 Yew Ann Leong
Yew Ann Leong Anurag Atnerkar1
Anurag Atnerkar1 Di Yu
Di Yu- 1Infection and Immunity Program, Department of Biochemistry and Molecular Biology, Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
- 2Department of Immunology and Infectious Disease, John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia
Human immunodeficiency virus (HIV) infects millions of people worldwide, and new cases continue to emerge. Once infected, the virus cannot be cleared by the immune system and causes acquired immunodeficiency syndrome. Combination antiretroviral therapeutic regimen effectively suppresses viral replication and halts disease progression. The treatment, however, does not eliminate the virus-infected cells, and interruption of treatment inevitably leads to viral rebound. The rebound virus originates from a group of virus-infected cells referred to as the cellular reservoir of HIV. Identifying and eliminating the HIV reservoir will prevent viral rebound and cure HIV infection. In this review, we focus on a recently discovered HIV reservoir in a subset of CD4+ T cells called the follicular helper T (TFH) cells. We describe the potential mechanisms for the emergence of reservoir in TFH cells, and the strategies to target and eliminate this viral reservoir.
Overview
Chronic infection caused by human immunodeficiency virus (HIV) is a global epidemic that leads to lifelong diseases and imposes significant health burdens (1). HIV targets the immune system by inducing the death of CD4+ T helper cells, a critical cellular component of the adaptive immune system. The death of CD4+ T cells cripples the immune system and increases the susceptibility of the host to opportunistic infections—a condition known as acquired immunodeficiency syndrome (AIDS). Combination antiretroviral therapeutic regimen (cART) effectively inhibits the life cycle of the virus to prevent viral dissemination in the body (2–4). Nevertheless, despite the effective suppression of viremia by cART, the underlying inflammation and dysfunctional immune response continue to pose significant health challenges to HIV-infected individuals (5–8). Most importantly, the ongoing administration of drugs is required; otherwise, the interruption of therapy inevitably leads to viral rebound and progression to AIDS (9–11). Viral rebound originates from cells harboring HIV that escape eradication by cART. These cells form the HIV viral reservoir (12). Identifying the reservoir and devising effective strategies for its elimination are the keys to realizing the goal of curing HIV. Follicular T helper (TFH) cells are a specialized subset of CD4+ T cells that reside in B cell follicles to assist the humoral immune response (13). Recently, TFH cells have been identified as the major cellular reservoir of HIV among CD4+ T helper cells (14–25). In this review, we focus on understanding the emergence of the TFH reservoir and propose strategies to target and eliminate the reservoir.
HIV Life Cycle and Stages of Disease
Human immunodeficiency virus is a single-stranded enveloped RNA virus that utilizes CD4 as the primary receptor for viral entry into the cells (26). HIV further subdivides to CC chemokine receptor 5 (CCR5) and CXC chemokine receptor-4 (CXCR4) tropic strains, which recognize the corresponding molecules on host cells as co-receptors for viral entry (27). As CD4 is the primary receptor for viral entry, the major target cells for productive HIV infection are CD4+ T cells (28, 29) and, to a lesser extent, macrophages (30). After viral entry, HIV releases a single-stranded RNA genome into the host cytoplasm. The RNA is synthesized into double-stranded complementary DNA (cDNA) by the viral reverse transcriptase. The cDNA is imported into the nucleus and integrated into host chromatin via viral integrase. The integrated cDNA—the provirus—is transcribed to produce viral RNA and proteins to form new virus to infect other cells (2). After HIV infection, viremia increases, with concomitant depletion of CD4+ T cells (31). The peak of viremia coincides with the activation of an anti-HIV immune response that leads to a brief reduction of viremia, which accompanies a transient recovery in the number of CD4+ T cells. This phase is the acute stage of the infection. The transient recovery of CD4+ T cells is then followed by their gradual depletion and a progressive increase of viremia, which constitute the chronic phase of the infection (31). If the infection is left untreated, the number of CD4+ T cells eventually falls below a critical level and the immunocompromised patient may die from AIDS-related complications (31). The changes in the number of CD4+ T cells are believed to be caused by virally induced direct or indirect cytopathic effect, which is mediated by both caspase-dependent and caspase-independent pathways (32–34). Cytotoxic CD8+ T lymphocytes (CTLs) are also implicated in the control of viremia and the death of infected CD4+ T cells (35, 36), and are described in more detail below.
cART and Disease Controllers
The administration of cART suppresses plasma viremia to an undetectable level in a majority of HIV-infected patients (2). A typical cART uses small molecule inhibitors that target different components of the virus replication cycle, such as reverse transcriptase, viral protease, and integrase, while additional drugs can be employed to target host components such as the co-receptor for viral entry, CCR5 (2). Nevertheless, cART is unable to remove the provirus that has been integrated into the host genome. This is the major limitation of cART: even after the successful suppression of plasma viremia, new virus can be regenerated from the integrated provirus when treatment is interrupted. These cells together form the HIV cellular reservoir (12). Therefore, novel therapies that target and eliminate the viral reservoir are needed to prevent viral rebound from those cells—that is, a cure for HIV [reviewed by Katlama et al. (37)].
There are two strategies for the cure of HIV: the sterilizing cure and functional cure (37). The sterilizing cure involves the removal from the body of every integrated provirus that is able to spawn virus, while the functional cure aims to suppress viral rebound using the body’s immune system without the complete removal of provirus (37).
So far, the only case of a sterilizing cure is referred to as the “Berlin patient” case. In that case, an HIV-infected patient who suffered acute myelogenous leukemia received myeloablative chemotherapy and irradiation, which was followed by the transplantation of bone marrow cells from a CCR5Δ32 donor (38, 39). CCR5Δ32 is a deleterious mutation that abrogates CCR5 expression on the cell surface (38, 39). cART was discontinued after engraftment of the CCR5Δ32 bone marrow cells, and viral rebound has not yet been observed 8 years after the procedures, implicating a sterilizing cure of HIV. Although this case renewed interest in the search for a sterilizing cure, this method would be invasive to an otherwise healthy patient and expensive to implement on a larger scale.
However, a functional cure has occurred “naturally” in a few individuals (<5% of those infected) who have the ability to spontaneously suppress viremia without antiretroviral therapy (40). These patients are referred to as “elite controllers” or “long-term non-progressors” (40). They possess protective HLA haplotypes and potent anti-HIV CTL responses, which may contribute to their smaller viral reservoirs compared with disease progressors (35, 36). Some other patients (<1% of cART-treated), known as post-interruption viremia controllers (PIVCs) (41), are able to spontaneously suppress virus after treatment is interrupted. Interestingly, PIVC patients are not distinguished by a protective HLA subtype: their recovery is correlated more with their low viral load at the time cART is commenced (42, 43) and with the very early initiation of cART (41). Understanding the mechanisms of viral suppression in these individuals will provide important insight that may enable functional cure in disease progressors.
Viral Replication in the Follicles of Lymphoid Tissues
Understanding the tissue site of viral replication will provide clues for the identification of HIV cellular reservoirs. The major replication site of HIV has been found to be in the follicular structure of lymph nodes (LNs) (16, 29, 30, 44–47). LNs are organized into cortex and paracortex areas. Cortex areas containing follicular structures consist mainly of B cells and follicular dendritic cells (FDCs), which are primarily responsible for the humoral immunity. While the paracortex areas contain predominantly T cells and conventional dendritic cells, which are primarily responsible for cellular immunity. Immunohistochemical analysis of LN tissue from patients not treated with antiretroviral drugs has revealed two distinct patterns of viral RNA staining: a diffuse staining confined within the follicles, and cell-associated staining that is scattered throughout the tissue (44). The diffused follicular presence comes from virus-immune complexes trapped on FDCs, while the cell-associated viral RNA comes from virus-infected CD4+ T cells (29, 43). FDCs, although not directly infected by the virus (48), can harbor the virus-immune complexes and serve as a significant reservoir, as studies have shown that this trapped virus can effectively infect CD4+ T cells in vitro even in the presence of viral neutralizing antibodies (49, 50).
While productively infected CD4+ T cells were found throughout the LN tissues, B cell follicles contain 31 times more infected CD4+ T cells than extrafollicular regions, indicating that B cell follicles are the preferred site of viral infection, replication, or both in CD4+ T cells (29). Upon cART, the numbers of viruses bound on FDCs and virus-infected CD4+ T cells are reduced dramatically (51, 52). Nevertheless, some diffused RNA (51) as well as virus-infected CD4+ T cells are still detected in follicles but not in extrafollicular regions (17, 51). These findings suggest that B cell follicles within LN tissues are a hotspot for viral replication in both untreated and cART-treated patients. Understanding the etiology of the B cell follicles as a replication hotspot and targeting this sanctuary site may be a feasible strategy to achieve a functional or sterilizing cure for HIV infection.
Introduction to TFH Cells
CD4+ T cells that reside in B cell follicles, termed TFH cells, follow a distinct differentiation program that leads to a unique transcriptional profile and specialized function (13, 53). TFH cells localize to B cell follicles via a high surface expression of CXC chemokine receptor 5 (CXCR5) and a low level of CC chemokine receptor 7 and are able to promote a humoral response due to their close proximity to B cells and FDCs (54–56). In addition to their strategic location, TFH cells also express co-stimulatory receptors and cytokines that are involved in assisting B cell functioning for the humoral response (57). The upregulated genes include accessory proteins such as ICOS and SAP, which assist T–B interaction (13, 58–62), and the signature cytokine interleukin (IL)-21, which enhances the B cell response (13, 63–65). Within the CXCR5+ TFH cells, a subpopulation that expresses the highest level of programmed death-1 (PD1) is called germinal center TFH cells. These cells are able to produce the most IL-21 with the most potent B cell helper activity compared with the PD1 low counterparts (66).
