioli M, Urra X, Behr S, Brait VH, Gelderblom M, et al. Dendritic cells in brain diseases. Biochim Biophys Acta (2016) 1862:352–67. doi:10.1016/j.bbadis.2015.11.003
3. Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol (2005) 26:485–95. doi:10.1016/j.it.2005.07.004
4. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol (2002) 61:1013–21. doi:10.1093/jnen/61.11.1013
5. Dalpke AH, Schafer MK, Frey M, Zimmermann S, Tebbe J, Weihe E, et al. Immunostimulatory CpG-DNA activates murine microglia. J Immunol (2002) 168:4854–63. doi:10.4049/jimmunol.168.10.4854
6. Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘on’ and ‘off’ signals control microglia. Trends Neurosci (2007) 30:596–602. doi:10.1016/j.tins.2007.08.007
7. Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, et al. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol (2008) 508:687–710. doi:10.1002/cne.21668
8. de Rivero Vaccari JP, Brand F III, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, et al. Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem (2016) 136(Suppl 1):39–48. doi:10.1111/jnc.13036
9. Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol (2016) 132:317–38. doi:10.1007/s00401-016-1606-5
10. Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am J Pathol (1996) 148:1819–38.
11. Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci U S A (2013) 110:2264–9. doi:10.1073/pnas.1211270110
12. Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A (2002) 99:15620–5. doi:10.1073/pnas.232565399
13. Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med (1999) 5:49–55. doi:10.1038/4734
14. Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J (2014) 33:7–22. doi:10.1002/embj.201386609
15. Deczkowska A, Baruch K, Schwartz M. Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol (2016) 37:181–92. doi:10.1016/j.it.2016.01.006
16. Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, et al. IFN-gamma-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain (2013) 136:3427–40. doi:10.1093/brain/awt259
17. Wolf SA, Fisher J, Bechmann I, Steiner B, Kwidzinski E, Nitsch R. Neuroprotection by T-cells depends on their subtype and activation state. J Neuroimmunol (2002) 133:72–80. doi:10.1016/S0165-5728(02)00367-3
18. Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A (2004) 101:8180–5. doi:10.1073/pnas.0402268101
19. Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci (2006) 9:268–75. doi:10.1038/nn1629
20. Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microsc Res Tech (2001) 54:47–58. doi:10.1002/jemt.1120
21. Spleiss O, Appel K, Boddeke HW, Berger M, Gebicke-Haerter PJ. Molecular biology of microglia cytokine and chemokine receptors and microglial activation. Life Sci (1998) 62:1707–10. doi:10.1016/S0024-3205(98)00132-5
22. Alvarez JI, Saint-Laurent O, Godschalk A, Terouz S, Briels C, Larouche S, et al. Focal disturbances in the blood-brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol Dis (2015) 74:14–24. doi:10.1016/j.nbd.2014.09.016
23. Laman JD, Weller RO. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol (2013) 8:840–56. doi:10.1007/s11481-013-9470-8
24. O’Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol (2007) 179:958–66. doi:10.4049/jimmunol.179.2.958
25. Zhang Z, Zhang ZY, Wu Y, Schluesener HJ. Lesional accumulation of CD163+ macrophages/microglia in rat traumatic brain injury. Brain Res (2012) 1461:102–10. doi:10.1016/j.brainres.2012.04.038
26. Wang L, Xie Y, Zhu LJ, Chang TT, Mao YQ, Li J. An association between immunosenescence and CD4(+)CD25(+) regulatory T cells: a systematic review. Biomed Environ Sci (2010) 23:327–32. doi:10.1016/S0895-3988(10)60072-4
27. Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature (2011) 477:90–4. doi:10.1038/nature10357
28. McGeer PL, McGeer EG, Kawamata T, Yamada T, Akiyama H. Reactions of the immune system in chronic degenerative neurological diseases. Can J Neurol Sci (1991) 18:376–9. doi:10.1017/S0317167100032479
29. Anderson G, Maes M. How immune-inflammatory processes link CNS and psychiatric disorders: classification and treatment implications. CNS Neurol Disord Drug Targets (2017) 16(3):266–78. doi:10.2174/1871527315666161122144659
30. Stuart MJ, Singhal G, Baune BT. Systematic review of the neurobiological relevance of chemokines to psychiatric disorders. Front Cell Neurosci (2015) 9:357. doi:10.3389/fncel.2015.00357
31. Lenart N, Brough D, Denes A. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J Cereb Blood Flow Metab (2016) 36:1668–85. doi:10.1177/0271678X16662043
32. Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci (2014) 8:315. doi:10.3389/fnins.2014.00315
33. Su F, Bai F, Zhou H, Zhang Z. Microglial toll-like receptors and Alzheimer’s disease. Brain Behav Immun (2016) 52:187–98. doi:10.1016/j.bbi.2015.10.010
34. Iwata M, Ota KT, Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun (2013) 31:105–14. doi:10.1016/j.bbi.2012.12.008
35. Kim HK, Chen W, Andreazza AC. The potential role of the NLRP3 inflammasome as a link between mitochondrial complex I dysfunction and inflammation in bipolar disorder. Neural Plast (2015) 2015:408136. doi:10.1155/2015/408136
36. Zhang Y, Liu L, Peng YL, Liu YZ, Wu TY, Shen XL, et al. Involvement of inflammasome activation in lipopolysaccharide-induced mice depressive-like behaviors. CNS Neurosci Ther (2014) 20:119–24. doi:10.1111/cns.12170
37. Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A (2004) 101:2173–8. doi:10.1073/pnas.0308512100
38. Hoozemans JJ, Veerhuis R, Rozemuller JM, Eikelenboom P. Neuroinflammation and regeneration in the early stages of Alzheimer’s disease pathology. Int J Dev Neurosci (2006) 24:157–65. doi:10.1016/j.ijdevneu.2005.11.001
39. Greene-Schloesser D, Robbins ME, Peiffer AM, Shaw EG, Wheeler KT, Chan MD. Radiation-induced brain injury: a review. Front Oncol (2012) 2:73. doi:10.3389/fonc.2012.00073
40. Ballesteros-Zebadua P, Chavarria A, Celis MA, Paz C, Franco-Perez J. Radiation-induced neuroinflammation and radiation somnolence syndrome. CNS Neurol Disord Drug Targets (2012) 11:937–49. doi:10.2174/1871527311201070937
41. Mandell LR, Walker RW, Steinherz P, Fuks Z. Reduced incidence of the somnolence syndrome in leukemic children with steroid coverage during prophylactic cranial radiation therapy. Results of a pilot study. Cancer (1989) 63:1975–8.
42. Behin A, Delattre JY. Complications of radiation therapy on the brain and spinal cord. Semin Neurol (2004) 24:405–17. doi:10.1055/s-2004-861535
43. Duffner PK. Long-term effects of radiation therapy on cognitive and endocrine function in children with leukemia and brain tumors. Neurologist (2004) 10:293–310. doi:10.1097/01.nrl.0000144287.35993.96
44. Eaton BR, Yock T. The use of proton therapy in the treatment of benign or low-grade pediatric brain tumors. Cancer J (2014) 20:403–8. doi:10.1097/PPO.0000000000000079
45. Sklar CA, Constine LS. Chronic neuroendocrinological sequelae of radiation therapy. Int J Radiat Oncol Biol Phys (1995) 31:1113–21. doi:10.1016/0360-3016(94)00427-M
46. Sundgren PC, Cao Y. Brain irradiation: effects on normal brain parenchyma and radiation injury. Neuroimaging Clin N Am (2009) 19:657–68. doi:10.1016/j.nic.2009.08.014
47. Klein M, Heimans JJ, Aaronson NK, van der Ploeg HM, Grit J, Muller M, et al. Effect of radiotherapy and other treatment-related factors on mid-term to long-term cognitive sequelae in low-grade gliomas: a comparative study. Lancet (2002) 360:1361–8. doi:10.1016/S0140-6736(02)11398-5
