 Benoit Stijlemans
Benoit Stijlemans Magdalena Radwanska3†
Magdalena Radwanska3† Carl De Trez
Carl De Trez- 1Laboratory of Cellular and Molecular Immunology, Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 2Myeloid Cell Immunology Lab, VIB-UGent Center for Inflammation Research, Ghent, Belgium
- 3Laboratory for Biomedical Research, Ghent University Global Campus, Yeonsu-Gu, Incheon, South Korea
- 4Structural Biology Research Centre (SBRC), VIB, Brussels, Belgium
African trypanosomosis is a debilitating disease of great medical and socioeconomical importance. It is caused by strictly extracellular protozoan parasites capable of infecting all vertebrate classes including human, livestock, and game animals. To survive within their mammalian host, trypanosomes have evolved efficient immune escape mechanisms and manipulate the entire host immune response, including the humoral response. This report provides an overview of how trypanosomes initially trigger and subsequently undermine the development of an effective host antibody response. Indeed, results available to date obtained in both natural and experimental infection models show that trypanosomes impair homeostatic B-cell lymphopoiesis, B-cell maturation and survival and B-cell memory development. Data on B-cell dysfunctioning in correlation with parasite virulence and trypanosome-mediated inflammation will be discussed, as well as the impact of trypanosomosis on heterologous vaccine efficacy and diagnosis. Therefore, new strategies aiming at enhancing vaccination efficacy could benefit from a combination of (i) early parasite diagnosis, (ii) anti-trypanosome (drugs) treatment, and (iii) anti-inflammatory treatment that collectively might allow B-cell recovery and improve vaccination.
Introduction
African trypanosomes are strictly extracellular single-celled protozoan parasites belonging to the genus Trypanosoma, which cause debilitating diseases in humans and livestock and consequently significantly affect the socioeconomic development of sub-Saharan Africa (1). About 70 million people distributed over a surface of one and a half million square kilometers are estimated to be at risk for contracting sleeping sickness in Africa (2). The distribution of African trypanosomes coincides mostly with the distribution of the habitat of the hematophagic insect vector, i.e., the tsetse “fly” (Glossina sp.), with tsetse meaning “fly” in the Tswana language of Southern Africa (3). Human African trypanosomosis (HAT) or sleeping sickness is caused by Trypanosoma brucei gambiense (west and central Africa) and Trypanosoma brucei rhodesiense (eastern and southern Africa) (4, 5). Both parasites cause infections that exhibit clinically diverse patterns and hence require different patient management, with the less prevalent T. b. rhodesiense HAT considered to be the more acute and virulent/lethal form of the disease (6, 7). HAT mainly affects remote rural communities where the health infrastructure is often minimal. In general, the disease is characterized by two stages: the early hemolymphatic stage whereby parasites proliferate in the blood and lymphatic system and the late meningoencephalitic stage whereby parasites penetrate the blood–brain barrier and proliferate in the cerebral spinal fluid (8). When patients in the meningoencephalitic stage remain untreated, an encephalitic reaction can occur resulting in coma and subsequent death (9–11). However, it is important to mention that in recent years a number of reports have indicated that HAT is not always lethal and that both T. b. gambiense and T. b. rhodesiense can result in chronic human infections with little or no symptoms (12, 13). Limited surveillance in particular of non-symptomatic cases, however, make it hard to assess how widespread these non-lethal cases are, or what the molecular and genetic underlying factors are that account for HAT resistance in certain individuals (14).
According to WHO, recent successes in the fight against HAT have brought the annual new cases to less than 10,000 (5, 7, 8). To design and maintain future control strategies, it is important to indicate that T. b. gambiense is an anthroponotic disease with a minor role for animal reservoirs that accounts for 98% of the reported HAT cases and causes a chronic, gradually progressing disease, whereby the late meningoencephalitic stage is not reached before months or even years of infection (10, 15). T. b. rhodesiense on the other hand is a zoonotic disease affecting mainly animals (livestock and wildlife), with humans being only accidentally infected, and represents only 2% of the reported HAT cases, whereby the infections are acute and progress rapidly (within weeks) to the late meningoencephalitic stage (10, 16). The zoonotic nature of T. b. rhodesiense infections make them more difficult to control compared to T. b. gambiense infections (15, 17, 18). Animal African trypanosomosis (AAT) also known as Nagana is a second form of trypanosomosis that affects sub-Saharan Africa. It is mainly caused by Trypanosoma congolense, Trypanosoma vivax, and to a lesser extent Trypanosoma brucei brucei, while surra and dourine are also forms of AAT caused by Trypanosoma evansi and Trypanosoma equiperdum, respectively (19–21). Of note, some parasites acquired a mechanical transmission mode (hence, they can reside outside the tsetse/vector belt) and are also found in South/Latin America (T. vivax and T. evansi) and Asia (T. evansi and T. equiperdum) (19, 21–23). Yet, T. congolense forms a major constraint on livestock production and remains the leading cause of livestock morbidity and mortality in sub-Saharan Africa. Hereby, cattle succumb to infection primarily due to parasite-induced anemia or complications resulting from secondary, opportunistic infections (24). Progressive disease for a prolonged time will weaken these animals, thereby preventing them to be used as draft animals or for food/milk production. As a result, farming in the tsetse belt remains challenging and hampers the development of poor societies, leading to great economic losses in terms of productivity (25, 26). Indeed, AAT accounts for an estimated annual loss of about US$5 billion, whereby Africa invests every year at least US$30 million to control cattle trypanosomosis in term of curative and prophylactic treatments (27, 28). The total losses for the total tsetse-infested lands in terms of agricultural gross domestic product are US$4.75 billion per year (1). In fact, the impact of AAT on the affected areas is the combined result of environmental, political, sociocultural, entomological, and livestock management factors (29), whereby (i) the political instability of the areas hampers controlled intervention strategies and subsequently discourages commercial investment in control strategies, (ii) pharmaceutical companies are less prone to engage/invest in drug discovery/development against diseases that affect the poorest people, (iii) wild animals function as reservoir of the parasite and therefore hamper the control of the disease, and (iv) the inappropriate use of the available drugs resulting in the emergence of drug resistance (30, 31). Up till now, not a single-field applicable vaccine exists, and chemotherapy is the only strategy available to treat the disease, which is associated with high drug toxicity. Nevertheless, so far chemotherapy remains the only therapeutic choice for these diseases, whereby they target unique organelles of trypanosomes such as glycosomes and the kinetoplast that are absent in the mammalian host or trypanosome metabolic pathways that differ from the host counterparts [carbohydrate metabolism, protein and lipid modifications, and programmed cell death (PCD)] (32–34). Unlike the situation with HAT, where the nifurtimox–eflornithine combination therapy is the preferred first-line treatment for second-stage disease (35, 36), no drug combinations are currently used for AAT (27). Instead, alternating use of compounds, particularly diminazene and isometamidium (called a “sanative pair”), with low risk of cross-resistance, is recommended where possible. Hence, there is an urgent need to optimize trypanocide usage/delivery such as extending the half-life of current trypanocides to use lower quantities of trypanocide in a more effective way and, consequently, pose a decreased risk of toxicity and possibly decreased resistance development (37, 38). However, there is some optimism since progress in HAT/AAT control measures were made over the past decade due to the establishment of the Pan-African tsetse and trypanosome eradication campaign, funded by the African Development Bank, which was established in the year 2000. This organization has set tsetse elimination as its goal and has strengthened renewed interest in the research and development of control/intervention options (29, 39). Overall, “elimination” of T. b. gambiense HAT has been targeted for 2020 under leadership of the WHO (40).
One crucial factor that stands in the way of total eradication of trypanosomosis in general is inefficient diagnosis of the infection. To date, microscopy detection of the parasite remains the only available tool to diagnose AAT and T. b. rhodesiense HAT in a reliable way. Only for T. b. gambiense, monitoring tools are available for both detection of exposure and staging of the disease (41). The latter is important to reduce the risk of treatment-associated complications occurring during treatment of the second stage of the disease (42). In this context, improvement in staging diagnosis and early screening methods are current challenges, which would avoid delayed patient treatment. Diagnosis is often hampered due to lack of positive predictive value of existing field applicable techniques and the fact that antibody (Ab)-based detection cannot differentiate between active or passed—but cured—infections (41). Immunodiagnostics based on antigen detection in this case would be preferable but are currently non-existent for trypanosomosis in the field (41, 43). An additional complication resides in the recent finding that tsetse-transmissible T. b. gambiense parasites can be found in human skin biopsies from undiagnosed individuals (44). Hence, this suggests that the current diagnostic methods and control policies need to be reevaluated.
In the next sections, we will give an overview of (i) the different escape mechanisms used by African trypanosomes to survive within their mammalian host and (ii) their strategies to undermine the entire host immune response, including the humoral response, which in turn hampers vaccine development. This review will focus on two most relevant AAT species T. brucei and T. congolense, given that for both parasites, established murine models and field studies in the economically and clinically relevant host (cattle) are available (45). While for T. vivax, field study information is scanty, hardly any representative experimental data are available as these parasites do not grow in mice unless they are carefully adapted (23, 46, 47). However, there is prospect since Minoprio and coworkers were able to establish a murine model for T. vivax (48). Although rodents are not natural hosts for these pathogens, murine models can be considered valuable tools to unravel the interactions and the immune evasion mechanisms of these parasites with their mammalian host.
Host–Parasite Interactions
Life Cycle of African Trypanosomes
African trypanosomes have a digenetic/heteroxenous complex life cycle alternating between the intestine of the tsetse fly vector and the blood/tissues of the mammalian host, whereby they progress through different developmental stages, i.e., procyclic or trypomastigote forms, respectively (49, 50). Yet, to survive in each of these hosts, they undergo essential changes at the level of morphology, energy metabolism, and surface coat protein expression (51, 52). Hereby, trypanosomes feed by absorbing nutrients (proteins, carbohydrates, and fats) as well as iron and oxygen from the body fluids of the host to generate the energy necessary for the vital processes (53). Within the bloodstream of the mammalian host, they subsist as bloodstream forms (BSFs) that are ingested by tsetse flies during a blood meal, wherein they differentiate into procyclic forms in the insect midgut. Next, they migrate to the proboscis (mouth parts) where they differentiate into epimastigote forms and finally into infective metacyclic forms (MCFs) that can be transmitted to a new mammalian host during the next blood meal. Although within the tsetse fly both T. brucei and T. congolense parasites have a similar migratory life cycle (i.e., initial establishment of midgut infection and invasion of the proventriculus), they exhibit differences in transitional developmental stages with production of infective MCFs in the proboscis for T. congolense and in the salivary glands for T. brucei (54–56). Within the mammalian host, T. congolense is a strictly intravascular parasite, whereby they bind to circulating erythrocytes and endothelial cells through their flagellum, causing damage at the adhesion site (57, 58). In contrast, T. brucei can also extravasate blood vessels and invade tissues and cause severe tissue injury (44, 59, 60). Hence, this implies differences in virulence mechanisms, host–pathogen relationships, and pathogenic effects between the two species (61). In addition, T. congolense exists strictly as a long slender (LS) dividing form, whereas T. brucei parasites are pleomorphic (i.e., can exhibit two forms); a LS dividing form and a short stumpy (SS) non-dividing form that is preadapted for transmission to the fly (62, 63). This transition, which involves a quorum sensing factor (i.e., an enigmatic stumpy inducing factor), is suggested to help control parasitemia and to increase the host survival time, thereby increasing the probability for successful transmission of the trypanosomes to a new host (50, 64). It is suggested that this removal of the majority of the population is an altruistic form of PCD and the counterpart of apoptosis in metazoan (65, 66). Hereby, increased intracellular reactive oxygen species and prostaglandin D2, which is produced principally by stumpy forms, are promoting this PCD that can be considered as a second control point in terminal differentiation to the SS form (67, 68). Moreover, it is suggested that the SS form is heterogeneous, whereby one part is altruistic and undergoes apoptosis-like events, thereby stimulating the host’s immune response and eliminating the major LS and SS antigen population, while the other is tsetse infective (69). Both the LS and SS forms are covered by a dense variant surface glycoprotein (VSG) coat, which protects them from both the innate and the adaptive host immune systems. Of note, within the tsetse fly, the parasites are covered by a procyclin coat, and only when they differentiate into the MCF (infective form), they express a metacyclic VSG coat (70).