The differentiation of TFH cells has been an area of intensive study because of their significance in multiple human diseases (67). A complex network of key transcriptional factors that imprints the transcriptional profiles of TFH cells has been identified and shown to tightly coordinate TFH cell differentiation by multiple pathways (13). Cytokines, such as IL-6 (68), IL-7 (69), IL-12 (70, 71), IL-21 (72), type 1 interferons (73, 74), and transforming growth factor-β (74), have been shown to enhance TFH differentiation, while IL-2 (75) inhibits differentiation by the induction of downstream signaling molecules, including signal transducer and activator of transcription proteins and Janus kinases (73, 74, 76–80). Dendritic cells (70, 81) and B cells (61, 82) are also important for TFH differentiation and maintenance in vivo by providing antigenic stimulation, together with many of the cytokines and costimulatory signals.
TFH as a HIV Reservoir in CD4+ T Cells
To realize a cure for HIV infection, the major subset of CD4+ T cells that function as the viral reservoir must be identified and eliminated. The colocalization of HIV RNA and TFH cells in B cell follicles suggests that TFH cells are the major HIV reservoir within CD4+ T cells. Indeed, a number of studies have found that, in untreated humans and non-human primate models, TFH cells harbor higher levels of viral RNA and DNA than non-TFH cells (14–25). To determine whether these cells contain replication-competent virus, TFH cells were sorted and activated in vitro and found to produce significantly more infectious virus than their non-TFH counterparts (20). Importantly, in cART-treated patients who have achieved aviremia, TFH cells remain the major subset with active viral transcription and produce the highest amount of replication-competent virus compared with other subsets (21, 83). Together, these findings show that TFH cells are a major cellular reservoir for HIV in both untreated and cART-treated patients.
In addition to TFH cells, several other subsets of CD4+ T cells have been demonstrated to be preferentially infected in peripheral blood, such as central memory CD4+ T (TCM), transitional memory CD4+ T (TTM) (84), and stem cell-like memory CD4+ T (TSCM) cells (85). In these studies, CD4+ T cell subsets were purified using fluorophore-conjugated antibodies targeting a unique set of surface molecules that are expressed on these subsets. The purified cells were then subjected to the measurement of viral content using different assays. Although these studies identified different CD4+ T cell subsets as the major contributors to the HIV reservoir, the antibodies used in these studies may not have mutually excluded the different subsets of CD4+ T cells. For example, the antibodies and gating strategy used to identify TCM and TTM cells in Chomont’s studies (84) may also have included TFH cells (Figure 1), which have been identified to be the major contributors for HIV reservoir in Banga et al.’s studies (83). Therefore, these studies may not necessarily be conflicting, on the contrary, the studies by Banga et al. may indeed support the early discoveries in Chomont’s studies.
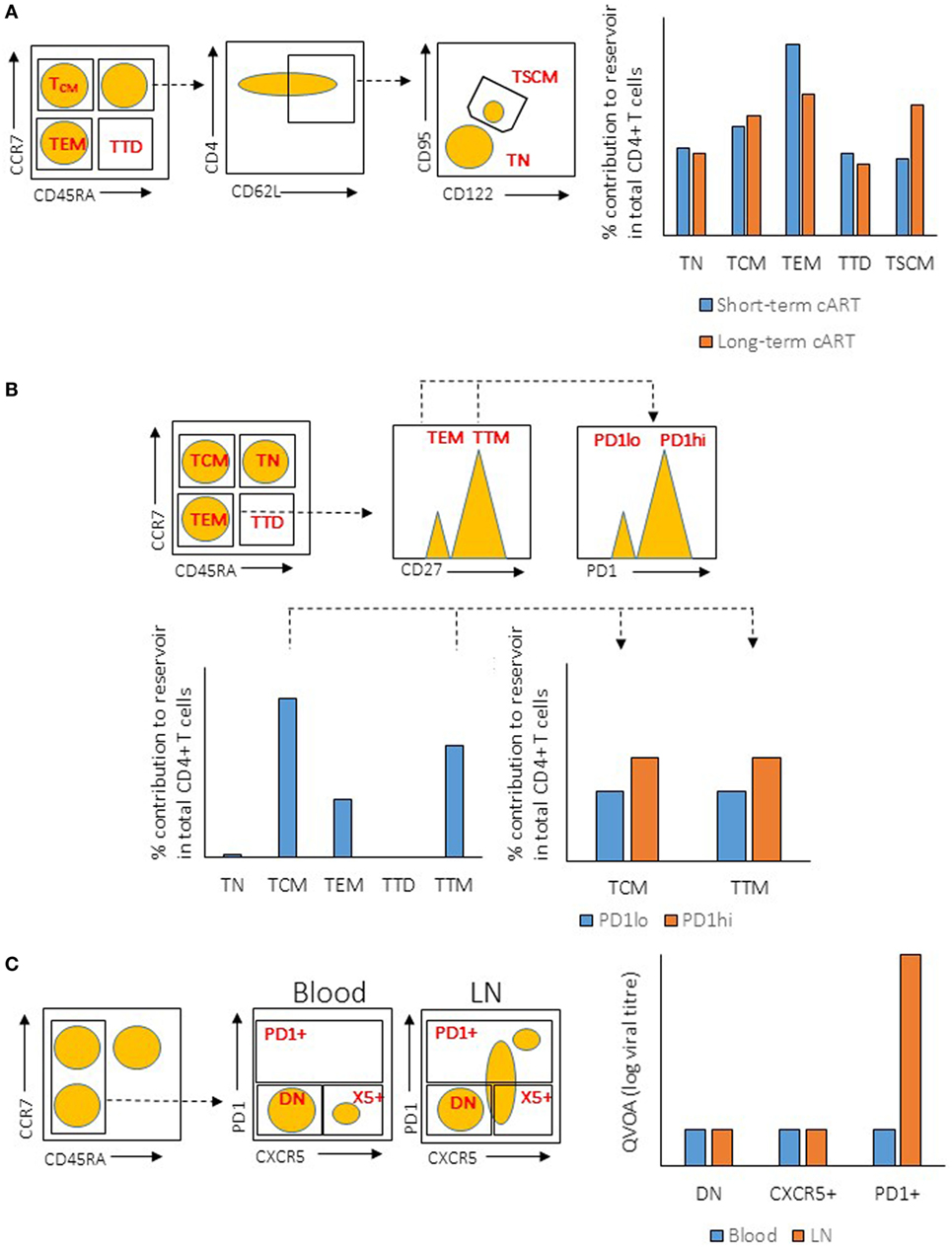
Figure 1. Illustration of gating strategies for CD4+ T cells subsets and their relative viral reservoir among different studies. (A) Gating strategy and CD4+ T cells reservoir characteristic from the study by Buzon et al. (Left) Plots show the gating strategy used to isolate the different population of CD4+ T cells from peripheral blood mononuclear cell (PBMC) (sorted population indicated in red). Dotted line indicates the subgating of the different subsets. (Right) Level of integrated proviral DNA from the sorted CD4+ T cells subsets was determined by PCR after short-term or long-term combination antiretroviral therapeutic regimen (cART). (B) Gating strategy and CD4+ T cells reservoir characteristic from the study by Chomont et al. (Left) Plots show the gating strategy used to isolate the different population of CD4+ T cells from PBMC (sorted population indicated in red). Dotted line indicates the subgating of the different subsets. (Right) Level of integrated proviral DNA from the sorted CD4+ T cells subsets was determined by PCR. (C) Gating strategy and CD4+ T cells reservoir characteristic from the study by Banga et al. (Left) Plots show the gating strategy used to isolate the different population of CD4+ T cells from PBMC or lymph node (LN) tissues (sorted population indicated in red). Dotted line indicates the subgating of the different subsets. (Right) ELISA for viral antigen P24 was used to quantify the level of virus production from the sorted CD4+ T cells subsets after stimulation by anti-CD3/CD28 antibodies [quantitative viral outgrowth assay (QVOA)]. TN, naïve T cells; TTD, terminally differentiated T cells; TEM, effector memory T cells; DN, double-negative cells.
On a different note, different assays have been used to assess the size of the cellular reservoir in these studies, such as PCR-based quantification of proviral DNA and quantification of cells that produce competent virus in vitro [namely, quantitative viral outgrowth assay (QVOA)]. Although PCR quantification is a quick, simple, and cost-effective method, that method is problematic and does not reflect the true reservoir that is responsible for viral rebound (86). The level of viral DNA in infected cells does not correlate with the production of replication-competent viruses, as the majority (approximately 98%) of viral DNA in the cells are replication defective (86–89). This could undermine studies in which viral DNA measurement is the only method used to determine the size of the reservoir, and future studies should utilize multiple quantification methods to accurately measure the true size of HIV reservoir.
In addition to the assay method, the site of tissue sampling may also affect the conclusions in the studies. It has recently been shown that HIV replication is ongoing in tissues while it is absent in peripheral blood mononuclear cells (PBMCs) (90, 91)—a phenomenon that could stem from the insufficient penetration of cART drugs in the tissues (92). Indeed, studies have found that rebound virus originates from lymphoid tissues (47, 93), demonstrating the importance and relevance of studying the tissue viral reservoir instead of the reservoir in PBMCs. A study by Banga et al. (83) demonstrated that TFH cells in LNs, but not PBMCs, are the only subset in CD4+ T cells that produce infectious virus after in vitro stimulation (or QVOA). This is a robust evidence demonstrating that TFH cells are the major reservoir in long-term cART-treated patients. The ability of TFH cells to produce infectious virus in vitro suggests that this reservoir should be eliminated or controlled in order to prevent HIV rebound. Multiple mechanisms may contribute to the establishment of TFH cells as the major reservoir, and each of those mechanisms can be exploited to target and eliminate the reservoir (Figure 2). The mechanisms are discussed in the following sections.
