 Julia Pollmann
Julia Pollmann Alexander Rölle
Alexander Rölle Maike Hofmann3
Maike Hofmann3 Adelheid Cerwenka
Adelheid Cerwenka- 1Research Group Innate Immunity, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 2Clinical Cooperation Unit Applied Tumor-Immunity, German Cancer Research Center (DKFZ), National Center for Tumor Diseases (NCT), Heidelberg, Germany
- 3Faculty of Medicine, Department of Medicine II, University Hospital Freiburg, University of Freiburg, Freiburg im Breisgau, Germany
- 4Medical Faculty Mannheim, Division of Immunbiochemistry, University Heidelberg, Heidelberg, Germany
Hepatitis C virus (HCV) and human cytomegalovirus (HCMV) are prominent examples of RNA and DNA viruses, respectively, that establish a persistent infection in their host. HCV affects over 185 million patients worldwide, who are at high risk for developing liver fibrosis, liver cirrhosis, and ultimately hepatocellular carcinoma. Recent breakthroughs in HCV therapy, using direct-acting antivirals have provided the opportunity to monitor natural killer (NK) cells after clearance of a chronic infection. There is now increasing evidence that the individual NK cell repertoire before infection is predictive for the course of disease. HCMV affects the majority of the global population. While being asymptomatic in healthy individuals, HCMV represents a severe clinical challenge in immunocompromised patients. Both viral infections, HCV and HCMV, lead to long-lasting and profound alterations within the entire NK cell compartment. This review article, will discuss the diverse range of changes in the NK cell compartment as well as potential consequences for the course of disease.
Introduction
A wide range of viral infections challenge the immune system throughout the lifetime of its host exerting a substantial and often long-lasting impact on multiple immune parameters. Natural killer (NK) cells, vital players in the antiviral immune defense, have been shown to undergo substantial changes in phenotype, function, and subset distribution during persistent viral infections. Specific NK subsets have been associated with both efficient clearance of viruses and immune dysfunction.
Persistent viral infections can be latent or chronic. Latent infection is characterized by long periods of viral inactivity with no replication or production of new virions although stress stimuli can trigger episodes of reactivations. Prominent examples of viruses establishing latent infection are the herpes viruses [human cytomegalovirus (HCMV), herpes simplex virus (HSV), Epstein–Barr virus (EBV), varicella-zoster-virus (VZV)]. Other viruses, such as the majority of hepatitis viruses [hepatitis C Virus (HCV), hepatitis B Virus (HBV), hepatitis D Virus (HDV)] and human immunodeficiency virus (HIV), establish chronic infections in which constant replication takes place. This drives chronic inflammation, often resulting in severe tissue damage of the infected organ (1). In this review, we will focus primarily on the effects of latent HCMV and chronic HCV infection on NK cells.
Hepatitis C Virus is a hepatotropic, enveloped, (+)-strand RNA virus that is transmitted person-to-person via blood and establishes chronic infection in 55–85% of patients. The probability for spontaneous viral clearance depends on several factors such as age, sex, host genetic factors, coinfection with other viruses, and viral genotype (2, 3).
Currently, seven genotypes and multiple subgenotypes are described with distinct global distribution patterns. In developed countries, genotype 1 is the most common, accounting for around 50% of all HCV infections, even though it has the most favorable prognosis (4). Worldwide an estimated 2.5% of the world’s population is chronically infected with HCV. Throughout the decade-long infection, the liver suffers from immunopathology, resulting in fibrosis, cirrhosis, often progressing to hepatocellular carcinoma. Each year around 500,000 people die from HCV-related liver diseases (5). To establish chronicity, the virus interferes with several innate and adaptive immune pathways, such as recognition by retinoic acid inducible gene I (RIG-I), the primary sensor for HCV-RNA in the host cell’s cytoplasm (6). The emergence of viral escape variants facilitates evasion from recognition by CD8 T cells, which are the main effector cells against HCV (7).
Until 2011, standard therapy for HCV consisted of pegylated IFN-α/ribavirin. However, only around half of the patients achieved a sustained virological response (SVR) defined by no detectable HCV-RNA 24 weeks after treatment and side effects were drastic. In 2011, the first direct-acting antivirals (DAAs) were approved targeting essential viral proteins which revolutionized therapy by reaching SVR rates of >90% (4, 8, 9).
Human cytomegalovirus has a linear double-stranded DNA genome of 236 kbp. The virus spreads vertically and horizontally via bodily fluids by infecting epithelial and endothelial cells, macrophages, and DCs wherein it establishes life-long latency. This leads to high global prevalence rates of 60–85%, depending on socioeconomic factors, geographical location, and age. HCMV is an opportunistic pathogen, causing disease only in immunocompromised people, e.g., during transplantations or in HIV patients. Furthermore, transplacental transmission of HCMV can cause severe, primarily neurological, damage to the fetus (10).
Human cytomegalovirus has developed a plethora of strategies and dedicates a large portion of its genome to interfere with the host immune system. Many of these escape mechanisms have evolved to avoid recognition by NK cells (11–13).
Natural killer cells are important effector cells in the antiviral immune response in during HCMV and HCV infection (14–17).
The importance of NK cells in human Herpes virus infections was initially highlighted in a patient with a very rare NK cell deficiency and his enhanced susceptibility to recurrent infections (18), a clinical phenotype corroborated in later reports (19, 20). Moreover, it is also indirectly evident in the multiple immune evasion strategies that HCMV utilizes to prevent NK cell recognition (11, 12).
