 Franz Rödel1*
Franz Rödel1* Claudia Fournier2
Claudia Fournier2 Julia Wiedemann2
Julia Wiedemann2 Felicitas Merz2
Felicitas Merz2 Udo S. Gaipl3
Udo S. Gaipl3 Benjamin Frey3
Benjamin Frey3 Ludwig Keilholz4M. Heinrich Seegenschmiedt5
Ludwig Keilholz4M. Heinrich Seegenschmiedt5 Claus Rödel1
Claus Rödel1 Stephanie Hehlgans1
Stephanie Hehlgans1
- 1Department of Radiotherapy and Oncology, University Hospital of Frankfurt, Goethe-Universität, Frankfurt am Main, Germany
- 2Department of Biophysics, GSI Helmholtz Centre for Heavy Ion Research, Darmstadt, Germany
- 3Department of Radiation Oncology, Universitätsklinikum Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 4Department of Radiotherapy, Clinical Center Bayreuth, Bayreuth, Germany
- 5Strahlenzentrum Hamburg MVZ, Hamburg, Germany
For decades, low- and moderate-dose radiation therapy (RT) has been shown to exert a beneficial therapeutic effect in a multitude of non-malignant conditions including painful degenerative muscoloskeletal and hyperproliferative disorders. Dupuytren and Ledderhose diseases are benign fibroproliferative diseases of the hand/foot with fibrotic nodules and fascial cords, which determine debilitating contractures and deformities of fingers/toes, while keloids are exuberant scar formations following burn damage, surgery, and trauma. Although RT has become an established and effective option in the management of these diseases, experimental studies to illustrate cellular composites and factors involved remain to be elucidated. More recent findings, however, indicate the involvement of radiation-sensitive targets like mitotic fibroblasts/myofibroblasts as well as inflammatory cells. Radiation-related molecular mechanisms affecting these target cells include the production of free radicals to hamper proliferative activity and interference with growth factors and cytokines. Moreover, an impairment of activated immune cells involved in both myofibroblast proliferative and inflammatory processes may further contribute to the clinical effects. We here aim at briefly describing mechanisms contributing to a modulation of proliferative and inflammatory processes and to summarize current concepts of treating hyperproliferative diseases by low and moderate doses of ionizing radiation.
Introduction
The capacity of ionizing radiation to inhibit proliferation of malignant cancer cells are well explored (1–3) and widely used in clinical practice. By contrast, application of radiation therapy (RT) for non-malignant conditions is not a fully accepted practice in medicine. In line with that, the use of RT in the management of hyperproliferative non-cancerous disorders is controversially discussed and inadequately recognized by doctors from disciplines others than RT. However, long-term experiences impressively indicated a clinical benefit for patients (4, 5). Accordingly, treatment with irradiation concepts not exceeding a single dose of 5 Gy and total doses of 30 Gy [low- or intermediate-dose RT (LD-RT)] is an established and effective modality in the management of a variety of non-cancerous inflammatory, degenerative, and hyperproliferative/fibroproliferative disorders (4–6). The latter include, among others, heterotopic ossifications, symptomatic vertebral hemangiomas, Gorham–Stout syndrome, prophylaxis of keloid relapse after surgical excision (7), and, most prominent, palmar and plantar fibromatosis also known as Dupuytren disease (DD) and Ledderhose disease (LD) (8). The most effective treatment schedule, the radiobiological basis, and molecular/cellular mechanisms contributing to the modulation by ionizing radiation of these benign hyperproliferative disorders are far from being fully explored. Consequently, this review aims at summarizing current clinical concepts and antiproliferative as well as immune modulating properties of low- and moderate-dose irradiation focusing on DD, LD, and keloids. This may display a prerequisite for future systematic investigations to enhance clinical irradiation protocols.
Use of RT to Treat Benign Disorders
Non-malignant indications for LD-RT comprise about 10–30% of all patient cases treated in most academic, public, and private RT facilities in Germany (4, 9). In total, more than 50,000 patients per year are treated by LD-RT with the largest group suffering from painful degenerative musculoskeletal diseases, followed by symptomatic functional and hyperproliferative disorders with the latter to increase in numbers by 28.8% from 1999 to 2004 (8).
In 1831, Baron Guillaume Dupuytren described for the first time a fibrotic contracture of the palmar fascia of the hand, while fibrotic contractures of the plantar fascia of the foot were initially described by the German physician Georg Ledderhose in 1897 (4). DD is a prevalent disease with incidences varying between populations with up to 29% in the Western countries (10). Men are affected more often and earlier in life as women with a gender ratio from 3:1 to 6:1 (11) and with an onset of symptoms usually in the third to fourth decade of life (12). Concerning the etiology and pathogenesis of DD, several studies report on a strong genetic background (13, 14) apart from environmental risk factors including alcohol, smoking, hand trauma, and manual work (15–17).
In spite of a documented occurrence of 1.75 cases per 100,000 hospital admissions, the precise incidence of LD remains not exactly specified (18). It is known that men are affected twice as often as women, and in 25% of patients, both feet are involved. In 9–25% of patients, concomitant DD has been described (19, 20), while a coincidence with knuckle pads or Peyronies’s disease has been observed in 4% of cases (21).
Another clinically relevant example of benign hyperproliferative diseases are keloids, which are considered as dermal disorders in predisposed individuals caused by injuries to the deep dermis, including burn damage, surgery, and trauma. The classic description of a keloid is “an exuberant scar formation that extends beyond the borders of the original wound.” Keloids are relatively common diseases occurring in 5–15% of wounds (22) and tend to affect both sexes equally. The frequency of keloid occurrence in persons with highly pigmented skin is 15 times elevated compared to those with less pigmented skins (23). Surgical resection is the standard in treating keloid patients, but excision alone results in unacceptably high recurrence rates of 45–100% (24).
According to a recent guideline from the German Society of Radiation Therapy and Oncology (DEGRO), single doses of 0.5–1.0 Gy (total doses of 3.0–6.0 Gy) and two or three fractions per week are recommended in patients with painful degenerative and inflammatory diseases (6, 8). By contrast, different schedules are advised when treating hyperproliferative diseases like DD, LD, and keloids (5, 25). So far, total doses exceeding 20 Gy applied in single fractions of 3 Gy have been shown to comprise the most clinically relevant schedules. However, at present, only a few controlled studies have reported on alternative fractionation concepts. Against this background, a randomized study comparing no treatment versus either 21 or 30 Gy applied in 3-Gy single fractions over 2 weeks (7 Gy × 3 Gy) or by repeated 5 Gy × 3 Gy at intervals of 12 weeks has been conducted in patients with DD. After a median follow-up of 8 years, both regimes were significantly superior regarding disease progression and avoidance of preceding surgery compared to the control group (9). In a huge retrospective cohort, Betz et al. further analyzed a total of 135 DD patients (208 hands) treated with a total dose of 30 Gy, in two intervals of 5 daily fractions of 3.0 Gy, separated by 6–8 weeks. At a median follow-up of 13 years, early-stage disease was more likely to respond to treatment in terms of prevention of progression (26), and 66% of the patients showed a long-term relief of symptoms, while RT was not associated with increased complications following salvage surgery in case of progression and late skin toxicity (atrophy, dry desquamation).