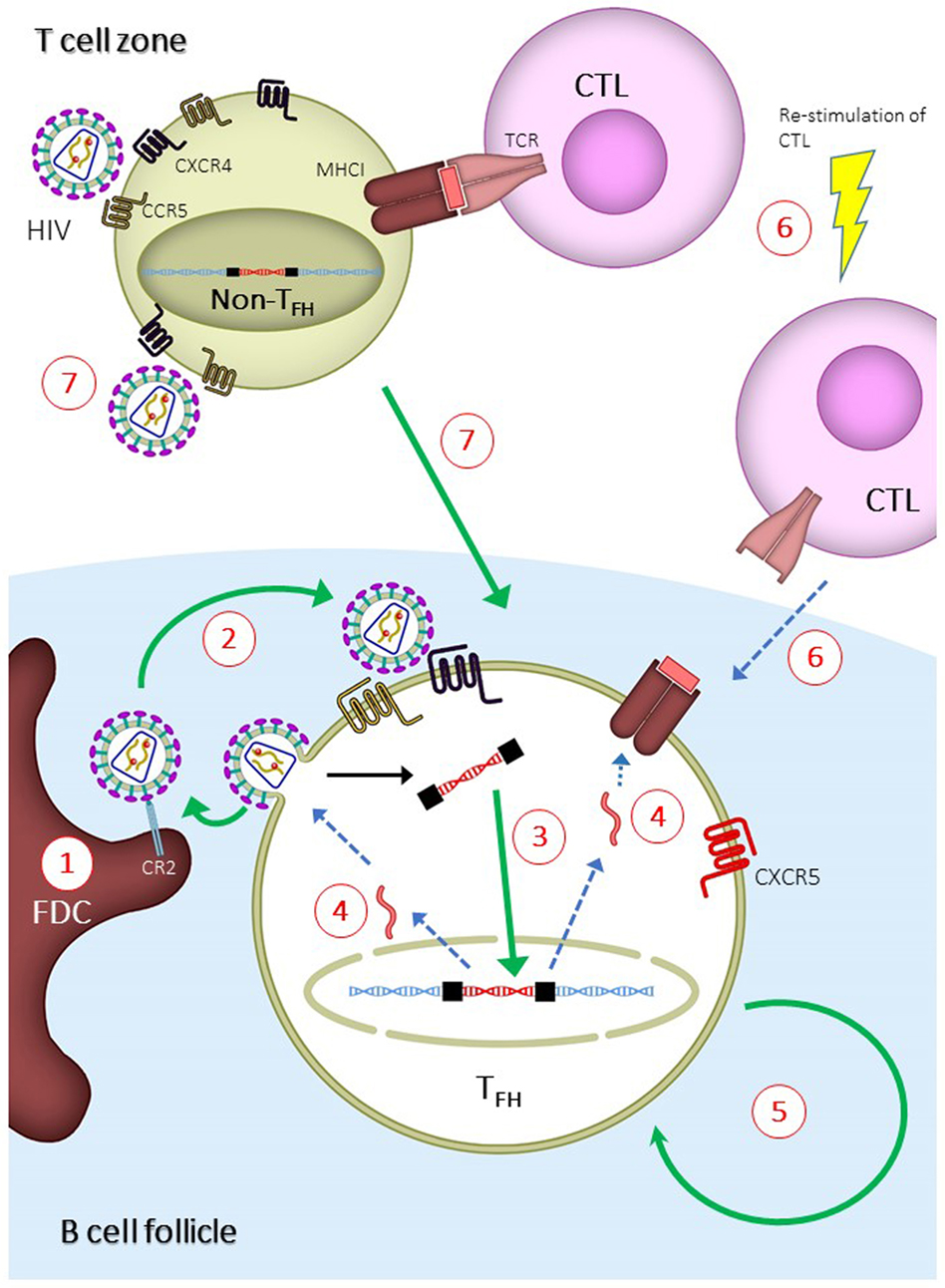
Figure 2. Mechanisms for the establishment of follicular T helper (TFH) cells as major human immunodeficiency virus (HIV) reservoir and proposed strategies to eliminate TFH reservoir. Green or blue arrows indicate pathways that are enhanced or inhibited, respectively, during the establishment of TFH reservoir. (1) Virus-immune complexes deposition on follicular dendritic cells (FDCs) via complement receptor type 2 (CR2) binding. Anti-CR2 can be used to displace virus-immune complexes. (2) Infection of TFH due to suboptimal antiretroviral drug penetration, which can be overcome by development of drugs with higher potency. (3) Increased integration of viral DNA in TFH cells. This can be overcome by inducing host restricting factors, such as SAMHD1, or treatment intensification with integrase inhibitor. (4) Reduced expression of viral genes, which can be overcome by latency reversal agent. (5) Long half-life and homeostatic renewal of latently infected TFH cells. Long-term survival can be inhibited by targeting specific pathways, such as modulation with cytokines and auranofin. (6) Reduced infiltration of CD8+ T lymphocytes (CTLs) into B cell follicles. Potency and follicular infiltration of CTLs can be boosted to control TFH infection. (7) Infection of non-TFH and differentiation of infected non-TFH to TFH. This can be blocked by preventing the differentiation of TFH cells.
Virus-Immune Complexes on FDCs Facilitate TFH Infection
In untreated patients, infectious virus-immune complexes displayed by FDCs, along with the close proximity to TFH cells, allow B cell follicles to become a fertile ground for viral replication. Therefore, understanding the mechanism by which virus-immune complexes are loaded onto FDCs will enable the elimination of this extracellular viral reservoir, which may in turn reduce the infection of TFH cells. Virus-immune complexes can form by opsonization with complement proteins (94). This is demonstrated by the trapping of virus-immune complexes on FDCs that occurs as early as 2 days after infection and before the development of an anti-HIV antibody response (95). This process is mediated by complement component 3 (C3) independent of anti-HIV antibodies (96–98). After deposition on the virus, C3 is then cleaved to iC3b and C3d, which bind to complement receptor type 2 (CR2) on FDCs (99, 100). Interestingly, during the chronic stages of the disease, B cells from untreated patients also harbor virus-immune complexes via CR2-binding (101). Most importantly, after 24 months of cART, by which time the viral loading on FDCs has almost fully disappeared (51, 52), FDCs isolated from these virally suppressed individuals are still able to infect CD4+ T cells in vitro, demonstrating that FDCs do contain infectious virus even under fully suppressed conditions (102). At this stage, virus is retained in the non-degrading cycling endosomes via binding to CR2 (100). These results demonstrate the essential role of complement and CR2 in the formation and maintenance of the extracellular viral reservoir on FDCs in B cell follicles under both before and during cART regimes. Anti-HIV antibodies generated during the later stage of the disease are able to form virus-antibody complexes and are involved in the control of the infection (103). Nevertheless, the deposition of virus-antibody complexes on FDCs has been demonstrated only in an in vitro study (104), and animal models are needed to examine whether this virus-antibody complex can be found on FDCs in vivo.
The complement system is an innate immune mechanism that induces the lysis of enveloped virus and infected cells via the formation of a membrane attacking complex (MAC) (105). The MAC can be activated by a classical complement pathway mediated by antibody opsonized on the targets, or an alternative pathway mediated by molecules such as complement protein C3 on the targets (105). Uninfected cells inhibit the MAC’s activity by expressing cell surface molecules that prevent the activation of the complement cascade, such as decay-accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) (106). The activation of the MAC is therefore the balance of complement activation and inhibition, such that opsonization by complement proteins or antibodies pushes the balance toward complement activation and the induction of the MAC (106). HIV hijacks this host inhibitory system by incorporating CD55 and CD59 molecules into the viral envelope, which then inhibits the MAC-mediated lysis of the virus (107–109). This is demonstrated by the more effective MAC-mediated lysis of virus in the presence of CD59 inhibitors (94). The fact that the extracellular reservoir on FDCs has been found to contain infectious and intact virus further demonstrates that the virus evades MAC-mediated lysis. Purging this extracellular reservoir may eventually reduce the infection of TFH cells, and therefore constrict the TFH reservoir.
Intrinsic TFH Factors That Facilitate Viral Persistence
Note that TFH cells have been found to be a major compartment for HIV production in cART-naïve patients, among whom virus production is active and de novo infection is prevalent. Therefore, in addition to proximity to FDCs as discussed above, two more factors may determine the level of infection in TFH cells: the intrinsic susceptibility of TFH cells to viral infection and the susceptibility of the infected TFH cells to apoptosis.
First, TFH cells isolated from peripheral blood are more permissive to HIV infection in vitro (15, 20, 21, 110), suggesting that intrinsic TFH factors enhance the infection of TFH cells. The enhanced infection could be due to a lack of intracellular host restriction factors, such as SAMHD1, allowing high degree of viral replication (110). Further studies are required to measure the expression level of other host restriction factors (111, 112) in TFH cells and to determine the role of these factors in restricting viral infection. Of note, CCR5 and CXCR4—the co-receptors of HIV—are differentially expressed on TFH cells that are isolated from different tissues (19, 21, 24, 67). To what degree the expression level of co-receptors or intracellular restriction factors might affect the viral infection in TFH cells remains to be determined.
Second, the accumulation of highly infected TFH cells in HIV patients suggests that these cells manage to evade virus-induced direct or indirect cell death. The evasion could be due to host mechanisms that “silence” the viral genome, resulting in minimal viral gene expression (113). Several mechanisms mediate the silencing of the viral genome. Specific cellular activation, such as during the resting memory state, can prevent the full expression of the viral genes because of incomplete host transcriptional machinery (113, 114). In addition, some transcription factors and epigenetic modifications have been shown to actively suppress the expression of viral genes in resting memory T cells (113–115). The transcriptional suppressor BCL6—a critical factor that initiates the TFH cell differentiation program (13)—has been shown to suppress the expression of viral genes in TFH cells (116). Conversely, silencing viral gene expression allows the survival of infected cells via two mechanisms. First, the diminished viral gene expression downregulates viral production, which in turn prevents the virus-induced cytopathic effect (117). Second, the reduced antigen presentation on MHC-I prevents recognition by CTLs (118) or natural killer cells (119) and therefore prevents cell-mediated cytotoxic killing. Both of these mechanisms contribute to reduced apoptosis and the accumulation of infected TFH cells. Vigorous studies are undertaking to understand the mechanism of viral gene suppression and to determine if inducing the expression of viral genes could lead to apoptosis of the infected cells, and therefore the reduction of viral reservoir.
