 Fabiana Maria das Graças Corsi-Zuelli1*
Fabiana Maria das Graças Corsi-Zuelli1* Fernanda Brognara2
Fernanda Brognara2 Gustavo Fernando da Silva Quirino3
Gustavo Fernando da Silva Quirino3 Carlos Hiroji Hiroki4
Carlos Hiroji Hiroki4 Rafael Sobrano Fais4
Rafael Sobrano Fais4 Cristina Marta Del-Ben1
Cristina Marta Del-Ben1 Luis Ulloa5
Luis Ulloa5 Helio Cesar Salgado2
Helio Cesar Salgado2 Alexandre Kanashiro6
Alexandre Kanashiro6 Camila Marcelino Loureiro7*
Camila Marcelino Loureiro7*
- 1Department of Neuroscience and Behavior, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
- 2Department of Physiology, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
- 3Department of Cellular and Molecular Biology, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
- 4Department of Pharmacology, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
- 5Department of Surgery, Center of Immunology and Inflammation, Rutgers University-New Jersey Medical School, Newark, NJ, United States
- 6Department of Physiological Sciences, Federal University of São Carlos, São Carlos, Brazil
- 7Department of Internal Medicine, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
Schizophrenia is one of the most debilitating mental disorders and is aggravated by the lack of efficacious treatment. Although its etiology is unclear, epidemiological studies indicate that infection and inflammation during development induces behavioral, morphological, neurochemical, and cognitive impairments, increasing the risk of developing schizophrenia. The inflammatory hypothesis of schizophrenia is also supported by clinical studies demonstrating systemic inflammation and microglia activation in schizophrenic patients. Although elucidating the mechanism that induces this inflammatory profile remains a challenge, mounting evidence suggests that neuroimmune interactions may provide therapeutic advantages to control inflammation and hence schizophrenia. Recent studies have indicated that vagus nerve stimulation controls both peripheral and central inflammation via alpha-7 nicotinic acetylcholine receptor (α7nAChR). Other findings have indicated that vagal stimulation and α7nAChR-agonists can provide therapeutic advantages for neuropsychiatric disorders, such as depression and epilepsy. This review analyzes the latest results regarding: (I) the immune-to-brain pathogenesis of schizophrenia; (II) the regulation of inflammation by the autonomic nervous system in psychiatric disorders; and (III) the role of the vagus nerve and α7nAChR in schizophrenia.
Introduction
Mental disorders are a major clinical and scientific challenge in modern medicine with an estimated prevalence of approximately 17% of the population (1, 2). Schizophrenia is one of the most debilitating psychotic disorders due to the lack of effective treatment (3, 4). Schizophrenia is a chronic psychiatric disorder characterized by faulty perception and withdrawal from reality. Schizophrenia symptomatology comprises positive (delusions, hallucinations), negative symptoms (social withdrawal, apathy) cognitive alterations, disorganized thinking, and psychomotor disturbances (2). The average life expectancy of schizophrenia patients is 10–25 years less than the normal population due to health problems and a higher suicide rate (5–7). Despite its significant social implications, schizophrenia is neglected worldwide (3, 4, 8).
Current treatments for schizophrenia are inefficacious, and there is an unmet clinical need for new and safe therapeutic strategies (9–12). Schizophrenia is usually treated with typical or atypical antipsychotics. Typical antipsychotics often induce significant psychomotor side effects. Atypical antipsychotics are the usual first-line treatment, although they are associated with metabolic syndrome and an increased cardiovascular risk of death (11, 12). An explanation for the inefficacious treatments is the insufficient knowledge about the etiology of schizophrenia. Both groups of antipsychotics are believed to be antagonists for dopamine receptors in the brain, and thus, previous studies mostly focused on the dopaminergic system (13). Although dopaminergic dysfunction contributes to schizophrenia, the mechanisms leading to this dysfunction are unknown. Recent studies demonstrate an abnormal inflammatory profile that can cause neurotransmission dysfunction in schizophrenia (14, 15). Early infections and other immune alterations during pregnancy and development can contribute to schizophrenia and other neurological disorders (16–19). These studies are contemporary with recent investigations demonstrating that vagal stimulation controls both central and peripheral inflammation (20–24) and that vagal stimulation can provide therapeutic advantages for neuropsychiatric disorders, such as depression and epilepsy (25, 26). However, little is known about the potential of this mechanism for treating schizophrenia (27). We reasoned that vagal stimulation may control inflammation and provide novel therapeutic advantages for schizophrenia. In this article, we evaluate this hypothesis by reviewing autonomic vagal dysfunction in psychiatric disorders and discussing the potential of vagal stimulation and alpha-7 cholinergic receptor (α7nAChR) agonists for treating schizophrenia.
Immune-to-Brain Pathogenesis: From Homeostasis to Inflammation
Unregulated inflammation induced by infection or trauma results in excessive production of inflammatory cytokines, such as tumor necrosis factor (TNF), interferon-γ (IFN-γ), and interleukins (IL-1β, IL-6, etc.). These cytokines influence the homeostasis of several organs, as well as the central nervous system (CNS) (28).
Despite the traditional view of the brain as an immunologically privileged site, multiple studies have demonstrated that the CNS interacts with peripheral inflammatory cytokines through several pathways, described as follows (29). First, the humoral pathway: peripheral cytokines diffuse into the CNS through circumventricular organs and structures lacking the blood–brain barrier (BBB). Second, the cellular pathway: peripheral immune cells enter the CNS due to alterations in the BBB permeability and through the actions of chemoattractant mediators. Third, the microbiota–gut–brain axis: the microbiota–gut can transmit signals to the brain via the vagus nerve, immune mediators, and microbial metabolites, thereby altering neurotransmission in the CNS (30, 31). Fourth, the recently discovered central lymphatic pathway or the glymphatic system: mediated by functional lymphatic vessels in the CNS (32). In this pathway, extracellular fluids (the cerebrospinal fluid and interstitial fluid) draining from the brain parenchyma to the cervical and lumbar lymph nodes facilitate the traffic of antigens and immune cells affecting peripheral and central inflammation (33). Finally, the neural pathway: the afferent vagus nerve detects peripheral inflammatory cytokines (TNF, IL-1β, IL-6) and transmits signals to the nucleus tractus solitarius, and thereby to the hypothalamus (29, 34). All these pathways serve as immune-to-brain cross talk that facilitate central inflammation and behavioral changes.
The Inflammatory Hypotheses of Schizophrenia
A balance between the pro- and anti-inflammatory cytokines is critical for proper brain development (35). Epidemiological studies indicate that infections during development increase the risk of schizophrenia in adulthood (36–39). These studies report an association between elevated maternal inflammatory cytokines levels (especially IL-8 and TNF) and risk of schizophrenia in adult offspring (16, 37). It has been observed in preclinical studies that maternal immune activation in rodents induces inflammatory cytokines (IL-1β, IL-6, TNF) and reduces anti-inflammatory cytokines (IL-10) in both the maternal fluids and in the fetal brain, inducing schizophrenia-like behaviors in the offspring (35, 40). Likewise, direct IL-6 inoculation into pregnant rodents also induces schizophrenia-like abnormalities in the offspring. This effect is prevented by neutralizing IL-6 antibodies, genetic depletion of the IL-6 gene (IL-6 knockout) (35), or overexpression of anti-inflammatory cytokines (IL-10) in the macrophages of pregnant dams (41).
Genetic studies have demonstrated the implications of immune-related genes in schizophrenia (42). A Danish cohort study reported a significant relationship between severe infections and the risk of schizophrenia. A previous history of autoimmune disorders increases the risk of schizophrenia by 36%. This risk of schizophrenia increases up to 60% in patients with a previous history of infection and hospitalization (19). Several clinical studies demonstrate a chronic low-grade inflammation in schizophrenia (43–46). Early studies suggested that this chronic low-grade inflammation may be due to chronically activated macrophages that fail to properly control T-lymphocytes in the so called “macrophage-T-lymphocyte hypothesis” (47). Thereafter, Schwarz et al. (48) suggested that psychotic patients have a T helper cells type 2-profile (Th2) characterized by increased Th2-produced IL-4 and decreased T helper cells type 1 (Th1)-produced IFN-γ (48). In contrast, a shift away from Th2-produced IL-4 and toward Th1-produced IFN-γ was later highlighted, suggesting the involvement of transforming growth factor (TGF)-β in the Th1/Th2 regulation of schizophrenia. Although contradictory, these hypotheses concur that an inflammatory imbalance is involved in schizophrenia (49).