In contrast to DD, only a few clinical investigations have been published concerning RT of LD. After a median follow-up of 22 months, Heyd et al. reported a complete remission of the nodes in 33.3% of cases and a decrease or numerical reduction in 54.5% of the cases following weekly fractions of 3.0 Gy (15 Gy), repeated after 6 weeks. About 70% of the patients indicated a reduction of pain and an improvement of their gait pattern (18).
As mentioned before, keloid scars tend to display high recurrence rates of 45–100% following surgical debulking or resection (24). By contrast, adjuvant RT has been shown to result in the avoidance of renewed excessive scar formation and good cosmetic outcome with a 60–90% success rate (22, 27, 28). There is conclusive evidence that single doses of 2.0–5.0 Gy and total doses of 16–20 Gy/series with five fractions per week are effective for the prevention of local relapses after surgical excision of keloids (5). RT can be applied with low-energy X-rays (150–200 kV), low-energy electrons (4–10 MeV), or brachytherapy (29). To obtain the optimal antiproliferative effect, radiation should be initiated immediately after the surgical excision, preferably within the first 24 h.
In conclusion, the clinical/empirical experience of different dose requirements and treatment schedules to treat degenerative and hyperproliferative benign diseases may indicate distinctive cellular components and mechanisms to be affected in response to ionizing radiation. In case of hyperproliferative disorders, both antiproliferative and anti-inflammatory effects may account for elevated dose requirements that will be reviewed below.
Basic Mechanisms of Radiation Exposure and Cancer Risk Assessment after RT of Benign Diseases
In the last decades, there has been increasing interest in the physical and molecular cellular response following exposure to ionizing radiation. Initial events cover damage to DNA by direct hits of photons or electrons or generation of radicals, e.g., reactive oxygen species (ROS), that indirectly cause DNA double-strand beaks (DSBs), the most severe kind of damage (30, 31). Induction of these lesions promptly results in the activation of DSB damage repair processes, most importantly non-homologous end joining or homologous recombination, and subsequently triggers execution of a multitude of cellular signaling pathways referred to as the DNA damage response (DDR) (2, 32). These responses cover posttranslational modifications and/or altered gene expression of proteins to initiate cell cycle alterations (e.g., radiation-induced arrest) or execute cell death by mitotic catastrophy, apoptosis, autophagy, or induction of senescence (2, 3, 32). Importantly, the classical paradigm in radiobiology on (nuclear) targeted effects, indicating that DNA DSBs are solely responsible for the biological consequences of radiation exposure, is now challenged by reports on non-(DNA) targeted effects. These effects cover, among others, bystander or abscopal effects and adaptive responses and are considered to be involved in the regulation of intercellular communication and modulation of the activity of a multitude of immune components by low- or intermediate-dose ionizing radiation [reviewed in Ref. (33)]. Accordingly, although not proven experimentally at present, one may assume that RT of hyperproliferative disorders may include both targeted (cell proliferation/death) and non-targeted effects of ionizing radiation (modulation of immune components).
Due to reports from the sixties of the last century on increased mortality from leukemia and anemia (34), LD-RT is still considered unfashionable in some countries. However, risk assessment of carcinogenesis after low-dose radiation treatment of benign diseases is challenging due to a relatively small number of patients treated worldwide, latency of carcinogenesis, which requires a long-term follow-up, and different treatment regimes and techniques that are not directly comparable with the present advanced methodology (35, 36). In general, the risk to develop radiation-induced cancer can be estimated by calculation of the equivalent dose of a specific tissue or organ using the effective dose (E) concept as proposed by the International Commission of Radiological Protection (37). These estimations, however, are controversially discussed and problematic in cases where organs receive heterogenous exposure, and calculation of the effective dose might overestimate the true probability in some cases and underestimate it in others (38). An alternative and more accurate approach for the estimation of the risk to develop malignancies is a direct assessment from epidemiological data of patients who have undergone radiotherapy for benign diseases (35, 38). However, these data are still scarcely available, and follow-up times are often too short. In summary, estimation of cancer risk after radiation treatment for benign diseases is challenging, but for current clinical protocols regarded to be small especially for older patients (36). By contrast, the balance of risk and benefit has to be considered carefully for younger patients, and children should not be subjected to LD-RT at all.
Cellular and Molecular Basis of Hyperproliferative Diseases
Dupuytren disease and LD are among the best-described diseases with proliferation of fibrous tissue to form two structurally distinct elements, nodules and cords, which have features in common with benign fibromatosis (39, 40). Aberrant cellular proliferation is involved in the formation of these elements, which are induced by a genuine unknown reason, injury, or a variety of trigger mechanisms (41). Histologically, nodules present a highly vascularized tissue with a high percentage of fibroblasts and myofibroblasts, while cords are more avascular, acellular, and collagen-rich tissues. As mentioned before, the prominent cellular components in the nodules are fibroblasts/myofibroblasts. The latter comprise differentiated cells that share characteristics of fibroblasts and, by the expression of α-smooth muscle actin, contractile properties similar to those of smooth muscle cells (15, 42, 43). These myofibroblasts originate from several sources including quiescent tissue fibroblasts, circulating cluster of differentiation (CD)34+ fibrocytes, and a phenotypic conversion of various cell types including epithelial and endothelial cells.
Several studies further indicated infiltration of multiple immune cells in Dupuytren’s contractures. These cover different lineages of lymphocytes including CD3, CD4, CD8, CD45RA+ naïve and CD45RO+ activated cells, CD68- and S100-positive macrophages (44), and Langerhans cells. Further, compared to peripheral blood detection, transcription factor FOXP3-positive regulatory T-cells were more abundant in fibrotic tissue. Notably, immunoscope analysis indicated a restricted T-cell receptor αβ repertoire, indicating an (auto)antigen-driven expansion of intralesional T-cell clones with Th1-/Th17-weighted immune responses (44). Finally, in favor of a causal involvement of inflammatory processes in DD, elevated levels of the pro-inflammatory cytokines interleukin (IL)-6 and an abundant expression of transforming growth factor-β1 (TGF-β1) have been reported (44).
In contrast, keloids present reddish tumor-like lesions extending beyond a surgical scar (28), which do not respect the borders of the original wound area. Functionally, keloids arise from either insufficient degradation and remodeling of extracellular matrix (ECM) components due to an imbalance in expression of matrix metalloproteinases or excessive ECM deposition by an increased activity of fibroblasts and myofibroblasts (45). Furthermore, keloid stem cells have been described, which share characteristics with skin progenitor cells and are transformed from dermal progenitor cells in a pathological niche of keloid tissues. These keloid stem cells are self-renewal and, by asynchronous divisions, continually generate new keloid cells, thus leading to overgrowth of keloid tissue and posttherapy recurrences (46).
Recently, to assess characteristics of cellular composition, tissue specimens from 28 keloid patients were subjected to immunohistochemical analyses (47). An increased number of CD20- and CD3-positive lymphocytes, CD68-positive macrophages, and CD1α+ Langerhans cells were recorded, indicating characteristics in keloid tissue similar to autoimmune diseases (47). This notion was further strengthen by the detection of elevated levels of TGF-β1; vascular endothelial growth factor (VEGF); platelet-derived growth factor-α in line with inflammatory cytokines IL-6, IL-8, and IL-18; and chemokine-like factor 1 (48, 49).