Exclusion of CTLs in B Cell Follicles
CD8+ T lymphocytes have a well-characterized role in controlling HIV infection by inducing the death of cells harboring provirus—a feat that cannot be accomplished by cART (35, 36, 120, 121). Indeed, as mentioned above, elite controllers and long-term non-progressors have the ability to control viremia in the absence of cART (40). These patients have a strong genetic link to protective MHC-I alleles, which are the MHC-I alleles that present specific viral peptides that induce a strong anti-HIV CTL response (122). Similarly, in non-human primate models, a macaque genotype that possesses a protective MHC-I allele is often used as a model to study the role of CTLs in controlling HIV infection (123). In addition to their role in non-treated elite controllers, CTLs are also important for viral control in cART-treated subjects. This was demonstrated by a recent study in macaques showing that cART-mediated viral suppression is lost when CTLs are experimentally depleted (124). In addition to the control of active HIV infection, the non-redundant role of CTLs is also implicated in the establishment of the TFH reservoir.
Another plausible hypothesis for the establishment of TFH cells as a major reservoir comes from the relatively reduced prevalence of CTLs in B cell follicles. In SIV-infected elite controller macaques with a strong CTL response, the infection is confined to TFH cells (16, 46). In experiments where CTLs were depleted, non-TFH cells became strongly infected, while the infection in TFH cells increased only marginally (16). These results demonstrate that the potent CTL response in elite controllers is able to control infection in non-TFH cells, but is less able to do so in TFH cells within B cell follicles (16). Immunohistochemistry confirmed the accumulation of infected TFH cells in B cell follicles and, importantly, demonstrated low numbers of CD8+ T cells in the same anatomical location (16, 46, 125). A lack of CD8+ T cells in B cell follicles was also found in HIV-infected human patients (28, 50, 126). Together, these experiments show that a potent CTL response is able to eliminate the extrafollicular infection of non-TFH cells but is unable to effectively control the intrafollicular infection of TFH cells in both SIV-infected animal models and HIV-infected patients, probably because of the low number of CTLs in B cell follicles.
Nevertheless, our recent study has found that the CTLs in B cell follicles, although lower in number than in extrafollicular regions, have the ability to control infection in the follicles (127). Our first evidence came from the immunohistochemistry staining of LNs from untreated HIV-infected patients (127). Compared with uninfected controls, these patients have significantly increased numbers of CTLs in their B cell follicles (127). We found that, when co-stained with HIV+ RNA, the CTLs are juxtaposed to the infected CD4+ T cells in B cell follicles, indicating the ability of these follicle-infiltrating CTLs to control the infection of TFH cells (127). As mentioned above, CXCR5 is the chemokine receptor expressed on TFH cells that facilitates the positioning of TFH in B cell follicles. Along with other groups, we found a population of CTLs expressing CXCR5 (127–132) that localizes to B cell follicles in both chronically infected mice and HIV+ patients (127, 129). This population—follicular cytotoxic T (TFC) cells—has a transcriptional profile that is distinct from non-TFC CD8+ T cells (127–130). Notably, the TFC population was found to correlate negatively with the level of viremia in untreated HIV-infected patients, demonstrating the ability of these cells to control HIV infection (129).
To determine whether TFC cells are required for the control of infection in B cell follicles, we employed two mouse models of persistent viral infections: the lymphocytic choriomeningitis virus (LCMV) that infects hematopoietic cells and murid-gammaherpesvirus-4 (MuHV-4), a model virus for Epstein–Barr virus in humans who establish latent infection in B cells (127). Interestingly, we found an increased infection of TFH cells compared with non-TFH cells in LCMV infection, suggesting that B cell follicles are common sanctuary sites for lymphotropic virus (127). We then prevented the follicular infiltration of TFC cells by deleting CXCR5 expression in CTLs and found a further accumulation of infected TFH cells (127). A similar accumulation of infected B cells was found in MuHV-4 infection in the absence of CXCR5 on TFC cells (127). Together, these results demonstrate that TFC cells, although low in quantity, are able to control infection in B cell follicles.
The ability of TFC cells to control HIV/SIV infections is further demonstrated in recent studies (128, 131). In these studies, CXCR5+ CD8+ T cells from SIV-infected macaques (131) and HIV-infected patients (128) were found to eliminate virus-infected TFH cells when cocultured in vitro. Moreover, the anti-HIV activity of CXCR5+ CD8+ T cells from HIV-infected patients was significantly enhanced by a bispecific antibody (128). The bispecific antibody is a fusion antibody that contains two covalently linked monoclonal antibodies (mAbs): an anti-CD3 mAb that activates polyclonal CTLs and an anti-HIV mAb (VRC07) that recognizes viral antigen on infected cells (128). The use of the bispecific antibody is a clever design as majority of the CXCR5+ CD8+ T cells in the follicles were found to be non-HIV specific (128), and with the use of this antibody, one can “borrow” the cytotoxicity of these polyclonal CXCR5+ CD8+ T cells to eliminate TFH infection in B cell follicles. All in all, the discovery of TFC cells in chronic infections and their ability to control infection in follicles are important steps toward understanding the follicle sanctuary for HIV infection, and devising strategies to eliminate infection by boosting the number and cytotoxicity of TFC cells.
Long-term Survival of TFH Cells during cART
Combination antiretroviral therapeutic regimen suppresses viral replication, but cannot eliminating cells with integrated provirus. While the provirus-harboring cells evade viral cytopathic effect and CTL-mediated cell death, these infected cells can persist in the body via homeostatic proliferation due to the ability of the integrated provirus to propagate via mitotic division. The steady maintenance of this viral reservoir is therefore dependent on the infected cells’ rate of homeostatic proliferation and susceptibility to apoptosis, and the duration of these cells to remain in the interphase of the cell-division cycle (133). Indeed, the half-life of virus-infected resting memory T cells during cART is approximately 44 months, reflecting the long-term homeostatic maintenance of memory T cells (134, 135). Among the resting memory T cells, infected TCM and TTM cells persist via T cell receptor-driven and IL-7-mediated homeostatic proliferation, respectively (84). TFH cells, as described above, contain more infected cells than TCM and TTM cell populations, constituting the major reservoir within memory T cells during cART (21, 83). While the half-life of infected TFH cells during cART is not known, TFH cells are able to form long-term memory (136–138) and require IL-7 signaling to maintain cell numbers in vivo (68, 139), indicating a mechanism similar to TTM cells for the persistence of the viral reservoir. TFH cells from HIV-infected patients were found to express higher levels of an anti-apoptotic gene, BCL2, which may further facilitate the persistence of the TFH reservoir by suppressing apoptosis (140). In addition to maintaining cell survival and reducing apoptosis, TFH cells also appear to be preferentially multiplied compared with their non-TFH counterparts (17, 20, 22, 24, 141). The expansion of TFH cells is positively correlated with the level of IL-6, a cytokine that promotes TFH differentiation in the plasma (7, 17, 22). The molecular mechanisms for the long-term persistence and expansion of TFH cells remain poorly understood; however, targeting these pathways is a plausible strategy to reduce the viral reservoir in this compartment.
How to Eliminate the TFH Reservoir in B Cell Follicles
In this review, we define the HIV reservoir as the cellular populations that are able to generate infectious virus after treatment interruption. According to this definition, the HIV reservoir would encompass the reservoirs with the following characteristics: (1) latently infected cells with integrated proviruses that do not express viral genes during ART, yet are able to generate infectious virus after treatment interruption. This reservoir includes cells that contain HIV DNA but not HIV RNA (142); (2) infected cells with integrated proviruses expressing viral genes, which contain both cell-associated HIV DNA and RNA (142); (3) active viral replication in tissues reservoir, which is identified by the evolution of HIV DNA at tissue sanctuary sites (see below); (4) extracellular viral reservoir, which consist of infectious viruses bound to FDCs in B cell follicles. All of the abovementioned reservoirs are either found in TFH cells (point 1 and 2) or are related to tissues’ sanctuary sites (point 3) and B cell follicles (point 4). Therefore, targeting the B cell follicles in lymphoid tissues might significantly impact all types of reservoirs. In this section, we discuss the potential strategies for targeting the B cell follicles to eliminate the HIV reservoir (Figure 2).
Eliminating Residual Replication and Purging the FDC Reservoir
As discussed above, FDCs may facilitate the infection of TFH cells due to the proximity of the two within B cell follicles. Nonetheless, it is disputable whether active viral infection and replication occur in vivo under cART that blocks the de novo infection of CD4+ T cells [see the review by Eisele and Siliciano (143)]. Residual viremia after prolonged cART has been detected at below 50 copies/mL using sensitive methods, indicating a continuous production of virus. The source of this viremia—whether it is produced from infected cells with integrated provirus or is due to continual viral replication that escapes cART suppression—is still debatable. Some studies have shown conclusive evidence of continual viral replication in lymphoid tissues (90, 92), while contradictory evidence was observed in PBMCs (144–147). For lymphoid tissues, insufficient penetration of antiretroviral agents into the lymphoid follicles could be one of the mechanisms that cause residual viral replication (92). For PBMCs, a definitive study on Raltegravir, an antiretroviral drug that inhibits the integration of linear cDNA into chromosomes, showed that viral replication is continual in the PBMCs of some cART patients (148, 149). During active viral infection, the reversed transcribed linear cDNA must be integrated into the chromosome to allow the production of new virus. At this stage, the host DNA repair mechanisms are able to induce the self-ligation of linear DNA to form circular 2-long terminal repeat (2-LTR) DNA as a way of preventing chromosomal integration (148, 149). After Raltegravir administration to cART-treated patients, the level of 2-LTR increased in 29% of the individuals, suggesting that active viral replication and infection continue to occur in these individuals (149). This is likely to be due to the suboptimal suppression of viral replication by standard cART.