Parasite Escape Mechanisms in the Mammalian Host
To survive as extracellular parasites within the mammalian host environment (i.e., blood or extravascular tissues), African trypanosomes have developed efficient immune evasion mechanisms, at both the parasite level and the level of modulating host responses. Indeed, during millions of years of coevolution with their mammalian host, these parasites have “learned” to divert and sculpture the host immune system to prevent the generation of an effective response. The most predominant changes at the level of the host occurring during African trypanosomosis are massive splenomegaly coinciding with destruction of the lymphoid architecture and hepatomegaly. These modulations are followed by lymphadenopathy and hypergammaglobulinemia, leading to systemic multiple organ failure and death in experimental mouse models (71). In this section, we will give an overview of the most prominent escape mechanisms trypanosomes developed to allow successful infection within their mammalian host.
Parasite-Associated Escape Mechanisms
Already at the onset of infection, i.e., inoculation of trypanosome-containing saliva upon the bite of a tsetse fly, components present in the saliva are able to (i) dampen local host inflammatory immune responses characterized by the release of trypanolytic molecules, i.e., tumor necrosis factor (TNF) and nitric oxide (NO), thereby favoring parasite development and (ii) trigger mast cell degranulation resulting in release of histamine and increased vasodilatation, thereby allowing parasite dissemination/extravasation into the blood circulation [reviewed in Stijlemans et al. (72)]. Within the mammalian host, trypanosomes are very proficient in avoiding and subsequently reorchestrating host immune responses. Being extracellular parasites, they are confronted with the host’s humoral immune response; hence, to allow infection to occur, they have to overcome this major obstacle. In first instance, these parasites are covered with a very dense coat composed of approximately 5 × 106 identical VSG homodimers of 50–60 kDa subunits that are anchored in the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor, which functions as a ~15-nm thick barrier and protects the cell from Abs that might bind to buried conserved proteins (73, 74). Second, to prevent Ab-mediated elimination by Abs raised against the immunodominant/immunogenic VSGs, these parasites acquired a system of antigenic variation, whereby they are equipped with a battery of more than 1,000 different VSG genes and pseudogenes in their genome that in turn can undergo segmental gene recombination to encode an estimated 10,000 different VSG surface coats during infection (75). Hence, at regular time points [i.e., upon recognition by the host’s humoral response or when a maximal density is reached (Quorum sensing)], they switch their coat into a different variable antigen type, thereby allowing escaping Ab-mediated elimination (76). This antigenic variation is accomplished by (i) in situ switching of transcriptional control (i.e., changing the VSG expression site) or (ii) gene replacement resulting in a switch of the terminal telomeric VSG gene itself (77, 78). Besides antigenic variation, these parasites were shown to express a mosaic VSG during the process of VSG switching (i.e., changing from metacyclic to BSFs, or during the course of infection), which in turn might be an efficient way to prevent effective Ab recognition (79, 80). Also the infective MCFs use this differential VSG expression to generate diversity and counter existing partial immunity/enhance transmission, while BSF use this to prolong infection (see Figure 1). Interestingly, the MCFs initiate VSG expression by each cell, activating at random one from a small subset of metacyclic VSG (M-VSG) genes, resulting in a heterogenous population, whereby each trypanosome expresses a single VSG (81, 82). Hereby, the M-VSG expression is regulated exclusively at the transcriptional level, while the bloodstream VSG expression is regulated mainly at the posttranscriptional levels and transcribed polycistronically (83). Third, trypanosomes also exhibit a very high endocytosis rate as an efficient way to acquire nutrients and at the same time to remove Ab-bound VSG molecules and thereby prevent Ab-mediated or even complement-mediated opsonization/elimination (84, 85). This might allow parasites to transiently escape T-cell-independent B-cell-mediated elimination, which is the first line of defense. This immunological escape also gives time to transform into trypomastigote forms, which are adapted to survive in the mammalian host during the initiation of infection and gives the parasites an immunological advantage during the process of antigenic variation.
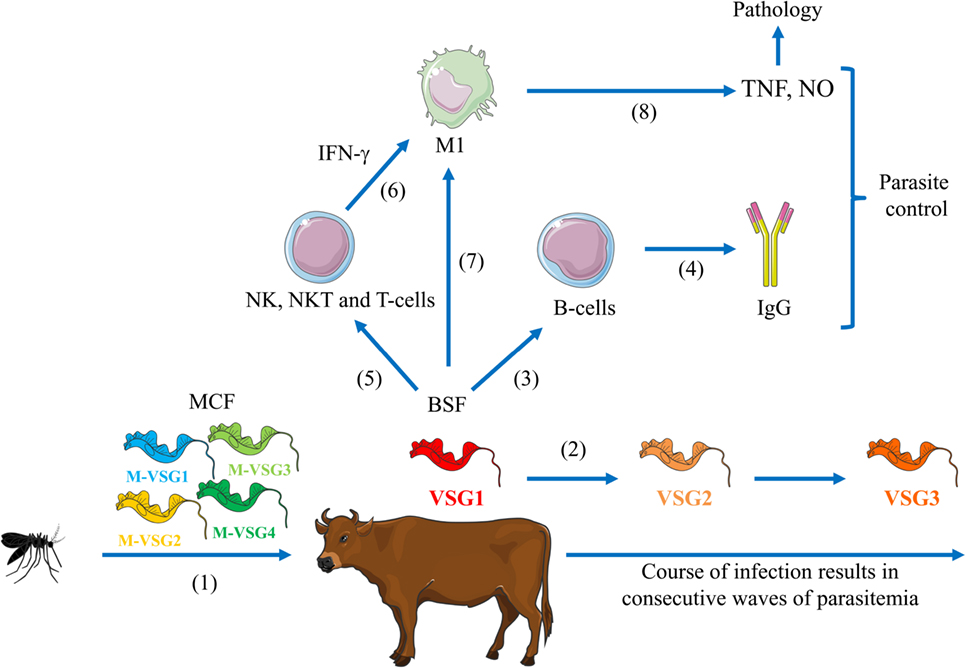
Figure 1. Model for African trypanosomosis within the mammalian host. (1) Upon the bite of a trypanosome-infected tsetse fly, metacyclic form (MCF) parasites are inoculated within the mammalian host. (2) These parasites differentiate into bloodstream forms (BSFs) and switch their metacyclic VSG (M-VSG) into a bloodstream uniform VSG (VSG1) giving rise to the first parasitemia peak. During the course of infection, there is antigenic variation (VSG2, VSG3, etc.) giving rise to different peaks of parasitemia. (3,4) Parasites-derived components trigger B-cell activation and production of antiparasite IgG needed for parasitemia control. (5,6) Parasite-derived components trigger NK, NKT, and T-cell activation resulting in the production of interferon-gamma (IFN-γ). (7) Parasite-derived components in concert with IFN-γ trigger the induction of classically activated macrophages (M1 cells). (8) These M1 cells release pro-inflammatory cytokines [like tumor necrosis factor (TNF)] that are needed for parasite control, but at the same time contribute to pathology development if maintained during the course of infection.
Although mainly IgMs play a key role during T. brucei infection, while in T. congolense infections, IgGs are mostly important (86, 87), it could be assumed that complement-mediated lysis is also an important innate defense mechanism. However, trypanosomes have developed efficient mechanisms to avoid complement-mediated elimination. First, trypanosomes are able to avoid elimination via the complement pathway, which is typically activated via immune complexes with Abs. Indeed, by releasing vast amounts of soluble VSG (mainly at the peak of parasitemia), Abs and complement factors will be scavenged and thereby induce a state of hypocomplementemia that can favor the survival of the parasites (88). Second, besides undermining the classical activation of the complement pathway that could contribute to trypanosome clearance through Ab-mediated trypanolysis and/or phagocytosis, the alternative pathway of complement activation occurring in the absence of specific Abs (i.e., during early stages of infection) is also impaired. Indeed, by masking sites on the VSG plasma membrane, which are capable of promoting alternative pathway activation, the cascade is blocked at the C3 convertase stage, thereby impairing the generation of the terminal complex (C5–C9) that normally induces trypanolysis (89, 90). However, it seems that the later stages of the complement activation cascade do not play a detrimental role in parasite control. Indeed, in AKR mice, which are natural C5 KO mice (91), the absence of the complement lysis pathway does not prevent periodic trypanosome clearance and does not hamper long-term survival in case of T. congolense infections (92, 93). However, soluble complement molecules, such as C3a and C5a, secreted during early stages of trypanosome infection, can contribute to the initiation of the early inflammatory immune response and also act as (i) chemotactic agents attracting phagocytes to the site of infection and (ii) release histamine from mast cells, thereby increasing microvascular permeability (94), which would allow/enable parasite extravasation into the blood circulation.
Parasite-Induced Escape Mechanisms
Besides being equipped with tools to avoid elimination by the host’s “innate” humoral response, trypanosomes also undermine the host cellular immune responses to allow chronic infection. Moreover, the data in literature suggest that the efficiency to modulate the innate immune response is crucial for the progression of trypanosomosis (95). Hereby, the suppression of cellular immune responses is an efficient mechanism to evade host defense mechanisms and a general feature of trypanosomosis in bovine, human, and murine hosts. To this end, these parasites are equipped with a battery of molecules able to modulate early antiparasite responses to allow establishment. It is important to mention that the course of an African trypanosome infection can be characterized by an early release of interferon-gamma (IFN-γ) by activated NK-, NKT- and T-cells required to induce classically activated macrophages (M1) (see Figure 1). In turn, these activated M1 develop upon exposure to parasite-derived molecules such as VSG and CpG a type-1 inflammatory immune response leading to the production of the potential trypanocidal molecules such as TNF and NO that in conjunction with Abs will contribute to parasite control (96, 97). Yet, persistence of this type-1 immune response and hyperactivated M1 cells will culminate in trypanosusceptible animals into immunopathological features such as the systemic immune response syndrome and anemia (98). Trypanotolerant animals on the other hand are able to switch to a more type-2 immune response and the induction of alternatively activate macrophages (M2), whereby the anti-inflammatory cytokine interleukin (IL)-10 was shown to play a pivotal “dampening” role (99, 100).