The two main strategies employed by various viruses to evade NK cells are preventing the upregulation of activating ligands or enhancing the expression of ligands for inhibitory NK cell receptors. Upon viral infection, a variety of stress-induced molecules are expressed on the surface of infected cells, which can be recognized by activating receptors on NK cells. Important activating receptors belong to the natural cytotoxicity receptor (NCR) family, including NKp30, NKp44, and NKp46, which can recognize cellular as well as viral ligands. However, many of the NCR ligands still remain elusive. MHC class I molecules, in particular HLA-C, provide the main inhibitory signals for NK cells via interacting with killer immunoglobulin-like receptors (KIRs) (21). The KIRs, like their MHC ligands, are genetically highly polymorphic and expressed in a stochastic manner, leaving every NK cell with 0–4 KIR receptors (22). Furthermore, different KIR haplotypes—group A and B—have been identified. While the group A haplotype comprises almost exclusively inhibitory, the group B haplotypes also encode activating KIRs (23). Indeed, most receptors are expressed only on subsets of NK cells. Therefore, NK cells are not a uniform cell population but composed of many different subsets that differ in their mode of activation and their functional properties. In this review, we will discuss some of the NK cell subsets that have been studied in HCV and HCMV infections (Figure 1).
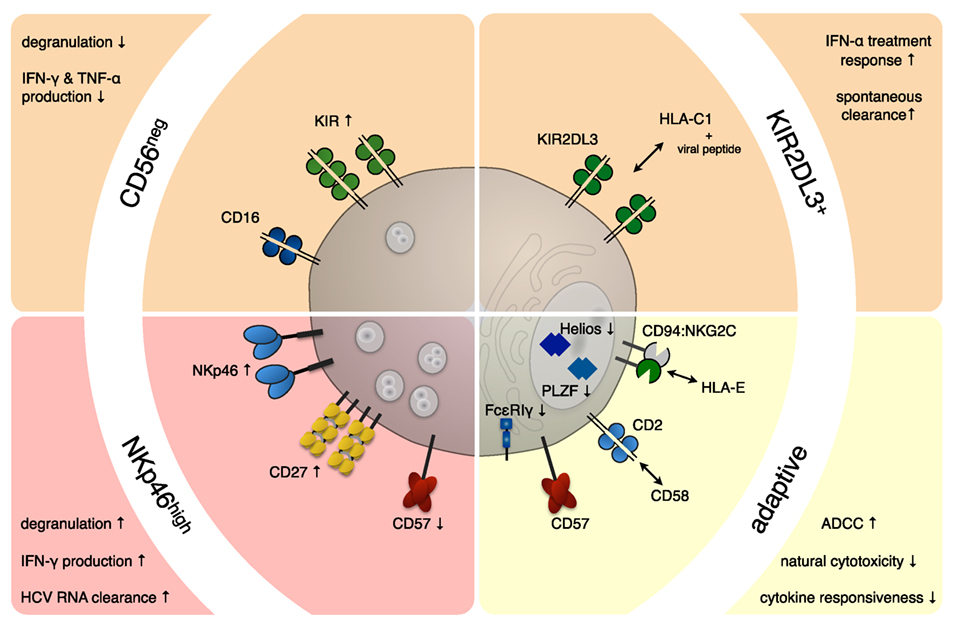
Figure 1. NK cell subsets in chronic viral infections. Persistent infections exert a profound and long-lasting impact on the NK cell compartment. In hepatitis C virus (HCV) (red background) and human cytomegalovirus (HCMV) (yellow background) infection, a number of NK cell subsets have been shown to expand and/or correlate with disease progression and outcome. During chronic HCV infection, an NK cell subset displaying increased expression of the natural cytotoxicity receptor NKp46 was observed, which proved superior in clearing HCV-RNA from infected hepatocytes. On the other hand, expansions of functionally impaired CD56neg NK cells (CD56−CD16+CD3−CD14−CD19−) were described during chronic HCV and human immunodeficiency virus and sporadically during reactivation episodes of HCMV. HCV patients with high numbers of NK cells expressing the inhibitory receptor KIR2DL3 on an HLA-C1/C1 background are more likely to spontaneously clear the virus or respond favorably to IFN-α treatment. Furthermore, certain killer immunoglobulin-like receptors (KIRs), including KIR2DL3 seem to be overrepresented on expanded adaptive NK cell subsets in HCMV infection. These adaptive NK cell subsets often express NKG2C and CD57 and are characterized by low/absent levels of key signaling molecules (e.g., FcεRIγ) and transcription factors (e.g., PLZF and/or Helios).
NK Cell Subsets Carrying Specific KIRs
One receptor that has repeatedly been implicated in the anti-HCV immune response is the inhibitory KIR2DL3. In 2004, Khakoo and colleagues (24) reported a genetic association between the expression of KIR2DL3 and HCV clearance. They studied more than thousand patients infected with HCV, of whom 685 developed a chronic infection while 352 cleared the virus. Those patients, whose NK cells expressed the inhibitory KIR2DL3 homozygously on an HLA-C1/C1 background, were more likely to clear the virus spontaneously. Of note, when stratified in terms of route of infection, this association was only observed in patients with presumed low-dose viral inoculum, e.g., after a needle-stick, suggesting that the antiviral NK cell response is most efficient in situations with low viral load. Other studies in cohorts of injection drug users confirmed that exposed but uninfected individuals had a higher frequency of KIR2DL3 expression than drug users with chronic and resolved infections or healthy donors (25). This association was also reflected in the response to treatment, in which KIR2DL3–HLA-C1 expressing patients had a higher chance to achieve SVR after IFN-α-based treatment (26). However, in other studies, a correlation between KIR2DL3 and outcome of infection was not observed, albeit with smaller and different patient cohorts (27, 28). The current hypothesis for this association is that NK cells expressing KIR2DL3, which has a rather weak binding affinity to HLA-C (29), tend to receive less inhibitory signals and are, therefore, more easily activated than NK cells expressing KIRs with higher binding affinities.