In summary, patients enrolled for reservoir purging trials should have minimal residual viral replication in the body in order to prevent the replenishment of the reservoir after purging. FDCs, on the other hand, can store virus-immune complexes that facilitate the reinfection of TFH cells in B cell follicles. Virus-immune complexes can be displaced from FDCs and B cells using anti-CR2 antibodies (150), or directly destroyed by inducing the formation of a MAC using anti-CD55 antibody or the bacterial toxin intermedilysin that blocks host inhibitor CD55 (94). Minimizing the residual replication and loading of the virus-immune complex on FDCs would be an important consideration to prevent the accumulation of infected TFH cells before attempting to purge the provirus-containing TFH cells.
Reduce the Number of Memory TFH Cells
The long half-life of resting memory T cells enables the persistence of the HIV reservoir. Purging total memory cells, including memory TFH cells, could be a plausible method of reducing the TFH reservoir. A proof of concept for this strategy is the use of auranofin, a gold-based chemical that targets the metabolic profile of TCM and TTM cells (151) to reduce the size of the latent reservoir (152). In combination with cART, auranofin has been found to reduce viral rebound and spontaneously control SIV in macaques (153), which resemble human PIVCs. A similar strategy can be used to target TFH cells, as TFH cells have been shown to possess a metabolic profile different from other CD4+ T subtypes (57, 154–156). Other survival pathways can also be targeted, such as by blocking the activity of IL-7, which maintains the long-term survival of TFH cells (69), as well as by blocking the activity of the antiapoptotic BCL2 gene that is highly expressed on TFH cells (140). Similarly, modulating the differentiation of TFH cells may also reduce the total number of TFH cells. We have shown that low-dose IL-2 treatment is able to reduce the number of TFH cells in patients with systemic lupus erythematosus (157). Treatment with IL-2 or anti-IL-6 may inhibit TFH cell differentiation and prevent the accumulation of infected TFH cells. Other novel reagents can also be developed to target the complex regulation of TFH differentiation (158). However, it is important to acknowledge that a gross reduction of TFH cells may negatively impact the humoral immune response, including the anti-HIV antibody response that might have been established over the course of infection. Therefore, more specific targeting of latently infected TFH cells instead of total TFH reduction may be a superior strategy.
Specific Targeting of Latently Infected TFH Cells: Shock and Kill
A new method, “shock and kill,” has been proposed for the elimination of the latent HIV reservoir (159). As mentioned above, silencing viral gene expression prevents the presentation of viral peptides on MHC-I and their recognition by CTLs. To overcome this hurdle, it was proposed to induce the expression of viral genes, which could in turn expose latently infected cells to virus-induced cytopathic effect and immune system-mediated killing (160). In a clinical trial in which cART patients were treated with a histone deacetylase inhibitor—an epigenetic modifier drug belonging to a group of latency reversal agents (LRAs)—viral gene expression was induced in latently infected cells in cART-treated patients (160, 161). Nevertheless, even with the increased viral transcription, treatment with LRAs in vivo did not reduce the size of the viral reservoir (160), and viral rebound was not affected after cART regime was interrupted (161). This observation suggests that “shocking” the latently infected cells to express viral gene products alone is not sufficient, and that the simultaneous induction of the immune system to “kill” is also required (159, 162). To that end, the enhancement of CTL function may be the most suitable candidate in this strategy to kill the latently infected cells (159, 162).
Cytotoxic killing by CTLs is needed to control and eliminate the latent cellular reservoir; however, CTLs in HIV-infected patients are often defective due to exhaustion (163) and mutations in viral genes that allow infected cells to escape elimination by CTLs (164). Exhaustion is a state of CTLs that is characterized by reduced effector functions and failure to control infections, which is typically observed in chronic infections such as with HIV and hepatitis C and B viruses (163). Persistent antigenic signaling and inflammatory signals contribute to the exhaustion of CTLs, and revitalizing them has been shown to restore immunity (163). Indeed, in vitro LRAs showed that latently infected CD4+ T cells can only be killed by IL-2-activated allogenic CTLs, but not by unstimulated CTLs, demonstrating that the restimulation of exhausted CTLs can be useful to eliminate the latent reservoir (162). In addition to CTL exhaustion, viral CTL escape mutations also play a significant role in viral persistence (164). In patients treated early with cART, CTL clones are able to recognize non-escaped mutations and kill infected CD4+ T cells (165). In patients who did not receive treatment early, CTLs failed to recognize virus-infected cells due to emergence of escape mutations (165). These studies demonstrate that revitalizing CTL clones in addition to targeting non-escape mutations are pre-requisites for the shock-and-kill strategy.
Finally, to target the TFH cell reservoir, CTLs need to penetrate the B cell follicular sanctuary to reach the latently infected TFH cells. The depletion of B cell follicles using depleting antibodies has been proposed to achieve this. However, depleting B cell follicles can result in a dysfunctional humoral response and a compromised anti-HIV antibody response. To specifically target infected TFH cells, promoting the infiltration of CTLs into B cell follicles is a preferred strategy. Recent publications highlighted the critical role of CXCR5+ TFC cells in the control of follicular infections (127–129, 131), suggesting possible therapeutic benefits of boosting the number and antiviral activity of TFC cells in B cell follicles. Finally, to enable CTLs’ detection of infected TFH cells, special consideration might be needed to “shock” viral gene expression in TFH cells due to the ability of the TFH-specific transcription factor BCL6 to suppress viral gene expression.
All in all, multiple strategies are needed to reduce and control the viral reservoir in TFH cells. This may eventually bring the reservoir under control to achieve a functional cure, which may in turn eliminate viral rebound and viral transmission in the absence of cART.
Conclusion
This review originates from the recent discoveries demonstrating that TFH cells are the major viral reservoir among CD4+ T cells. We have proposed hypotheses to explain the formation of this reservoir and strategies for its elimination. The major HIV reservoir in TFH cells is a concrete observation that agrees well with the long-observed hotspot for HIV replication, the B cell follicles. Nonetheless, more studies are needed to validate the significance of this cellular reservoir and to determine whether removing the reservoir can considerably delay or eliminate viral rebound. While viral DNA and RNA in CD4+ T cells have been the strongest predictor of the “time to viral rebound” [that is, higher levels of cell-associated RNA and DNA lead to a shorter time to viral rebound after treatment is interrupted (166)], it would be interesting to learn whether the level of provirus in TFH cells can be used as a superior predictor for the time to rebound. Hence, in addition to helping us understand HIV pathogenesis, the TFH reservoir can also be developed as a biomarker to assess the efficacy of therapeutic interventions in HIV cure studies.
At least two types of reservoir exist in B cell follicles during cART: the virus-immune complexes retained on the surface of antigen presenting cells and the intracellular proviral reservoir that produces infectious virus when treatment is interrupted. Both of these reservoirs should be eliminated to prevent viral rebound, and extra effort is needed to target these reservoirs in B cell follicles. The hypotheses and strategies proposed here ought to be empirically verified in animal models to determine the true cause(s) for the establishment of the TFH reservoir and to determine the most rational and promising therapeutic interventions. After some great successes but also some limitations in using SIV-infected non-human primate models to study human HIV infection, humanized mice are also being developed as excellent tools for the study of HIV pathogenesis. The successful grafting of TFH cells into the mice would allow us to study the role of the TFH cell reservoir in chronic HIV disease (24). The role of TFH cells as the major reservoir among CD4+ T cells warrants further investigation into its role in HIV pathogenesis, and such crucial findings should not be overlooked in the development of therapeutic strategies to cure HIV infection.
Author Contributions
YL and DY wrote the manuscript. AA revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the National Health and Medical Research Council of Australia (YL; GNT1085509 to DY), the amfAR Research Consortium on HIV Eradication (109327-59-RGRL, DY), The Creative and Novel Ideas in HIV Research Program of The International AIDS Society (DY), and Australian Centre for HIV and Hepatitis Virology Research (2015-69 to DY).
References
1. Hemelaar J. The origin and diversity of the HIV-1 pandemic. Trends Mol Med (2012) 18(3):182–92. doi: 10.1016/j.molmed.2011.12.001
2. Pau AK, George JM. Antiretroviral therapy: current drugs. Infect Dis Clin North Am (2014) 28(3):371–402. doi:10.1016/j.idc.2014.06.001
3. Kabbara WK, Ramadan WH. Emtricitabine/rilpivirine/tenofovir disoproxil fumarate for the treatment of HIV-1 infection in adults. J Infect Public Health (2015) 8(5):409–17. doi:10.1016/j.jiph.2015.04.020
4. Hong FF, Mellors JW. Impact of antiretroviral therapy on HIV-1 persistence: the case for early initiation. AIDS Rev (2015) 17(2):71–82.
5. Deeks SG. Immune dysfunction, inflammation, and accelerated aging in patients on antiretroviral therapy. Top HIV Med (2009) 17(4):118–23.