Undermining Macrophage Functionality
To sustain the development of the first (most prominent) peak of parasitemia in the blood and its control by the host, some parasite-derived molecules are able to dampen pro-inflammatory responses (TNF, NO) by these M1. Most research so far has been performed using T. brucei infections and indicate that these parasites release components such as adenylate cyclase (AdC) and kinesin heavy chain (TbKHC-1) to dampen initial host responses, thereby allowing early parasite establishment (101, 102). Indeed, AdC released by altruistic parasites upon parasite phagocytosis by liver-associated myeloid cells prevents production of the trypanolytic cytokine TNF (via a protein kinase A pathway), which promotes early establishment of trypanosomes within the mammalian host (101). On the other hand, the release of TbKHC-1 by parasites induces IL-10 and arginase release by myeloid cells in a SIGN-R1-dependent manner and favors initial parasite seeding by inducing the production of polyamines, which constitute trypanosome essential nutrients (102, 103). Recently, it was also shown that metabolites produced by trypanosomes such as indolepyruvate (i.e., a transamination product of tryptophan) can dampen macrophage pro-inflammatory responses that prevent elimination (104). Finally, the order of exposure to parasite-derived versus host-derived macrophage-activating components as well as the relative concentration of these mediators may influence the ability of the host to respond to trypanosome infections. Indeed, early during infection, exposure of macrophages to soluble VSG (encompassing the glycosylinositolphosphate substituent) before IFN-γ priming downregulated the level of signal transducer and activator of transcription 1 phosphorylation, which in turn reduced transcription of pro-inflammatory cytokines such as TNF (105). So far, nothing is known about such mechanisms for T. congolense. In summary, it seems that trypanosomes have developed a system whereby altruistic parasites are phagocytosed, thereby disabling the M1-mediated innate immune response required for parasite control and paving the way for initiation and establishment of the first wave of parasitemia.
Modulation of T-Cell Functionality
Besides undermining the antitrypanosomal potential of the myeloid system, the parasite is also impairing T-cell help required to mount a more efficient response during the course of infection. Early during T. brucei and T. congolense infection, T-cell suppression is occurring via suppressive myeloid cells by inhibiting IL-2 secretion and downregulation of IL-2 receptor expression (106–108), whereby prostaglandins were found to play an important role in the murine model, but not in the bovine model (109). In addition, early data on T cell regulation and trypanosomosis showed that both IFN-γ and TNF play a key role in the suppressive effects on CD4 and CD8 T-cells (107). Furthermore, this suppressive phenotype of the host cells during the early stages of T. brucei infection is due to a combination of (i) trypanosome-released macrophage-activating factors leading to secretion of immunosuppressive factors such as NO, prostaglandins, and TNF and (ii) host-derived IFN-γ needed for optimal macrophage activation (110, 111). Moreover, this work also showed that within the T. brucei model, there is a compartmentalization of the suppressive effect in murine models during the later stages of infection, whereby NO plays a key role in macrophage-mediated splenic suppression, whereas the macrophage-mediated lymph node suppression occurred in an IFN-γ-dependent manner (110). Hence, at this stage of infection, an IFN-γ-independent suppressive mechanism is elicited in the spleen, whereas in the lymph nodes, IFN-γ is required yet not sufficient to inhibit T cell proliferation. In this context, it was shown that the trypanosome suppression-inducing factor (TSIF) released by T. brucei during the course of infection induces TNF and NO secretion by classically activated macrophages (i.e., M1), which is a prerequisite for parasite control. However, at the same time, it blocks T-cell proliferation in a NO, IFN-γ, and cell contact-dependent manner as well as downregulates type-2 immune responses required to dampen M1-mediated pathogenicity (112). Moreover, TSIF was shown to be essential for parasite biology given that TSIF knock-down parasites die within 2 days. In the T. congolense model, it was shown that besides IFN-γ, the anti-inflammatory cytokine IL-10 also contributed to T-cell suppression (113). Finally, although both murine and bovine African trypanosomosis induce suppression, it seems that NO does not play a role in the loss of T-cell proliferative function in the bovine trypanosomosis model and that, in contrast to the mouse model, the capacity of monocytes and macrophages to produce NO is actually downregulated in infected cattle (114). In summary, these results suggest that the T-cell suppression is multifactorial, tissue and infection stage dependent, and host/parasite dependent. However, T-cells are dispensable for parasite control, which was evidenced by the fact that mice lacking a functional T-cell compartment are as efficient as immune-competent animals in controlling trypanosome infection (86). These data indicate that T-cell-independent B-cell-mediated elimination is the driving factor implicated in controlling parasitemia. Nevertheless, T-cells play a key role in the development of African trypanosomosis-associated pathogenicity, such as anemia (115).
Undermining B-Cell Functionality
Given that trypanosomes are extracellular parasites, it is not surprising that the host–parasite coevolution resulted in a subtle equilibrium between suppression of B-cell and Ab functionality and parasite persistence. Indeed, besides suppression of myeloid cells and T-cells, B-cells were also found to be negatively affected during the early stages of trypanosome infection. Accordingly, trypanosomes exert full control of the different types of host immune responses to establish chronic infection. In this context, in cattle, it was found that there are also differences in humoral responses between T. congolense-infected trypano-resistant (N’Dama) and trypanosusceptible Boran cattle, further highlighting the importance of the humoral immune response in parasitemia control (116). In the following section, the effect of African trypanosome infections at the level of the B-cell compartment will be scrutinized.
In homeostatic conditions, B-cells develop from bone marrow (BM)-derived hematopoietic stem cells (HSCs) that initially differentiate into multipotent progenitor cells and subsequently into common lymphoid progenitor cells (117). Next, B-cell lymphopoiesis occurs through several developmental stages, such as pre-pro-B, pro-B, pre-B, and, finally immature B-cells, which is a highly regulated process with alternating phases of cell proliferation and differentiation (118, 119). During this process, these different B-cell subsets rearrange their immunoglobulin heavy-chain and light-chain gene loci and express different surface markers that can be identified via flow cytometry (see Table 1; Figure 2, left panel). Within the BM, these B-cells also undergo a positive and negative selection procedure, whereby the B-cell receptor (BCR) plays a checkpoint role (120). If the BCRs do not bind their antigen, they stop their development, i.e., during positive selection, while during negative selection, binding of self-antigens to the BCR triggers either clonal deletion, receptor editing, anergy, or ignorance, resulting in central tolerance (121). At the last stage of differentiation within the BM, these immature B-cells exhibit a high IgM expression and low or no expression of the IgD maturation marker. To complete their development, immature B cells migrate to the spleen via the blood as transitional B cells (T1 type). In the spleen, these transitional B (T1) cells differentiate into T2 cells before they develop into two types of mature naïve B-cells (122), namely follicular B (FoB) or marginal zone B (MZB) cells (see Figure 2, right panel). In homeostatic conditions, B (FoB) cells are mainly located in the white pulp area of the spleen where they form primary B-cell follicles, preferentially undergo T-cell-dependent activation (upon activation via proteins and glycoproteins), and can give rise to both short-lived plasma cells (i.e. plasmablasts) for immediate protection and high-affinity class-switched IgG long-lived plasma cells and memory B cells for persistent protection. In contrast, MZB cells are concentrated outside the splenic marginal sinus surrounding the white pulp. Most of the time, they initiate a fast and preferentially T-cell-independent activation (upon activation via polysaccharides or unmethylated CpG DNA) giving rise to not only short-lived plasma cells that rapidly produce low-affinity Abs of IgM isotype but also some populations of long-lived plasma cells (123). Overall, B-cell activation is considered a very efficient defense system against invading “extracellular/blood-borne” pathogens. However, African trypanosomes have developed efficient ways to undermine the host’s humoral response to establish chronic infection and allow completion of its life cycle/transmission. Indeed, using murine models, it was shown that African trypanosomes (both T. brucei and T. congolense) already during the early stages of infection trigger polyclonal B-cell activation in an attempt to dilute-out VSG-specific Abs during the course of infection. For example, it was shown that the CpG motifs of the T. brucei trypanosomal genomic DNA triggers TLR-9 signaling events and contributes to polyclonal B-cell activation (96). This phenomenon might contribute to parasite immune evasion by driving unselective differentiation of B cells into short-lived plasma cells. In addition, Fcγ-receptors on phagocytes become saturated by polyspecific Abs, thereby reducing the efficiency of opsonization-mediated parasite clearance. Besides polyclonal B-cell activation, trypanosomes also undermine the “protective” humoral response by ablating B cell lymphopoiesis in primary and secondary lymphoid organs during both T. brucei and T. congolense infections. This was reflected by a depletion of all developmental B-cell stages in the BM and the spleen as well as previous effector B cells, such as memory B cells (124–126), thereby preventing the development of a B-cell memory required for permanent elimination (Figure 3). Similar results were obtained in the experimental T. vivax model (127). In addition, experimental results obtained in mice and livestock animals have shown that trypanosome infections exert detrimental effects on non-pathogen-related vaccines, by preventing the occurrence of memory recall responses (126, 128–130), or on the maintenance of the antigen-specific plasma B cell pool driving the development of collagen-induced arthritis (CIA) in DBA/1 prone mice (131). This destruction of the B-cell compartment at the level of both the BM and the spleen could be attributed to either parasite-derived components and/or host-derived (infection-induced) components. Interestingly, a recent work by Cnops et al. (132), revealed that during murine infection with a chronic low-virulent T. b. gambiense field isolate, FoB cells are retained, which coincided with reduced production of TNF and IFN-γ pro-inflammatory cytokines during the acute stage of infection compared to T. brucei and T. congolense infections. This finding was paralleled by the finding of Lejon et al. (133), which showed that in T. b. gambiense, HAT patients’ low parasite levels seem to be associated with limited B cell dysfunction, whereby B-cell memory responses are only slightly reduced; however, the functionality of these memory B cells was not verified in rechallenge studies. These findings indicate that in both experimental trypanosomosis and natural infection, the inflammation stage linked to the acuteness of infection could be a major determinant in the processes that drive B-cell compartment destruction. This hypothesis is reinforced by the fact that both IFN-γ(−/−) and IFN-γR(−/−) mice are protected from early trypanosomosis-associated FoB cell depletion (134). This phenotype coincided with a drastic inhibition of B-cell apoptosis and a reduced activation of FoB cells and inflammatory responses during the first week postinfection. These data demonstrated that IFN-γ is an important cytokine involved in undermining trypanosomosis-associated B-cell responses. So far, the cellular source of early IFN-γ production involved in triggering impaired B-cell lymphopoiesis remains to be fully elucidated. However, it was recently suggested that NK-cells, an important early source of IFN-γ, are involved in B-cell killing and suppressing humoral immunity within the T. brucei model (115, 135). Yet, it cannot be excluded that other sources of IFN-γ, such as NKT, CD8+, and CD4+ T-cells, are also involved in IFN-γ-mediated B-cell apoptosis given that there is a transition of IFN-γ production by these cells during the course of T. brucei infection (115). Interestingly, upon drug treatment [suramin and diminazene aceturate (Berenil)] of T. brucei- and T. congolense-infected mice, the BM B-cell lymphopoiesis is reinitiated, and the splenic B-cell subsets are repopulated, suggesting that an active chronic infection (i.e., parasite–host interaction) is involved in undermining the humoral responses via either parasite-released components and/or inflammatory-based mechanism(s) (136, 137). However, Uzonna and coworkers (137) showed that Berenil besides exerting trypanolytic effects could also modulate the host immune response to the parasite by dampening excessive immune activation and production of pathology-promoting pro-inflammatory cytokines. Hence, it cannot be excluded that the beneficial effects of Berenil for treatment of AT are multifactorial: (i) eliminate parasites thereby resulting in reduced triggering of host inflammatory immune responses and (i) reduce the host’s pro-inflammatory potential to respond to pro-inflammatory/parasite-derived components. In both cases, this will result in a more efficient host-mediated parasite control due to a recovery from the impaired B-cell lymphopoiesis and protection from infection-associated pathogenicity due to lower inflammatory responses (138). In this context, it was recently shown within the T. congolense experimental model that also host molecules such as macrophage migration inhibitory factor (MIF) can play a key role in regulating trypanosomosis-associated pro-inflammatory responses and B-cell homeostasis (139). In this work, it was shown that T. congolense-infected MIF-deficient mice exhibited increased Ab titers that correlated with reduced B-cell apoptosis. Hence, MIF could be considered as a target to alleviate the impaired B-cell lymphopoiesis.