48. Surma-aho O, Niemela M, Vilkki J, Kouri M, Brander A, Salonen O, et al. Adverse long-term effects of brain radiotherapy in adult low-grade glioma patients. Neurology (2001) 56:1285–90. doi:10.1212/WNL.56.10.1285
49. Douw L, Klein M, Fagel SS, van den Heuvel J, Taphoorn MJ, Aaronson NK, et al. Cognitive and radiological effects of radiotherapy in patients with low-grade glioma: long-term follow-up. Lancet Neurol (2009) 8:810–8. doi:10.1016/S1474-4422(09)70204-2
50. Taphoorn MJ. Neurocognitive sequelae in the treatment of low-grade gliomas. Semin Oncol (2003) 30:45–8. doi:10.1053/j.seminoncol.2003.11.023
51. Jalali R, Mallick I, Dutta D, Goswami S, Gupta T, Munshi A, et al. Factors influencing neurocognitive outcomes in young patients with benign and low-grade brain tumors treated with stereotactic conformal radiotherapy. Int J Radiat Oncol Biol Phys (2010) 77:974–9. doi:10.1016/j.ijrobp.2009.06.025
52. Reimers TS, Ehrenfels S, Mortensen EL, Schmiegelow M, Sonderkaer S, Carstensen H, et al. Cognitive deficits in long-term survivors of childhood brain tumors: identification of predictive factors. Med Pediatr Oncol (2003) 40:26–34. doi:10.1002/mpo.10211
53. Reimers TS, Mortensen EL, Schmiegelow K. Memory deficits in long-term survivors of childhood brain tumors may primarily reflect general cognitive dysfunctions. Pediatr Blood Cancer (2007) 48:205–12. doi:10.1002/pbc.20818
54. Tang Y, Luo D, Rong X, Shi X, Peng Y. Psychological disorders, cognitive dysfunction and quality of life in nasopharyngeal carcinoma patients with radiation-induced brain injury. PLoS One (2012) 7:e36529. doi:10.1371/journal.pone.0036529
55. Meyers CA, Geara F, Wong PF, Morrison WH. Neurocognitive effects of therapeutic irradiation for base of skull tumors. Int J Radiat Oncol Biol Phys (2000) 46:51–5. doi:10.1016/S0360-3016(99)00376-4
56. Paulino AC, Simon JH, Zhen W, Wen BC. Long-term effects in children treated with radiotherapy for head and neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys (2000) 48:1489–95. doi:10.1016/S0360-3016(00)00799-9
57. Cull A, Gregor A, Hopwood P, Macbeth F, Karnicka-Mlodkowska H, Thatcher N, et al. Neurological and cognitive impairment in long-term survivors of small cell lung cancer. Eur J Cancer (1994) 30A:1067–74. doi:10.1016/0959-8049(94)90458-8
58. Gondi V, Paulus R, Bruner DW, Meyers CA, Gore EM, Wolfson A, et al. Decline in tested and self-reported cognitive functioning after prophylactic cranial irradiation for lung cancer: pooled secondary analysis of Radiation Therapy Oncology Group randomized trials 0212 and 0214. Int J Radiat Oncol Biol Phys (2013) 86:656–64. doi:10.1016/j.ijrobp.2013.02.033
59. Meadows AT, Gordon J, Massari DJ, Littman P, Fergusson J, Moss K. Declines in IQ scores and cognitive dysfunctions in children with acute lymphocytic leukaemia treated with cranial irradiation. Lancet (1981) 2:1015–8. doi:10.1016/S0140-6736(81)91216-2
60. Waber DP, Tarbell NJ, Fairclough D, Atmore K, Castro R, Isquith P, et al. Cognitive sequelae of treatment in childhood acute lymphoblastic leukemia: cranial radiation requires an accomplice. J Clin Oncol (1995) 13:2490–6. doi:10.1200/JCO.1995.13.10.2490
61. Adeberg S, Harrabi SB, Bougatf N, Bernhardt D, Rieber J, Koerber SA, et al. Intensity-modulated proton therapy, volumetric-modulated arc therapy, and 3D conformal radiotherapy in anaplastic astrocytoma and glioblastoma: a dosimetric comparison. Strahlenther Onkol (2016) 192:770–9. doi:10.1007/s00066-016-1007-7