Additionally, there is now increasing evidence that peptides presented in MHC class I complexes also influence binding affinities of KIRs. This has already been shown for HIV peptides (30–32) and could in 2016 for the first time be shown for an HCV peptide presented by an HLA-C1 molecule to KIR2DL3 on NK cells (33). Lunemann et al. (33) identified peptides derived from HCV NS3 and Core protein that stabilized expression of HLA-C*03:04 on transfected 721.221 cells and facilitated binding of KIR2DL3–Fc proteins. One of the peptides derived from HCV Core protein was furthermore able to inhibit degranulation of primary KIR2DL3+ NK cells. When comparing core peptide sequences from different HCV genotypes, the genotype 1-derived peptide provided the strongest inhibitory signal, while peptides from other genotypes were much less effective. These results encourage speculations about possible viral escape mechanisms by modulation of KIR binding and the implication that NK cells exert evolutionary pressure on the HCV genome. Future studies should address if the identified peptides can also be presented on primary hepatocytes and if other KIRs/HLA interactions are involved as well.
Moreover, there is substantial evidence for an overrepresentation of certain KIRs in expanded adaptive NK cell subpopulations (see below) in HCMV. The currently available data yield a complex picture with some reports highlighting KIR2DL2/3 (34–36) others KIR3DL1 (37) or activating KIRs (38).
The CD56neg NK Cell Subset
Traditionally, NK cells are classified as CD3−CD56+ lymphocytes, which are further divided into a CD56dimCD16+ and a CD56brightCD16− subset. During chronic viral infections, especially in HIV and HCV, a subset of CD3−CD56−CD16+ NK cells is detectable (39). These cells miss expression of lineage markers, such as CD14 and CD19 or markers of other cell types positive for CD16, while expressing a variety of NK cell receptors (NCR, KIR, and NKG2). They are, therefore, classified as CD56neg NK cells. This NK cell subset is found at low percentages (around 5% of all NK cells) in healthy adults and even in neonates, but can expand to 10–40% of all NK cells during HIV, acute, chronic, and resolved HCV infections or HIV/HCV coinfection. Concurrently, a drop in the percentage of CD56dim NK cells is observed (40–42).
Phenotypically, CD56neg NK cells from healthy donors and chronic HCV patients are similar and expression of many receptors is comparable between CD56neg and CD56dim NK cells from chronic HCV patients. Only CD57 and to a lesser extent NKp30 were found to be expressed at lower levels in the CD56neg NK subset (40). Functionally, however, CD56neg NK cells appear impaired. In response to different stimuli, the CD56neg NK subset of HCV patients failed to secrete significant amounts of IFN-γ and TNF-α and displayed low perforin expression and degranulation (40, 42). Yet, they showed higher TRAIL expression compared to CD56neg NK cells from healthy controls (41). This functional impairment seems to be a general feature of the CD56neg NK cell subset, as it was observed in various chronic inflammatory situations. However, overnight in vitro culture of CD56neg NK cells with IL-2, IL-12, or IL-15 resulted in cytotoxicity levels comparable to stimulated CD56+ NK cells, indicating that upon stimulation CD56neg NK cells can effectively function (43). It was shown in HCV, as well as in HIV/HCV coinfection, that high pre-treatment levels of CD56neg NK cells correlated with treatment failure (40, 44). Furthermore, after successful IFN-α/ribavirin treatment of HIV/HCV coinfected patients, absolute levels and percentage of CD56neg NK cells normalized after 4 weeks (45).
So far, the role of this particular NK cell subset was not evaluated during DAA treatment of patients, but several lines of evidence suggest that the decline in CD56neg NK cells after treatment is not induced by IFN-α, but results from a decrease in viral load (39). Until now, it was shown that the CD56dim and CD56bright NK cell subsets quickly normalize after DAA treatment in regard to numbers and functionality (46–48). This observation could indicate that the CD56neg subset also normalizes. However, further studies are needed to address this and other open questions: does the CD56neg subset represent a terminally differentiated or exhausted cell type or rather a specific lineage? What is the degree of plasticity in terms of other NK cell subsets becoming CD56neg or CD56neg NK cells acquiring CD56pos phenotypes? The fact that neonates and healthy individuals already have this CD56neg subset, might argue against an exhausted phenotype, although many of their properties resemble exhausted T cells.
In contrast to HCV and HIV, increased levels of CD56neg cells have only been reported sporadically in HCMV infection and were observed only in a subset of patients experiencing viral reactivation (49).
The NKp46high NK Cell Subset
Expression of NK cell receptors was analyzed in different HCV patient cohorts (e.g., in acute or chronic infection and during treatment), but the obtained results have been highly controversial (50, 51). With regards to NKp46, however, several independent studies observed the involvement of an NKp46high NK cell subset expressing multiple markers of immature NK cells (52) in protection from infection, spontaneous clearance, liver inflammation, progression of fibrosis, and outcome of treatment.
In prospective studies of injection drug users, a higher percentage of CD56dim NK cells with increased levels of NKp46 was correlated with a higher percentage of individuals remaining seronegative, suggesting that high expression of NKp46 might be predictive for protection from infection (25). However, Alter et al. (42) reported lower expression of activating receptors, including NKp46, in patients with acute infection who subsequently cleared the virus than in those, who progressed to chronic infection.