Table 1. B-cell surface marker expression used to track cellular alterations during infection.
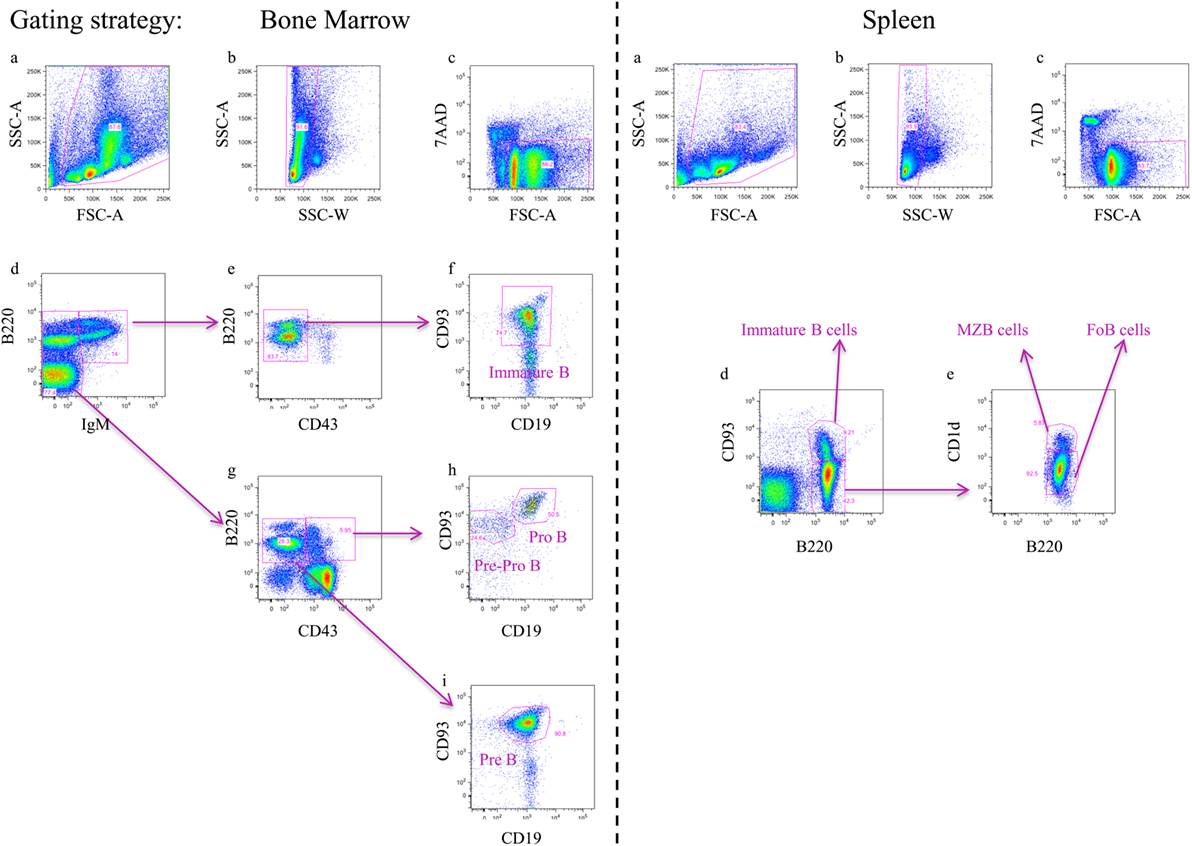
Figure 2. FACS gating strategy to track B-cell alterations during infection within the bone marrow (BM) and spleen. BM (left panel): (A,B) In an FSC-A versus SSC-A plot, a life gate was selected followed by gating on singlets in an SSC-A versus SSC-W plot, respectively. (C,D) Following gating on 7AAD− cells and plotting in a B220 versus IgM allow identification of IgM+ and IgM− cells. (E,F) The IgM+ cells are plotted in a B220 versus CD43 plot to identify B220+CD43− cells and subsequently plotted in a CD93 versus CD19 to identify immature B-cells (CD93+CD19+). (G) The IgM− cells are plotted in a B220 versus CD43 plot to identify B220+CD43− cells and B220+CD43+ cells. (H) The B220+CD43− cells are plotted in a CD93 versus CD19 to identify pre B-cells (CD93+CD19+). (I) The B220+CD43+ cells are plotted in a CD93 versus CD19 to identify pro B-cells (CD93+CD19+) and pre-pro B-cells (CD93+CD19−). Spleen (right panel): (A,B) In an FSC-A versus SSC-A plot, a life gate was selected followed by gating on singlets in an SSC-A versus SSC-W plot, respectively. (C,D) Following gating on 7AAD− cells and plotting in a CD93 versus B220 allow identification of immature B-cells (CD93+B220+). (E) The CD93+B220− cells are subsequently plotted in a CD1d versus B220 plot to identify marginal zone B (MZB) cells (CD1d+B220+) and follicular B (FoB) cells (CD1d−B220+). It is important to mention that during infection, the expression levels of typical splenic B-cell subset markers can be modulated, which substantially complicates the identification of the different B-lymphocyte subsets.
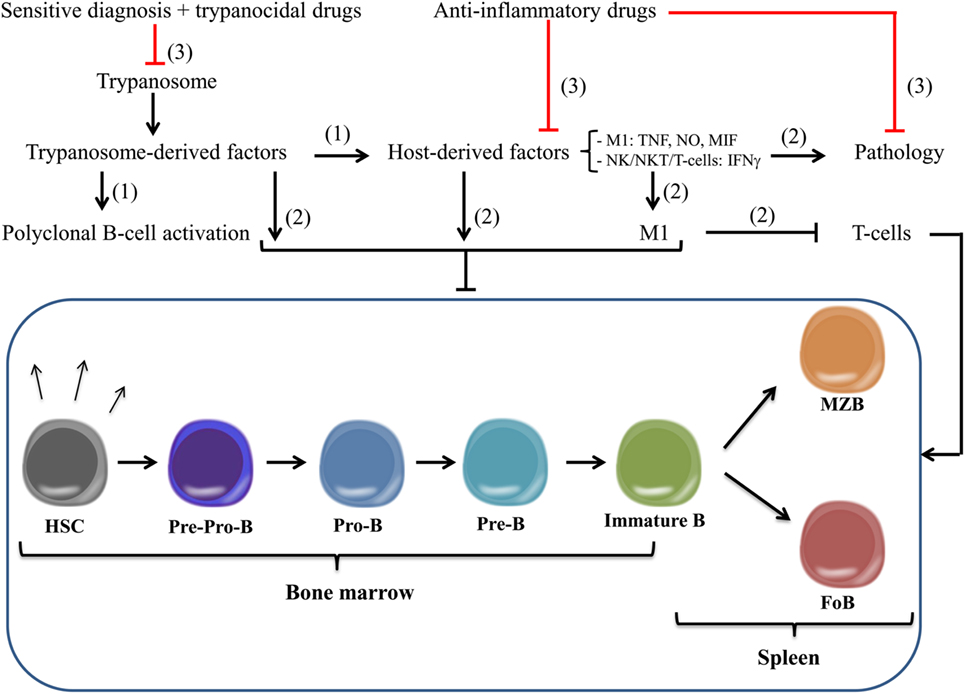
Figure 3. Model for African trypanosomosis-associated impaired B-cell lymphopoiesis and improved vaccine development. (1) During the course of African trypanosomosis (AT), parasite-derived components are released that trigger besides polyclonal B-cell activation also the production of host-derived pro-inflammatory factors (i.e., NK-, NKT-, and T-cell-derived IFN-γ, M1-cell-derived TNF, NO, MIF) needed directly/indirectly for early parasite control. Yet, following control of the first parasitemia peak, polyclonal B-cell activation leads to dilution of parasite-specific antibodies, whereas the persistent pro-inflammatory response contributes to suppression of host responses and pathology. (2) Both parasite- and host-derived components can lead to a general state of impaired B-cell lymphopoiesis in (i) the bone marrow (BM), ranging from pre-pro-B-cell, pro-B-cell, pre-B-cell, and immature B-cell and (ii) the spleen, ranging from immature B-cell till MZB cell and FoB cell. In addition, also host-derived factors (involving M1) can contribute to T-cell suppression that in turn can affect B-cell homeostasis. (3) Hence, a more efficient therapeutic intervention strategy for AT should consist of a combination of (i) more reliable/sensitive diagnosis systems allowing early-stage parasite detection, (ii) more efficient trypanocidal/toxic drugs allowing improved parasite treatment, and (iii) pro-inflammatory-blocking molecules that could lead to a reduced pathology and a restoration of normal B-cell responses, thereby allowing more efficient/optimal vaccination. M1, classically activated myeloid cells; MIF, macrophage migration inhibitory factor; IFN, interferon; TNF, tumor necrosis factor; NO, nitric oxide; MZB, marginal zone B; FoB, follicular B.