62. Greenberger BA, Pulsifer MB, Ebb DH, MacDonald SM, Jones RM, Butler WE, et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys (2014) 89:1060–8. doi:10.1016/j.ijrobp.2014.04.053
63. Indelicato DJ, Flampouri S, Rotondo RL, Bradley JA, Morris CG, Aldana PR, et al. Incidence and dosimetric parameters of pediatric brainstem toxicity following proton therapy. Acta Oncol (2014) 53:1298–304. doi:10.3109/0284186X.2014.957414
64. Kahalley LS, Ris MD, Grosshans DR, Okcu MF, Paulino AC, Chintagumpala M, et al. Comparing intelligence quotient change after treatment with proton versus photon radiation therapy for pediatric brain tumors. J Clin Oncol (2016) 34:1043–9. doi:10.1200/JCO.2015.62.1383
65. Yock TI, Bhat S, Szymonifka J, Yeap BY, Delahaye J, Donaldson SS, et al. Quality of life outcomes in proton and photon treated pediatric brain tumor survivors. Radiother Oncol (2014) 113:89–94. doi:10.1016/j.radonc.2014.08.017
66. Casciati A, Dobos K, Antonelli F, Benedek A, Kempf SJ, Belles M, et al. Age-related effects of X-ray irradiation on mouse hippocampus. Oncotarget (2016) 7:28040–58. doi:10.18632/oncotarget.8575
67. Gondi V, Pugh SL, Tome WA, Caine C, Corn B, Kanner A, et al. Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): a phase II multi-institutional trial. J Clin Oncol (2014) 32:3810–6. doi:10.1200/JCO.2014.57.2909
68. Rao AA, Ye H, Decker PA, Howe CL, Wetmore C. Therapeutic doses of cranial irradiation induce hippocampus-dependent cognitive deficits in young mice. J Neurooncol (2011) 105:191–8. doi:10.1007/s11060-011-0582-9
69. Tome WA, Gokhan S, Brodin NP, Gulinello ME, Heard J, Mehler MF, et al. A mouse model replicating hippocampal sparing cranial irradiation in humans: a tool for identifying new strategies to limit neurocognitive decline. Sci Rep (2015) 5:14384. doi:10.1038/srep14384
70. Pereira Dias G, Hollywood R, Bevilaqua MC, da Luz AC, Hindges R, Nardi AE, et al. Consequences of cancer treatments on adult hippocampal neurogenesis: implications for cognitive function and depressive symptoms. Neuro Oncol (2014) 16:476–92. doi:10.1093/neuonc/not321
71. Pinkham MB, Bertrand KC, Olson S, Zarate D, Oram J, Pullar A, et al. Hippocampal-sparing radiotherapy: the new standard of care for World Health Organization grade II and III gliomas? J Clin Neurosci (2014) 21:86–90. doi:10.1016/j.jocn.2013.04.005
72. Kalm M, Fukuda A, Fukuda H, Ohrfelt A, Lannering B, Bjork-Eriksson T, et al. Transient inflammation in neurogenic regions after irradiation of the developing brain. Radiat Res (2009) 171:66–76. doi:10.1667/RR1269.1
73. Dong XR, Luo M, Fan L, Zhang T, Liu L, Dong JH, et al. Corilagin inhibits the double strand break-triggered NF-kappaB pathway in irradiated microglial cells. Int J Mol Med (2010) 25:531–6. doi:10.3892/ijmm_00000374
74. Chen H, Chong ZZ, De Toledo SM, Azzam EI, Elkabes S, Souayah N. Delayed activation of human microglial cells by high dose ionizing radiation. Brain Res (2016) 1646:193–8. doi:10.1016/j.brainres.2016.06.002
75. Hong JH, Chiang CS, Campbell IL, Sun JR, Withers HR, McBride WH. Induction of acute phase gene expression by brain irradiation. Int J Radiat Oncol Biol Phys (1995) 33:619–26. doi:10.1016/0360-3016(95)00279-8
76. Hwang SY, Jung JS, Kim TH, Lim SJ, Oh ES, Kim JY, et al. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol Dis (2006) 21:457–67. doi:10.1016/j.nbd.2005.08.006
77. Lee WH, Sonntag WE, Mitschelen M, Yan H, Lee YW. Irradiation induces regionally specific alterations in pro-inflammatory environments in rat brain. Int J Radiat Biol (2010) 86:132–44. doi:10.3109/09553000903419346
78. Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res (2003) 63:4021–7.