6. d’Ettorre G, Ceccarelli G, Pavone P, Vittozzi P, De Girolamo G, Schietroma I, et al. What happens to cardiovascular system behind the undetectable level of HIV viremia? AIDS Res Ther (2016) 13:21. doi:10.1186/s12981-016-0105-z
7. Neuhaus J, Jacobs DR Jr, Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis (2010) 201(12):1788–95. doi:10.1086/652749
8. Dube MP, Sattler FR. Inflammation and complications of HIV disease. J Infect Dis (2010) 201(12):1783–5. doi:10.1086/652751
9. Frost SD, Martinez-Picado J, Ruiz L, Clotet B, Brown AJ. Viral dynamics during structured treatment interruptions of chronic human immunodeficiency virus type 1 infection. J Virol (2002) 76(3):968–79. doi:10.1128/JVI.76.3.968-979.2002
10. Oxenius A, Price DA, Günthard HF, Dawson SJ, Fagard C, Perrin L, et al. Stimulation of HIV-specific cellular immunity by structured treatment interruption fails to enhance viral control in chronic HIV infection. Proc Natl Acad Sci U S A (2002) 99(21):13747–137452. doi:10.1073/pnas.202372199
11. Ruiz L, Martinez-Picado J, Romeu J, Paredes R, Zayat MK, Marfil S, et al. Structured treatment interruption in chronically HIV-1 infected patients after long-term viral suppression. AIDS (2000) 14(4):397–403. doi:10.1097/00002030-200003100-00013
12. Barton K, Winckelmann A, Palmer S. HIV-1 reservoirs during suppressive therapy. Trends Microbiol (2016) 24(5):345–55. doi:10.1016/j.tim.2016.01.006
13. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi:10.1146/annurev-immunol-041015-055605
14. Hufert FT, van Lunzen J, Janossy G, Bertram S, Schmitz J, Haller O, et al. Germinal centre CD4+ T cells are an important site of HIV replication in vivo. AIDS (1997) 11(7):849–57. doi:10.1097/00002030-199707000-00003
15. Kohler SL, Pham MN, Folkvord JM, Arends T, Miller SM, Miles B, et al. Germinal center T follicular helper cells are highly permissive to HIV-1 and alter their phenotype during virus replication. J Immunol (2016) 196(6):2711–22. doi:10.4049/jimmunol.1502174
16. Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med (2015) 21(2):132–9. doi:10.1038/nm.3781
17. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest (2012) 122(9):3281–94. doi:10.1172/JCI63039
18. Brenchley JM, Vinton C, Tabb B, Hao XP, Connick E, Paiardini M, et al. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood (2012) 120(20):4172–81. doi:10.1182/blood-2012-06-437608
19. Mylvaganam GH, Velu V, Hong JJ, Sadagopal S, Kwa S, Basu R, et al. Diminished viral control during simian immunodeficiency virus infection is associated with aberrant PD-1hi CD4 T cell enrichment in the lymphoid follicles of the rectal mucosa. J Immunol (2014) 193(9):4527–36. doi:10.4049/jimmunol.1401222
20. Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med (2013) 210(1):143–56. doi:10.1084/jem.20121932
21. Pallikkuth S, Sharkey M, Babic DZ, Gupta S, Stone GW, Fischl MA, et al. Peripheral T follicular helper cells are the major HIV reservoir within central memory CD4 T cells in peripheral blood from chronically HIV-infected individuals on combination antiretroviral therapy. J Virol (2016) 90(6):2718–28. doi:10.1128/JVI.02883-15
22. Xu H, Wang X, Malam N, Aye PP, Alvarez X, Lackner AA, et al. Persistent simian immunodeficiency virus infection drives differentiation, aberrant accumulation, and latent infection of germinal center follicular T helper cells. J Virol (2016) 90(3):1578–87. doi:10.1128/JVI.02471-15
23. Klatt NR, Vinton CL, Lynch RM, Canary LA, Ho J, Darrah PA, et al. SIV infection of rhesus macaques results in dysfunctional T- and B-cell responses to neo and recall Leishmania major vaccination. Blood (2011) 118(22):5803–12. doi:10.1182/blood-2011-07-365874
24. Allam A, Majji S, Peachman K, Jagodzinski L, Kim J, Ratto-Kim S, et al. TFH cells accumulate in mucosal tissues of humanized-DRAG mice and are highly permissive to HIV-1. Sci Rep (2015) 5:10443. doi:10.1038/srep10443
25. Boritz EA, Darko S, Swaszek L, Wolf G, Wells D, Wu X, et al. Multiple origins of virus persistence during natural control of HIV infection. Cell (2016) 166(4):1004–15. doi:10.1016/j.cell.2016.06.039
26. Sattentau QJ, Moore JP. The role of CD4 in HIV binding and entry. Philos Trans R Soc Lond B Biol Sci (1993) 342(1299):59–66. doi:10.1098/rstb.1993.0136
27. Vicenzi E, Lio P, Poli G. The puzzling role of CXCR4 in human immunodeficiency virus infection. Theranostics (2013) 3(1):18–25. doi:10.7150/thno.5392
28. Embretson J, Zupancic M, Ribas JL, Burke A, Racz P, Tenner-Racz K, et al. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature (1993) 362(6418):359–62. doi:10.1038/362359a0
29. Folkvord JM, Armon C, Connick E. Lymphoid follicles are sites of heightened human immunodeficiency virus type 1 (HIV-1) replication and reduced antiretroviral effector mechanisms. AIDS Res Hum Retroviruses (2005) 21(5):363–70. doi:10.1089/aid.2005.21.363
30. Spiegel H, Herbst H, Niedobitek G, Foss HD, Stein H. Follicular dendritic cells are a major reservoir for human immunodeficiency virus type 1 in lymphoid tissues facilitating infection of CD4+ T-helper cells. Am J Pathol (1992) 140(1):15–22.
32. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505(7484):509–14. doi:10.1038/nature12940
34. Doitsh G, Greene WC. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe (2016) 19(3):280–91. doi:10.1016/j.chom.2016.02.012
35. McIlroy D. Do HIV-specific CTL continue to have an antiviral function during antiretroviral therapy? If not, why not, and what can be done about it? Front Immunol (2013) 4:52. doi:10.3389/fimmu.2013.00052
36. Jones RB, Walker BD. HIV-specific CD8(+) T cells and HIV eradication. J Clin Invest (2016) 126(2):455–63. doi:10.1172/JCI80566
37. Katlama C, Deeks SG, Autran B, Martinez-Picado J, van Lunzen J, Rouzioux C, et al. Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet (2013) 381(9883):2109–17. doi:10.1016/S0140-6736(13)60104-X
38. Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med (2009) 360(7):692–8. doi:10.1056/NEJMoa0802905
39. Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5 Delta32/Delta32 stem cell transplantation. Blood (2011) 117(10):2791–9. doi:10.1182/blood-2010-09-309591
41. Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog (2013) 9(3):e1003211. doi:10.1371/journal.ppat.1003211
42. Goujard C, Girault I, Rouzioux C, Lécuroux C, Deveau C, Chaix ML, et al. HIV-1 control after transient antiretroviral treatment initiated in primary infection: role of patient characteristics and effect of therapy. Antivir Ther (2012) 17(6):1001–9. doi:10.3851/IMP2273
43. Lodi S, Meyer L, Kelleher AD, Rosinska M, Ghosn J, Sannes M, et al. Immunovirologic control 24 months after interruption of antiretroviral therapy initiated close to HIV seroconversion. Arch Intern Med (2012) 172(16):1252–5. doi:10.1001/archinternmed.2012.2719
44. Biberfeld P, Chayt KJ, Marselle LM, Biberfeld G, Gallo RC, Harper ME. HTLV-III expression in infected lymph nodes and relevance to pathogenesis of lymphadenopathy. Am J Pathol (1986) 125(3):436–42.