Conclusions and Perspectives
The extracellular African trypanosomes have acquired efficient immune evasion mechanisms to undermine protective host immune responses and allow survival in the host’s extracellular environment. Hereby, they are proficient in avoiding elimination via the host’s humoral immune response by destroying the B-cell compartment/memory and shielding-off conserved epitope, thereby paving the way for chronic infection. This destruction of B-cell memory already very early during infection might explain the failure of developing an effective vaccine. Indeed, immunization with the immunodominant VSG did not yield any universal protection. So far, attempts to immunize with trypanosome molecules (VSG, beta tubulin, etc.), such as conserved membrane proteins or receptors for uptake of nutrients, have resulted in limited protective effects because such molecules are either concealed beneath the surface coat or are expressed at a to low level to induce protective host immunity (140, 141). However, some protection against AT-associated pathological features (i.e., anemia, tissue injury) has been achieved upon vaccination with pathology-inducing factors such as the VSG-derived GPI moiety (T. brucei, T. congolense) or the cysteine proteinase congopain (T. congolense), yet the animals were never fully cured (142, 143). Moreover, as far as the GPI-based strategy in the murine model was concerned, there was no effect on parasitemia but rather the protective effect correlated with reduced pro-inflammatory immune responses and was independent of the Ab response. This is in line with the observation that pathogenicity did not correlate with Ab levels at least for the experimental murine T. brucei model (86). In contrast, for the congopain vaccination strategy in experimental bovine T. congolense models, there was a correlation between reduced pathogenicity and increased Ab titers (143, 144). This is in line with the observation that T. congolense-infected N’Dama cattle (a trypanotolerant breed showing natural resistance to trypanosomosis) exhibited higher antiparasite Ab titers than the susceptible Boran breeds (145, 146), suggesting that there are differences in the frequency of trypanosome-specific Ab-secreting cells in the spleen and in the activation state of B-cells in the blood between both cattle breeds during infection. Interestingly, the sera from T. congolense-infected N’Dama cattle specifically recognized dimer-associated epitopes on the congopain antigen (147).
The current research on vaccine development has switched toward identification of invariant surface glycoproteins or conserved c-terminal VSG epitopes/peptides that are predicted to contain several MHC II recognition sites (148–150). Whether these latter approaches will lead to the development of an effective protective and antipathology vaccine will be challenging and possibly not achievable given that African trypanosomes undermine B-cell memory responses. In addition, also the whole genome transcriptome analysis (i.e., SAGE technique) that enables to (i) explore the full transcriptome of trypanosusceptible and trypanotolerant cattle might lead to the identification of interesting gene variations linked to the trypanotolerance status of the animal (151–153) and (ii) understand the molecular aspects of the trypanosome dialog with its tsetse and mammalian hosts (i.e., interaction with the salivary glands and LS versus SS differentiation, respectively) might pave the way to develop novel diagnostic/therapeutic intervention strategies (154, 155). Furthermore, although the loss of B-cell responses/memory during AT might rely on either a parasite-induced or a host-induced effect or a combination of both, understanding the molecular mechanisms used by the trypanosomes to dampen B cell responses might lead to the development of new therapeutics not only for AT but also for other diseases such as autoimmune diseases (i.e., CIA) or malaria, where B-cell dysfunction is contributing to the disease outcome (131, 156–158). In this context, it was shown that the B-cell adaptor molecule Bam32 plays a pivotal role in optimal Ab responses and resistance during T. congolense infections in mice (159). Besides parasite-derived molecules, also host-derived molecule could be considered as a potential target for intervention strategies. In this context, MIF can be proposed as potential candidate given that it can play a role both in innate as adaptive immunity via interaction with its main receptor CD74 to regulate the host inflammatory response (160, 161). Indeed, MIF was shown to play a pivotal role in stimulating/inflammatory responses and regulating T- and B-cell recruitment as well as B-cell proliferation/survival and thereby contribute to pathology development (162–164). Hence, blocking MIF-signaling could reduce inflammatory responses, thereby alleviating suppression of B-cell lymphopoiesis, which in turn might favor vaccine efficacy.
In the future, most likely, a combination of (i) more sensitive/reliable diagnosis techniques needed for early-stage parasite detection and (ii) anti-parasite intervention strategies (trypanocidal/trypanotoxic drugs) and (iii) antidisease/pathology (anti-inflammatory) intervention strategies will be required to combat AT (see Figure 3).
Author Contributions
All authors contributed to writing the manuscript. However, BS and MR are co-first and CT and SM share co-last authorship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, HG, and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We acknowledge the financial support of the Interuniversity Attraction Pole Program (PAI-IAP N. P7/41, http://www.belspo.be/belspo/iap/index_en.stm) and grants from the FWO (FWO G015016N and G.0.028.10.N.10) and the Bill and Melinda Gates Foundation grant 641K760. BS is a research fellow supported by the Strategic Research Program (SRP3, VUB): Targeting inflammation linked to infectious diseases and cancer (Nanobodies for Health). We would like to thank Prof. Dr. Patrick De Baetselier for the constructive discussions and help in writing the manuscript. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Yaro M, Munyard KA, Stear MJ, Groth DM. Combatting African Animal Trypanosomiasis (AAT) in livestock: the potential role of trypanotolerance. Vet Parasitol (2016) 225:43–52. doi:10.1016/j.vetpar.2016.05.003
2. Simarro PP, Cecchi G, Franco JR, Paone M, Diarra A, Ruiz-Postigo JA, et al. Estimating and mapping the population at risk of sleeping sickness. PLoS Negl Trop Dis (2012) 6:e1859. doi:10.1371/journal.pntd.0001859
3. Welburn SC, Molyneux DH, Maudlin I. Beyond tsetse – implications for research and control of Human African trypanosomiasis epidemics. Trends Parasitol (2016) 32:230–41. doi:10.1016/j.pt.2015.11.008
4. World Health Organization. Trypanosomiasis, Human African (Sleeping Sickness). World Health Organization (2013). 259 p. Available from: http://www.who.int/mediacentre/factsheets/fs259/en/
5. Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet (2010) 375:148–59. doi:10.1016/S0140-6736(09)60829-1
6. Simarro PP, Diarra A, Ruiz Postigo JA, Franco JR, Jannin JG. The Human African trypanosomiasis control and surveillance programme of the world health organization 2000–2009: the way forward. PLoS Negl Trop Dis (2011) 5(2):e1007. doi:10.1371/journal.pntd.0001007
7. Franco JR, Simarro PP, Diarra A, Jannin JG. Epidemiology of Human African trypanosomiasis. Clin Epidemiol (2014) 6:257–75. doi:10.2147/CLEP.S39728
8. Kennedy PG. Clinical features, diagnosis, and treatment of Human African trypanosomiasis (sleeping sickness). Lancet Neurol (2013) 12:186–94. doi:10.1016/S1474-4422(12)70296-X
9. Rodgers J. Trypanosomiasis and the brain. Parasitology (2010) 137:1995–2006. doi:10.1017/S0031182009991806
10. MacLean L, Reiber H, Kennedy PG, Sternberg JM. Stage progression and neurological symptoms in Trypanosoma brucei rhodesiense sleeping sickness: role of the CNS inflammatory response. PLoS Negl Trop Dis (2012) 6:e1857. doi:10.1371/journal.pntd.0001857
11. Tiberti N, Matovu E, Hainard A, Enyaru JC, Lejon V, Robin X, et al. New biomarkers for stage determination in Trypanosoma brucei rhodesiense sleeping sickness patients. Clin Transl Med (2013) 2:1. doi:10.1186/2001-1326-2-1
12. Jamonneau V, Ilboudo H, Kabore J, Kaba D, Koffi M, Solano P, et al. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Negl Trop Dis (2012) 6:e1691. doi:10.1371/journal.pntd.0001691
13. MacLean LM, Odiit M, Chisi JE, Kennedy PG, Sternberg JM. Focus-specific clinical profiles in Human African trypanosomiasis caused by Trypanosoma brucei rhodesiense. PLoS Negl Trop Dis (2010) 4:e906. doi:10.1371/journal.pntd.0000906
14. Bucheton B, Macleod A, Jamonneau V. Human host determinants influencing the outcome of Trypanosoma brucei gambiense infections. Parasite Immunol (2011) 33:438–47. doi:10.1111/j.1365-3024.2011.01287.x
15. WHO. Control and surveillance of Human African trypanosomiasis. World Health Organ Tech Rep Ser (2013) (984):1–237.
16. Lejon V, Bentivoglio M, Franco JR. Human African trypanosomiasis. Handb Clin Neurol (2013) 114:169–81. doi:10.1016/B978-0-444-53490-3.00011-X
17. Ruiz-Postigo JA, Franco JR, Lado M, Simarro PP. Human African trypanosomiasis in South Sudan: how can we prevent a new epidemic? PLoS Negl Trop Dis (2012) 6:e1541. doi:10.1371/journal.pntd.0001541
18. Hasker E, Mpanya A, Makabuza J, Mbo F, Lumbala C, Kumpel J, et al. Treatment outcomes for Human African trypanosomiasis in the Democratic Republic of the Congo: analysis of routine program data from the world’s largest sleeping sickness control program. Trop Med Int Health (2012) 17:1127–32. doi:10.1111/j.1365-3156.2012.03042.x
19. Desquesnes M, Dargantes A, Lai DH, Lun ZR, Holzmuller P, Jittapalapong S. Trypanosoma evansi and surra: a review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed Res Int (2013) 2013:321237. doi:10.1155/2013/321237
20. Brun R, Hecker H, Lun ZR. Trypanosoma evansi and T. equiperdum: distribution, biology, treatment and phylogenetic relationship (a review). Vet Parasitol (1998) 79:95–107. doi:10.1016/S0304-4017(98)00146-0
21. Claes F, Büscher P, Touratier L, Goddeeris BM. Trypanosoma equiperdum: master of disguise or historical mistake? Trends Parasitol (2005) 21:316–21. doi:10.1016/j.pt.2005.05.010
22. Jones TW, Dávila AMR. Trypanosoma vivax – out of Africa. Trends Parasitol (2001) 17:99–101. doi:10.1016/S1471-4922(00)01777-3
23. Osório AL, Madruga CR, Desquesnes M, Soares CO, Ribeiro LR, Costa SC. Trypanosoma (Duttonella) vivax: its biology, epidemiology, pathogenesis, and introduction in the New World – a review. Mem Inst Oswaldo Cruz (2008) 103:1–13. doi:10.1590/S0074-02762008000100001
24. Naessens J. Bovine trypanotolerance: a natural ability to prevent severe anaemia and haemophagocytic syndrome? Int J Parasitol (2006) 36:521–8. doi:10.1016/j.ijpara.2006.02.012
25. Disease T. African Animal trypanosomiasis African Animal trypanosomiasis. In Vitro (2009):1–5. Available from: http://www.cfsph.iastate.edu/Factsheets/pdfs/trypanosomiasis_african.pdf
26. Swallow B. Impacts of Trypanosomiasis on African Agriculture. Nairobi, Kenya: Int Livest Res Institute (1999). p. 1–46. Available from: http://www.cabdirect.org/abstracts/20003010139.html
27. Giordani F, Morrison LJ, Rowan TG, de Koning HP, Barrett MP. The animal trypanosomiases and their chemotherapy: a review. Parasitology (2016) 143(14):1862–89. doi:10.1017/S0031182016001268
28. Gilbert M, Jenner C, Pender J, Rogers D, Slingenbergh J, Wint W. The programme against African trypanosomiasis information system (PAATIS). The African Trypanosomes, Vol. 1 (2002). p. 11–24.