Recent meta-analyses indicate that acute and chronically ill patients demonstrate a low-grade inflammatory profile that correlates with the clinical symptoms of schizophrenia (43, 45, 46) (Table 1). This inflammatory profile was also reported in drug-naïve patients in the first episode of psychosis (45). Since these patients were drug-naïve and in the first manifestation of the disease, it is unlikely that inflammation was related to antipsychotics or duration of illness. Thus, inflammatory cytokines in the peripheral blood were suggested to be either state or trait biomarkers. State biomarkers refer to specific cytokines elevated in schizophrenia and normalized with antipsychotics. Trait biomarkers are cytokines that are elevated in schizophrenia and are not normalized following antipsychotic treatment (43).
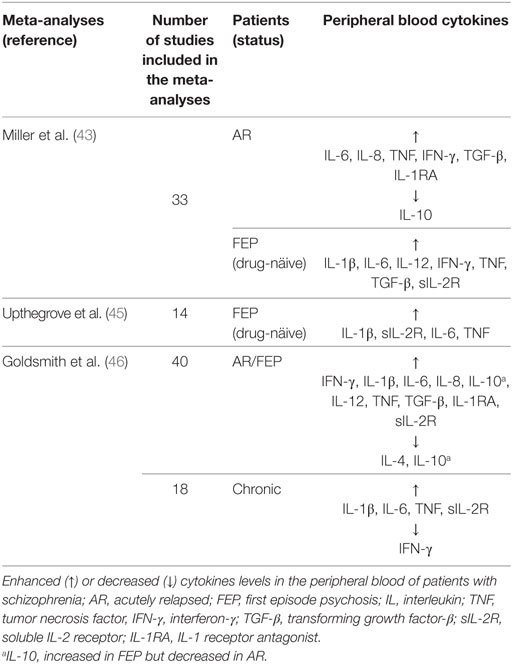
Table 1. Cytokine profile in schizophrenia.
The association between biological and environmental factors can have significant implications in schizophrenia (50). In this respect, Monji et al. (51) shed light on the microglia hypothesis of schizophrenia (51). Microglia are the resident macrophages in the CNS (52), and similar to peripheral macrophages, they show different activation states. Basal state microglia (M0) perform phagocytosis and promote neurite outgrowth (53–55). However, both physical (infections) (56) or psychological (early life stress) stressors induce microglial activation (57–62). In response to these events, microglial polarization is triggered, resulting in an inflammatory state (microglia type 1; M1) (63, 64). M1 microglia produce large amounts of inflammatory cytokines (TNF, IL-1, IL-6, IL-12) inducing neuronal cytotoxicity (57, 61, 62). In contrast, anti-inflammatory cytokines (IL-4, IL-10) induce microglial polarization toward an anti-inflammatory state (microglia type 2; M2), critical for homeostasis. The imbalance between these factors affects neurite outgrowth, neuronal connections, and neurotransmitter formation and induces neuronal cytotoxicity, contributing to neuropsychiatric disorders (57, 65–67). Indeed, increased microglial density and microglial activation have been demonstrated in the hippocampus and gray matter of schizophrenic patients, as demonstrated by postmortem and in vivo studies (68–73), and microglial activation has been linked to the pre-suicidal stress associated with schizophrenia (74).
Microglia-produced TNF induces neurotoxicity and neurodegeneration as demonstrated both in vitro (54, 75) and in vivo (76, 77). A typical example is that abnormal microglia activation alters tryptophan metabolism along the kynurenine pathway, producing metabolites that act as N-methyl-d-aspartate receptor (NMDAR)-agonists (quinolinic acid) or -antagonists, such as kynurenic acid (KYNA) (29, 78, 79). NMDAR dysfunction is associated with schizophrenia (80) and NMDAR-antagonists induce positive, negative, and cognitive symptoms in healthy volunteers, similar to those observed in schizophrenia (81, 82). Delusions and hallucinations related to autoantibodies blocking NMDARs were reported in schizophrenic and healthy controls (83, 84). The kynurenine pathway is also linked to oxidative stress. Neuronal apoptosis and structural changes in specific areas of the brain, such as the amygdala, hippocampus, and prefrontal cortex, are related to several psychiatric disorders, including schizophrenia (78). Together these studies demonstrate that inflammation of the CNS can contribute to schizophrenia (43, 45, 46).
The efficacy of antipsychotics may be due to microglial suppression and subsequent neuroprotection (85–87). Atypical antipsychotics inhibit TNF production by the IFN-γ-stimulated microglia (86, 87). Minocycline, a non-psychotic medication with potent effects in inhibiting microglia, has been suggested as an adjuvant in the treatment of schizophrenia (86). However, atypical antipsychotics induce metabolic and cardiovascular dysfunctions (11, 12). Thus, there is an unmet clinical need for new therapeutic strategies to control inflammation and the progression of schizophrenia.
Does Autonomic Immunomodulation Contribute to the Inflammatory Component of Schizophrenia?
The Autonomic Nervous System
The autonomic nervous system regulates the immune system through both the sympathetic and parasympathetic networks (21, 88, 89). This regulation is not only critical for physiological homeostasis, such as that in the gastrointestinal tract (90, 91), but also in pathological conditions that range from infection to trauma (22, 92–94). Briefly, sympathetic preganglionic neurons that originate from the thoracic and lumbar spinal segments synapse with postganglionic neurons in pre- or paravertebral ganglia. Parasympathetic preganglionic neurons originate from the brainstem and the sacral spinal cord and synapse with postganglionic neurons in terminal ganglia located near target organs. Both preganglionic sympathetic and parasympathetic neurons release acetylcholine (ACh). While all parasympathetic postganglionic neurons release ACh, most sympathetic postganglionic neurons release norepinephrine. Overall, sympathetic activity predominates during the “fight-or-flight” reactions, while parasympathetic activity predominates during “quiet” resting conditions (95). The vagus nerve—the major component of the parasympathetic system—plays a critical role in the communication between the brain and peripheral organs, such as the heart, lungs, and intestine (96).
The Autonomic Nervous System Regulation of Inflammation in Schizophrenia
Dysfunction of the autonomic nervous system may contribute to the inflammatory profile reported in schizophrenia. The balance between the sympathetic and parasympathetic systems can be determined by the heart rate variability (HRV), which represents the variation of the intervals between heartbeats (97). Parasympathetic nerves slow heart rate and increase HRV by releasing ACh. Sympathetic nerves accelerate heart rate and decrease HRV by releasing epinephrine and norepinephrine (98, 99). Lower HRV is a predictor of cardiac morbidity and mortality (100–102). Psychiatric patients tend to have an autonomic imbalance with low HRV suggesting a reduced parasympathetic and increased sympathetic tone (103–105). Low HRV has also been related to psychotic symptoms and depression (106–108); and thus, the vagal tone could serve as an index of the treatment response (109).
The polyvagal theory associates the autonomic neuronal system with affective experiences and contingent social behavior (110). Low vagal activity is associated with reduced social involvement and a less flexible behavioral response to environmental conditions (110). In agreement with this theory, Bylsma and coworkers suggested that “the cardiac autonomic balance may be a useful index that reflects the balance of the autonomic nervous system to respond to aspects of the environment that may be sensitive to psychophysiological abnormalities” (111). Thus, autonomic neuronal dysfunction and low vagal activity could contribute to schizophrenia.