In chronically infected patients, the majority of studies report elevated NKp46 expression on peripheral blood NK cells compared to healthy donors (51). Of note, this is even more pronounced in the liver (52). NKp46high peripheral NK cells from healthy donors and HCV patients perform better in reducing HCV-RNA from in vitro infected hepatocytes, produce higher levels of IFN-γ, and degranulate more ex vivo in response to different stimuli (52, 53). Likewise, intrahepatic NKp46high NK cells from HCV patients were shown to degranulate more ex vivo than NKp46dim cells (54). Furthermore, staining with an NKp46-Ig fusion protein revealed higher expression of a yet unknown NKp46-ligand on HCV-infected Huh7.5 hepatoma cells than on uninfected cells (53).
Accordingly, NKp46 levels in patients correlate positively with liver inflammation scores (54) and inversely with HCV serum levels (52), suggesting that NKp46high NK cells can kill infected hepatocytes and contribute to viral control during chronic infection. The NKp46high subset correlated with low fibrosis stages, possibly due to NKp46-dependent killing of hepatic stellate cells, the main drivers of fibrosis (54–56).
Even though the NKp46high NK cell subset might be beneficial in reducing viral load and liver fibrosis during chronic infection, it also predicts failure to IFN-α therapy (54, 57). After successful DAA treatment, previously elevated NKp46 levels in liver and blood normalize, concomitantly with a normalization of many other NK cell receptors (47).
In contrast, in HCMV infection, there is little evidence for a direct modulation or involvement of NCRs. One report observed the dissociation of the CD3zeta chain from NKp30 after engaging the HCMV tegument protein pp65, leading to greatly reduced NKp30-mediated killing (58).
Adaptive NK Cell Subsets
The most striking example for a long-lasting impact of a pathogenic challenge on distinct NK subsets (59) was initially identified in two key reports by Miguel Lopez-Botet’s group (60, 61). In HCMV-seropositive individuals, a higher proportion of NK cells expressing the activating receptor CD94/NKG2C was detected. This expanded subset displays lower NCR levels and increased expression of CD85j/LIR-1 (60) and CD2 (38). Similar observations were made in transplant recipients who suffered from acute CMV infection/reactivation (34, 37, 49, 62, 63). In vitro studies recapitulated subset expansion suggesting that exposure of NKG2C+ NK cells to infected cells was critical for this process (61). Moreover, the interaction between HLA-E and CD94/NKG2C was defined as a critical event for subset expansion (64, 65). To date, several additional factors have been reported to contribute to the expansion and activation of NKG2C+ NK cells in response to HCMV infection, such as IL-15 (61), IL-12 (64), and CD14+ monocytes (64, 66), as well as the interaction between CD2 and upregulated CD58 on infected cells (67). As expansion of subpopulations and their subsequent longevity resemble hallmarks of adaptive immune responses, the term “adaptive NK cells” was coined for human NK cells displaying these characteristics. From here on, we will use the term in this broadly defined sense, comprising multiple subsets.
Intriguingly, while a large number of studies describe NK subset expansions in other infections, e.g., HIV (68–70) Hantavirus (65), Chikungunya virus (71), EBV (72), and HBV/HCV (35, 73), seropositivity for HCMV seems to be a necessary pre-requirement. Altered HLA-E levels and/or a certain inflammatory cytokine milieu could be common denominators permitting the (re)expansion of NK cells “primed” initially by HCMV. The initial events, however, underlying the formation of this NK cell subpopulation in primary HCMV infection, remain enigmatic and represent a field of intense interest.
In recent years, it became clear that the initial definition as NKG2C+ (and CD57+) was not sufficient to encompass all adaptive NK cell subsets.
A study by Hwang and coworkers in 2012 (74) identified NK cell subpopulations with low or absent expression of the adaptor protein FcεRIγ in about one-third of all individuals tested. Zhang et al. (75) then established that the presence of FcεRIγ-deficient NK cells was strictly associated with prior exposure to HCMV. Expansion of an FcεRIγ− subset was also observed in HCMV+ chronic HCV patients and correlated with low liver damage and fibrosis levels, possibly implying an involvement of this subset in protection from immunopathology (76). Further reports extended the concept of HCMV driving the expansion of adaptive NK cell populations with deficiencies in key signaling molecules to Syk, EAT-2, and DAB2 and the transcription factors PLZF and Helios (77, 78). These features are not necessarily combined at a single-cell level and instead found in different combinations creating a previously unappreciated heterogeneity among adaptive NK cells.
Intriguingly, this molecular signature partially resembles exhausted T cells and in fact a recent study described high PD-1 expression on a subset of CD57+ NK cells also displaying increased LIR-1 levels as well as higher NKG2C expression in some donors (79). Together with lower NCR expression in adaptive NK subsets, these features suggest a decreased functionality. While this seems to be the case for classical tumor targets (78), superior antibody-dependent cellular cytotoxicity (ADCC) responses are emerging as a prominent and distinct characteristic of adaptive NK cells (38, 75, 77, 80–82), augmented by CD2 co-stimulation (67, 83). This functional specialization is accompanied by broad epigenetic modifications, including better accessibility of the ifn-γ locus (77, 78, 84). Latent HCMV infection might, therefore, be a worthy trade-off for the host if the interplay of CMV-induced adaptive NK cell populations and antigen-specific humoral immunity via ADCC results in elevated resistance to heterologous infection.
Besides deciphering the generation and function of adaptive NK cell subsets, several studies focused on their localization. In mice, a subset of NK cells endowed with antigen-specific memory has been shown to reside specifically in the liver (85, 86). In human liver samples, a subset, phenotypically similar to memory NK cells in the mouse (CD49a+T-bet+Eomes−) that also displayed high NKG2C expression, was identified. Yet, in contrast to peripheral NKG2C+ NK cells, they had an immature phenotype (CD57−, CD16−CD56bright), existed in high numbers in HCMV negative donors, and were incapable of mounting ADCC responses due to lack of CD16 expression (85, 87). Therefore, besides their striking resemblance to murine liver-resident memory NK cells and some shared features with human peripheral blood adaptive NK cells, the function of this unique human liver NK subset needs to be further defined.