29. Bouyer J, Bouyer F, Donadeu M, Rowan T, Napier G. Community- and farmer-based management of animal African trypanosomosis in cattle. Trends Parasitol (2013) 29:519–22. doi:10.1016/j.pt.2013.08.003
30. Baker N, de Koning HP, Maser P, Horn D. Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol (2013) 29:110–8. doi:10.1016/j.pt.2012.12.005
31. Delespaux V, de Koning HP. Drugs and drug resistance in African trypanosomiasis. Drug Resist Updat (2007) 10:30–50. doi:10.1016/j.drup.2007.02.004
32. Naula C, Burchmore R. A plethora of targets, a paucity of drugs: progress towards the development of novel chemotherapies for human African trypanosomiasis. Expert Rev Anti Infect Ther (2003) 1:157–65. doi:10.1586/14787210.1.1.157
33. Wenzler T, Schumann Burkard G, Schmidt RS, Mäser P, Bergner A, Roditi I, et al. A new approach to chemotherapy: drug-induced differentiation kills African trypanosomes. Sci Rep (2016) 6:22451. doi:10.1038/srep22451
34. Haanstra JR, Gerding A, Dolga AM, Sorgdrager FJH, Buist-homan M, Toit F, et al. Targeting pathogen metabolism without collateral damage to the host. Sci Rep (2017) 7:40406. doi:10.1038/srep40406
35. Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet (2009) 374:56–64. doi:10.1016/S0140-6736(09)61117-X
36. Alirol E, Schrumpf D, Amici Heradi J, Riedel A, de Patoul C, Quere M, et al. Nifurtimox-eflornithine combination therapy for second-stage gambiense Human African trypanosomiasis: medecins Sans Frontieres experience in the Democratic Republic of the Congo. Clin Infect Dis (2013) 56:195–203. doi:10.1093/cid/cis886
37. Unciti-Broceta JD, Arias JL, Maceira J, Soriano M, Ortiz-Gonzalez M, Hernandez-Quero J, et al. Specific cell targeting therapy bypasses drug resistance mechanisms in African trypanosomiasis. PLoS Pathog (2015) 11:e1004942. doi:10.1371/journal.ppat.1004942
38. Kroubi M, Karembe H, Betbeder D. Drug delivery systems in the treatment of African trypanosomiasis infections. Expert Opin Drug Deliv (2011) 8:735–47. doi:10.1517/17425247.2011.574122
39. Holt HR, Selby R, Mumba C, Napier GB, Guitian J. Assessment of animal African trypanosomiasis (AAT) vulnerability in cattle-owning communities of sub-Saharan Africa. Parasit Vectors (2016) 9:53. doi:10.1186/s13071-016-1336-5
40. Simarro PP, Cecchi G, Franco JR, Paone M, Diarra A, Priotto G, et al. Monitoring the progress towards the elimination of gambiense Human African trypanosomiasis. PLoS Negl Trop Dis (2015) 9:e0003785. doi:10.1371/journal.pntd.0003785
41. Bonnet J, Boudot C, Courtioux B. Overview of the diagnostic methods used in the field for Human African trypanosomiasis: what could change in the next years? Biomed Res Int (2015) 2015:583262. doi:10.1155/2015/583262
42. Kennedy PG. Human African trypanosomiasis-neurological aspects. J Neurol (2006) 253:411–6. doi:10.1007/s00415-006-0093-3
43. Odongo S, Sterckx YG, Stijlemans B, Pillay D, Baltz T, Muyldermans S, et al. An anti-proteome nanobody library approach yields a specific immunoassay for Trypanosoma congolense diagnosis targeting glycosomal aldolase. PLoS Negl Trop Dis (2016) 10:e0004420. doi:10.1371/journal.pntd.0004420
44. Capewell P, Cren-Travaillé C, Marchesi F, Johnston P, Clucas C, Benson RA, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife (2016) 5:e17716. doi:10.7554/eLife.17716
45. Morrison LJ, Vezza L, Rowan T, Hope JC. Animal African trypanosomiasis: time to increase focus on clinically relevant parasite and host species. Trends Parasitol (2016) 32:599–607. doi:10.1016/j.pt.2016.04.012
46. Delafosse A, Thébaud E, Desquesnes M, Michaux Y. Epidemiology of Trypanosoma vivax infection in cattle in the tsetse free area of Lake Chad. Prev Vet Med (2006) 74:108–19. doi:10.1016/j.prevetmed.2005.10.006
47. La Greca F, Haynes C, Stijlemans B, De Trez C, Magez S. Antibody-mediated control of Trypanosoma vivax infection fails in the absence of tumour necrosis factor. Parasite Immunol (2014) 36:271–6. doi:10.1111/pim.12106
48. Blom-Potar MC, Chamond N, Cosson A, Jouvion G, Droin-Bergere S, Huerre M, et al. Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. II. Immunobiological dysfunctions. PLoS Negl Trop Dis (2010) 4:e793. doi:10.1371/journal.pntd.0000793
49. Rotureau B, Van Den Abbeele J. Through the dark continent: African trypanosome development in the tsetse fly. Front Cell Infect Microbiol (2013) 3:53. doi:10.3389/fcimb.2013.00053
50. Achcar F, Kerkhoven EJ, Barrett MP. Trypanosoma brucei: meet the system. Curr Opin Microbiol (2014) 20:162–9. doi:10.1016/j.mib.2014.06.007
51. MacGregor P, Szoor B, Savill NJ, Matthews KR. Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat Rev Microbiol (2012) 10:431–8. doi:10.1038/nrmicro2779
52. Fenn K, Matthews KR. The cell biology of Trypanosoma brucei differentiation. Curr Opin Microbiol (2007) 10:539–46. doi:10.1016/j.mib.2007.09.014
53. Stijlemans B, Beschin A, Magez S, Van Ginderachter JA, De Baetselier P. Iron homeostasis and Trypanosoma brucei associated immunopathogenicity development: a battle/quest for iron. Biomed Res Int (2015) 2015:819389. doi:10.1155/2015/819389
54. Peacock L, Cook S, Ferris V, Bailey M, Gibson W. The life cycle of Trypanosoma (Nannomonas) congolense in the tsetse fly. Parasit Vectors (2012) 5:109. doi:10.1186/1756-3305-5-109
55. Ooi C-P, Bastin P. More than meets the eye: understanding Trypanosoma brucei morphology in the tsetse. Front Cell Infect Microbiol (2013) 3:71. doi:10.3389/fcimb.2013.00071
56. Van Den Abbeele J, Rotureau B. New insights in the interactions between African trypanosomes and tsetse flies. Front Cell Infect Microbiol (2013) 3:63. doi:10.3389/fcimb.2013.00063
57. Banks KL. Injury induced by Trypanosoma congolense adhesion to cell membranes. J Parasitol (1980) 66:34–7. doi:10.2307/3280584
58. Hemphill A, Frame I, Ross CA. The interaction of Trypanosoma congolense with endothelial cells. Parasitology (1994) 109(Pt 5):631–41. doi:10.1017/S0031182000076514
59. Trindade S, Rijo-Ferreira F, Carvalho T, Pinto-Neves D, Guegan F, Aresta-Branco F, et al. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe (2016) 19:837–48. doi:10.1016/j.chom.2016.05.002
60. Mogk S, Meiwes A, Shtopel S, Schraermeyer U, Lazarus M, Kubata B, et al. Cyclical appearance of African trypanosomes in the cerebrospinal fluid: new insights in how trypanosomes enter the CNS. PLoS One (2014) 9:e91372. doi:10.1371/journal.pone.0091372
61. Beschin A, Van Den Abbeele J, De Baetselier P, Pays E. African trypanosome control in the insect vector and mammalian host. Trends Parasitol (2014) 30:538–47. doi:10.1016/j.pt.2014.08.006
62. MacGregor P, Szöőr B, Savill NJ, Matthews KR. Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat Rev Microbiol (2012) 10:431–8. doi:10.1038/nrmicro2779
63. Rico E, Rojas F, Mony BM, Szoor B, Macgregor P, Matthews KR. Bloodstream form pre-adaptation to the tsetse fly in Trypanosoma brucei. Front Cell Infect Microbiol (2013) 3:78. doi:10.3389/fcimb.2013.00078
64. Mony BM, Matthews KR. Assembling the components of the quorum sensing pathway in African trypanosomes. Mol Microbiol (2015) 96:220–32. doi:10.1111/mmi.12949
65. Welburn SC, Macleod E, Figarella K, Duzensko M. Programmed cell death in African trypanosomes. Parasitology (2006) 132(Suppl):S7–18. doi:10.1017/S0031182006000825
66. Duszenko M, Figarella K, Macleod ET, Welburn SC. Death of a trypanosome: a selfish altruism. Trends Parasitol (2006) 22:536–42. doi:10.1016/j.pt.2006.08.010
67. Vassella E, Reuner B, Yutzy B, Boshart M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J Cell Sci (1997) 110(Pt 2):2661–71.
68. Figarella K, Rawer M, Uzcategui NL, Kubata BK, Lauber K, Madeo F, et al. Prostaglandin D2 induces programmed cell death in Trypanosoma brucei bloodstream form. Cell Death Differ (2005) 12:335–46. doi:10.1038/sj.cdd.4401564
69. Seed JR, Wenck MA. Role of the long slender to short stumpy transition in the life cycle of the African trypanosomes. Kinetoplastid Biol Dis (2003) 2:3. doi:10.1186/1475-9292-2-3
70. Barry JD, Graham SV, Fotheringham M, Graham VS, Kobryn K, Wymer B. VSG gene control and infectivity strategy of metacyclic stage Trypanosoma brucei. Mol Biochem Parasitol (1998) 91:93–105. doi:10.1016/S0166-6851(97)00193-X
71. Takeya M, Reinwald E, Risse HJ. Pathobiochemical alterations in experimental chronic and acute trypanosomal infection in mice. Clin Chem Lab Med (1987) 25:665–74. doi:10.1515/cclm.1987.25.10.665
72. Stijlemans B, Caljon G, Van Den Abbeele J, Van Ginderachter JA, Magez S, De Trez C. Immune evasion strategies of Trypanosoma brucei within the mammalian host: progression to pathogenicity. Front Immunol (2016) 7:233. doi:10.3389/fimmu.2016.00233
73. Schwede A, Macleod OJ, MacGregor P, Carrington M. How does the VSG coat of bloodstream form African trypanosomes interact with external proteins? PLoS Pathog (2015) 11:e1005259. doi:10.1371/journal.ppat.1005259
74. Schwede A, Jones N, Engstler M, Carrington M. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol Biochem Parasitol (2011) 175:201–4. doi:10.1016/j.molbiopara.2010.11.004
75. Morrison LJ, Marcello L, McCulloch R. Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell Microbiol (2009) 11:1724–34. doi:10.1111/j.1462-5822.2009.01383.x
77. Pays E, Vanhamme L, Pérez-Morga D. Antigenic variation in Trypanosoma brucei: facts, challenges and mysteries. Curr Opin Microbiol (2004) 7:369–74. doi:10.1016/j.mib.2004.05.001
78. Borst P. Antigenic variation and allelic exclusion. Cell (2002) 109:5–8. doi:10.1016/S0092-8674(02)00711-0
79. Hall JPJ, Wang H, David Barry J. Mosaic VSGs and the scale of Trypanosoma brucei antigenic variation. PLoS Pathog (2013) 9:e1003502. doi:10.1371/journal.ppat.1003502
80. Dubois ME, Demick KP, Mansfield JM. Trypanosomes expressing a mosaic variant surface glycoprotein coat escape early detection by the immune system. Infect Immun (2005) 73:2690–7. doi:10.1128/IAI.73.5.2690-2697.2005
81. Graham SV, Barry JD. Transcriptional regulation of metacyclic variant surface glycoprotein gene expression during the life cycle of Trypanosoma brucei. Mol Cell Biol (1995) 15:5945–56. doi:10.1128/MCB.15.11.5945
82. Graham SV, Terry S, Barry JD. A structural and transcription pattern for variant surface glycoprotein gene expression sites used in metacyclic stage Trypanosoma brucei. Mol Biochem Parasitol (1999) 103:141–54. doi:10.1016/S0166-6851(99)00128-0
83. Barry JD, McCulloch R. Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite. Adv Parasitol (2001) 49:1–70. doi:10.1016/S0065-308X(01)49037-3
84. Engstler M, Pfohl T, Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, et al. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell (2007) 131:505–15. doi:10.1016/j.cell.2007.08.046
85. Field MC, Carrington M. The trypanosome flagellar pocket. Nat Rev Microbiol (2009) 7:775–86. doi:10.1038/nrmicro2221
86. Magez S, Schwegmann A, Atkinson R, Claes F, Drennan M, De Baetselier P, et al. The role of B-cells and IgM antibodies in parasitemia, anemia, and VSG switching in Trypanosoma brucei-infected mice. PLoS Pathog (2008) 4:e1000122. doi:10.1371/journal.ppat.1000122
87. Magez S, Radwanska M, Drennan M, Fick L, Baral TN, Brombacher F, et al. Interferon-gamma and nitric oxide in combination with antibodies are key protective host immune factors during Trypanosoma congolense Tc13 Infections. J Infect Dis (2006) 193:1575–83. doi:10.1086/503808
88. Balber AE, Bangs JD, Jones SM, Proia RL. Inactivation or elimination of potentially trypanolytic, complement-activating immune complexes by pathogenic trypanosomes. Infect Immun (1979) 24:617–27.