Electrical vagus nerve stimulation (VNS) was approved by the food and drug administration for treating several neuropsychiatric disorders including refractory epilepsy and depression (25, 112–114). However, few studies have explored VNS in schizophrenia (27). The only study that addresses VNS in schizophrenia examined the effects of transcutaneous vagal stimulation (tVNS) (115). tVNS is a non-invasive electrical stimulation of the external ear allowing stimulation of the auricular vagal branch (116). tVNS of the cymba conche results in the strongest activation of the vagal afferent pathway in the brainstem, as observed through functional magnetic resonance imaging (117). A bicentric, randomized, sham-controlled and double-blind clinical investigation was performed in 20 schizophrenic patients, who were randomly assigned to two groups: one received daily active stimulation of the left auricle for 26 weeks; the other group received sham stimulation daily. Regarding efficacy, there was no difference between the sham and tVNS groups (115). However, only half of the patients adhered to the protocol. Given that the vagal stimulation treatment depends on patient adherence, it was not possible to conclude a result due to non-adherence to the protocol and methodological limitations. In contrast, experimental studies demonstrated that VNS significantly reversed hippocampal hyperactivity, mesolimbic dopaminergic dysfunction, and schizophrenia-like symptoms, including cognitive deficits (118, 119).
Autonomic dysfunction facilitates immune alterations and increases the susceptibility to infectious and immunological disorders. The vagus nerve directs the “cholinergic anti-inflammatory pathway” modulating inflammation, as reported in preclinical and clinical studies (21, 23, 24, 26, 93, 118–124). In clinical studies, VNS inhibited cytokine production, improved HRV, and ameliorated low moods and emotional symptoms in depressive patients resistant to pharmacological treatment (26, 124). Recent studies demonstrated brain inflammation reduction with VNS applied at a low frequency, a protocol that favors the activation of efferent vagus nerve fibers (23, 24). Inhibition of CNS inflammation can be a consequence of peripheral inflammation inhibition (22, 24). The vagal anti-inflammatory signals are mediated by α7nAChR, suggesting that nicotinic agonists mimic vagal anti-inflammatory potential (22, 125, 126). α7nAChR were detected in several cell types, including neurons and immune cells. In the CNS, α7nAChR are expressed by pyramidal interneurons (127, 128), immature granule cells (129), astrocytes (130), and microglia (131, 132). In the periphery, this receptor is expressed in monocytes (133, 134), dendritic cells (135), macrophages (120, 136), T-cells (137), and B-cells (138). In this regard, the use of selective α7nAChR-agonists in the treatment of psychiatric and neurological patients has been reported (139). Remarkably, activation of α7nAChR in cultured microglia cells inhibits LPS-induced release of cytokines and promotes conversion of M1 microglia to M2 (132, 140).
Genetic studies demonstrated that α7nAChR activity is reduced, especially in the hippocampus, thalamus, frontal cortex, brainstem, ventral tegmental area, nucleus accumbens, and the cingulate cortex of schizophrenic patients (141–146). This reduced activity is more remarkable in gamma-aminobutyric acid (GABA) interneurons (142) that are key players in schizophrenia, especially in the cognitive domain (147). In addition, α7nAChR participate in NMDA and GABAA receptors activity, and similar to NMDAR, they modulate calcium influx facilitating neurotransmission (148–150). Accordingly, α7nAChR has been involved in a myriad of brain functions, including learning, memory, neuroprotection, and synaptic plasticity (151–153). Conversely, α7nAChR dysfunction leads to abnormal NMDAR/GABAA function and perturbation of glutamatergic and GABAergic neurotransmission (154).
Kynurenic acid, besides acting as an NMDAR inhibitor, is also a potent non-competitive α7nAChR-antagonist (155) and is associated with hypoglutamatergic and hypocholinergic neurotransmission, facilitating cognitive deficits and sensory gating disturbances in schizophrenia (155). α7nAChR-agonists restore dopamine signaling in the brain (156) and improve negative symptoms and cognitive function in schizophrenia (139, 157–161). Variation in brain KYNA may be related to the nicotinic cholinergic system. It has been observed that nicotine reduces levels of KYNA in clinical trials (162). In rodents, this effect was clear during a 5-day nicotine treatment; however, prolonged treatment enhanced central levels of KYNA (155). Notably, increased brain levels of KYNA are reported in schizophrenia (14); this concurs with data demonstrating a high rate of cigarette smokers with schizophrenia (163). For instance, over 80% of schizophrenic patients were smokers compared to 20% of the general population of the USA in 2006 (164). Accordingly, a recent meta-analysis reported that people who smoke are three times more likely to suffer psychosis (165); thus, high cigarette smoking in schizophrenia is suggested as a physiological basis on which patients try to correct cognitive deficits caused by α7nAChR dysfunction (155). In a recent study, chronic nicotine reversed hypofrontality in an animal model of addiction and schizophrenia (166). α7nAChR represents a potential therapeutic target for cognitive deficits and sensory gating disturbances; nevertheless, cigarette smoking is toxic and unspecific with deleterious side effects, and it is critical to find specific and safer therapeutic strategies for schizophrenia (163).
Essentially, the development of schizophrenia is more complex. This condition is influenced by genetic vulnerability interacting synergistically with multiple environmental risk factors, such as infections or stress in early life, drug abuse, besides other environmental adversities occurring at critical periods of neurodevelopment (167–169). This gene–environmental interaction could produce a latent immune vulnerability. Thus, when this vulnerability is manifested, the individuals become more susceptible to immune dysfunctions, increasing their risk of developing schizophrenia (170).
Notably, stressful situations can induce an impairment of the α7nAChR (171–173). Animal models demonstrate an interaction between α7nAChR and the hypothalamic–pituitary–adrenal axis, a primary system responsible for the stress response (172). Prenatal restraint stress decreases α7nAChR expression in the hippocampus and prefrontal cortex in adult rats (173), while VNS reduces conditioned fear in rodents with posttraumatic stress disorder (174). A recent review stated that α7nAChR-agonists induce beneficial effects in patients with psychiatric disorders (139) ameliorating cognitive deficits, negative symptoms, and sensory gating disturbances in both preclinical and clinical trials of schizophrenia (139, 157–161). Advantageous effects for the negative symptoms have been reported repeatedly, while improvements in the cognitive domain remain controversial, deserving further exploration (175, 176). Together, these studies indicate that the vagus nerve and the α7nAChR may be involved in the inflammatory hypothesis of schizophrenia (Figure 1). Thus, future investigations are critical to determine their clinical potential in schizophrenia and other neurological disorders. Moreover, the consideration of stressful events in future investigations would be of interest. This would help to reduce the discrepancy regarding inflammatory processes in schizophrenia that are observed in data from several studies.
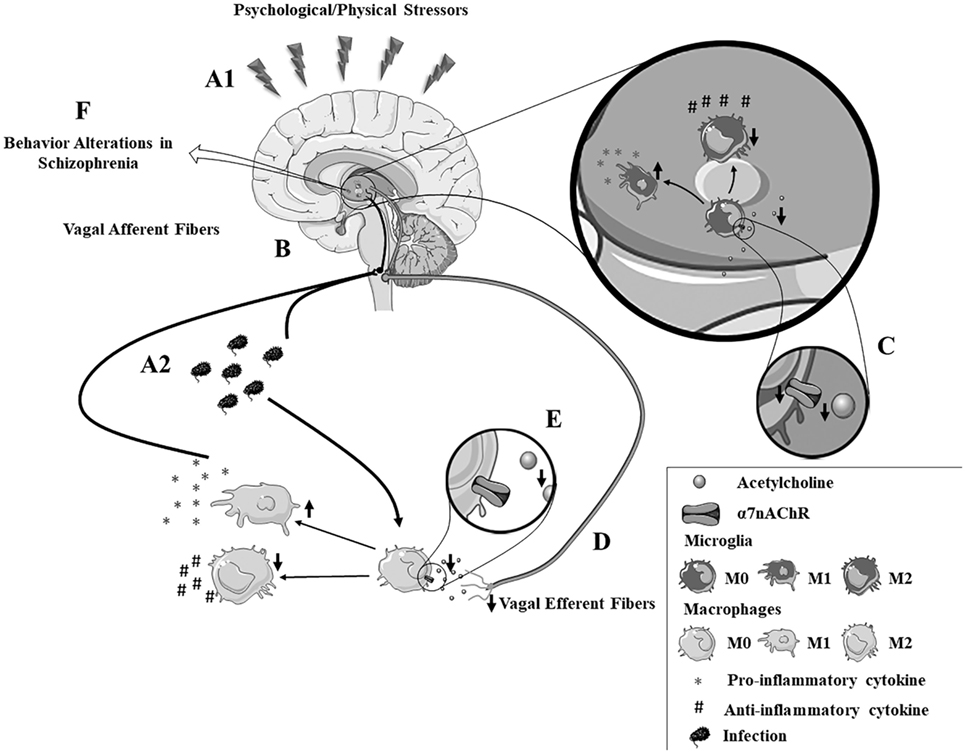
Figure 1. The autonomic hypothesis of schizophrenia. (A1,A2) Psychological or physical stressors contribute to enhanced production of inflammatory cytokines by both peripheral and brain immune cells. (B) The afferent vagus nerve facilitates immune-to-brain communication, by transmitting signals from the periphery to the brain. (C) Patients with schizophrenia have an intense autonomic imbalance characterized by α7nAChR dysfunction and reduced vagal tone. (D) The efferent vagus nerve plays a key role in the “cholinergic anti-inflammatory pathway,” a mechanism dependent on (E) acetylcholine binding to the α7nAChR, a pathway that is dysfunctional in schizophrenia patients. This impairment in the inflammatory reflex may contribute to (F) neuroinflammation and disrupted synthesis of neurotransmitters in schizophrenia. Note: the vagus nerve is constituted by both efferent and afferent fibers. The division shown in this figure is merely illustrative to explain the afferent and efferent neuroimmune routes.