45. Orenstein JM. HIV expression in surgical specimens. AIDS Res Hum Retroviruses (2008) 24(7):947–55. doi:10.1089/aid.2008.0265
46. Connick E, Folkvord JM, Lind KT, Rakasz EG, Miles B, Wilson NA, et al. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of rhesus macaques is linked to disease stage and inversely related to localization of virus-specific CTL. J Immunol (2014) 193(11):5613–25. doi:10.4049/jimmunol.1401161
47. Horiike M, Iwami S, Kodama M, Sato A, Watanabe Y, Yasui M, et al. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology (2012) 423(2):107–18. doi:10.1016/j.virol.2011.11.024
48. Reinhart TA, Rogan MJ, Viglianti GA, Rausch DM, Eiden LE, Haase AT. A new approach to investigating the relationship between productive infection and cytopathicity in vivo. Nat Med (1997) 3(2):218–21. doi:10.1038/nm0297-218
49. Heath SL, Tew JG, Tew JG, Szakal AK, Burton GF. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature (1995) 377(6551):740–4. doi:10.1038/377740a0
50. Smith BA, Gartner S, Liu Y, Perelson AS, Stilianakis NI, Keele BF, et al. Persistence of infectious HIV on follicular dendritic cells. J Immunol (2001) 166(1):690–6. doi:10.4049/jimmunol.166.1.690
51. Tenner-Racz K, Stellbrink HJ, van Lunzen J, Schneider C, Jacobs JP, Raschdorff B, et al. The unenlarged lymph nodes of HIV-1-infected, asymptomatic patients with high CD4 T cell counts are sites for virus replication and CD4 T cell proliferation: the impact of highly active antiretroviral therapy. J Exp Med (1998) 187(6):949–59. doi:10.1084/jem.187.6.949
52. Cavert W, Notermans DW, Staskus K, Wietgrefe SW, Zupancic M, Gebhard K, et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science (1997) 276(5314):960–4. doi:10.1126/science.276.5314.960
53. Yu D, Batten M, Mackay CR, King C. Lineage specification and heterogeneity of T follicular helper cells. Curr Opin Immunol (2009) 21(6):619–25. doi:10.1016/j.coi.2009.09.013
54. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med (2000) 192(11):1553–62. doi:10.1084/jem.192.11.1553
55. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
56. Arnold CN, Campbell DJ, Lipp M, Butcher EC. The germinal center response is impaired in the absence of T cell-expressed CXCR5. Eur J Immunol (2007) 37(1):100–9. doi:10.1002/eji.200636486
57. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
58. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med (2015) 212(2):217–33. doi:10.1084/jem.20141432
59. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote TFH cell differentiation. Immunity (2015) 42(2):239–51. doi:10.1016/j.immuni.2015.01.017
60. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor BCL6. Immunity (2011) 34(6):932–46. doi:10.1016/j.immuni.2011.03.023
61. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature (2013) 496(7446):523–7. doi:10.1038/nature12058
62. Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol (2010) 185(1):190–202. doi:10.4049/jimmunol.0903505
63. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med (2010) 207(2):353–63. doi:10.1084/jem.20091738
64. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi:10.1084/jem.20091777
65. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (2014) 345(6200):1058–62. doi:10.1126/science.1257861
66. Xu H, Wang X, Lackner AA, Veazey RS. PD-1(HIGH) Follicular CD4 T helper cell subsets residing in lymph node germinal centers correlate with B cell maturation and IgG production in rhesus macaques. Front Immunol (2014) 5:85. doi:10.3389/fimmu.2014.00085
67. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly: TFH cells in human health and disease. Nat Rev Immunol (2013) 13(6):412–26. doi:10.1038/nri3447
68. Chavele KM, Merry E, Ehrenstein MR. Cutting edge: circulating plasmablasts induce the differentiation of human T follicular helper cells via IL-6 production. J Immunol (2015) 194(6):2482–5. doi:10.4049/jimmunol.1401190
69. Seo YB, Im SJ, Namkoong H, Kim SW, Choi YW, Kang MC, et al. Crucial roles of interleukin-7 in the development of T follicular helper cells and in the induction of humoral immunity. J Virol (2014) 88(16):8998–9009. doi:10.1128/JVI.00534-14
70. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity (2009) 31(1):158–69. doi:10.1016/j.immuni.2009.04.016
71. Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol (2009) 87(8):590–600. doi:10.1038/icb.2009.64
72. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29(1):127–37. doi:10.1016/j.immuni.2008.06.001
73. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity (2014) 40(3):367–77. doi:10.1016/j.immuni.2014.02.005
74. Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, et al. Type I IFN induces binding of STAT1 to Bcl6: divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol (2014) 192(5):2156–66. doi:10.4049/jimmunol.1300675
75. Ballesteros-Tato A, León B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity (2012) 36(5):847–56. doi:10.1016/j.immuni.2012.02.012
76. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol (2014) 15(9):856–65. doi:10.1038/ni.2947
77. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood (2012) 119(17):3997–4008. doi:10.1182/blood-2011-11-392985
78. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol (2013) 190(7):3049–53. doi:10.4049/jimmunol.1203032
79. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med (2012) 209(2):243–50. doi:10.1084/jem.20111174
80. Nurieva RI, Podd A, Chen Y, Alekseev AM, Yu M, Qi X, et al. STAT5 protein negatively regulates T follicular helper (Tfh) cell generation and function. J Biol Chem (2012) 287(14):11234–9. doi:10.1074/jbc.M111.324046
81. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature (2016) 533(7601):110–4. doi:10.1038/nature17947
82. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38(3):596–605. doi:10.1016/j.immuni.2012.11.020
83. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med (2016) 22(7):754–61. doi:10.1038/nm.4113
84. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med (2009) 15(8):893–900. doi:10.1038/nm.1972
85. Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med (2014) 20(2):139–42. doi:10.1038/nm.3445
86. Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol (2016) 14(1):55–60. doi:10.1038/nrmicro.2015.5
87. Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell (2013) 155(3):540–51. doi:10.1016/j.cell.2013.09.020
88. Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med (2016) 22(9):1043–9. doi:10.1038/nm.4156
89. Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, et al. HIV-1 integration landscape during latent and active infection. Cell (2015) 160(3):420–32. doi:10.1016/j.cell.2015.01.020
90. Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Kosakovsky Pond SL, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature (2016) 530(7588):51–6. doi:10.1038/nature16933
91. Wong JK, Yukl SA. Tissue reservoirs of HIV. Curr Opin HIV AIDS (2016) 11(4):362–70. doi:10.1097/COH.0000000000000293
92. Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A (2014) 111(6):2307–12. doi:10.1073/pnas.1318249111
93. Rothenberger MK, Keele BF, Wietgrefe SW, Fletcher CV, Beilman GJ, Chipman JG, et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc Natl Acad Sci U S A (2015) 112(10):E1126–34. doi:10.1073/pnas.1414926112
94. Yu Q, Yu R, Qin X. The good and evil of complement activation in HIV-1 infection. Cell Mol Immunol (2010) 7(5):334–40. doi:10.1038/cmi.2010.8
95. Schacker T, Little S, Connick E, Gebhard-Mitchell K, Zhang ZQ, Krieger J, et al. Rapid accumulation of human immunodeficiency virus (HIV) in lymphatic tissue reservoirs during acute and early HIV infection: implications for timing of antiretroviral therapy. J Infect Dis (2000) 181(1):354–7. doi:10.1086/315178
96. Bánki Z, Kacani L, Rusert P, Pruenster M, Wilflingseder D, Falkensammer B, et al. Complement dependent trapping of infectious HIV in human lymphoid tissues. AIDS (2005) 19(5):481–6. doi:10.1097/01.aids.0000162336.20439.8d
97. Boyer V, Desgranges C, Trabaud MA, Fischer E, Kazatchkine MD. Complement mediates human immunodeficiency virus type 1 infection of a human T cell line in a CD4- and antibody-independent fashion. J Exp Med (1991) 173(5):1151–8. doi:10.1084/jem.173.5.1151
98. Doepper S, Stoiber H, Kacani L, Sprinzl G, Steindl F, Prodinger WM, et al. B cell-mediated infection of stimulated and unstimulated autologous T lymphocytes with HIV-1: role of complement. Immunobiology (2000) 202(3):293–305. doi:10.1016/S0171-2985(00)80035-2
99. Bánki Z, Wilflingseder D, Ammann CG, Pruenster M, Müllauer B, Holländer K, et al. Factor I-mediated processing of complement fragments on HIV immune complexes targets HIV to CR2-expressing B cells and facilitates B cell-mediated transmission of opsonized HIV to T cells. J Immunol (2006) 177(5):3469–76. doi:10.4049/jimmunol.177.5.3469
100. Ho J, Moir S, Kulik L, Malaspina A, Donoghue ET, Miller NJ, et al. Role for CD21 in the establishment of an extracellular HIV reservoir in lymphoid tissues. J Immunol (2007) 178(11):6968–74. doi:10.4049/jimmunol.178.11.6968
101. Moir S, Malaspina A, Li Y, Chun TW, Lowe T, Adelsberger J, et al. B cells of HIV-1-infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. J Exp Med (2000) 192(5):637–46. doi:10.1084/jem.192.5.637
102. Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, et al. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J Virol (2008) 82(11):5548–61. doi:10.1128/JVI.00124-08
103. Huber M, Trkola A. Humoral immunity to HIV-1: neutralization and beyond. J Intern Med (2007) 262(1):5–25. doi:10.1111/j.1365-2796.2007.01819.x
104. Smith-Franklin BA, Keele BF, Tew JG, Gartner S, Szakal AK, Estes JD, et al. Follicular dendritic cells and the persistence of HIV infectivity: the role of antibodies and Fcgamma receptors. J Immunol (2002) 168(5):2408–14. doi:10.4049/jimmunol.168.5.2408
105. Rus H, Cudrici C, Niculescu F. The role of the complement system in innate immunity. Immunol Res (2005) 33(2):103–12. doi:10.1385/IR:33:2:103
106. Morgan BP. Regulation of the complement membrane attack pathway. Crit Rev Immunol (1999) 19(3):173–98. doi:10.1615/CritRevImmunol.v19.i3.10
107. Saifuddin M, Parker CJ, Peeples ME, Gorny MK, Zolla-Pazner S, Ghassemi M, et al. Role of virion-associated glycosylphosphatidylinositol-linked proteins CD55 and CD59 in complement resistance of cell line-derived and primary isolates of HIV-1. J Exp Med (1995) 182(2):501–9. doi:10.1084/jem.182.2.501
108. Schmitz J, Zimmer JP, Kluxen B, Aries S, Bögel M, Gigli I, et al. Antibody-dependent complement-mediated cytotoxicity in sera from patients with HIV-1 infection is controlled by CD55 and CD59. J Clin Invest (1995) 96(3):1520–6. doi:10.1172/JCI118190
109. Stoiber H, Pintér C, Siccardi AG, Clivio A, Dierich MP. Efficient destruction of human immunodeficiency virus in human serum by inhibiting the protective action of complement factor H and decay accelerating factor (DAF, CD55). J Exp Med (1996) 183(1):307–10. doi:10.1084/jem.183.1.307
110. Ruffin N, Brezar V, Ayinde D, Lefebvre C, Schulze Zur Wiesch J, van Lunzen J, et al. Low SAMHD1 expression following T-cell activation and proliferation renders CD4+ T cells susceptible to HIV-1. AIDS (2015) 29(5):519–30. doi:10.1097/QAD.0000000000000594
111. Raposo RA, Abdel-Mohsen M, Deng X, Hecht FM, Pilcher CD, Pillai SK, et al. Dynamic regulation of host restriction factor expression over the course of HIV-1 infection in vivo. J Virol (2014) 88(19):11624–9. doi:10.1128/JVI.01771-14
112. Abdel-Mohsen M, Wang C, Strain MC, Lada SM, Deng X, Cockerham LR, et al. Select host restriction factors are associated with HIV persistence during antiretroviral therapy. AIDS (2015) 29(4):411–20. doi:10.1097/QAD.0000000000000572
113. Trono D, Van Lint C, Rouzioux C, Verdin E, Barré-Sinoussi F, Chun TW, et al. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science (2010) 329(5988):174–80. doi:10.1126/science.1191047
114. Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med (2011) 1(1):a007096. doi:10.1101/cshperspect.a007096
115. Kaczmarek K, Morales A, Henderson AJ. T cell transcription factors and their impact on HIV expression. Virology (Auckl) (2013) 2013(4):41–7.