Proliferating Mitotic Fibroblasts/Myofibroblasts are Radiosensitive Cells
Concerning radiation responsiveness, the course of DD and LD comprises three consecutive phases. These include a radiosensitive initial hyperproliferative period characterized by increased numbers of fibroblasts/myofibroblasts in line with an excessive deposition of ECM components, especially collagen, fibronectin, elastin, and proteoglycans (50, 51). The initial period is followed by an involutional phase with decreased radiation sensitivity in line with the formation of fiber bundles causing contractures. Finally, this phase is followed by a non-RT responsive residual phase with a predominant establishment of collagen filaments in the connective tissue (4, 42). Thus, the clinical implementation and clinical efficacy of RT to treat hyperproliferative DD and LD are strictly stage dependent, with a clinical efficacy most pronounced in the early nodular stage. With regard to target cells, the proliferative phase is characterized by the presence of radiation-responsive fibroblasts and/or myofibroblasts preceding the formation of nodular contractures (52, 53). These myofibroblasts differentiate from fibroblasts triggered by activation with fibrogenic cytokines secreted by macrophages or other cellular compounds (15, 51). This differentiation/activation process results in proliferation and excessive production of ECM components as mentioned before (54). The cellular source(s) of these myofibroblasts are still not entirely clear; however, they may be multiple (55). In addition to resident mesenchymal cells, myofibroblasts are derived from epithelial or endothelial cells in a process termed epithelial–mesenchymal transition or endothelial–mesenchymal transition (56–58). Moreover, a unique circulating fibroblast-like cell derived from bone marrow stem cells (59, 60) further accounts for myofibroblast development. These blood-born mesenchymal progenitors have a fibroblast/myofibroblast-like phenotype as they express CD34, CD45, and type I collagen and are commonly called fibrocytes.
Notably, in the field of radiation biology, an alternative definition of fibrocytes exists that differs from the immunological one given above that may cause some confusion. In their reports, Bayreuther and Rodemann indicated fibrocytes to constitute terminally differentiated postmitotic fibroblasts (PMF) with downregulation of transcription factor c-fos and a specific capacity for the synthesis of collagen types I, III, and V and proteoglycans (39, 61, 62). Taking this definition into account, single-dose irradiation in the range of 1–8 Gy has been shown to induce terminal differentiation of these cells into senescent fibrocytes at a high percentage level. By contrast, irradiation of long-term cultures with repeated doses of 10 times 0.6 Gy or 10 times 1.0 Gy revealed a marked reduction of their proliferative capacity (63, 64). This has even been demonstrated for densely ionizing irradiation (65). In line with that, the life span of non-proliferating PMF is limited and shortened by more than 40% following irradiation in comparison to physiological conditions (66). Moreover, these populations require a permanent renewal from a mitotically active progenitor fibroblast pool (67). Consequently, interference with the differentiation processes in line with eradicating mitotic precursor fibroblasts may display a substantial fundament for the clinical effects of antiproliferative low-dose irradiation.
From a mechanistic point of view, RT results in reduction of fibroblast proliferation, cell cycle arrest, and induction of cellular senescence as has been shown in irradiated long-term cultures of healthy human fibroblasts. Following an immediate cell cycle arrest, a period of a few weeks with premature differentiation and senescence was observed (68). Inhibition of cell proliferation and induction of cellular senescence were mediated by interruption of the cell cycle with an extended GO/G1 phase, in line with upregulation of cell cycle regulators TP53 and CDKN1A (p21) and senescence-associated genes p16 and p27 at protein levels (68, 69). Notably, concerning radiation-induced cell death, primary lung fibroblasts were able to prevent radiation-induced apoptosis by activation of protein kinase C (PKC), while PKC inhibition or attenuation results in downregulation of prosurvival and antiapoptotic signaling proteins and apoptosis induction (70).
Another study investigated the effect of irradiation on primary keloid fibroblasts (KFb) (71). X-ray exposure inhibited KFb proliferation and induced cell senescence in a dose- and time-dependent manner. On a molecular basis, mRNA and protein expression of senescence-associated genes p16, p21, and p27 increased after 4 Gy irradiation in a time-dependent manner. Responsible for this is considered a dynamic feedback-loop, triggered by activation of p21, followed by mitochondrial dysfunction and increased levels of ROS, resulting in elevated DNA damage and ongoing DDR (72). However, the fate of the fibroblast after irradiation-induced cell cycle arrest is not only determined by persistent DNA damage and p21 levels but also essentially depends on cellular Cdk2/p21 ratio (73).
Impairment of Proliferative Activity of Fibroblasts/Myofibroblasts by Free Radicals
It is a well-established fact that levels of ROS including superoxide (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (⋅OH) dramatically increase following exposure to ionizing radiation, resulting in damage to macromolecules and DNA in line with disturbance of a multitude of signal transduction pathways (74–77). These pathways, in a direct way, stimulate production of inflammatory and fibrogenic mediators that include chemotactic cytokines, mitogens, and mediators to modulate differentiation of the fibroblast/myofibroblast/fibrocyte axis (78, 79). Accordingly, the microenvironment in contracture tissue is characterized by the presence of a multilevel network of inflammatory/fibrogenic cytokines, ROS, and antioxidants that in sum may interfere with the clinical effectiveness of LD-RT. A close connection between ROS production and local ischemia was further confirmed in an early study showing elevated quantities of hypoxanthine and xanthine oxidase activity to catalyze elevated levels of O2− and H2O2 in palmar fascia of patients with DD (80). Besides this, addition of free oxygen radicals to cultures of fibroblasts derived from DD palmar fascia dose dependently increases collagen type III expression at low concentrations or inhibits proliferation at higher doses (81). This possibly may indicate that ionizing radiation induces a level of ROS production that exceed a threshold to inhibit proliferation of fibroblasts and/or myofibroblasts.
Cytokines and Growth Factors Comprise Targets of Radiation in Hyperproliferative Diseases
Analogous to inflammatory diseases and fibrotic disorders, levels of cytokines and growth factors secreted by a multitude of cell types including platelets and macrophages have extensively been analyzed in DD, LD, and keloid specimens (82–84). These molecules cover fibroblast growth factor, PDGF, epidermal growth factor, connective tissue growth factor, TGF-β1, IL-1, IL-6, VEGF, and tumor necrosis factor-α (TNF-α) (41, 83, 85–87). TGF-β1 is well documented to constitute a key player (84, 88), which is undoubtedly among the cytokines most implicated in both the process of fibrosis induction and radiation response. TGF-β1, which is produced by a wide range of inflammatory, mesenchymal, and epithelial cells, is critical in many facets of the fibrogenic process, such as ROS generation and conversion of fibroblasts into myofibroblasts (43, 86, 89). The factor transduces its signal by a heteromeric complex formation of related type I and type II transmembrane receptors, resulting in phosphorylation and activation of receptor-regulated mother against decapentaplegic homolog 2 (Smad2) and Smad3 molecules (R-Smads). These R-Smads in turn associate with Smad4 (Co-Smad) to form a heteromeric Smad transcription factor complex that regulates expression of a large array of target genes (90). All of these components were reported to have increased expression patterns in DD, resulting in accelerated TGF-β signaling (88, 91). Importantly, Wong and Mudera further reported on a negative feedback inhibition of TGF-β1 in Dupuytren’s fibroblasts. In their study, the group reported on lower doses (1–10 ng/ml) to increase myofibroblast activation in an experimental collagen model, whereas higher concentrations (20–30 ng/ml) impaired contraction in DD fibroblasts (92). Accordingly, it is convincible to assume that increased TGF-β1 transcription and secretion triggered by ionizing radiation in endothelial cells and fibroblasts/fibrocytes (18, 63, 64) may result in inhibition of fibroblast/myofibroblast proliferation and ECM deposition in irradiated tissue.