79. Jenrow KA, Brown SL, Lapanowski K, Naei H, Kolozsvary A, Kim JH. Selective inhibition of microglia-mediated neuroinflammation mitigates radiation-induced cognitive impairment. Radiat Res (2013) 179:549–56. doi:10.1667/RR3026.1
80. Schindler MK, Forbes ME, Robbins ME, Riddle DR. Aging-dependent changes in the radiation response of the adult rat brain. Int J Radiat Oncol Biol Phys (2008) 70:826–34. doi:10.1016/j.ijrobp.2007.10.054
81. Li MD, Burns TC, Kumar S, Morgan AA, Sloan SA, Palmer TD. Aging-like changes in the transcriptome of irradiated microglia. Glia (2015) 63:754–67. doi:10.1002/glia.22782
82. Dietrich J, Monje M, Wefel J, Meyers C. Clinical patterns and biological correlates of cognitive dysfunction associated with cancer therapy. Oncologist (2008) 13:1285–95. doi:10.1634/theoncologist.2008-0130
83. Hua K, Schindler MK, McQuail JA, Forbes ME, Riddle DR. Regionally distinct responses of microglia and glial progenitor cells to whole brain irradiation in adult and aging rats. PLoS One (2012) 7:e52728. doi:10.1371/journal.pone.0052728
84. Belarbi K, Jopson T, Arellano C, Fike JR, Rosi S. CCR2 deficiency prevents neuronal dysfunction and cognitive impairments induced by cranial irradiation. Cancer Res (2013) 73:1201–10. doi:10.1158/0008-5472.CAN-12-2989
85. Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. J Neuroimmunol (2010) 224:80–4. doi:10.1016/j.jneuroim.2010.05.015
86. Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab (2010) 30:459–73. doi:10.1038/jcbfm.2009.240
87. Acharya MM, Patel NH, Craver BM, Tran KK, Giedzinski E, Tseng BP, et al. Consequences of low dose ionizing radiation exposure on the hippocampal microenvironment. PLoS One (2015) 10:e0128316. doi:10.1371/journal.pone.0128316
88. Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron (2006) 49:489–502. doi:10.1016/j.neuron.2006.01.022
89. Burrell K, Hill RP, Zadeh G. High-resolution in vivo analysis of normal brain response to cranial irradiation. PLoS One (2012) 7:e38366. doi:10.1371/journal.pone.0038366
90. Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci (2007) 10:1544–53. doi:10.1038/nn2015
91. Lampron A, Lessard M, Rivest S. Effects of myeloablation, peripheral chimerism, and whole-body irradiation on the entry of bone marrow-derived cells into the brain. Cell Transplant (2012) 21:1149–59. doi:10.3727/096368911X593154
92. Morganti JM, Jopson TD, Liu S, Gupta N, Rosi S. Cranial irradiation alters the brain’s microenvironment and permits CCR2+ macrophage infiltration. PLoS One (2014) 9:e93650. doi:10.1371/journal.pone.0093650
93. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci (2007) 10:1538–43. doi:10.1038/nn2014
94. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
95. Garg AD, Dudek-Peric AM, Romano E, Agostinis P. Immunogenic cell death. Int J Dev Biol (2015) 59:131–40. doi:10.1387/ijdb.150061pa
96. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med (2010) 2:63ra94. doi:10.1126/scitranslmed.3001375
97. Griffiths MR, Gasque P, Neal JW. The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: a brief review. Int J Inflam (2010) 2010:151097. doi:10.4061/2010/151097
98. Fricker M, Oliva-Martin MJ, Brown GC. Primary phagocytosis of viable neurons by microglia activated with LPS or abeta is dependent on calreticulin/LRP phagocytic signalling. J Neuroinflammation (2012) 9:196. doi:10.1186/1742-2094-9-196
99. Lu F, Li YQ, Aubert I, Wong CS. Endothelial cells regulate p53-dependent apoptosis of neural progenitors after irradiation. Cell Death Dis (2012) 3:e324. doi:10.1038/cddis.2012.59
100. Ramsamooj P, Notario V, Dritschilo A. Enhanced expression of calreticulin in the nucleus of radioresistant squamous carcinoma cells in response to ionizing radiation. Cancer Res (1995) 55:3016–21.
101. Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ (2007) 14:1848–50. doi:10.1038/sj.cdd.4402201
102. Lv Z, Bian Z, Shi L, Niu S, Ha B, Tremblay A, et al. Loss of cell surface CD47 clustering formation and binding avidity to SIRPalpha facilitate apoptotic cell clearance by macrophages. J Immunol (2015) 195:661–71. doi:10.4049/jimmunol.1401719
103. Frank MG, Adhikary S, Sobesky JL, Weber MD, Watkins LR, Maier SF. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav Immun (2016) 51:99–108. doi:10.1016/j.bbi.2015.08.001
104. Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, et al. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci (2006) 26:6413–21. doi:10.1523/JNEUROSCI.3815-05.2006
105. Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia (2014) 62:26–38. doi:10.1002/glia.22581
106. D’Avella D, Cicciarello R, Albiero F, Mesiti M, Gagliardi ME, Russi E, et al. Quantitative study of blood-brain barrier permeability changes after experimental whole-brain radiation. Neurosurgery (1992) 30:30–4. doi:10.1227/00006123-199201000-00006
107. Diserbo M, Agin A, Lamproglou I, Mauris J, Staali F, Multon E, et al. Blood-brain barrier permeability after gamma whole-body irradiation: an in vivo microdialysis study. Can J Physiol Pharmacol (2002) 80:670–8. doi:10.1139/y02-070
108. Sandor N, Walter FR, Bocsik A, Santha P, Schilling-Toth B, Lener V, et al. Low dose cranial irradiation-induced cerebrovascular damage is reversible in mice. PLoS One (2014) 9:e112397. doi:10.1371/journal.pone.0112397
109. Fauquette W, Amourette C, Dehouck MP, Diserbo M. Radiation-induced blood-brain barrier damages: an in vitro study. Brain Res (2012) 1433:114–26. doi:10.1016/j.brainres.2011.11.022
110. Li YQ, Chen P, Haimovitz-Friedman A, Reilly RM, Wong CS. Endothelial apoptosis initiates acute blood-brain barrier disruption after ionizing radiation. Cancer Res (2003) 63:5950–6.