Very recently, another liver-resident NK cell population, characterized as Eomeshi, CXCR6+ (88–90), and CD49e− (91) was described, following up HLA-mismatched human liver-transplants. The authors demonstrated that these Eomeshi NK cells survive for up to 13 years (90). This remarkable longevity makes an involvement of this subset in tissue homeostasis or antiviral responses against chronic infections a plausible scenario, which awaits further investigation.
Conclusion
Virus infections, especially with persistent viruses, have a remarkable impact on the NK cell compartment, shape the overall NK cell repertoire, and profoundly affect their effector functions. However, vice versa, the different NK subset composition and receptor distribution before infection can also be decisive how well infections can be combated. The diversity of human NK cell subsets is one of the emerging topics in the field. Especially, tissue-resident NK cells and other subsets of helper innate lymphoid cells (ILCs), their development, regulation, antiviral functions, and plasticity in tissues, such as in the liver, are currently an area of intense research. A better understanding of the development and dynamics of ILCs comprising both NK cells and helper ILCs subsets in affected tissues during chronic viral infection might help the design of improved targeted strategies for therapeutic intervention.
Author Contributions
JP and AR wrote the manuscript. MH revised the manuscript. AC revised the manuscript and provided conceptual input.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, RJ, and handling editor declared their shared affiliation, and the handling editor states that the process, nevertheless, met the standards of a fair and objective review.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (TRR 179 to AC, JP, and MH).
References
1. Boldogh I, Albrecht T, Porter DD. Persistent viral infections. In: Baron S, editor. Medical Microbiology. Chapter 46, 4th ed. Galveston, TX: University of Texas Medical Branch at Galveston (1996).
2. Harris HE, Eldridge KP, Harbour S, Alexander G, Teo C-G, Ramsay ME. HCV National Register Steering Group*. Does the clinical outcome of hepatitis C infection vary with the infecting hepatitis C virus type? J Viral Hepat (2007) 14:213–20. doi:10.1111/j.1365-2893.2006.00795.x
3. Grebely J, Page K, Sacks-Davis R, van der Loeff MS, Rice TM, Bruneau J, et al. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology (2014) 59:109–20. doi:10.1002/hep.26639
4. Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology (2015) 61:77–87. doi:10.1002/hep.27259
5. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380:2095–128. doi:10.1016/S0140-6736(12)61728-0
6. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature (2005) 437:1167–72. doi:10.1038/nature04193
7. Petrovic D, Dempsey E, Doherty DG, Kelleher D, Long A. Hepatitis C virus – T-cell responses and viral escape mutations. Eur J Immunol (2012) 42:17–26. doi:10.1002/eji.201141593
8. European Association for Study of Liver. EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol (2014) 60:392–420. doi:10.1016/j.jhep.2013.11.003
9. Shahid I, ALMalki WH, Hafeez MH, Hassan S. Hepatitis C virus infection treatment: an era of game changer direct acting antivirals and novel treatment strategies. Crit Rev Microbiol (2016) 42:535–47. doi:10.3109/1040841X.2014.970123
10. Mocarski ES Jr, Pass RF. Human cytomegalovirus: general features. Encyclopedia of Virology (Third Edition). Oxford: Academic Press (2008). p. 474–85.
11. Noriega V, Redmann V, Gardner T, Tortorella D. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res (2012) 54:140–51. doi:10.1007/s12026-012-8304-8
12. Rossini G, Cerboni C, Santoni A, Landini MP, Landolfo S, Gatti D, et al. Interplay between human cytomegalovirus and intrinsic/innate host responses: a complex bidirectional relationship. Mediators Inflamm (2012) 2012:607276. doi:10.1155/2012/607276
13. Wills MR, Poole E, Lau B, Krishna B, Sinclair JH. The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol (2015) 12:128–38. doi:10.1038/cmi.2014.75
14. Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med (2013) 19:859–68. doi:10.1038/nm.3251
15. Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol (2013) 31:163–94. doi:10.1146/annurev-immunol-032712-100001
16. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol (2016) 16:112–23. doi:10.1038/nri.2015.9
17. Rölle A, Pollmann J, Cerwenka A. Memory of infections: an emerging role for natural killer cells. PLoS Pathog (2013) 9:e1003548. doi:10.1371/journal.ppat.1003548
18. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med (1989) 320:1731–5. doi:10.1056/NEJM198906293202605
19. Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. Fatal varicella associated with selective natural killer cell deficiency. J Pediatr (2005) 146:423–5. doi:10.1016/j.jpeds.2004.11.022
20. Eidenschenk C, Dunne J, Jouanguy E, Fourlinnie C, Gineau L, Bacq D, et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am J Hum Genet (2006) 78:721–7. doi:10.1086/503269
21. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol (2011) 89:216–24. doi:10.1038/icb.2010.78
22. Cichocki F, Miller JS, Anderson SK. Killer immunoglobulin-like receptor transcriptional regulation: a fascinating dance of multiple promoters. J Innate Immun (2011) 3:242–8. doi:10.1159/000323929
23. Rajalingam R. Overview of the killer cell immunoglobulin-like receptor system. Methods Mol Biol (2012) 882:391–414. doi:10.1007/978-1-61779-842-9_23
24. Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science (2004) 305:872–4. doi:10.1126/science.1097670
25. Thoens C, Berger C, Trippler M, Siemann H, Lutterbeck M, Broering R, et al. KIR2DL3+NKG2A natural killer cells are associated with protection from productive hepatitis C virus infection in people who inject drugs. J Hepatol (2014) 61(3):475–81. doi:10.1016/j.jhep.2014.04.020
26. Knapp S, Warshow U, Hegazy D, Brackenbury L, Guha IN, Fowell A, et al. Consistent beneficial effects of killer cell immunoglobulin-like receptor 2DL3 and group 1 human leukocyte antigen-C following exposure to hepatitis C virus. Hepatology (2010) 51:1168–75. doi:10.1002/hep.23477
27. Montes-Cano MA, Caro-Oleas JL, Romero-Gómez M, Diago M, Andrade R, Carmona I, et al. HLA-C and KIR genes in hepatitis C virus infection. Hum Immunol (2005) 66:1106–9. doi:10.1016/j.humimm.2006.02.001
28. Rauch A, Laird R, McKinnon E, Telenti A, Furrer H, Weber R, et al. Influence of inhibitory killer immunoglobulin-like receptors and their HLA-C ligands on resolving hepatitis C virus infection. Tissue Antigens (2007) 69(Suppl 1):237–40. doi:10.1111/j.1399-0039.2006.773_4.x
29. Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol (1998) 161:571–7.
30. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature (2011) 476:96–100. doi:10.1038/nature10237
31. Fadda L, Körner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw*0102-restricted HIV-1 p24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS Pathog (2012) 8:e1002805. doi:10.1371/journal.ppat.1002805
32. van Teijlingen NH, Hölzemer A, Körner C, Garcia-Beltran WF, Schafer JL, Fadda L, et al. Sequence variations in HIV-1 p24 Gag-derived epitopes can alter binding of KIR2DL2 to HLA-C*03:04 and modulate primary natural killer cell function. AIDS (2014) 28:1399–408. doi:10.1097/QAD.0000000000000284
33. Lunemann S, Martrus G, Hölzemer A, Chapel A, Ziegler M, Körner C, et al. Sequence variations in HCV core-derived epitopes alter binding of KIR2DL3 to HLA-C. J Hepatol (2016) 65(2):252–8. doi:10.1016/j.jhep.2016.03.016
34. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood (2012) 119:2665–74. doi:10.1182/blood-2011-10-386995
35. Béziat V, Dalgard O, Asselah T, Halfon P, Bedossa P, Boudifa A, et al. CMV drives clonal expansion of NKG2C+ NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol (2012) 42:447–57. doi:10.1002/eji.201141826
36. Djaoud Z, David G, Bressollette C, Willem C, Rettman P, Gagne K, et al. Amplified NKG2C+ NK cells in cytomegalovirus (CMV) infection preferentially express killer cell Ig-like receptor 2DL: functional impact in controlling CMV-infected dendritic cells. J Immunol (2013) 191:2708–16. doi:10.4049/jimmunol.1301138
37. Lopez-Vergès S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, et al. Expansion of a unique CD57+ NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A (2011) 108:14725–32. doi:10.1073/pnas.1110900108
38. Béziat V, Liu LL, Malmberg J-A, Ivarsson MA, Sohlberg E, Björklund AT, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood (2013) 121:2678–88. doi:10.1182/blood-2012-10-459545
39. Björkström NK, Ljunggren H-G, Sandberg JK. CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol (2010) 31:401–6. doi:10.1016/j.it.2010.08.003
40. Gonzalez VD, Falconer K, Björkström NK, Blom KG, Weiland O, Ljunggren H-G, et al. Expansion of functionally skewed CD56-negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN-alpha and ribavirin treatment. J Immunol (2009) 183:6612–8. doi:10.4049/jimmunol.0901437
41. Conry SJ, Meng Q, Hardy G, Yonkers NL, Sugalski JM, Hirsch A, et al. Genetically associated CD16(+)56(-) natural killer cell interferon (IFN)-αR expression regulates signaling and is implicated in IFN-α-induced hepatitis C virus decline. J Infect Dis (2012) 205:1131–41. doi:10.1093/infdis/jis027
42. Alter G, Jost S, Rihn S, Reyor LL, Nolan BE, Ghebremichael M, et al. Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+ NK cells in acute HCV infection may predict viral clearance. J Hepatol (2011) 55:278–88. doi:10.1016/j.jhep.2010.11.030
43. Gaddy J, Broxmeyer HE. Cord blood CD16+56- cells with low lytic activity are possible precursors of mature natural killer cells. Cell Immunol (1997) 180:132–42. doi:10.1006/cimm.1997.1175
44. Anthony DD, Conry SJ, Medvik K, Sandhya Rani MR, Falck-Ytter Y, Blanton RE, et al. Baseline levels of soluble CD14 and CD16+56- natural killer cells are negatively associated with response to interferon/ribavirin therapy during HCV-HIV-1 coinfection. J Infect Dis (2012) 206:969–73. doi:10.1093/infdis/jis434
45. Gonzalez VD, Falconer K, Michaëlsson J, Moll M, Reichard O, Alaeus A, et al. Expansion of CD56- NK cells in chronic HCV/HIV-1 co-infection: reversion by antiviral treatment with pegylated IFNalpha and ribavirin. Clin Immunol (2008) 128:46–56. doi:10.1016/j.clim.2008.03.521
46. Spaan M, van Oord G, Kreefft K, Hou J, Hansen BE, Janssen HLA, et al. Immunological analysis during interferon-free therapy for chronic hepatitis C virus infection reveals modulation of the natural killer cell compartment. J Infect Dis (2015) 213:216–23. doi:10.1093/infdis/jiv391
47. Serti E, Chepa-Lotrea X, Kim YJ, Keane M, Fryzek N, Liang TJ, et al. Successful interferon-free therapy of chronic hepatitis c virus infection normalizes natural killer cell function. Gastroenterology (2015) 149:190–200.e2. doi:10.1053/j.gastro.2015.03.004
48. Burchill MA, Golden-Mason L, Wind-Rotolo M, Rosen HR. Memory re-differentiation and reduced lymphocyte activation in chronic HCV-infected patients receiving direct-acting antivirals. J Viral Hepat (2015) 22:983–91. doi:10.1111/jvh.12465
49. Chiesa Della M, Falco M, Podestà M, Locatelli F, Moretta L, Frassoni F, et al. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus? Blood (2012) 119:399–410. doi:10.1182/blood-2011-08-372003