Future Perspectives
The inter-relationship between the nervous and the immune systems is critical to understand the pathogenesis of schizophrenia. In brief, a reduced parasympathetic tone could contribute to inflammation observed in schizophrenic patients. This mechanism combined with stress-mediated dysfunctions of the α7nAChR can enhance the impairment of the inflammatory reflex, contributing to schizophrenia’s symptoms. In the face of microglial hyperactivation, future investigations controlling microglial activation through innovative approaches, such as VNS and α7nAChR modulation, may provide clinical advantages for treating schizophrenia. As early exposure to stressors induces changes in the inflammatory reflex, a better understanding of the association between biological and environmental factors would potentially improve the diagnosis and treatment of schizophrenia. In this regard, public health interventions controlling stressful events, such as public education and comprehensive approaches to early treatment focusing on individual, social and environmental factors, might be beneficial for mental health promotion and prevention of future psychiatric disorders.
Author Contributions
AK proposed the review to the authors and together with HS, LU, and CD-B revised the manuscript. FC-Z and CL suggested the topic for this review, coordinated the research group, drafted and revised this manuscript. FB also drafted and revised the manuscript, and together with AK, HS, and LU was essential in the consideration of the autonomic nervous system. GQ and CH were helpful in providing general information about inflammation. RF participated in the elaboration of the figure. All authors approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This review originated from discussions between graduate students and researchers during the course: “Neural Control of the Inflammatory Response” (RFI5806) from the Department of Physiology, Ribeirão Preto Medical School that was coordinated by HS and AK.
Funding
FC-Z was a recipient from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/PROEX) and now receives a grant from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), grant number: 2016/12195-9; GQ is a recipient of a fellowship from FAPESP, grant number: 2015/10378-6 and RF receives grants from FAPESP: 2016/11988-5 and 2016/07641-0; FB and CH are recipients of fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), grant numbers: 154027/2016-2; 830699/1999-6. AK is recipient of a fellowship from CAPES/PNPD and FAPESP, grant number: 2011/20343-4; CL receives a grant from CAPES/PROEX. CD-B receives a grant from the Thematic Project FAPESP Process number 2012/05178-0. This work received financial support from the Department of Physiology, Ribeirão Preto Medical School (Thematic Project FAPESP Process number 2013/20549-7).
Abbreviations
ACh, acetylcholine; BBB, blood–brain barrier; CNS, central nervous system; GABA, gamma-aminobutyric acid; HRV, heart rate variability; IL, interleukin; IFN, interferon; KYNA, kynurenic acid; NMDAR, N-methyl-d-aspartate receptor; M1, microglia type 1; M2, microglia type 2; TGF, transforming growth factor; Th1, T helper cells type 1; Th2, T helper cells type 2; TNF, tumor necrosis factor; VNS, vagus nerve stimulation; tVNS, transcutaneous vagus nerve stimulation; α7nAChR, alpha-7 nicotinic acetylcholine receptor.
References
1. Steel Z, Marnane C, Iranpour C, Chey T, Jackson JW, Patel V, et al. The global prevalence of common mental disorders: a systematic review and meta-analysis 1980–2013. Int J Epidemiol (2014) 43(2):476–93. doi: 10.1093/ije/dyu038
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association (2013).
3. Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet (2013) 382(9904):1575–86. doi:10.1016/S0140-6736(13)61611-6
4. Collaborators GBoDS. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2015) 386(9995):743–800. doi:10.1016/S0140-6736(15)60692-4
5. Laursen TM, Munk-Olsen T, Vestergaard M. Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr Opin Psychiatry (2012) 25(2):83–8. doi:10.1097/YCO.0b013e32835035ca
6. Pompili M, Amador XF, Girardi P, Harkavy-Friedman J, Harrow M, Kaplan K, et al. Suicide risk in schizophrenia: learning from the past to change the future. Ann Gen Psychiatry (2007) 6:10. doi:10.1186/1744-859X-6-10
7. van Os J, Kapur S. Schizophrenia. Lancet (2009) 374(9690):635–45. doi:10.1016/S0140-6736(09)60995-8
8. Whiteford HA, Ferrari AJ, Degenhardt L, Feigin V, Vos T. The global burden of mental, neurological and substance use disorders: an analysis from the Global Burden of Disease Study 2010. PLoS One (2015) 10(2):e0116820. doi:10.1371/journal.pone.0116820
9. Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet (2009) 373(9657):31–41. doi:10.1016/S0140-6736(08)61764-X
10. Agid O, Kapur S, Remington G. Emerging drugs for schizophrenia. Expert Opin Emerg Drugs (2008) 13(3):479–95. doi:10.1517/14728214.13.3.479
11. Grundmann M, Kacirova I, Urinovska R. Therapeutic drug monitoring of atypical antipsychotic drugs. Acta Pharm (2014) 64(4):387–401. doi:10.2478/acph-2014-0036
12. Gardner-Sood P, Lally J, Smith S, Atakan Z, Ismail K, Greenwood KE, et al. Cardiovascular risk factors and metabolic syndrome in people with established psychotic illnesses: baseline data from the IMPaCT randomized controlled trial. Psychol Med (2015) 45(12):2619–29. doi:10.1017/S0033291715000562
13. Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol (2015) 29(2):97–115. doi:10.1177/0269881114563634
14. Kegel ME, Bhat M, Skogh E, Samuelsson M, Lundberg K, Dahl ML, et al. Imbalanced kynurenine pathway in schizophrenia. Int J Tryptophan Res (2014) 7:15–22. doi:10.4137/IJTR.S16800
15. Koola MM. Kynurenine pathway and cognitive impairments in schizophrenia: pharmacogenetics of galantamine and memantine. Schizophr Res Cogn (2016) 4:4–9. doi:10.1016/j.scog.2016.02.001
16. Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Bernstein D, Yolken RH. Maternal infections and subsequent psychosis among offspring. Arch Gen Psychiatry (2001) 58(11):1032–7. doi:10.1001/archpsyc.58.11.1032
17. Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, et al. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry (2004) 161(5):889–95. doi:10.1176/appi.ajp.161.5.889
18. Brown AS, Schaefer CA, Quesenberry CP, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry (2005) 162(4):767–73. doi:10.1176/appi.ajp.162.4.767
19. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry (2011) 168(12):1303–10. doi:10.1176/appi.ajp.2011.11030516
20. Pavlov VA, Tracey KJ. Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci (2017) 20(2):156–66. doi:10.1038/nn.4477