111. Zhao Z, Johnson MS, Chen B, Grace M, Ukath J, Lee VS, et al. Live-cell imaging to detect phosphatidylserine externalization in brain endothelial cells exposed to ionizing radiation: implications for the treatment of brain arteriovenous malformations. J Neurosurg (2016) 124:1780–7. doi:10.3171/2015.4.JNS142129
112. Ljubimova NV, Levitman MK, Plotnikova ED, Eidus L. Endothelial cell population dynamics in rat brain after local irradiation. Br J Radiol (1991) 64:934–40. doi:10.1259/0007-1285-64-766-934
113. Pena LA, Fuks Z, Kolesnick RN. Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res (2000) 60:321–7.
114. Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, et al. Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci Rep (2017) 7:39501. doi:10.1038/srep39501
115. Rochfort KD, Cummins PM. The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem Soc Trans (2015) 43:702–6. doi:10.1042/BST20140319
116. Ungvari Z, Podlutsky A, Sosnowska D, Tucsek Z, Toth P, Deak F, et al. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci (2013) 68:1443–57. doi:10.1093/gerona/glt057
117. Wang Y, Boerma M, Zhou D. Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res (2016) 186:153–61. doi:10.1667/RR14445.1
118. Kyrkanides S, Olschowka JA, Williams JP, Hansen JT, O’Banion MK. TNF alpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J Neuroimmunol (1999) 95:95–106. doi:10.1016/S0165-5728(98)00270-7
119. Olschowka JA, Kyrkanides S, Harvey BK, O’Banion MK, Williams JP, Rubin P, et al. ICAM-1 induction in the mouse CNS following irradiation. Brain Behav Immun (1997) 11:273–85. doi:10.1006/brbi.1997.0506
120. Sharp CD, Jawahar A, Warren AC, Elrod JW, Nanda A, Alexander JS. Gamma knife irradiation increases cerebral endothelial expression of intercellular adhesion molecule 1 and E-selectin. Neurosurgery (2003) 53:154–60; discussion 160–1. doi:10.1227/01.NEU.0000068840.84484.DA
121. Moravan MJ, Olschowka JA, Williams JP, O’Banion MK. Cranial irradiation leads to acute and persistent neuroinflammation with delayed increases in T-cell infiltration and CD11c expression in C57BL/6 mouse brain. Radiat Res (2011) 176:459–73. doi:10.1667/RR2587.1
122. de Vries HE, Hendriks JJ, Honing H, De Lavalette CR, van der Pol SM, Hooijberg E, et al. Signal-regulatory protein alpha-CD47 interactions are required for the transmigration of monocytes across cerebral endothelium. J Immunol (2002) 168:5832–9. doi:10.4049/jimmunol.168.11.5832
123. Martinelli R, Newton G, Carman CV, Greenwood J, Luscinskas FW. Novel role of CD47 in rat microvascular endothelium: signaling and regulation of T-cell transendothelial migration. Arterioscler Thromb Vasc Biol (2013) 33:2566–76. doi:10.1161/ATVBAHA.113.301903
124. Xing C, Arai K, Park KP, Lo EH. Induction of vascular endothelial growth factor and matrix metalloproteinase-9 via CD47 signaling in neurovascular cells. Neurochem Res (2010) 35:1092–7. doi:10.1007/s11064-010-0159-6
125. Moravan MJ, Olschowka JA, Williams JP, O’Banion MK. Brain radiation injury leads to a dose- and time-dependent recruitment of peripheral myeloid cells that depends on CCR2 signaling. J Neuroinflammation (2016) 13:30. doi:10.1186/s12974-016-0496-8