89. Ferrante A, Allison AC. Alternative pathway activation of complement by African trypanosomes lacking a glycoprotein coat. Parasite Immunol (1983) 5:491–8. doi:10.1111/j.1365-3024.1983.tb00763.x
90. Devine DV, Falk RJ, Balber AE. Restriction of the alternative pathway of human complement by intact Trypanosoma brucei subsp. gambiense. Infect Immun (1986) 52:223–9.
91. Ooi YM, Colten HR. Genetic defect in secretion of complement C5 in mice. Nature (1979) 282:207–8. doi:10.1038/282207a0
92. Jarvinen JA, Dalmasso A. Trypanosoma musculi infections in normocomplementemic, C5-deficient, and C3-depleted mice. Infect Immun (1977) 16:557–63.
93. Morrison WI, Roelants GE, Mayor-Withey KS, Murray M. Susceptibility of inbred strains of mice to Trypanosoma congolense: correlation with changes in spleen lymphocyte populations. Clin Exp Immunol (1978) 32:25–40.
94. Mekori YA, Metcalfe DD. Mast cells in innate immunity. Immunol Rev (2000) 173:131–40. doi:10.1016/j.jaci.2004.04.045
95. Paulnock DM, Freeman BE, Mansfield JM. Modulation of innate immunity by African trypanosomes. Parasitology (2010) 137:2051–63. doi:10.1017/S0031182010001460
96. Drennan MB, Stijlemans B, Van den Abbeele J, Quesniaux VJ, Barkhuizen M, Brombacher F, et al. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J Immunol (2005) 175:2501–9. doi:10.4049/jimmunol.175.4.2501
97. Mansfield JM, Paulnock DM. Regulation of innate and acquired immunity in African trypanosomiasis. Parasite Immunol (2005) 27:361–71. doi:10.1111/j.1365-3024.2005.00791.x
98. Stijlemans B, Guilliams M, Raes G, Beschin A, Magez S, De Baetselier P. African trypanosomosis: from immune escape and immunopathology to immune intervention. Vet Parasitol (2007) 148:3–13. doi:10.1016/j.vetpar.2007.05.005
99. Stijlemans B, Vankrunkelsven A, Caljon G, Bockstal V, Guilliams M, Bosschaerts T, et al. The central role of macrophages in trypanosomiasis-associated anemia: rationale for therapeutical approaches. Endocr Metab Immune Disord Drug Targets (2010) 10:71–82. doi:10.2174/187153010790827966
100. Bosschaerts T, Morias Y, Stijlemans B, Hérin M, Porta C, Sica A, et al. IL-10 limits production of pathogenic TNF by M1 myeloid cells through induction of nuclear NF-κB p50 member in Trypanosoma congolense infection-resistant C57BL/6 mice. Eur J Immunol (2011) 41:3270–80. doi:10.1002/eji.201041307
101. Salmon D, Vanwalleghem G, Morias Y, Denoeud J, Krumbholz C, Lhomme F, et al. Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science (2012) 337:463–6. doi:10.1126/science.1222753
102. De Muylder G, Daulouède S, Lecordier L, Uzureau P, Morias Y, Van Den Abbeele J, et al. A Trypanosoma brucei kinesin heavy chain promotes parasite growth by triggering host arginase activity. PLoS Pathog (2013) 9:e1003731. doi:10.1371/journal.ppat.1003731
103. Willert E, Phillips MA. Regulation and function of polyamines in African trypanosomes. Trends Parasitol (2012) 28:66–72. doi:10.1016/j.pt.2011.11.001
104. McGettrick AF, Corcoran SE, Barry PJ, McFarland J, Crès C, Curtis AM, et al. Trypanosoma brucei metabolite indolepyruvate decreases HIF-1α and glycolysis in macrophages as a mechanism of innate immune evasion. Proc Natl Acad Sci U S A (2016) 113(48):E7778–87. doi:10.1073/pnas.1608221113
105. Coller SP, Mansfield JM, Paulnock DM. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN-gamma-induced nitric oxide production via reduction in STAT1 phosphorylation in African trypanosomiasis. J Immunol (2003) 171:1466–72. doi:10.4049/jimmunol.171.3.1466
106. Sileghem M, Darji A, Hamers R, Van De Winkel M, De Baetselier P. Dual role of macrophages in the suppression of interleukin 2 production and interleukin 2 receptor expression in trypanosome-infected mice. Eur J Immunol (1989) 19:829–35. doi:10.1002/eji.1830190508
107. Darji A, Beschin A, Sileghem M, Heremans H, Brys L, De Baetselier P. In vitro simulation of immunosuppression caused by Trypanosoma brucei: active involvement of gamma interferon and tumor necrosis factor in the pathway of suppression. Infect Immun (1996) 64:1937–43.
108. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Immunoregulation in experimental murine Trypanosoma congolense infection: anti-IL-10 antibodies reverse trypanosome-mediated suppression of lymphocyte proliferation in vitro and moderately prolong the lifespan of genetically susceptible BALB/c mice. Parasite Immunol (1998) 20:293–302. doi:10.1046/j.1365-3024.1998.00156.x
109. Sileghem M, Norman Flynn J. Suppression of interleukin 2 secretion and interleukin 2 receptor expression during tsetse-transmitted trypanosomiasis in cattle. Eur J Immunol (1992) 22:767–73. doi:10.1002/eji.1830220321
110. Beschin A, Brys L, Magez S, Radwanska M, De Baetselier P. Trypanosoma brucei infection elicits nitric oxide-dependent and nitric oxide-independent suppressive mechanisms. J Leukoc Biol (1998) 63:429–39.
111. Millar AE, Sternberg J, McSharry C, Wei XQ, Liew FY, Turner CM. T-Cell responses during Trypanosoma brucei infections in mice deficient in inducible nitric oxide synthase. Infect Immun (1999) 67:3334–8.
112. Gomez-Rodriguez J, Stijlemans B, De Muylder G, Korf H, Brys L, Berberof M, et al. Identification of a parasitic immunomodulatory protein triggering the development of suppressive M1 macrophages during African trypanosomiasis. J Infect Dis (2009) 200:1849–60. doi:10.1086/648374
113. Uzonna JE, Kaushik RS, Zhang Y, Gordon JR, Tabel H. Experimental murine Trypanosoma congolense infections. II. Role of splenic adherent CD3+Thy1.2+ TCR-alpha beta gamma delta- CD4+8- and CD3+Thy1.2+ TCR-alpha beta gamma delta- CD4-8-cells in the production of IL-4, IL-10, and IFN-gamma and in trypanosom. J Immunol (1998) 161:6189–97.
114. Taylor K, Lutje V, Mertens B. Nitric oxide synthesis is depressed in Bos indicus cattle infected with Trypanosoma congolense and Trypanosoma vivax and does not mediate T-cell suppression. Infect Immun (1996) 64:4115–22.
115. Cnops J, De Trez C, Stijlemans B, Keirsse J, Kauffmann F, Barkhuizen M, et al. NK-, NKT- and CD8-derived IFNγ drives myeloid cell activation and erythrophagocytosis, resulting in trypanosomosis-associated acute anemia. PLoS Pathog (2015) 11:e1004964. doi:10.1371/journal.ppat.1004964
116. Taylor KA, Lutje V, Kennedy D, Authié E, Boulangé A, Logan-Henfrey L, et al. Trypanosoma congolense: B-lymphocyte responses differ between trypanotolerant and trypanosusceptible cattle. Exp Parasitol (1996) 83:106–16. doi:10.1006/expr.1996.0054
117. Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev (2010) 238:37–46. doi:10.1111/j.1600-065X.2010.00963.x
118. Boller S, Grosschedl R. The regulatory network of B-cell differentiation: a focused view of early B-cell factor 1 function. Immunol Rev (2014) 261:102–15. doi:10.1111/imr.12206
119. Reth M, Nielsen P. Signaling circuits in early B-cell development. Adv Immunol (2014) 122:129–75. doi:10.1016/B978-0-12-800267-4.00004-3
120. Mårtensson I-L, Almqvist N, Grimsholm O, Bernardi AI. The pre-B cell receptor checkpoint. FEBS Lett (2010) 584:2572–9. doi:10.1016/j.febslet.2010.04.057
121. Lebien TW, Tedder TF. B lymphocytes: how they develop and function. Blood (2008) 112:1570–80. doi:10.1182/blood-2008-02-078071
122. Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol (2008) 20:149–57. doi:10.1016/j.coi.2008.03.014
123. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15:160–71. doi:10.1038/nri3795
124. Bockstal V, Guirnalda P, Caljon G, Goenka R, Telfer JC, Frenkel D, et al. T. brucei infection reduces B lymphopoiesis in bone marrow and truncates compensatory splenic lymphopoiesis through transitional B-cell apoptosis. PLoS Pathog (2011) 7:e1002089. doi:10.1371/journal.ppat.1002089
125. Obishakin E, de Trez C, Magez S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol (2014) 36:187–98. doi:10.1111/pim.12099
126. Radwanska M, Guirnalda P, De Trez C, Ryffel B, Black S, Magez S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog (2008) 4:e1000078. doi:10.1371/journal.ppat.1000078
127. Chamond N, Cosson A, Blom-Potar MC, Jouvion G, D’Archivio S, Medina M, et al. Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. I. Parasitological, hematological and pathological parameters. PLoS Negl Trop Dis (2010) 4:e792. doi:10.1371/journal.pntd.0000792
128. Holland WG, Do TT, Huong NT, Dung NT, Thanh NG, Vercruysse J, et al. The effect of Trypanosoma evansi infection on pig performance and vaccination against classical swine fever. Vet Parasitol (2003) 111:115–23. doi:10.1016/S0304-4017(02)00363-1
129. Holland WG, My LN, Dung TV, Thanh NG, Tam PT, Vercruysse J, et al. The influence of T. evansi infection on the immuno-responsiveness of experimentally infected water buffaloes. Vet Parasitol (2001) 102:225–34. doi:10.1016/S0304-4017(01)00534-9
130. Sharpe RT, Langley AM, Mowat GN, Macaskill JA, Holmes PH. Immunosuppression in bovine trypanosomiasis: response of cattle infected with Trypanosoma congolense to foot-and-mouth disease vaccination and subsequent live virus challenge. Res Vet Sci (1982) 32:289–93.