More recently, TNF-α was identified as an additional key regulator involved in the fibrotic process and differentiation of fibroblasts into myofibroblasts in the palm of patients affected by DD, via activation of Wnt signaling pathway (13, 87). Moreover, TNF-α directly regulates TGF-β1 expression, as shown in lung fibroblasts (93). Finally, targeting TNF-α by the use of neutralizing antibodies diminished the contractile activity of myofibroblasts derived from DD patients, reduced the expression of α-SMA, and mediated disassembly of the contractile apparatus, thus qualifying the cytokine as a therapeutic target in DD.
Impact of Macrophage Activity and Endothelial Cells on Proliferation of Myofibroblasts
While factors affecting the beginning and development of DD and LD as well as keloids have been extensively studied (15, 25, 82, 94), the mechanistic basis for the regulation of proliferative elements remains not entirely resolved. These processes, however, may include several prominent elements: a fibrogenic/angiogenic element associated with proliferation and an immune cell component. Indeed, histological studies identified the presence of clusters of macrophages and T-lymphocytes in early DD and keloids and a correlation between the numbers of macrophages and the quantity of myofibroblasts (87, 95, 96).
Notably, with regard to cytokine production, a hampered pro-inflammatory TNF-α and IL-1 secretion from human RAW 264.7 or murine macrophages stimulated by lipopolysaccharides has been reported following LD-RT (97–99). Mechanistically, the hampered cytokine production was correlated to a diminished nuclear translocation of the immune relevant transcription factor nuclear factor kappaB (NF-κB) subunit RelA (p65) in line with a lowered induction of NF-κB upstream p38 mitogen-activated protein kinase and downstream protein kinase B (Akt) (99, 100). In addition, inflammatory macrophages revealed a reduction in their capacity to perform an oxidative burst and a diminished activity of the enzyme inducible nitric oxide synthase upon low-dose irradiation, resulting in lower levels of ROS and nitric oxide (NO) induction (101, 102). Considering the pivotal function of macrophages in inflammatory and fibrogenic cascades, a lowered production of cytokines, ROS, and NO may essentially contribute to a hampered myofibroblast proliferation and to the clinical benefit of low- and intermediate-dose irradiation in hyperproliferative disorders (Figure 1).
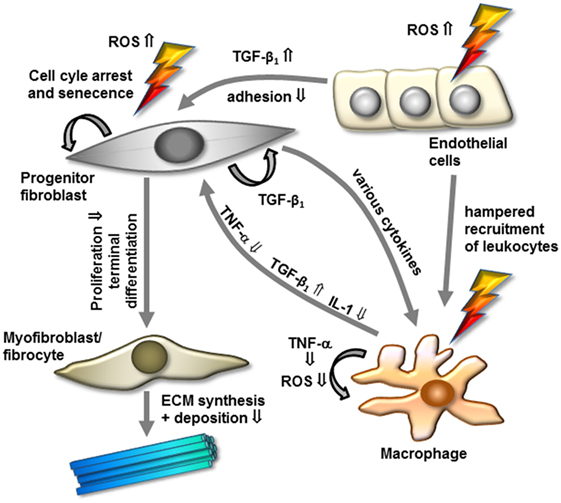
Figure 1. Model of modulation of cellular components and factors by low-dose radiotherapy for the treatment of hyperproliferative/fibrotic benign diseases. Progenitor mitotic fibroblasts are activated by transforming growth factor-β1 (TGF-β1) and additional factors to differentiate into myofibroblasts/fibrocytes, resulting in increased extracellular matrix (ECM) synthesis and deposition. In contrast, irradiation might interfere with these processes by increasing free radicals, inactivating radiosensitive mitotic fibroblasts/myofibroblasts, and promoting terminal differentiation into senescent fibrocytes. Further, low-dose irradiation modulates inflammatory components in modulating cytokine expression, macrophage, and endothelial cell activity. Abbreviations and details are given in the text.
It further has been shown that a clinically therapeutic effect of steroids if given in an early phase of DD, results from a reduction in leukocyte adhesion/diapedesis (103) as well as increased apoptosis of macrophages and fibroblasts (104). In a mechanistic manner, endothelial cells are critically implicated in the regulation of (pro-)inflammatory cascades, which are mediated by a locally restricted adhesion of immune components from the peripheral blood and secretion of an array of cytokines/growth factors including TGF-β1 and IL-6 (105–107). In that context, our group and others have shown a diminished leukocyte adhesion to 40–50% of the level of non-irradiated cells most pronounced at a 4- and 24-h period following LD-RT. This effect is mainly mediated and functionally attributed to the expression of TGF-β1 from endothelial cells (106, 108, 109). Accordingly, it is reasonable to speculate that a hampered recruitment of monocytes/macrophages from peripheral blood may promote antiproliferative/inflammatory properties of low- and intermediate-dose ionizing radiation and thus contributes to beneficial effects of LD-RT in DD, LD, and keloids.
Conclusion and Future Perspectives
The pathogenesis of hyperproliferative/fibrogenic disorders is complex, considered to evolve from system biology diseases based on a multitude of (patho)physiological networks (110), and still remains elusive despite extensive investigation. Accordingly, one may assume that the empirically proven beneficial efficacy of (low dose) RT is mediated by the modulation of a variety of pathways and cellular targets involved (Figure 1). Among these targets, the fibroblast/myofibroblast system originating from several sources comprises a characteristic connector, linking DD, LD, and keloid diseases. Radiation-related molecular mechanisms affecting these cellular components include a direct influence on cell cycle regulation, production of oxygen radicals to diminish their proliferative capacity, and interference with growth factor and cytokine expression (15). Moreover, reduced numbers of activated immune cells implicated in concomitant inflammatory processes, and proliferation of fibroblasts/myofibroblasts (111, 112) may further contribute to the therapeutic effects of radiation. Consequently, the use of low- or moderate-dose RT for early-stage DD and LD and postsurgical keloids not only covers a robust radiobiological rationale but also has been proven as low-cost and effective treatment with clinically acceptable acute and long-term toxicity (8). Even though remarkable progress has been achieved during the last years in the knowledge of radiobiological mechanisms most prominent after a low-dose exposure (33, 113), a therapeutic efficacy in hyperproliferative disorders may originate from an overlap of antiproliferative and immune-modulatory effects as documented by different dose requirements in daily clinical applications.
As stated before, the number of patients annually treated with low- and intermediate-dose irradiation at least in Germany continuously increases in line with a growing acceptance from other medical disciplines. Moreover, based on preclinical radiobiological considerations (113), recent trials confirmed a clinical isoeffect of single dose of 0.5 and 1 Gy irradiation (total dose 3 or 6 Gy) in terms of pain relief and long-term response at least in degenerative skeletal disorders (114, 115). Consequently, for radiation protection purposes and decreasing putative radiation risk, standard use of 0.5 Gy/3 Gy schedules is now recommended for the treatment of these diseases (6). Although comparable optimization studies are still lacking in hyperproliferative disorders, one may draw the conclusion by analogy that a dose reduction may further increase acceptance of RT in the clinical management of DD, LD, and keloids and increase numbers of patients treated for these indications worldwide. Moreover, in terms of a decrease in single and total doses, combined modality treatment with, e.g., anti-inflammatory drugs should be addressed in future clinical investigations to boost treatment routines including RT.