21. Martelli D, Farmer DG, Yao ST. The splanchnic anti-inflammatory pathway: could it be the efferent arm of the inflammatory reflex? Exp Physiol (2016) 101(10):1245–52. doi:10.1113/EP085559
22. Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov (2005) 4(8):673–84. doi:10.1038/nrd1797
23. Schweighöfer H, Rummel C, Roth J, Rosengarten B. Modulatory effects of vagal stimulation on neurophysiological parameters and the cellular immune response in the rat brain during systemic inflammation. Intensive Care Med Exp (2016) 4(1):19. doi:10.1186/s40635-016-0091-4
24. Meneses G, Bautista M, Florentino A, Díaz G, Acero G, Besedovsky H, et al. Electric stimulation of the vagus nerve reduced mouse neuroinflammation induced by lipopolysaccharide. J Inflamm (2016) 13:33. doi:10.1186/s12950-016-0140-5
25. Ardesch JJ, Buschman HP, Wagener-Schimmel LJ, van der Aa HE, Hageman G. Vagus nerve stimulation for medically refractory epilepsy: a long-term follow-up study. Seizure (2007) 16(7):579–85. doi:10.1016/j.seizure.2007.04.005
26. Nemeroff CB, Mayberg HS, Krahl SE, McNamara J, Frazer A, Henry TR, et al. VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology (2006) 31(7):1345–55. doi:10.1038/sj.npp.1301082
27. Cimpianu CL, Strube W, Falkai P, Palm U, Hasan A. Vagus nerve stimulation in psychiatry: a systematic review of the available evidence. J Neural Transm (2016) 124:145–58. doi:10.1007/s00702-016-1642-2
28. Corbin JG, Kelly D, Rath EM, Baerwald KD, Suzuki K, Popko B. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci (1996) 7(5):354–70. doi:10.1006/mcne.1996.0026
29. Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry (2015) 2(3):258–70. doi:10.1016/S2215-0366(14)00122-9
30. Desbonnet L, Clarke G, Traplin A, O’Sullivan O, Crispie F, Moloney RD, et al. Gut microbiota depletion from early adolescence in mice: implications for brain and behaviour. Brain Behav Immun (2015) 48:165–73. doi:10.1016/j.bbi.2015.04.004
31. De Palma G, Lynch MD, Lu J, Dang VT, Deng Y, Jury J, et al. Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice. Sci Transl Med (2017) 9(379):eaaf6397. doi:10.1126/scitranslmed.aaf6397
32. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523(7560):337–41. doi:10.1038/nature14432
33. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med (2015) 212(7):991–9. doi:10.1084/jem.20142290
34. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9(1):46–56. doi:10.1038/nrn2297
35. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci (2007) 27(40):10695–702. doi:10.1523/JNEUROSCI.2178-07.2007
36. McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev (2008) 30:67–76. doi:10.1093/epirev/mxn001
37. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry (2010) 167(3):261–80. doi:10.1176/appi.ajp.2009.09030361
38. Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychol Med (2013) 43(2):239–57. doi:10.1017/S0033291712000736
39. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol (2014) 10(11):643–60. doi:10.1038/nrneurol.2014.187
40. Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav Immun (2012) 26(4):623–34. doi:10.1016/j.bbi.2012.01.015
41. Meyer U. Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry (2014) 75(4):307–15. doi:10.1016/j.biopsych.2013.07.011
42. Andreassen OA, Harbo HF, Wang Y, Thompson WK, Schork AJ, Mattingsdal M, et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol Psychiatry (2015) 20(2):207–14. doi:10.1038/mp.2013.195
43. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry (2011) 70(7):663–71. doi:10.1016/j.biopsych.2011.04.013
44. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry (2008) 63(8):801–8. doi:10.1016/j.biopsych.2007.09.024
45. Upthegrove R, Manzanares-Teson N, Barnes NM. Cytokine function in medication-naive first episode psychosis: a systematic review and meta-analysis. Schizophr Res (2014) 155(1–3):101–8. doi:10.1016/j.schres.2014.03.005
46. Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry (2016) 21(12):1696–709. doi:10.1038/mp.2016.3
47. Smith RS, Maes M. The macrophage-T-lymphocyte theory of schizophrenia: additional evidence. Med Hypotheses (1995) 45(2):135–41. doi:10.1016/0306-9877(95)90062-4
48. Schwarz MJ, Müller N, Riedel M, Ackenheil M. The Th2-hypothesis of schizophrenia: a strategy to identify a subgroup of schizophrenia caused by immune mechanisms. Med Hypotheses (2001) 56(4):483–6. doi:10.1054/mehy.2000.1203
49. Kim YK, Myint AM, Lee BH, Han CS, Lee HJ, Kim DJ, et al. Th1, Th2 and Th3 cytokine alteration in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2004) 28(7):1129–34. doi:10.1016/j.pnpbp.2004.05.047
50. Modai S, Shomron N. Molecular risk factors for schizophrenia. Trends Mol Med (2016) 22(3):242–53. doi:10.1016/j.molmed.2016.01.006
51. Monji A, Kato TA, Mizoguchi Y, Horikawa H, Seki Y, Kasai M, et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry (2013) 42:115–21. doi:10.1016/j.pnpbp.2011.12.002
52. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci (2009) 29(13):3974–80. doi:10.1523/JNEUROSCI.4363-08.2009
53. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci (2009) 29(43):13435–44. doi:10.1523/JNEUROSCI.3257-09.2009
54. Cacci E, Claasen JH, Kokaia Z. Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res (2005) 80(6):789–97. doi:10.1002/jnr.20531
55. Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology (2012) 37(9):1491–505. doi:10.1016/j.psyneuen.2012.02.003
56. Häusler KG, Prinz M, Nolte C, Weber JR, Schumann RR, Kettenmann H, et al. Interferon-gamma differentially modulates the release of cytokines and chemokines in lipopolysaccharide- and pneumococcal cell wall-stimulated mouse microglia and macrophages. Eur J Neurosci (2002) 16(11):2113–22. doi:10.1046/j.1460-9568.2002.02287.x
57. de Pablos RM, Villarán RF, Argüelles S, Herrera AJ, Venero JL, Ayala A, et al. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci (2006) 26(21):5709–19. doi:10.1523/JNEUROSCI.0802-06.2006
58. Wang HT, Huang FL, Hu ZL, Zhang WJ, Qiao XQ, Huang YQ, et al. Early-life social isolation-induced depressive-like behavior in rats results in microglial activation and neuronal histone methylation that are mitigated by minocycline. Neurotox Res (2017) 31(4):505–20. doi:10.1007/s12640-016-9696-3
59. Roque A, Ochoa-Zarzosa A, Torner L. Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav Immun (2016) 55:39–48. doi:10.1016/j.bbi.2015.09.017
60. Gracia-Rubio I, Moscoso-Castro M, Pozo OJ, Marcos J, Nadal R, Valverde O. Maternal separation induces neuroinflammation and long-lasting emotional alterations in mice. Prog Neuropsychopharmacol Biol Psychiatry (2016) 65:104–17. doi:10.1016/j.pnpbp.2015.09.003
61. Blandino P, Barnum CJ, Deak T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol (2006) 173(1–2):87–95. doi:10.1016/j.jneuroim.2005.11.021