126. Mathews JD, Forsythe AV, Brady Z, Butler MW, Goergen SK, Byrnes GB, et al. Cancer risk in 680,000 people exposed to computed tomography scans in childhood or adolescence: data linkage study of 11 million Australians. BMJ (2013) 346:f2360. doi:10.1136/bmj.f2360
127. Orbach DB, Stamoulis C, Strauss KJ, Manchester J, Smith ER, Scott RM, et al. Neurointerventions in children: radiation exposure and its import. AJNR Am J Neuroradiol (2014) 35:650–6. doi:10.3174/ajnr.A3758
128. Pearce MS, Salotti JA, Little MP, McHugh K, Lee C, Kim KP, et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet (2012) 380:499–505. doi:10.1016/S0140-6736(12)60815-0
129. Karlsson P, Holmberg E, Lundell M, Mattsson A, Holm LE, Wallgren A. Intracranial tumors after exposure to ionizing radiation during infancy: a pooled analysis of two Swedish cohorts of 28,008 infants with skin hemangioma. Radiat Res (1998) 150:357–64. doi:10.2307/3579984
130. Rajaraman P, Doody MM, Yu CL, Preston DL, Miller JS, Sigurdson AJ, et al. Cancer risks in U.S. radiologic technologists working with fluoroscopically guided interventional procedures, 1994–2008. AJR Am J Roentgenol (2016) 206:1101–8; quiz 1109. doi:10.2214/AJR.15.15265
131. Blomstrand M, Holmberg E, Aberg MA, Lundell M, Bjork-Eriksson T, Karlsson P, et al. No clinically relevant effect on cognitive outcomes after low-dose radiation to the infant brain: a population-based cohort study in Sweden. Acta Oncol (2014) 53:1143–50. doi:10.3109/0284186X.2014.899434
132. Buratovic S, Stenerlow B, Fredriksson A, Sundell-Bergman S, Eriksson P. Developmental effects of fractionated low-dose exposure to gamma radiation on behaviour and susceptibility of the cholinergic system in mice. Int J Radiat Biol (2016) 92:371–9. doi:10.3109/09553002.2016.1164911
133. Eriksson P, Buratovic S, Fredriksson A, Stenerlow B, Sundell-Bergman S. Neonatal exposure to whole body ionizing radiation induces adult neurobehavioural defects: critical period, dose-response effects and strain and sex comparison. Behav Brain Res (2016) 304:11–9. doi:10.1016/j.bbr.2016.02.008
134. Lowe XR, Bhattacharya S, Marchetti F, Wyrobek AJ. Early brain response to low-dose radiation exposure involves molecular networks and pathways associated with cognitive functions, advanced aging and Alzheimer’s disease. Radiat Res (2009) 171:53–65. doi:10.1667/RR1389.1
135. Yin E, Nelson DO, Coleman MA, Peterson LE, Wyrobek AJ. Gene expression changes in mouse brain after exposure to low-dose ionizing radiation. Int J Radiat Biol (2003) 79:759–75. doi:10.1080/09553000310001610961
136. Katsura M, Cyou-Nakamine H, Zen Q, Zen Y, Nansai H, Amagasa S, et al. Effects of chronic low-dose radiation on human neural progenitor cells. Sci Rep (2016) 6:20027. doi:10.1038/srep20027
137. Shin SC, Lee KM, Kang YM, Kim K, Kim CS, Yang KH, et al. Alteration of cytokine profiles in mice exposed to chronic low-dose ionizing radiation. Biochem Biophys Res Commun (2010) 397:644–9. doi:10.1016/j.bbrc.2010.05.121
138. Suman S, Kallakury BV, Fornace AJ Jr, Datta K. Protracted upregulation of leptin and IGF1 is associated with activation of PI3K/Akt and JAK2 pathway in mouse intestine after ionizing radiation exposure. Int J Biol Sci (2015) 11:274–83. doi:10.7150/ijbs.10684
139. Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol (2000) 68:437–46.