Very recently, a modular assay for detailed immunophenotyping of peripheral whole blood samples of patients following low-dose radon spa therapy (RAD-ON01 study) (116, 117) and low-dose X-irradiation (IMMO-LDRT01: http://ClinicalTrials.gov identifier: NCT02653079) have been developed. These multicolor flow cytometry approaches may be well adapted for a detailed monitoring of immunological properties in patients with DD, LD, and keloids. Accordingly, to the author’s point of view, future research activities should concentrate on basic, translational, and clinical efforts (dose optimization studies, patient’s immunophenotyping, and combined modality treatment) and on the development of suitable preclinical models for hyperproliferative disorders to further characterize additional factors and mechanisms contributing to the clinical effects of LD-RT.
Author Contributions
FR, CF, JW, FM, UG, BF, CR, and SH: drafted the manuscript and wrote it together with the coauthors and performed final evaluation. LK and MS: drafted the manuscript (clinical part) and wrote it together with the coauthors and performed final evaluation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This manuscript was supported by a grant of the German Federal Ministry of Education and Research (BMBF, GREWIS, 02NUK017A, 02NUK017D, 02NUK017F, and 02NUK017G), in part by the European Commission (FP7-OPERRA-2014—Grant Agreement Number 604984; proposal VIBRATO) and by the research training group GRK1657 of the German Research Foundation (DFG).
References
1. Dent P, Yacoub A, Contessa J, Caron R, Amorino G, Valerie K, et al. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat Res (2003) 159(3):283–300. doi: 10.1667/0033-7587(2003)159[0283:SARIAO]2.0.CO;2
2. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: cell cycle and proliferation-dependent regulation. Semin Cancer Biol (2016) 3(7–38):51–64. doi:10.1016/j.semcancer.2016.03.003
3. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer (2016) 16(1):20–33. doi:10.1038/nrc.2015.2
4. Seegenschmiedt MH, Makoski HB, Trott KR, Brady LWE. Radiotherapy for Non-Malignant Disorders. Berlin, Heidelberg: Springer Verlag (2008).
5. Seegenschmiedt MH, Micke O, Niewald M, Mucke R, Eich HT, Kriz J, et al. DEGRO guidelines for the radiotherapy of non-malignant disorders: part III: hyperproliferative disorders. Strahlenther Onkol (2015) 191(7):541–8. doi:10.1007/s00066-015-0818-2
6. Ott OJ, Niewald M, Weitmann HD, Jacob I, Adamietz IA, Schaefer U, et al. DEGRO guidelines for the radiotherapy of non-malignant disorders. Part II: painful degenerative skeletal disorders. Strahlenther Onkol (2015) 191(1):1–6. doi:10.1007/s00066-014-0757-3
7. Suit H, Spiro I. Radiation treatment of benign mesenchymal disease. Semin Radiat Oncol (1999) 9(2):171–8. doi:10.1053/SRAO00900171
8. Seegenschmiedt MH, Micke O, Muecke R. Radiotherapy for non-malignant disorders: state of the art and update of the evidence-based practice guidelines. Br J Radiol (2015) 88(1051):20150080. doi:10.1259/bjr.20150080
9. Seegenschmiedt MH, Micke O. [Radiotherapy of non-malignant diseases. Past, present and future]. Strahlenther Onkol (2012) 188(Suppl 3):272–90. doi:10.1007/s00066-012-0195-z
10. Lanting R, Broekstra DC, Werker PM, van den Heuvel ER. A systematic review and meta-analysis on the prevalence of Dupuytren disease in the general population of Western countries. Plast Reconstr Surg (2014) 133(3):593–603. doi:10.1097/01.prs.0000438455.37604.0f
11. Hindocha S, McGrouther DA, Bayat A. Epidemiological evaluation of Dupuytren’s disease incidence and prevalence rates in relation to etiology. Hand (N Y) (2009) 4(3):256–69. doi:10.1007/s11552-008-9160-9
13. Dolmans GH, Werker PM, Hennies HC, Furniss D, Festen EA, Franke L, et al. Wnt signaling and Dupuytren’s disease. N Engl J Med (2011) 365(4):307–17. doi:10.1056/NEJMoa1101029
14. Hu FZ, Nystrom A, Ahmed A, Palmquist M, Dopico R, Mossberg I, et al. Mapping of an autosomal dominant gene for Dupuytren’s contracture to chromosome 16q in a Swedish family. Clin Genet (2005) 68(5):424–9. doi:10.1111/j.1399-0004.2005.00504.x
15. Shih B, Bayat A. Scientific understanding and clinical management of Dupuytren disease. Nat Rev Rheumatol (2010) 6(12):715–26. doi:10.1038/nrrheum.2010.180
16. Bovenzi M, Franzinelli A, Scattoni L, Vannuccini L. Hand-arm vibration syndrome among travertine workers: a follow up study. Occup Environ Med (1994) 51(6):361–5. doi:10.1136/oem.51.6.361
17. Godtfredsen NS, Lucht H, Prescott E, Sorensen TI, Gronbaek M. A prospective study linked both alcohol and tobacco to Dupuytren’s disease. J Clin Epidemiol (2004) 57(8):858–63. doi:10.1016/j.jclinepi.2003.11.015
18. Heyd R, Dorn AP, Herkstroter M, Rodel C, Muller-Schimpfle M, Fraunholz I. Radiation therapy for early stages of morbus Ledderhose. Strahlenther Onkol (2010) 186(1):24–9. doi:10.1007/s00066-009-2049-x
20. Fausto de Souza D, Micaelo L, Cuzzi T, Ramos ESM. Ledderhose disease: an unusual presentation. J Clin Aesthet Dermatol (2010) 3(9):45–7.
21. Allen RA, Woolner LB, Ghormley RK. Soft-tissue tumors of the sole; with special reference to plantar fibromatosis. J Bone Joint Surg Am (1955) 37-A(1):14–26. doi:10.2106/00004623-195537010-00002
22. Guix B, Andres A, Salort P. Keloids and hypertrophic scars. In: Seegenschmiedt MH, editor. Radiotherapy for Nonmalignat Disorders. Berlin: Springer (2008) 209–24
23. Alhady SM, Sivanantharajah K. Keloids in various races. A review of 175 cases. Plast Reconstr Surg (1969) 44(6):564–6. doi:10.1097/00006534-196912000-00006
24. Mustoe TA, Cooter RD, Gold MH, Hobbs FD, Ramelet AA, Shakespeare PG, et al. Panel on Scar: international clinical recommendations on scar management. Plast Reconstr Surg (2002) 110(2):560–71. doi:10.1097/00006534-200208000-00031
25. Werker P, Dias J, Eaton C, Reichert B, Wach BE. Dupuytren Disease and Related Diseases – The Cutting Edge. Berlin, Heidelberg: Springer Verlag (2016).