131. De Trez C, Katsandegwaza B, Caljon G, Magez S. Experimental African trypanosome infection by needle passage or natural tsetse fly challenge thwarts the development of collagen-induced arthritis in DBA/1 prone mice via an impairment of antigen specific B cell autoantibody titers. PLoS One (2015) 10:e0130431. doi:10.1371/journal.pone.0130431
132. Cnops J, Kauffmann F, De Trez C, Baltz T, Keirsse J, Radwanska M, et al. Maintenance of B cells during chronic murine Trypanosoma brucei gambiense infection. Parasite Immunol (2016) 38:642–7. doi:10.1111/pim.12344
133. Lejon V, Mumba Ngoyi D, Kestens L, Boel L, Barbé B, Kande Betu V, et al. Gambiense human African trypanosomiasis and immunological memory: effect on phenotypic lymphocyte profiles and humoral immunity. PLoS Pathog (2014) 10:e1003947. doi:10.1371/journal.ppat.1003947
134. Cnops J, De Trez C, Bulte D, Radwanska M, Ryffel B, Magez S. IFN-gamma mediates early B-cell loss in experimental African trypanosomosis. Parasite Immunol (2015) 37:479–84. doi:10.1111/pim.12208
135. Frenkel D, Zhang F, Guirnalda P, Haynes C, Bockstal V, Radwanska M, et al. Trypanosoma brucei co-opts NK cells to kill splenic B2 B cells. PLoS Pathog (2016) 12:e1005733. doi:10.1371/journal.ppat.1005733
136. Cnops J, Bockstal V, De Trez C, Miquel MC, Radwanska M, Magez S. Curative drug treatment of trypanosomosis leads to the restoration of B-cell lymphopoiesis and splenic B-cell compartments. Parasite Immunol (2015) 37:485–91. doi:10.1111/pim.12209
137. Kuriakose S, Muleme HM, Onyilagha C, Singh R, Jia P, Uzonna JE. Diminazene aceturate (Berenil) modulates the host cellular and inflammatory responses to Trypanosoma congolense infection. PLoS One (2012) 7:e48696. doi:10.1371/journal.pone.0048696
138. Wellde BT, Preston JM, Kovatch RM, Higgs J, Chumo DA. Trypanosoma congolense: erythrocyte indices, plasma iron turnover and effects of treatment in infected cattle. Ann Trop Med Parasitol (1989) 83(Suppl 1):201–6. doi:10.1080/00034983.1989.11812425
139. Stijlemans B, Brys L, Korf H, Bieniasz-Krzywiec P, Sparkes A, Vansintjan L, et al. MIF-mediated hemodilution promotes pathogenic anemia in experimental African trypanosomosis. PLoS Pathog (2016) 12:e1005862. doi:10.1371/journal.ppat.1005862
140. Magez S, Radwanska M. African trypanosomiasis and antibodies: implications for vaccination, therapy and diagnosis. Future Microbiol (2009) 4:1075–87. doi:10.2217/fmb.09.65
141. La Greca F, Magez S. Vaccination against trypanosomiasis: can it be done or is the trypanosome truly the ultimate immune destroyer and escape artist? Hum Vaccin (2011) 7:1225–33. doi:10.4161/hv.7.11.18203
142. Stijlemans B, Baral TN, Guilliams M, Brys L, Korf J, Drennan M, et al. A glycosylphosphatidylinositol-based treatment alleviates trypanosomiasis-associated immunopathology. J Immunol (2007) 179:4003–14. doi:10.4049/jimmunol.179.6.4003
143. Authié E, Boulangé A, Muteti D, Lalmanach G, Gauthier F, Musoke AJ. Immunisation of cattle with cysteine proteinases of Trypanosoma congolense: targetting the disease rather than the parasite. Int J Parasitol (2001) 31:1429–33. doi:10.1016/S0020-7519(01)00266-1
144. Lalmanach G, Boulange A, Serveau C, Lecaille F, Scharfstein J, Gauthier F, et al. Congopain from Trypanosoma congolense: drug target and vaccine candidate. Biol Chem (2002) 383:739–49. doi:10.1515/BC.2002.077
145. Authie E, Duvallet G, Robertson C, Williams DJ. Antibody responses to a 33 kDa cysteine protease of Trypanosoma congolense: relationship to “trypanotolerance” in cattle. Parasite Immunol (1993) 15:465–74. doi:10.1111/j.1365-3024.1993.tb00632.x
146. Taylor KA, Lutje V, Kennedy D, Authie E, Boulange A, Logan-Henfrey L, et al. Trypanosoma congolense: B-lymphocyte responses differ between trypanotolerant and trypanosusceptible cattle. Exp Parasitol (1996) 83:106–16. doi:10.1006/expr.1996.0054
147. Boulange AF, Khamadi SA, Pillay D, Coetzer TH, Authie E. Production of congopain, the major cysteine protease of Trypanosoma (Nannomonas) congolense, in Pichia pastoris reveals unexpected dimerisation at physiological pH. Protein Expr Purif (2011) 75:95–103. doi:10.1016/j.pep.2010.09.002
148. Lanca AS, de Sousa KP, Atouguia J, Prazeres DM, Monteiro GA, Silva MS. Trypanosoma brucei: immunisation with plasmid DNA encoding invariant surface glycoprotein gene is able to induce partial protection in experimental African trypanosomiasis. Exp Parasitol (2011) 127:18–24. doi:10.1016/j.exppara.2010.06.017
149. Dagenais TR, Demick KP, Bangs JD, Forest KT, Paulnock DM, Mansfield JM. T-cell responses to the trypanosome variant surface glycoprotein are not limited to hypervariable subregions. Infect Immun (2009) 77:141–51. doi:10.1128/IAI.00729-08
150. Radwanska M, Magez S, Dumont N, Pays A, Nolan D, Pays E. Antibodies raised against the flagellar pocket fraction of Trypanosoma brucei preferentially recognize HSP60 in cDNA expression library. Parasite Immunol (2000) 22:639–50. doi:10.1046/j.1365-3024.2000.00348.x
151. Berthier D, Chantal I, Thevenon S, Sakande H, Maillard J-C, Bengaly Z, et al. Study of bovine trypanotolerance by whole transcriptome analysis. Ann N Y Acad Sci (2008) 1149:71–6. doi:10.1196/annals.1428.062
152. O’Gorman GM, Park SD, Hill EW, Meade KG, Coussens PM, Agaba M, et al. Transcriptional profiling of cattle infected with Trypanosoma congolense highlights gene expression signatures underlying trypanotolerance and trypanosusceptibility. BMC Genomics (2009) 10:207. doi:10.1186/1471-2164-10-207
153. Auty H, Torr SJ, Michoel T, Jayaraman S, Morrison LJ. Cattle trypanosomosis: the diversity of trypanosomes and implications for disease epidemiology and control. Rev Sci Tech (2015) 34:587–98. doi:10.20506/rst.34.2.2382
154. Mulindwa J, Mercé C, Matovu E, Enyaru J, Clayton C. Transcriptomes of newly-isolated Trypanosoma brucei rhodesiense reveal hundreds of mRNAs that are co-regulated with stumpy-form markers. BMC Genomics (2015) 16:1118. doi:10.1186/s12864-015-2338-y
155. Telleria EL, Benoit JB, Zhao X, Savage AF, Regmi S, e Silva TLA, et al. Insights into the trypanosome-host interactions revealed through transcriptomic analysis of parasitized tsetse fly salivary glands. PLoS Negl Trop Dis (2014) 8:e2649. doi:10.1371/journal.pntd.0002649
156. Ng DH, Skehel JJ, Kassiotis G, Langhorne J. Recovery of an antiviral antibody response following attrition caused by unrelated infection. PLoS Pathog (2014) 10:e1003843. doi:10.1371/journal.ppat.1003843
157. Bockstal V, Geurts N, Magez S. Acute disruption of bone marrow B lymphopoiesis and apoptosis of transitional and marginal zone B cells in the spleen following a blood-stage plasmodium chabaudi infection in mice. J Parasitol Res (2011) 2011:11. doi:10.1155/2011/534697
158. Keitany GJ, Kim KS, Krishnamurty AT, Hondowicz BD, Hahn WO, Dambrauskas N, et al. Blood stage malaria disrupts humoral immunity to the pre-erythrocytic stage circumsporozoite protein. Cell Rep (2016) 17:3193–205. doi:10.1016/j.celrep.2016.11.060
159. Onyilagha C, Jia P, Jayachandran N, Hou S, Okwor I, Kuriakose S, et al. The B cell adaptor molecule Bam32 is critically important for optimal antibody response and resistance to Trypanosoma congolense infection in mice. PLoS Negl Trop Dis (2015) 9:e0003716. doi:10.1371/journal.pntd.0003716
160. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med (2003) 197:1467–76. doi:10.1084/jem.20030286
161. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol (2003) 3:791–800. doi:10.1038/nri1200
162. Stijlemans B, Leng L, Brys L, Sparkes A, Vansintjan L, Caljon G, et al. MIF contributes to Trypanosoma brucei associated immunopathogenicity development. PLoS Pathog (2014) 10:e1004414. doi:10.1371/journal.ppat.1004414
163. Klasen C, Ohl K, Sternkopf M, Shachar I, Schmitz C, Heussen N, et al. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. J Immunol (2014) 192:5273–84. doi:10.4049/jimmunol.1302209
Keywords: B-cell lymphopoiesis, African trypanosomosis, vaccination strategies, inflammation, T-cells, macrophage migration inhibitory factor (MIF)
Citation: Stijlemans B, Radwanska M, De Trez C and Magez S (2017) African Trypanosomes Undermine Humoral Responses and Vaccine Development: Link with Inflammatory Responses? Front. Immunol. 8:582. doi: 10.3389/fimmu.2017.00582
Received: 31 January 2017; Accepted: 01 May 2017;
Published: 24 May 2017
Edited by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilReviewed by:
Paul Cos, University of Antwerp, BelgiumHerbert Leonel de Matos Guedes, Federal University of Rio de Janeiro, Brazil
Copyright: © 2017 Stijlemans, Radwanska, De Trez and Magez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benoit Stijlemans, YmVub2l0LnN0aWpsZW1hbnNAdnViLmJl
†These authors share co-first authorship.
‡These authors share co-last authorship.