50. Heeg M, Thimme R. NK cells and hepatitis C: NKp46 expression linked to antiviral and antifibrotic activity. Hepatology (2012) 56(4):1197–200. doi:10.1002/hep.25858
51. Yoon JC, Yang CM, Song Y, Lee JM. Natural killer cells in hepatitis C: current progress. World J Gastroenterol (2016) 22:1449. doi:10.3748/wjg.v22.i4.1449
52. Krämer B, Körner C, Kebschull M, Glässner A, Eisenhardt M, Nischalke HD, et al. NKp46(High) expression defines a NK cell subset that is potentially involved in control of HCV replication and modulation of liver fibrosis. Hepatology (2012) 56(4):1201–13. doi:10.1002/hep.25804
53. Golden-Mason L, Stone AEL, Bambha KM, Cheng L, Rosen HR. Race- and gender-related variation in NKp46 expression associated with differential anti-HCV immunity. Hepatology (2012) 56(4):1214–22. doi:10.1002/hep.25771
54. Pembroke T, Christian A, Jones E, Hills RK, Wang ECY, Gallimore AM, et al. The paradox of NKp46 +natural killer cells: drivers of severe hepatitis C virus-induced pathology but in-vivo resistance to interferon α treatment. Gut (2014) 63:515–24. doi:10.1136/gutjnl-2013-304472
55. Cosgrove C, Berger CT, Kroy DC, Cheney PC, Ghebremichael M, Aneja J, et al. Chronic HCV infection affects the NK cell phenotype in the blood more than in the liver. PLoS One (2014) 9:e105950. doi:10.1371/journal.pone.0105950
56. Gur C, Doron S, Kfir-Erenfeld S, Horwitz E, Abu-Tair L, Safadi R, et al. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut (2012) 61:885–93. doi:10.1136/gutjnl-2011-301400
57. Bozzano F, Picciotto A, Costa P, Marras F, Fazio V, Hirsch I, et al. Activating NK cell receptor expression/function (NKp30, NKp46, DNAM-1) during chronic viraemic HCV infection is associated with the outcome of combined treatment. Eur J Immunol (2011) 41:2905–14. doi:10.1002/eji.201041361
58. Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol (2005) 6:515–23. doi:10.1038/ni1190
59. Rölle A, Brodin P. Immune adaptation to environmental influence: the case of NK cells and HCMV. Trends Immunol (2016) 37:233–43. doi:10.1016/j.it.2016.01.005
60. Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104:3664–71. doi:10.1182/blood-2004-05-2058
61. Guma M, Budt M, Sáez A, Brckalo T, Hengel H, Angulo A, et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood (2006) 107:3624–31. doi:10.1182/blood-2005-09-3682
62. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C+ NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol (2012) 189:5082–8. doi:10.4049/jimmunol.1201964
63. Davis ZB, Cooley SA, Cichocki F, Felices M, Wangen R, Luo X, et al. Adaptive natural killer cell and killer cell immunoglobulin-like receptor-expressing T cell responses are induced by cytomegalovirus and are associated with protection against cytomegalovirus reactivation after allogeneic donor hematopoietic cell transplantation. Biol Blood Marrow Transplant (2015) 21:1653–62. doi:10.1016/j.bbmt.2015.05.025
64. Rölle A, Pollmann J, Ewen E-M, Le VTK, Halenius A, Hengel H, et al. IL-12-producing monocytes and HLA-E control HCMV-driven NKG2C+ NK cell expansion. J Clin Invest (2014) 124:5305–16. doi:10.1172/JCI77440
65. Björkström NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med (2011) 208:13–21. doi:10.1084/jem.20100762
66. Cichocki F, Cooley S, Davis Z, DeFor TE, Schlums H, Zhang B, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia (2016) 30:456–63. doi:10.1038/leu.2015.260
67. Rölle A, Halenius A, Ewen E-M, Cerwenka A, Hengel H, Momburg F. CD2-CD58 interactions are pivotal for the activation and function of adaptive natural killer cells in human cytomegalovirus infection. Eur J Immunol (2016) 46:2420–5. doi:10.1002/eji.201646492
68. Gumá M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J Infect Dis (2006) 194:38–41. doi:10.1086/504719
69. Goodier MR, Mela CM, Steel A, Gazzard B, Bower M, Gotch F. NKG2C+ NK cells are enriched in AIDS patients with advanced-stage Kaposi’s sarcoma. J Virol (2007) 81:430–3. doi:10.1128/JVI.01567-06
70. Brunetta E, Fogli M, Varchetta S, Bozzo L, Hudspeth KL, Marcenaro E, et al. Chronic HIV-1 viremia reverses NKG2A/NKG2C ratio on natural killer cells in patients with human cytomegalovirus co-infection. AIDS (2010) 24:27–34. doi:10.1097/QAD.0b013e3283328d1f
71. Petitdemange C, Becquart P, Wauquier N, Béziat V, Debré P, Leroy EM, et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog (2011) 7:e1002268. doi:10.1371/journal.ppat.1002268