140. Tezapsidis N, Johnston JM, Smith MA, Ashford JW, Casadesus G, Robakis NK, et al. Leptin: a novel therapeutic strategy for Alzheimer’s disease. J Alzheimers Dis (2009) 16:731–40. doi:10.3233/JAD-2009-1021
141. Warren MW, Hynan LS, Weiner MF. Leptin and cognition. Dement Geriatr Cogn Disord (2012) 33:410–5. doi:10.1159/000339956
142. Witte AV, Kobe T, Graunke A, Schuchardt JP, Hahn A, Tesky VA, et al. Impact of leptin on memory function and hippocampal structure in mild cognitive impairment. Hum Brain Mapp (2016) 37:4539–49. doi:10.1002/hbm.23327
143. Rombouts C, Aerts A, Quintens R, Baselet B, El-Saghire H, Harms-Ringdahl M, et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int J Radiat Biol (2014) 90:560–74. doi:10.3109/09553002.2014.905724
144. Yentrapalli R, Azimzadeh O, Barjaktarovic Z, Sarioglu H, Wojcik A, Harms-Ringdahl M, et al. Quantitative proteomic analysis reveals induction of premature senescence in human umbilical vein endothelial cells exposed to chronic low-dose rate gamma radiation. Proteomics (2013) 13:1096–107. doi:10.1002/pmic.201200463
145. Cacao E, Cucinotta FA. Modeling heavy-ion impairment of hippocampal neurogenesis after acute and fractionated irradiation. Radiat Res (2016) 186:624–37. doi:10.1667/RR14569.1
146. Lee SH, Dudok B, Parihar VK, Jung KM, Zoldi M, Kang YJ, et al. Neurophysiology of space travel: energetic solar particles cause cell type-specific plasticity of neurotransmission. Brain Struct Funct (2016). doi:10.1007/s00429-016-1345-3
147. Parihar VK, Pasha J, Tran KK, Craver BM, Acharya MM, Limoli CL. Persistent changes in neuronal structure and synaptic plasticity caused by proton irradiation. Brain Struct Funct (2015) 220:1161–71. doi:10.1007/s00429-014-0709-9
148. Sweet TB, Panda N, Hein AM, Das SL, Hurley SD, Olschowka JA, et al. Central nervous system effects of whole-body proton irradiation. Radiat Res (2014) 182:18–34. doi:10.1667/RR13699.1
149. Impey S, Pelz C, Tafessu A, Marzulla T, Turker MS, Raber J. Proton irradiation induces persistent and tissue-specific DNA methylation changes in the left ventricle and hippocampus. BMC Genomics (2016) 17:273. doi:10.1186/s12864-016-2581-x
150. Khan SY, Tariq MA, Perrott JP, Brumbaugh CD, Kim HJ, Shabbir MI, et al. Distinctive microRNA expression signatures in proton-irradiated mice. Mol Cell Biochem (2013) 382:225–35. doi:10.1007/s11010-013-1738-z
151. Suman S, Kumar S, Fornace AJ, Datta K. Space radiation exposure persistently increased leptin and IGF1 in serum and activated leptin-IGF1 signaling axis in mouse intestine. Sci Rep (2016) 6:31853. doi:10.1038/srep31853
152. Baluchamy S, Ravichandran P, Periyakaruppan A, Ramesh V, Hall JC, Zhang Y, et al. Induction of cell death through alteration of oxidants and antioxidants in lung epithelial cells exposed to high energy protons. J Biol Chem (2010) 285:24769–74. doi:10.1074/jbc.M110.138099
153. Baluchamy S, Ravichandran P, Ramesh V, He Z, Zhang Y, Hall JC, et al. Reactive oxygen species mediated tissue damage in high energy proton irradiated mouse brain. Mol Cell Biochem (2012) 360:189–95. doi:10.1007/s11010-011-1056-2
154. Baulch JE, Craver BM, Tran KK, Yu L, Chmielewski N, Allen BD, et al. Persistent oxidative stress in human neural stem cells exposed to low fluences of charged particles. Redox Biol (2015) 5:24–32. doi:10.1016/j.redox.2015.03.001
155. Suman S, Rodriguez OC, Winters TA, Fornace AJ Jr, Albanese C, Datta K. Therapeutic and space radiation exposure of mouse brain causes impaired DNA repair response and premature senescence by chronic oxidant production. Aging (Albany NY) (2013) 5:607–22. doi:10.18632/aging.100587
156. Vlkolinsky R, Krucker T, Nelson GA, Obenaus A. (56)Fe-particle radiation reduces neuronal output and attenuates lipopolysaccharide-induced inhibition of long-term potentiation in the mouse hippocampus. Radiat Res (2008) 169:523–30. doi:10.1667/RR1228.1
Keywords: ionizing radiation, cognitive effects, neuroinflammation, immune reactions, low-dose radiation
Citation: Lumniczky K, Szatmári T and Sáfrány G (2017) Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 8:517. doi: 10.3389/fimmu.2017.00517
Received: 01 February 2017; Accepted: 18 April 2017;
Published: 05 May 2017
Edited by:
Sherven Sharma, VA Greater Los Angeles Healthcare System (VHA), USAReviewed by:
Lorenzo Manti, University of Naples Federico II, ItalyDaniel Olive, Institut national de la santé et de la recherche médicale (INSERM), France
Copyright: © 2017 Lumniczky, Szatmári and Sáfrány. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katalin Lumniczky, bHVtbmljemt5LmthdGFsaW5Ab3Nza2kuaHU=