62. Shimoda M, Jones VC, Kobayashi M, Suzuki F. Microglial cells from psychologically stressed mice as an accelerator of cerebral cryptococcosis. Immunol Cell Biol (2006) 84(6):551–6. doi:10.1111/j.1440-1711.2006.01466.x
63. Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun (2007) 21(1):47–59. doi:10.1016/j.bbi.2006.03.005
64. Johnson JD, O’Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun (2002) 16(4):461–76. doi:10.1006/brbi.2001.0638
65. Mahadevan J, Sundaresh A, Rajkumar RP, Muthuramalingam A, Menon V, Negi VS, et al. An exploratory study of immune markers in acute and transient psychosis. Asian J Psychiatr (2017) 25:219–23. doi:10.1016/j.ajp.2016.11.010
66. Lee EE, Hong S, Martin AS, Eyler LT, Jeste DV. Inflammation in schizophrenia: cytokine levels and their relationships to demographic and clinical variables. Am J Geriatr Psychiatry (2017) 25(1):50–61. doi:10.1016/j.jagp.2016.09.009
67. Szymona K, Zdzisińska B, Karakuła-Juchnowicz H, Kocki T, Kandefer-Szerszeń M, Flis M, et al. Correlations of kynurenic acid, 3-hydroxykynurenine, sIL-2R, IFN-α, and IL-4 with clinical symptoms during acute relapse of schizophrenia. Neurotox Res (2017). doi:10.1007/s12640-017-9714-0
68. Busse S, Busse M, Schiltz K, Bielau H, Gos T, Brisch R, et al. Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: further evidence for disease course-related immune alterations? Brain Behav Immun (2012) 26(8):1273–9. doi:10.1016/j.bbi.2012.08.005
69. Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett (1999) 271(2):126–8. doi:10.1016/S0304-3940(99)00545-5
70. Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol (2000) 59(2):137–50. doi:10.1093/jnen/59.2.137
71. Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol (2006) 112(3):305–16. doi:10.1007/s00401-006-0090-8
72. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med (2009) 50(11):1801–7. doi:10.2967/jnumed.109.066647
73. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry (2008) 64(9):820–2. doi:10.1016/j.biopsych.2008.04.025
74. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res (2008) 42(2):151–7. doi:10.1016/j.jpsychires.2006.10.013
75. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science (2003) 302(5651):1760–5. doi:10.1126/science.1088417
76. Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, et al. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci (2006) 26(38):9703–12. doi:10.1523/JNEUROSCI.2723-06.2006
77. Kaneko N, Kudo K, Mabuchi T, Takemoto K, Fujimaki K, Wati H, et al. Suppression of cell proliferation by interferon-alpha through interleukin-1 production in adult rat dentate gyrus. Neuropsychopharmacology (2006) 31(12):2619–26. doi:10.1038/sj.npp.1301137
78. Meyer U, Schwarz MJ, Müller N. Inflammatory processes in schizophrenia: a promising neuroimmunological target for the treatment of negative/cognitive symptoms and beyond. Pharmacol Ther (2011) 132(1):96–110. doi:10.1016/j.pharmthera.2011.06.003
79. Möller M, Du Preez JL, Emsley R, Harvey BH. Social isolation rearing in rats alters plasma tryptophan metabolism and is reversed by sub-chronic clozapine treatment. Neuropharmacology (2012) 62(8):2499–506. doi:10.1016/j.neuropharm.2012.02.021
80. Bressan RA, Pilowsky LS. [Glutamatergic hypothesis of schizophrenia]. Rev Bras Psiquiatr (2003) 25(3):177–83. doi:10.1590/S1516-44462003000300011
81. Gouzoulis-Mayfrank E, Heekeren K, Neukirch A, Stoll M, Stock C, Obradovic M, et al. Psychological effects of (S)-ketamine and N,N-dimethyltryptamine (DMT): a double-blind, cross-over study in healthy volunteers. Pharmacopsychiatry (2005) 38(6):301–11. doi:10.1055/s-2005-918695
82. Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug; sernyl. AMA Arch Neurol Psychiatry (1959) 81(3):363–9. doi:10.1001/archneurpsyc.1959.02340150095011
83. Steiner J, Walter M, Glanz W, Sarnyai Z, Bernstein HG, Vielhaber S, et al. Increased prevalence of diverse N-methyl-d-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-d-aspartate glutamate receptor encephalitis. JAMA Psychiatry (2013) 70(3):271–8. doi:10.1001/2013.jamapsychiatry.86
84. Lennox BR, Palmer-Cooper EC, Pollak T, Hainsworth J, Marks J, Jacobson L, et al. Prevalence and clinical characteristics of serum neuronal cell surface antibodies in first-episode psychosis: a case-control study. Lancet Psychiatry (2017) 4(1):42–8. doi:10.1016/S2215-0366(16)30375-3
85. Bian Q, Kato T, Monji A, Hashioka S, Mizoguchi Y, Horikawa H, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(1):42–8. doi:10.1016/j.pnpbp.2007.06.031
86. Seki Y, Kato TA, Monji A, Mizoguchi Y, Horikawa H, Sato-Kasai M, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-γ-stimulated microglia in co-culture model. Schizophr Res (2013) 151(1–3):20–8. doi:10.1016/j.schres.2013.09.011
87. Kato T, Monji A, Hashioka S, Kanba S. Risperidone significantly inhibits interferon-gamma-induced microglial activation in vitro. Schizophr Res (2007) 92(1–3):108–15. doi:10.1016/j.schres.2007.01.019
89. Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med (2012) 209(6):1057–68. doi:10.1084/jem.20120571
90. Pellissier S, Dantzer C, Canini F, Mathieu N, Bonaz B. Psychological adjustment and autonomic disturbances in inflammatory bowel diseases and irritable bowel syndrome. Psychoneuroendocrinology (2010) 35(5):653–62. doi:10.1016/j.psyneuen.2009.10.004
91. Bonaz B, Sinniger V, Pellissier S. Anti-inflammatory properties of the vagus nerve: potential therapeutic implications of vagus nerve stimulation. J Physiol (2016) 594(20):5781–90. doi:10.1113/JP271539
92. Cai B, Dong W, Sharpe S, Deitch EA, Ulloa L. Survival and inflammatory responses in experimental models of hemorrhage. J Surg Res (2011) 169(2):257–66. doi:10.1016/j.jss.2009.11.712
93. Ulloa L. The anti-inflammatory potential of selective cholinergic agonists. Shock (2011) 36(1):97–8. doi:10.1097/SHK.0b013e31821820d2
94. Bassi GS, Brognara F, Castania JA, Talbot J, Cunha TM, Cunha FQ, et al. Baroreflex activation in conscious rats modulates the joint inflammatory response via sympathetic function. Brain Behav Immun (2015) 49:140–7. doi:10.1016/j.bbi.2015.05.002
95. McCorry LK. Physiology of the autonomic nervous system. Am J Pharm Educ (2007) 71(4):78. doi:10.5688/aj710478
96. Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci (2000) 85(1–3):1–17. doi:10.1016/S1566-0702(00)00215-0
97. Heart Rate Variability. Standards of measurement, physiological interpretation, and clinical use. Task force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Eur Heart J (1996) 17(3):354–81.
98. Stauss HM. Physiologic mechanisms of heart rate variability. Rev Bras Hipertens (2007) 14(1):8–15.
99. Squires RW. Defining the high-risk cardiac patient. In: Robertson L, Enderle K, Hambly L, editors. Exercise Prescription for the High-Risk Cardiac Patient. Human Kinetics (1998). p. 1–16.