72. Saghafian-Hedengren S, Sohlberg E, Theorell J, Carvalho-Queiroz C, Nagy N, Persson J-O, et al. Epstein-Barr virus coinfection in children boosts cytomegalovirus-induced differentiation of natural killer cells. J Virol (2013) 87:13446–55. doi:10.1128/JVI.02382-13
73. Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology (2009) 137:1151–60, 1160.e1–7. doi:10.1053/j.gastro.2009.05.047
74. Hwang I, Zhang T, Scott JM, Kim AR, Lee T, Kakarla T, et al. Identification of human NK cells that are deficient for signaling adaptor FcRγ and specialized for antibody-dependent immune functions. Int Immunol (2012) 24:793–802. doi:10.1093/intimm/dxs080
75. Zhang T, Scott JM, Hwang I, Kim S. Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. J Immunol (2013) 190:1402–6. doi:10.4049/jimmunol.1203034
76. Oh JS, Ali AK, Kim S, Corsi DJ, Cooper CL, Lee S-H. NK cells lacking FcεRIγ are associated with reduced liver damage in chronic hepatitis C virus infection. Eur J Immunol (2016) 46:1020–9. doi:10.1002/eji.201546009
77. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity (2015) 42:431–42. doi:10.1016/j.immuni.2015.02.013
78. Schlums H, Cichocki F, Tesi B, Theorell J, Béziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity (2015) 42:443–56. doi:10.1016/j.immuni.2015.02.008
79. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol (2016) 139(1):335–46. doi:10.1016/j.jaci.2016.04.025
80. Wu Z, Sinzger C, Frascaroli G, Reichel J, Bayer C, Wang L, et al. Human cytomegalovirus-induced NKG2Chi CD57hi natural killer cells are effectors dependent on humoral antiviral immunity. J Virol (2013) 87:7717–25. doi:10.1128/JVI.01096-13
81. Costa-Garcia M, Vera A, Moraru M, Vilches C, López-Botet M, Muntasell A. Antibody-mediated response of NKG2Cbright NK cells against human cytomegalovirus. J Immunol (2015) 194:2715–24. doi:10.4049/jimmunol.1402281
82. Zhou J, Amran FS, Kramski M, Angelovich TA, Elliott J, Hearps AC, et al. An NK cell population lacking FcRγ is expanded in chronically infected HIV patients. J Immunol (2015) 194:4688–97. doi:10.4049/jimmunol.1402448
83. Liu LL, Landskron J, Ask EH, Enqvist M, Sohlberg E, Traherne JA, et al. Critical role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep (2016) 15:1088–99. doi:10.1016/j.celrep.2016.04.005
84. Luetke-Eversloh M, Hammer Q, Durek P, Nordström K, Gasparoni G, Pink M, et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog (2014) 10:e1004441. doi:10.1371/journal.ppat.1004441
85. Marquardt N, Béziat V, Nyström S, Hengst J, Ivarsson MA, Kekäläinen E, et al. Cutting edge: identification and characterization of human intrahepatic CD49a+ NK cells. J Immunol (2015) 194:2467–71. doi:10.4049/jimmunol.1402756
86. Paust S, Gill HS, Wang B-Z, Flynn MP, Moseman EA, Senman B, et al. Critical role for the chemokine receptor CXCR6 in NK cell–mediated antigen-specific memory of haptens and viruses. Nat Immunol (2010) 11:1127–35. doi:10.1038/ni.1953
87. Hydes T, Abuhilal M, Armstrong T, Primrose J, Takhar A, Khakoo S. Natural killer cell maturation markers in the human liver and expansion of an NKG2C+KIR+ population. Lancet (2015) 385(Suppl 1):S45. doi:10.1016/S0140-6736(15)60360-9
88. Stegmann KA, Robertson F, Hansi N, Gill U, Pallant C, Christophides T, et al. CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci Rep (2016) 6:26157. doi:10.1038/srep26157
89. Harmon C, Robinson MW, Fahey R, Whelan S, Houlihan DD, Geoghegan J, et al. Tissue-resident Eomes(hi) T-bet(lo) CD56(bright) NK cells with reduced proinflammatory potential are enriched in the adult human liver. Eur J Immunol (2016) 46:2111–20. doi:10.1002/eji.201646559
90. Cuff AO, Robertson FP, Stegmann KA, Pallett LJ, Maini MK, Davidson BR, et al. Eomeshi NK cells in human liver are long-lived and do not recirculate but can be replenished from the circulation. J Immunol (2016) 197:4283–91. doi:10.4049/jimmunol.1601424
Keywords: natural killer cells, hepatitis C virus, human cytomegalovirus, chronic infection, natural killer subsets
Citation: Pollmann J, Rölle A, Hofmann M and Cerwenka A (2017) Hepatitis C Virus and Human Cytomegalovirus—Natural Killer Cell Subsets in Persistent Viral Infections. Front. Immunol. 8:566. doi: 10.3389/fimmu.2017.00566
Received: 01 February 2017; Accepted: 27 April 2017;
Published: 17 May 2017
Edited by:
Ulrike Koehl, Hannover Medical School, GermanyReviewed by:
Roland Jacobs, Hannover Medical School, GermanyMiguel López-Botet, Institut Hospital del Mar d’Investigacions Mèdiques, Spain
Copyright: © 2017 Pollmann, Rölle, Hofmann and Cerwenka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adelheid Cerwenka, YS5jZXJ3ZW5rYUBka2Z6LWhlaWRlbGJlcmcuZGU=, YWRlbGhlaWQuY2Vyd2Vua2FAbWVkbWEudW5pLWhlaWRlbGJlcmcuZGU=