116. Baron BW, Desai M, Baber LJ, Paras L, Zhang Q, Sadhu A, et al. BCL6 can repress transcription from the human immunodeficiency virus type I promoter/enhancer region. Genes Chromosomes Cancer (1997) 19(1):14–21. doi:10.1002/(SICI)1098-2264(199705)19:1<14::AID-GCC3>3.0.CO;2-3
117. Cummins NW, Badley AD. Anti-apoptotic mechanisms of HIV: lessons and novel approaches to curing HIV. Cell Mol Life Sci (2013) 70(18):3355–63. doi:10.1007/s00018-012-1239-3
118. Petersen JL, Morris CR, Solheim JC. Virus evasion of MHC class I molecule presentation. J Immunol (2003) 171(9):4473–8. doi:10.4049/jimmunol.171.9.4473
119. Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity (1999) 10(6):661–71. doi:10.1016/S1074-7613(00)80065-5
120. Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, et al. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature (2012) 491(7422):129–33. doi:10.1038/nature11443
121. Friedrich TC, Valentine LE, Yant LJ, Rakasz EG, Piaskowski SM, Furlott JR, et al. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J Virol (2007) 81(7):3465–76. doi:10.1128/JVI.02392-06
122. Nomura T, Matano T. Association of MHC-I genotypes with disease progression in HIV/SIV infections. Front Microbiol (2012) 3:234. doi:10.3389/fmicb.2012.00234
123. Loffredo JT, Sidney J, Bean AT, Beal DR, Bardet W, Wahl A, et al. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol (2009) 182(12):7763–75. doi:10.4049/jimmunol.0900111
124. Cartwright EK, Spicer L, Smith SA, Lee D, Fast R, Paganini S, et al. CD8(+) Lymphocytes are required for maintaining viral suppression in SIV-infected macaques treated with short-term antiretroviral therapy. Immunity (2016) 45(3):656–68. doi:10.1016/j.immuni.2016.08.018
125. Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Spatial alterations between CD4(+) T follicular helper, B, and CD8(+) T cells during simian immunodeficiency virus infection: T/B cell homeostasis, activation, and potential mechanism for viral escape. J Immunol (2012) 188(7):3247–56. doi:10.4049/jimmunol.1103138
126. Connick E, Mattila T, Folkvord JM, Schlichtemeier R, Meditz AL, Ray MG, et al. CTL fail to accumulate at sites of HIV-1 replication in lymphoid tissue. J Immunol (2007) 178(11):6975–83. doi:10.4049/jimmunol.178.11.6975
127. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol (2016) 17(10):1187–96. doi:10.1038/ni.3543
128. Petrovas C, Ferrando-Martinez S, Gerner MY, Casazza JP, Pegu A, Deleage C, et al. Follicular CD8 T cells accumulate in HIV infection and can kill infected cells in vitro via bispecific antibodies. Sci Transl Med (2017) 373(9):1–14.
129. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature (2016) 537(7620):412–28. doi:10.1038/nature19317
130. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi:10.1038/nature19330
131. Miles B, Miller SM, Folkvord JM, Levy DN, Rakasz EG, Skinner PJ, et al. Follicular regulatory CD8 T cells impair the germinal center response in SIV and ex vivo HIV infection. PLoS Pathog (2016) 12(10):e1005924. doi:10.1371/journal.ppat.1005924
132. Mylvaganam GH, Rios D, Abdelaal HM, Iyer S, Tharp G, Mavinger M, et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. PNAS (2017) 114(8):1976–81. doi:10.1073/pnas.1621418114
133. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity (2008) 29(6):848–62. doi:10.1016/j.immuni.2008.11.002
135. Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med (2003) 9(6):727–8. doi:10.1038/nm880
136. Tsai LM, Yu D. Follicular helper T-cell memory: establishing new frontiers during antibody response. Immunol Cell Biol (2014) 92(1):57–63. doi:10.1038/icb.2013.68
137. Ise W, Inoue T, McLachlan JB, Kometani K, Kubo M, Okada T, et al. Memory B cells contribute to rapid Bcl6 expression by memory follicular helper T cells. Proc Natl Acad Sci U S A (2014) 111(32):11792–7. doi:10.1073/pnas.1404671111
138. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity (2015) 42(4):704–18. doi:10.1016/j.immuni.2015.03.002
139. Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, et al. BCL6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol (2013) 190(8):4014–26. doi:10.4049/jimmunol.1202963
140. Haas MK, Levy DN, Folkvord JM, Connick E. Distinct patterns of Bcl-2 expression occur in R5- and X4-tropic HIV-1-producing lymphoid tissue cells infected ex vivo. AIDS Res Hum Retroviruses (2015) 31(3):298–304. doi:10.1089/aid.2014.0155
141. Vinuesa CG. HIV and T follicular helper cells: a dangerous relationship. J Clin Invest (2012) 122(9):3059–62. doi:10.1172/JCI65175
142. Yukl SA, Shergill AK, Ho T, Killian M, Girling V, Epling L, et al. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J Infet Dis (2013) 208(8):1212–20. doi:10.1093/infdis/jit308
143. Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity (2012) 37(3):377–88. doi:10.1016/j.immuni.2012.08.010
144. Kieffer TL, Finucane MM, Nettles RE, Quinn TC, Broman KW, Ray SC, et al. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J Infect Dis (2004) 189(8):1452–65. doi:10.1086/382488
145. Persaud D, Siberry GK, Ahonkhai A, Kajdas J, Monie D, Hutton N, et al. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J Virol (2004) 78(2):968–79. doi:10.1128/JVI.78.2.968-979.2004
146. Mens H, Pedersen AG, Jørgensen LB, Hue S, Yang Y, Gerstoft J, et al. Investigating signs of recent evolution in the pool of proviral HIV type 1 DNA during years of successful HAART. AIDS Res Hum Retroviruses (2007) 23(1):107–15. doi:10.1089/aid.2006.0089
147. Evering TH, Mehandru S, Racz P, Tenner-Racz K, Poles MA, Figueroa A, et al. Absence of HIV-1 evolution in the gut-associated lymphoid tissue from patients on combination antiviral therapy initiated during primary infection. PLoS Pathog (2012) 8(2):e1002506. doi:10.1371/journal.ppat.1002506
149. Buzón MJ, Massanella M, Llibre JM, Esteve A, Dahl V, Puertas MC, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med (2010) 16(4):460–5. doi:10.1038/nm.2111
150. Kacani L, Prodinger WM, Sprinzl GM, Schwendinger MG, Spruth M, Stoiber H, et al. Detachment of human immunodeficiency virus type 1 from germinal centers by blocking complement receptor type 2. J Virol (2000) 74(17):7997–8002. doi:10.1128/JVI.74.17.7997-8002.2000
151. Chirullo B, Sgarbanti R, Limongi D, Shytaj IL, Alvarez D, Das B, et al. A candidate anti-HIV reservoir compound, auranofin, exerts a selective ‘anti-memory’ effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis (2013) 4:e944. doi:10.1038/cddis.2013.473
152. Lewis MG, DaFonseca S, Chomont N, Palamara AT, Tardugno M, Mai A, et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS (2011) 25(11):1347–56. doi:10.1097/QAD.0b013e328347bd77
153. Shytaj IL, Norelli S, Chirullo B, Della Corte A, Collins M, Yalley-Ogunro J, et al. A highly intensified ART regimen induces long-term viral suppression and restriction of the viral reservoir in a simian AIDS model. PLoS Pathog (2012) 8(6):e1002774. doi:10.1371/journal.ppat.1002774
154. Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep (2015) 12(12):2099–110. doi:10.1016/j.celrep.2015.08.049
155. Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, Krishnamoorthy V, et al. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat Immunol (2014) 15(10):957–64. doi:10.1038/ni.2985
156. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi:10.1126/science.1176676
157. He J, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22(9):991–3. doi:10.1038/nm.4148
158. Pratama A, Vinuesa CG. Control of TFH cell numbers: why and how? Immunol Cell Biol (2014) 92(1):40–8. doi:10.1038/icb.2013.69
160. Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature (2012) 487(7408):482–5. doi:10.1038/nature11286
161. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV (2014) 1(1):e13–21. doi:10.1016/S2352-3018(14)70014-1
162. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity (2012) 36(3):491–501. doi:10.1016/j.immuni.2012.01.014
163. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi:10.1038/nri3862
164. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med (2004) 10(3):282–9. doi:10.1038/nm992
165. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature (2015) 517(7534):381–5. doi:10.1038/nature14053
Keywords: human immunodeficiency virus reservoir, cytotoxic T lymphocytes, follicular helper T cells, TFH reservoir, B cell follicle sanctuary
Citation: Leong YA, Atnerkar A and Yu D (2017) Human Immunodeficiency Virus Playing Hide-and-Seek: Understanding the TFH Cell Reservoir and Proposing Strategies to Overcome the Follicle Sanctuary. Front. Immunol. 8:622. doi: 10.3389/fimmu.2017.00622
Received: 06 October 2016; Accepted: 10 May 2017;
Published: 31 May 2017
Edited by:
Smita S. Iyer, Emory University, United StatesReviewed by:
Cristian Apetrei, University of Pittsburgh, United StatesGail Skowron, Boston University School of Medicine, United States
Copyright: © 2017 Leong, Atnerkar and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yew Ann Leong, ZXdhbi5sZW9uZ0Btb25hc2guZWR1;
Di Yu, ZGkueXVAYW51LmVkdS5hdQ==