100. Ponikowski P, Anker SD, Chua TP, Szelemej R, Piepoli M, Adamopoulos S, et al. Depressed heart rate variability as an independent predictor of death in chronic congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol (1997) 79(12):1645–50. doi:10.1016/S0002-9149(97)00215-4
101. La Rovere MT, Pinna GD, Maestri R, Mortara A, Capomolla S, Febo O, et al. Short-term heart rate variability strongly predicts sudden cardiac death in chronic heart failure patients. Circulation (2003) 107(4):565–70. doi:10.1161/01.CIR.0000047275.25795.17
102. Hemingway H, Shipley M, Brunner E, Britton A, Malik M, Marmot M. Does autonomic function link social position to coronary risk? The Whitehall II study. Circulation (2005) 111(23):3071–7. doi:10.1161/CIRCULATIONAHA.104.497347
103. Bassett D, Bear N, Nutt D, Hood S, Bassett S, Hans D. Reduced heart rate variability in remitted bipolar disorder and recurrent depression. Aust N Z J Psychiatry (2016) 50(8):793–804. doi:10.1177/0004867416652734
104. Malaspina D, Bruder G, Dalack GW, Storer S, Van Kammen M, Amador X, et al. Diminished cardiac vagal tone in schizophrenia: associations to brain laterality and age of onset. Biol Psychiatry (1997) 41(5):612–7. doi:10.1016/S0006-3223(96)00161-8
105. Bär KJ, Letzsch A, Jochum T, Wagner G, Greiner W, Sauer H. Loss of efferent vagal activity in acute schizophrenia. J Psychiatr Res (2005) 39(5):519–27. doi:10.1016/j.jpsychires.2004.12.007
106. Bär KJ, Boettger MK, Berger S, Baier V, Sauer H, Yeragani VK, et al. Decreased baroreflex sensitivity in acute schizophrenia. J Appl Physiol (1985) (2007) 102(3):1051–6. doi:10.1152/japplphysiol.00811.2006
107. Toichi M, Kubota Y, Murai T, Kamio Y, Sakihama M, Toriuchi T, et al. The influence of psychotic states on the autonomic nervous system in schizophrenia. Int J Psychophysiol (1999) 31(2):147–54. doi:10.1016/S0167-8760(98)00047-6
108. Zahn TP, Pickar D. Autonomic activity in relation to symptom ratings and reaction time in unmedicated patients with schizophrenia. Schizophr Res (2005) 79(2–3):257–70. doi:10.1016/j.schres.2005.05.025
109. Chambers AS, Allen JJ. Vagal tone as an indicator of treatment response in major depression. Psychophysiology (2002) 39(6):861–4. doi:10.1111/1469-8986.3960861
110. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol (2001) 42(2):123–46. doi:10.1016/S0167-8760(01)00162-3
111. Bylsma LM, Yaroslavsky I, Rottenberg J, Jennings JR, George CJ, Kiss E, et al. Juvenile onset depression alters cardiac autonomic balance in response to psychological and physical challenges. Biol Psychol (2015) 110:167–74. doi:10.1016/j.biopsycho.2015.07.003
112. De Benedictis A, Freri E, Rizzi M, Franzini A, Ragona F, Specchio N, et al. Vagus nerve stimulation for drug-resistant epilepsia partialis continua: report of four cases. Epilepsy Res (2013) 107(1–2):163–71. doi:10.1016/j.eplepsyres.2013.07.010
113. Rush AJ, Sackeim HA, Marangell LB, George MS, Brannan SK, Davis SM, et al. Effects of 12 months of vagus nerve stimulation in treatment-resistant depression: a naturalistic study. Biol Psychiatry (2005) 58(5):355–63. doi:10.1016/j.biopsych.2005.05.024
114. Bajbouj M, Merkl A, Schlaepfer TE, Frick C, Zobel A, Maier W, et al. Two-year outcome of vagus nerve stimulation in treatment-resistant depression. J Clin Psychopharmacol (2010) 30(3):273–81. doi:10.1097/JCP.0b013e3181db8831
115. Hasan A, Wolff-Menzler C, Pfeiffer S, Falkai P, Weidinger E, Jobst A, et al. Transcutaneous noninvasive vagus nerve stimulation (tVNS) in the treatment of schizophrenia: a bicentric randomized controlled pilot study. Eur Arch Psychiatry Clin Neurosci (2015) 265(7):589–600. doi:10.1007/s00406-015-0618-9
116. Frangos E, Komisaruk BR. Access to vagal projections via cutaneous electrical stimulation of the neck: fMRI evidence in healthy humans. Brain Stimul (2017) 10(1):19–27. doi:10.1016/j.brs.2016.10.008
117. Yakunina N, Kim SS, Nam EC. Optimization of transcutaneous vagus nerve stimulation using functional MRI. Neuromodulation (2016) 20:290–300. doi:10.1111/ner.12541
118. Smucny J, Visani A, Tregellas JR. Could vagus nerve stimulation target hippocampal hyperactivity to improve cognition in schizophrenia? Front Psychiatry (2015) 6:43. doi:10.3389/fpsyt.2015.00043
119. Perez SM, Carreno FR, Frazer A, Lodge DJ. Vagal nerve stimulation reverses aberrant dopamine system function in the methylazoxymethanol acetate rodent model of schizophrenia. J Neurosci (2014) 34(28):9261–7. doi:10.1523/JNEUROSCI.0588-14.2014
120. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature (2000) 405(6785):458–62. doi:10.1038/35013070
121. Ondicova K, Pecenák J, Mravec B. The role of the vagus nerve in depression. Neuro Endocrinol Lett (2010) 31(5):602–8.
122. Vonck K, Raedt R, Naulaerts J, De Vogelaere F, Thiery E, Van Roost D, et al. Vagus nerve stimulation…25 years later! What do we know about the effects on cognition? Neurosci Biobehav Rev (2014) 45:63–71. doi:10.1016/j.neubiorev.2014.05.005
123. Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, et al. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ Heart Fail (2009) 2(6):692–9. doi:10.1161/CIRCHEARTFAILURE.109.873968
124. Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry (2014) 71(12):1381–91. doi:10.1001/jamapsychiatry.2014.1611
125. Ulloa L. The cholinergic anti-inflammatory pathway meets microRNA. Cell Res (2013) 23(11):1249–50. doi:10.1038/cr.2013.128
127. Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther (2013) 137(1):22–54. doi:10.1016/j.pharmthera.2012.08.012
128. Dineley KT, Pandya AA, Yakel JL. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci (2015) 36(2):96–108. doi:10.1016/j.tips.2014.12.002
129. John D, Shelukhina I, Yanagawa Y, Deuchars J, Henderson Z. Functional alpha7 nicotinic receptors are expressed on immature granule cells of the postnatal dentate gyrus. Brain Res (2015) 1601:15–30. doi:10.1016/j.brainres.2014.12.041
130. Shen JX, Yakel JL. Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci (2012) 48(1):14–21. doi:10.1007/s12031-012-9719-3
131. Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem (2004) 89(2):337–43. doi:10.1046/j.1471-4159.2004.02347.x
132. Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res (2006) 83(8):1461–70. doi:10.1002/jnr.20850
133. Hamano R, Takahashi HK, Iwagaki H, Yoshino T, Nishibori M, Tanaka N. Stimulation of alpha7 nicotinic acetylcholine receptor inhibits CD14 and the toll-like receptor 4 expression in human monocytes. Shock (2006) 26(4):358–64. doi:10.1097/01.shk.0000228168.86845.60
134. Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdés-Ferrer SI, Patel NB, et al. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med (2009) 15(7–8):195–202. doi:10.2119/molmed.2009.00039
135. Sato K, Nagayama H, Tadokoro K, Juji T, Takahashi TA. Extracellular signal-regulated kinase, stress-activated protein kinase/c-Jun N-terminal kinase, and p38mapk are involved in IL-10-mediated selective repression of TNF-alpha-induced activation and maturation of human peripheral blood monocyte-derived dendritic cells. J Immunol (1999) 162(7):3865–72.
136. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature (2003) 421(6921):384–8. doi:10.1038/nature01339
137. Razani-Boroujerdi S, Boyd RT, Dávila-García MI, Nandi JS, Mishra NC, Singh SP, et al. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol (2007) 179(5):2889–98. doi:10.4049/jimmunol.179.5.2889
138. Skok MV, Kalashnik EN, Koval LN, Tsetlin VI, Utkin YN, Changeux JP, et al. Functional nicotinic acetylcholine receptors are expressed in B lymphocyte-derived cell lines. Mol Pharmacol (2003) 64(4):885–9. doi:10.1124/mol.64.4.885
139. Kalkman HO, Feuerbach D. Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol Life Sci (2016) 73(13):2511–30. doi:10.1007/s00018-016-2175-4
140. Zhang Q, Lu Y, Bian H, Guo L, Zhu H. Activation of the α7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am J Transl Res (2017) 9(3):971–85.