26. Betz N, Ott OJ, Adamietz B, Sauer R, Fietkau R, Keilholz L. Radiotherapy in early-stage Dupuytren’s contracture. Long-term results after 13 years. Strahlenther Onkol (2010) 186(2):82–90. doi:10.1007/s00066-010-2063-z
27. Kal HB, Veen RE. Biologically effective doses of postoperative radiotherapy in the prevention of keloids. Dose-effect relationship. Strahlenther Onkol (2005) 181(11):717–23. doi:10.1007/s00066-005-1407-6
28. Kutzner J, Schneider L, Seegenschmiedt MH. [Radiotherapy of keloids. Patterns of care study – results]. Strahlenther Onkol (2003) 179(1):54–8. doi:10.1007/s00066-003-1023-2
29. Arnault JP, Peiffert D, Latarche C, Chassagne JF, Barbaud A, Schmutz JL. Keloids treated with postoperative Iridium 192* brachytherapy: a retrospective study. J Eur Acad Dermatol Venereol (2009) 23(7):807–13. doi:10.1111/j.1468-3083.2009.03190.x
30. Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol (1988) 35:95–125. doi:10.1016/S0079-6603(08)60611-X
31. O’Driscoll M, Jeggo PA. The role of double-strand break repair – insights from human genetics. Nat Rev Genet (2006) 7(1):45–54. doi:10.1038/nrg1746
32. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature (2009) 461(7267):1071–8. doi:10.1038/nature08467
33. Rodel F, Frey B, Multhoff G, Gaipl U. Contribution of the immune system to bystander and non-targeted effects of ionizing radiation. Cancer Lett (2015) 356(1):105–13. doi:10.1016/j.canlet.2013.09.015
34. Brown WM, Doll R. Mortality from cancer and other causes after radiotherapy for ankylosing spondylitis. Br Med J (1965) 2(5474):1327–32. doi:10.1136/bmj.2.5474.1327
35. Mazonakis M, Damilakis J. Cancer risk after radiotherapy for benign diseases. Phys Med (2017). doi:10.1016/j.ejmp.2017.01.014
36. McKeown SR, Hatfield P, Prestwich RJ, Shaffer RE, Taylor RE. Radiotherapy for benign disease; assessing the risk of radiation-induced cancer following exposure to intermediate dose radiation. Br J Radiol (2015) 88(1056):20150405. doi:10.1259/bjr.20150405
37. The 2007 Recommendations of the International Commission on Radiological Protection. ICRP publication 103. Ann ICRP (2007) 37(2–4):1–332. doi:10.1016/j.icrp.2007.10.003
38. Trott KR, Kamprad F. Estimation of cancer risks from radiotherapy of benign diseases. Strahlenther Onkol (2006) 182(8):431–6. doi:10.1007/s00066-006-1542-8
39. Rodemann HP, Bamberg M. Cellular basis of radiation-induced fibrosis. Radiother Oncol (1995) 35(2):83–90. doi:10.1016/0167-8140(95)01540-W
40. Grenfell S, Borg M. Radiotherapy in fascial fibromatosis: a case series, literature review and considerations for treatment of early-stage disease. J Med Imaging Radiat Oncol (2014) 58(5):641–7. doi:10.1111/1754-9485.12178
41. Cordova A, Tripoli M, Corradino B, Napoli P, Moschella F. Dupuytren’s contracture: an update of biomolecular aspects and therapeutic perspectives. J Hand Surg Br (2005) 30(6):557–62. doi:10.1016/j.jhsb.2005.07.002
42. Fitzgerald AM, Kirkpatrick JJ, Naylor IL. Dupuytren’s disease. The way forward? J Hand Surg Br (1999) 24(4):395–9. doi:10.1054/jhsb.1999.0207
43. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol (2002) 3(5):349–63. doi:10.1038/nrm809
44. Mayerl C, Del Frari B, Parson W, Boeck G, Piza-Katzer H, Wick G, et al. Characterisation of the inflammatory response in Dupuytren’s disease. J Plast Surg Hand Surg (2016) 50(3):171–9. doi:10.3109/2000656X.2016.1140054
45. Shih B, Brown JJ, Armstrong DJ, Lindau T, Bayat A. Differential gene expression analysis of subcutaneous fat, fascia, and skin overlying a Dupuytren’s disease nodule in comparison to control tissue. Hand (N Y) (2009) 4(3):294–301. doi:10.1007/s11552-009-9164-0
46. Qu M, Song N, Chai G, Wu X, Liu W. Pathological niche environment transforms dermal stem cells to keloid stem cells: a hypothesis of keloid formation and development. Med Hypotheses (2013) 81(5):807–12. doi:10.1016/j.mehy.2013.08.033
47. Jiao H, Fan J, Cai J, Pan B, Yan L, Dong P, et al. Analysis of characteristics similar to autoimmune disease in keloid patients. Aesthetic Plast Surg (2015) 39(5):818–25. doi:10.1007/s00266-015-0542-4
48. Do DV, Ong CT, Khoo YT, Carbone A, Lim CP, Wang S, et al. Interleukin-18 system plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. Br J Dermatol (2012) 166(6):1275–88. doi:10.1111/j.1365-2133.2011.10721.x
49. Zhang M, Xu Y, Liu Y, Cheng Y, Zhao P, Liu H, et al. Chemokine-like factor 1 (CKLF-1) is overexpressed in keloid patients: a potential indicating factor for keloid-predisposed individuals. Medicine (Baltimore) (2016) 95(11):e3082. doi:10.1097/MD.0000000000003082
50. Abraham DJ, Eckes B, Rajkumar V, Krieg T. New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma. Curr Rheumatol Rep (2007) 9(2):136–43. doi:10.1007/s11926-007-0008-z
51. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol (2007) 170(6):1807–16. doi:10.2353/ajpath.2007.070112
52. Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia (1971) 27(5):549–50. doi:10.1007/BF02147594
53. Rudolph R, Vande Berg J. The myofibroblast in Dupuytren’s contracture. Hand Clin (1991) 7(4):683–92.
54. Berndt A, Kosmehl H, Katenkamp D, Tauchmann V. Appearance of the myofibroblastic phenotype in Dupuytren’s disease is associated with a fibronectin, laminin, collagen type IV and tenascin extracellular matrix. Pathobiology (1994) 62(2):55–8. doi:10.1159/000163879
55. Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep (2006) 8(2):145–50. doi:10.1007/s11926-006-0055-x
56. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med (2007) 13(8):952–61. doi:10.1038/nm1613
57. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc (2006) 3(4):377–82. doi:10.1513/pats.200601-004TK
58. Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med (2016) 5(4):E45. doi:10.3390/jcm5040045
59. Iqbal SA, Hayton MJ, Watson JS, Szczypa P, Bayat A. First identification of resident and circulating fibrocytes in Dupuytren’s disease shown to be inhibited by serum amyloid P and Xiapex. PLoS One (2014) 9(6):e99967. doi:10.1371/journal.pone.0099967
60. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med (1994) 1(1):71–81.
61. Bayreuther K, Rodemann HP, Francz PI, Maier K. Differentiation of fibroblast stem cells. J Cell Sci Suppl (1988) 10:115–30. doi:10.1242/jcs.1988.Supplement_10.9
62. Bayreuther K, Rodemann HP, Hommel R, Dittmann K, Albiez M, Francz PI. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc Natl Acad Sci U S A (1988) 85(14):5112–6. doi:10.1073/pnas.85.14.5112
63. Rodemann HP, Peterson HP, Schwenke K, von Wangenheim KH. Terminal differentiation of human fibroblasts is induced by radiation. Scanning Microsc (1991) 5(4):1135–42.
64. Bumann J, Santo-Holtje L, Loffler H, Bamberg M, Rodemann HP. Radiation-induced alterations of the proliferation dynamics of human skin fibroblasts after repeated irradiation in the subtherapeutic dose range. Strahlenther Onkol (1995) 171(1):35–41.
65. Fournier C, Scholz M, Weyrather WK, Rodemann HP, Kraft G. Changes of fibrosis-related parameters after high- and low-LET irradiation of fibroblasts. Int J Radiat Biol (2001) 77(6):713–22. doi:10.1080/095530000110045025
66. Bayreuther K, Francz PI, Rodemann HP. Fibroblasts in normal and pathological terminal differentiation, aging, apoptosis and transformation. Arch Gerontol Geriatr (1992) 15(Suppl 1):47–74. doi:10.1016/S0167-4943(05)80006-8
67. Herskind C, Rodemann HP. Spontaneous and radiation-induced differentiation of fibroblasts. Exp Gerontol (2000) 35(6–7):747–55. doi:10.1016/S0531-5565(00)00168-6
68. Fournier C, Winter M, Zahnreich S, Nasonova E, Melnikova L, Ritter S. Interrelation amongst differentiation, senescence and genetic instability in long-term cultures of fibroblasts exposed to different radiation qualities. Radiother Oncol (2007) 83(3):277–82. doi:10.1016/j.radonc.2007.04.022
69. Fournier C, Wiese C, Taucher-Scholz G. Accumulation of the cell cycle regulators TP53 and CDKN1A (p21) in human fibroblasts after exposure to low- and high-LET radiation. Radiat Res (2004) 161(6):675–84. doi:10.1667/RR3182
70. Bluwstein A, Kumar N, Leger K, Traenkle J, Oostrum J, Rehrauer H, et al. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis (2013) 4:e498. doi:10.1038/cddis.2013.15
71. Ji J, Tian Y, Zhu YQ, Zhang LY, Ji SJ, Huan J, et al. Ionizing irradiation inhibits keloid fibroblast cell proliferation and induces premature cellular senescence. J Dermatol (2015) 42(1):56–63. doi:10.1111/1346-8138.12702
72. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol (2010) 6:347. doi:10.1038/msb.2010.5
73. Kollarovic G, Studencka M, Ivanova L, Lauenstein C, Heinze K, Lapytsko A, et al. To senesce or not to senesce: how primary human fibroblasts decide their cell fate after DNA damage. Aging (Albany NY) (2016) 8(1):158–77. doi:10.18632/aging.100883
74. Travis EL. Organizational response of normal tissues to irradiation. Semin Radiat Oncol (2001) 11(3):184–96. doi:10.1053/srao.2001.25243
75. Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet (2011) 20(20):3997–4004. doi:10.1093/hmg/ddr327
76. Dettmering T, Zahnreich S, Colindres-Rojas M, Durante M, Taucher-Scholz G, Fournier C. Increased effectiveness of carbon ions in the production of reactive oxygen species in normal human fibroblasts. J Radiat Res (2015) 56(1):67–76. doi:10.1093/jrr/rru083
77. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol (2013) 13(5):349–61. doi:10.1038/nri3423
78. Robbins ME, Zhao W. Chronic oxidative stress and radiation-induced late normal tissue injury: a review. Int J Radiat Biol (2004) 80(4):251–9. doi:10.1080/09553000410001692726
79. Zhao W, Robbins ME. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Curr Med Chem (2009) 16(2):130–43. doi:10.2174/092986709787002790
80. Murrell GA, Francis MJ, Bromley L. Free radicals and Dupuytren’s contracture. Br Med J (Clin Res Ed) (1987) 295(6610):1373–5. doi:10.1136/bmj.295.6610.1373
81. Murrell GA, Francis MJ, Bromley L. Modulation of fibroblast proliferation by oxygen free radicals. Biochem J (1990) 265(3):659–65. doi:10.1042/bj2650659
82. Baird KS, Crossan JF, Ralston SH. Abnormal growth factor and cytokine expression in Dupuytren’s contracture. J Clin Pathol (1993) 46(5):425–8. doi:10.1136/jcp.46.5.425
83. Bianchi E, Taurone S, Bardella L, Signore A, Pompili E, Sessa V, et al. Involvement of pro-inflammatory cytokines and growth factors in the pathogenesis of Dupuytren’s contracture: a novel target for a possible future therapeutic strategy? Clin Sci (Lond) (2015) 129(8):711–20. doi:10.1042/CS20150088
84. Kloen P, Jennings CL, Gebhardt MC, Springfield DS, Mankin HJ. Transforming growth factor-beta: possible roles in Dupuytren’s contracture. J Hand Surg Am (1995) 20(1):101–8. doi:10.1016/S0363-5023(05)80067-X
85. Alman BA, Naber SP, Terek RM, Jiranek WA, Goldberg MJ, Wolfe HJ. Platelet-derived growth factor in fibrous musculoskeletal disorders: a study of pathologic tissue sections and in vitro primary cell cultures. J Orthop Res (1995) 13(1):67–77. doi:10.1002/jor.1100130111
86. Badalamente MA, Sampson SP, Hurst LC, Dowd A, Miyasaka K. The role of transforming growth factor beta in Dupuytren’s disease. J Hand Surg Am (1996) 21(2):210–5. doi:10.1016/S0363-5023(96)80102-X
87. Verjee LS, Verhoekx JS, Chan JK, Krausgruber T, Nicolaidou V, Izadi D, et al. Unraveling the signaling pathways promoting fibrosis in Dupuytren’s disease reveals TNF as a therapeutic target. Proc Natl Acad Sci U S A (2013) 110(10):E928–37. doi:10.1073/pnas.1301100110
88. Krause C, Kloen P, Ten Dijke P. Elevated transforming growth factor beta and mitogen-activated protein kinase pathways mediate fibrotic traits of Dupuytren’s disease fibroblasts. Fibrogenesis Tissue Repair (2011) 4(1):14. doi:10.1186/1755-1536-4-14
89. Alioto RJ, Rosier RN, Burton RI, Puzas JE. Comparative effects of growth factors on fibroblasts of Dupuytren’s tissue and normal palmar fascia. J Hand Surg Am (1994) 19(3):442–52. doi:10.1016/0363-5023(94)90059-0
90. Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol (2007) 8(12):970–82. doi:10.1038/nrm2297
91. Kloen P. New insights in the development of Dupuytren’s contracture: a review. Br J Plast Surg (1999) 52(8):629–35. doi:10.1054/bjps.1999.3187
92. Wong M, Mudera V. Feedback inhibition of high TGF-beta1 concentrations on myofibroblast induction and contraction by Dupuytren’s fibroblasts. J Hand Surg Br (2006) 31(5):473–83. doi:10.1016/j.jhsb.2006.05.007
93. Sullivan DE, Ferris M, Pociask D, Brody AR. Tumor necrosis factor-alpha induces transforming growth factor-beta1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am J Respir Cell Mol Biol (2005) 32(4):342–9. doi:10.1165/rcmb.2004-0288OC