141. Leonard S, Freedman R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psychiatry (2006) 60(2):115–22. doi:10.1016/j.biopsych.2006.03.054
142. Freedman R, Adams CE, Leonard S. The alpha7-nicotinic acetylcholine receptor and the pathology of hippocampal interneurons in schizophrenia. J Chem Neuroanat (2000) 20(3–4):299–306. doi:10.1016/S0891-0618(00)00109-5
143. Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, et al. Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: alpha-bungarotoxin and nicotine binding in the thalamus. J Neurochem (1999) 73(4):1590–7. doi:10.1046/j.1471-4159.1999.0731590.x
144. Guan ZZ, Zhang X, Blennow K, Nordberg A. Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport (1999) 10(8):1779–82. doi:10.1097/00001756-199906030-00028
145. Woo TU, Kim AM, Viscidi E. Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia. Brain Res (2008) 1218:267–77. doi:10.1016/j.brainres.2008.03.092
146. Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, et al. Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J Chem Neuroanat (2001) 22(1–2):115–26. doi:10.1016/S0891-0618(01)00117-X
147. Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci (2012) 35(1):57–67. doi:10.1016/j.tins.2011.10.004
148. Li S, Nai Q, Lipina TV, Roder JC, Liu F. α7nAchR/NMDAR coupling affects NMDAR function and object recognition. Mol Brain (2013) 6:58. doi:10.1186/1756-6606-6-58
149. Shen L, Cui WY, Chen RZ, Wang H. Differential modulation of GABAA and NMDA receptors by α7-nicotinic receptor desensitization in cultured rat hippocampal neurons. Acta Pharmacol Sin (2016) 37(3):312–21. doi:10.1038/aps.2015.106
150. Liu Z, Conroy WG, Stawicki TM, Nai Q, Neff RA, Berg DK. EphB receptors co-distribute with a nicotinic receptor subtype and regulate nicotinic downstream signaling in neurons. Mol Cell Neurosci (2008) 38(2):236–44. doi:10.1016/j.mcn.2008.02.013
151. Yakel JL. Nicotinic ACh receptors in the hippocampal circuit; functional expression and role in synaptic plasticity. J Physiol (2014) 592(19):4147–53. doi:10.1113/jphysiol.2014.273896
152. Nordman JC, Phillips WS, Kodama N, Clark SG, Del Negro CA, Kabbani N. Axon targeting of the alpha 7 nicotinic receptor in developing hippocampal neurons by Gprin1 regulates growth. J Neurochem (2014) 129(4):649–62. doi:10.1111/jnc.12641
153. Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J Mol Neurosci (2006) 30(1–2):137–40. doi:10.1385/JMN:30:1:137
154. Lin H, Hsu FC, Baumann BH, Coulter DA, Lynch DR. Cortical synaptic NMDA receptor deficits in α7 nicotinic acetylcholine receptor gene deletion models: implications for neuropsychiatric diseases. Neurobiol Dis (2014) 63:129–40. doi:10.1016/j.nbd.2013.11.021
155. Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci (2001) 21(19):7463–73.
156. Pichat P, Bergis OE, Terranova JP, Urani A, Duarte C, Santucci V, et al. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (II) efficacy in experimental models predictive of activity against cognitive symptoms of schizophrenia. Neuropsychopharmacology (2007) 32(1):17–34. doi:10.1038/sj.npp.1301188
157. Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry (2006) 63(6):630–8. doi:10.1001/archpsyc.63.6.630
158. Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Johnson L, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry (2008) 165(8):1040–7. doi:10.1176/appi.ajp.2008.07071135
159. Sinkus ML, Lee MJ, Gault J, Logel J, Short M, Freedman R, et al. A 2-base pair deletion polymorphism in the partial duplication of the alpha7 nicotinic acetylcholine gene (CHRFAM7A) on chromosome 15q14 is associated with schizophrenia. Brain Res (2009) 1291:1–11. doi:10.1016/j.brainres.2009.07.041
160. Egea J, Buendia I, Parada E, Navarro E, León R, Lopez MG. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol (2015) 97(4):463–72. doi:10.1016/j.bcp.2015.07.032
161. Keefe RS, Meltzer HA, Dgetluck N, Gawryl M, Koenig G, Moebius HJ, et al. Randomized, double-blind, placebo-controlled study of encenicline, an α7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology (2015) 40(13):3053–60. doi:10.1038/npp.2015.176
162. Milart P, Młynarczyk M, Sikorski R. Influence of maternal smoking on kynurenic acid concentrations in umbilical cord blood. Ginekol Pol (2000) 71(8):843–7.
163. Sagud M, Mihaljević-Peles A, Mück-Seler D, Pivac N, Vuksan-Cusa B, Brataljenović T, et al. Smoking and schizophrenia. Psychiatr Danub (2009) 21(3):371–5.
164. Keltner NL, Grant JS. Smoke, smoke, smoke that cigarette. Perspect Psychiatr Care (2006) 42(4):256–61. doi:10.1111/j.1744-6163.2006.00085.x
165. Gurillo P, Jauhar S, Murray RM, MacCabe JH. Does tobacco use cause psychosis? Systematic review and meta-analysis. Lancet Psychiatry (2015) 2(8):718–25. doi:10.1016/S2215-0366(15)00152-2
166. Koukouli F, Rooy M, Tziotis D, Sailor KA, O’Neill HC, Levenga J, et al. Nicotine reverses hypofrontality in animal models of addiction and schizophrenia. Nat Med (2017) 23(3):347–54. doi:10.1038/nm.4274
167. Baharnoori M, Bhardwaj SK, Srivastava LK. Neonatal behavioral changes in rats with gestational exposure to lipopolysaccharide: a prenatal infection model for developmental neuropsychiatric disorders. Schizophr Bull (2012) 38(3):444–56. doi:10.1093/schbul/sbq098
168. Karl T, Arnold JC. Schizophrenia: a consequence of gene-environment interactions? Front Behav Neurosci (2014) 8:435. doi:10.3389/fnbeh.2014.00435
169. Tournier M. [First-episodes psychosis: clinical and epidemiological news]. Encephale (2013) 39(Suppl 2):S74–8. doi:10.1016/S0013-7006(13)70099-X
170. Kinney DK, Hintz K, Shearer EM, Barch DH, Riffin C, Whitley K, et al. A unifying hypothesis of schizophrenia: abnormal immune system development may help explain roles of prenatal hazards, post-pubertal onset, stress, genes, climate, infections, and brain dysfunction. Med Hypotheses (2010) 74(3):555–63. doi:10.1016/j.mehy.2009.09.040
171. Holgate JY, Bartlett SE. Early life stress, nicotinic acetylcholine receptors and alcohol use disorders. Brain Sci (2015) 5(3):258–74. doi:10.3390/brainsci5030258
172. Okada S, Yamaguchi-Shima N, Shimizu T, Arai J, Lianyi L, Wakiguchi H, et al. Role of brain nicotinic acetylcholine receptor in centrally administered corticotropin-releasing factor-induced elevation of plasma corticosterone in rats. Eur J Pharmacol (2008) 587(1–3):322–9. doi:10.1016/j.ejphar.2008.03.005
173. Baier CJ, Pallarés ME, Adrover E, Monteleone MC, Brocco MA, Barrantes FJ, et al. Prenatal restraint stress decreases the expression of alpha-7 nicotinic receptor in the brain of adult rat offspring. Stress (2015) 18(4):435–45. doi:10.3109/10253890.2015.1022148
174. Peña DF, Engineer ND, McIntyre CK. Rapid remission of conditioned fear expression with extinction training paired with vagus nerve stimulation. Biol Psychiatry (2013) 73(11):1071–7. doi:10.1016/j.biopsych.2012.10.021
175. Boggs DL, Carlson J, Cortes-Briones J, Krystal JH, D’Souza DC. Going up in smoke? A review of nAChRs-based treatment strategies for improving cognition in schizophrenia. Curr Pharm Des (2014) 20(31):5077–92. doi:10.2174/1381612819666131216121019
Keywords: schizophrenia, immune system, cytokines, inflammation, microglia, vagus nerve stimulation, alpha-7 nicotinic acetylcholine receptor, cholinergic anti-inflammatory pathway
Citation: Corsi-Zuelli FMG, Brognara F, Quirino GFS, Hiroki CH, Fais RS, Del-Ben CM, Ulloa L, Salgado HC, Kanashiro A and Loureiro CM (2017) Neuroimmune Interactions in Schizophrenia: Focus on Vagus Nerve Stimulation and Activation of the Alpha-7 Nicotinic Acetylcholine Receptor. Front. Immunol. 8:618. doi: 10.3389/fimmu.2017.00618
Received: 08 February 2017; Accepted: 10 May 2017;
Published: 31 May 2017
Edited by:
Valentin A. Pavlov, Northwell Health at Manhasset, United StatesReviewed by:
Bruno Bonaz, Grenoble Faculty of Medicine and Hospital, FranceMauricio Rosas-Ballina, University of Basel, Switzerland
Maryna Skok, Palladin Institute of Biochemistry (NAN Ukraine), Ukraine
Copyright: © 2017 Corsi-Zuelli, Brognara, Quirino, Hiroki, Fais, Del-Ben, Ulloa, Salgado, Kanashiro and Loureiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabiana Maria das Graças Corsi-Zuelli, ZmFiaWFuYS56dWVsbGlAdXNwLmJy;
Camila Marcelino Loureiro, Y2FtaWxhLmxvdXJlaXJvQHVzcC5icg==