94. Zhu Z, Ding J, Tredget EE. The molecular basis of hypertrophic scars. Burns Trauma (2016) 4:2. doi:10.1186/s41038-015-0026-426
95. Andrew JG, Andrew SM, Ash A, Turner B. An investigation into the role of inflammatory cells in Dupuytren’s disease. J Hand Surg Br (1991) 16(3):267–71. doi:10.1016/0266-7681(91)90051-O
96. Baird KS, Alwan WH, Crossan JF, Wojciak B. T-cell-mediated response in Dupuytren’s disease. Lancet (1993) 341(8861):1622–3. doi:10.1016/0140-6736(93)90760-E
97. Conrad S, Ritter S, Fournier C, Nixdorff K. Differential effects of irradiation with carbon ions and X-rays on macrophage function. J Radiat Res (2009) 50(3):223–31. doi:10.1269/jrr.08115
98. Tsukimoto M, Homma T, Mutou Y, Kojima S. 0.5 Gy gamma radiation suppresses production of TNF-alpha through up-regulation of MKP-1 in mouse macrophage RAW264.7 cells. Radiat Res (2009) 171(2):219–24. doi:10.1667/RR1351.1
99. Lodermann B, Wunderlich R, Frey S, Schorn C, Stangl S, Rodel F, et al. Low dose ionising radiation leads to a NF-kappaB dependent decreased secretion of active IL-1beta by activated macrophages with a discontinuous dose-dependency. Int J Radiat Biol (2012) 88(10):727–34. doi:10.3109/09553002.2012.689464
100. Wunderlich R, Ernst A, Rodel F, Fietkau R, Ott O, Lauber K, et al. Low and moderate doses of ionizing radiation up to 2 Gy modulate transmigration and chemotaxis of activated macrophages, provoke an anti-inflammatory cytokine milieu, but do not impact upon viability and phagocytic function. Clin Exp Immunol (2015) 179(1):50–61. doi:10.1111/cei.12344
101. Hildebrandt G, Loppnow G, Jahns J, Hindemith M, Anderegg U, Saalbach A, et al. Inhibition of the iNOS pathway in inflammatory macrophages by low-dose X-irradiation in vitro. Is there a time dependence? Strahlenther Onkol (2003) 179(3):158–66. doi:10.1007/s00066-003-1044-x
102. Schaue D, Marples B, Trott KR. The effects of low-dose X-irradiation on the oxidative burst in stimulated macrophages. Int J Radiat Biol (2002) 78(7):567–76. doi:10.1080/09553000210126457
103. Meek RM, McLellan S, Crossan JF. Dupuytren’s disease. A model for the mechanism of fibrosis and its modulation by steroids. J Bone Joint Surg Br (1999) 81(4):732–8. doi:10.1302/0301-620X.81B4.9163
104. Meek RM, McLellan S, Reilly J, Crossan JF. The effect of steroids on Dupuytren’s disease: role of programmed cell death. J Hand Surg Br (2002) 27(3):270–3. doi:10.1054/jhsb.2001.0742
105. Speyer CL, Ward PA. Role of endothelial chemokines and their receptors during inflammation. J Invest Surg (2011) 24(1):18–27. doi:10.3109/08941939.2010.521232
106. Roedel F, Kley N, Beuscher HU, Hildebrandt G, Keilholz L, Kern P, et al. Anti-inflammatory effect of low-dose X-irradiation and the involvement of a TGF-beta1-induced down-regulation of leukocyte/endothelial cell adhesion. Int J Radiat Biol (2002) 78(8):711–9. doi:10.1080/09553000210137671
107. Rödel F, Schaller U, Schultze-Mosgau S, Beuscher HU, Keilholz L, Herrmann M, et al. The induction of TGF-beta(1) and NF-kappaB parallels a biphasic time course of leukocyte/endothelial cell adhesion following low-dose X-irradiation. Strahlenther Onkol (2004) 180(4):194–200. doi:10.1007/s00066-004-1237-y
108. Kern PM, Keilholz L, Forster C, Hallmann R, Herrmann M, Seegenschmiedt MH. Low-dose radiotherapy selectively reduces adhesion of peripheral blood mononuclear cells to endothelium in vitro. Radiother Oncol (2000) 54(3):273–82. doi:10.1016/S0167-8140(00)00141-9
109. Hildebrandt G, Radlingmayr A, Rosenthal S, Rothe R, Jahns J, Hindemith M, et al. Low-dose radiotherapy (LD-RT) and the modulation of iNOS expression in adjuvant-induced arthritis in rats. Int J Radiat Biol (2003) 79(12):993–1001. doi:10.1080/09553000310001636639
110. Rehman S, Goodacre R, Day PJ, Bayat A, Westerhoff HV. Dupuytren’s: a systems biology disease. Arthritis Res Ther (2011) 13(5):238. doi:10.1186/ar3438
111. Rubin P, Soni A, Williams JP. The molecular and cellular biologic basis for the radiation treatment of benign proliferative diseases. Semin Radiat Oncol (1999) 9(2):203–14. doi:10.1053/SRAO00900203
112. Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol (2010) 97(1):149–61. doi:10.1016/j.radonc.2010.09.002
113. Rödel F, Frey B, Manda K, Hildebrandt G, Hehlgans S, Keilholz L, et al. Immunomodulatory properties and molecular effects in inflammatory diseases of low-dose X-irradiation. Front Oncol (2012) 2:120. doi:10.3389/fonc.2012.00120
114. Ott OJ, Hertel S, Gaipl US, Frey B, Schmidt M, Fietkau R. The Erlangen dose optimization trial for low-dose radiotherapy of benign painful elbow syndrome. Long-term results. Strahlenther Onkol (2014) 190(3):293–7. doi:10.1007/s00066-013-0504-1
115. Ott OJ, Hertel S, Gaipl US, Frey B, Schmidt M, Fietkau R. The Erlangen dose optimization trial for radiotherapy of benign painful shoulder syndrome. Long-term results. Strahlenther Onkol (2014) 190(4):394–8. doi:10.1007/s00066-013-0520-1
116. Rühle PF, Fietkau R, Gaipl US, Frey B. Development of a modular assay for detailed immunophenotyping of peripheral human whole blood samples by multicolor flow cytometry. Int J Mol Sci (2016) 17(8):E1316. doi:10.3390/ijms17081316
Keywords: low-dose radiation therapy, hyperproliferative diseases, fibroblasts/myofibroblast, cytokines, antiproliferative effect, anti-inflammatory effect
Citation: Rödel F, Fournier C, Wiedemann J, Merz F, Gaipl US, Frey B, Keilholz L, Seegenschmiedt MH, Rödel C and Hehlgans S (2017) Basics of Radiation Biology When Treating Hyperproliferative Benign Diseases. Front. Immunol. 8:519. doi: 10.3389/fimmu.2017.00519
Received: 10 January 2017; Accepted: 18 April 2017;
Published: 03 May 2017
Edited by:
Sherven Sharma, VA Greater Los Angeles Healthcare System (VHA), USACopyright: © 2017 Rödel, Fournier, Wiedemann, Merz, Gaipl, Frey, Keilholz, Seegenschmiedt, Rödel and Hehlgans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franz Rödel, ZnJhbnoucm9lZGVsQGtndS5kZQ==