 Mohammed Amir
Mohammed Amir Mohammad Aqdas
Mohammad Aqdas Sajid Nadeem
Sajid Nadeem Kaneez F. Siddiqui
Kaneez F. Siddiqui Nargis Khan
Nargis Khan Javaid A. Sheikh
Javaid A. Sheikh Javed N. Agrewala*
Javed N. Agrewala*
- Immunology Laboratory, CSIR-Institute of Microbial Technology, Chandigarh, India
It is instrumental for the Mycobacterium tuberculosis (Mtb) to persist within its host in dormancy. Mtb represses most of its metabolic machinery during latency, but upregulates the expression of latency-associated protein alpha-crystallin protein (Acr1). Therefore, it is imperative to understand how throughout dormancy, Mtb employs Acr1 to regulate the host immunity. This study reveals that Acr1 exhibits divergent effect on the pre- and post-maturation stages of dendritic cells (DCs). In the current study, we demonstrate that early encounter of bone marrow cells with Acr1 while differentiating into DCs (AcrDCpre), leads to impairment in their maturation. In contrast, when exposed to Acr1 after maturation (AcrDCpost), DCs show augmentation in their activity, secretion of TNF-α, IL-12, IL-6, and activation of T cells. Additionally, AcrDCpost promoted the polarization of naïve CD4 T cells to Th1 cells and Th17 cells and restricted the intracellular growth of Mtb. Furthermore, these DCs upregulated the expression of CCR7 and exhibited enhanced migratory capabilities. The discrete impact of Acr1 on DCs is mediated through a mechanism involving STAT-1, SOCS-3, ERK, TLR-4, and NF-κB signaling pathways. This study reveals the unprecedented role of Acr1 in distinctly modulating the function of DCs at different stages of maturation.
Introduction
One of the most striking features of Mycobacterium tuberculosis (Mtb) is its ability to survive in a state of dormancy within the hostile environment of host (1). Therefore, it is of the utmost importance to understand the factors that govern the bug’s survival and manage to subvert the host’s immunity. Some of the suggested strategies that Mtb exploits to tune host immunity are inhibition of phagolysosome fusion, impairment of innate immunity by obstructing pattern recognition receptors signaling, hampering the adaptive immunity by modulating the expression of costimulatory molecules, and production of cytokines along with exhaustion and apoptosis of T cells (2–7). Among the various proteins, alpha-crystallin protein (Acr1) is most dramatically upregulated during dormancy (8). Mtb devoid of Acr1 fails to maintain its latency (9), indicating that Acr1 is indispensable for the persistence of mycobacterium in dormant state.
Dendritic cells (DCs) play a cardinal role in imparting immunity against Mtb by activating and differentiating naïve T cells to effector and memory T cells. Upon infection with Mtb, DCs can initiate either inflammatory or regulatory responses that determine whether a pathogen will be cleared or retained (10). In response, Mtb can exploit its own machinery to inhibit T cell, B cell, and macrophage functions (6, 11).
Several proteins of Mtb like CFP-10 and ESAT-6 are reported to elicit the maturation of DCs and prime the T cell response (12, 13). In contrast, other investigators suggest that DCs differentiated in the presence of 10 kDa secretory antigen (MTSA) may provide a niche for the survival of mycobacterium (14). Recently, we have demonstrated that 16 kDa protein of Mtb, also known as Acr1/Rv2031c, inhibited the maturation of bone marrow cells (BMCs) to DCs (6). These DCs exhibited tolerogenic function and phenotype. Further, an interesting finding suggested an immunomodulatory role of Acr1 on differentiated DCs by suppressing tumor growth (15). This prompted us to investigate the impact of Acr1 on the differentiated DCs (AcrDCpost) and their ability to stimulate T cells as well as to restrict the growth of Mtb. Furthermore, we compared AcrDCpost functionality with DC precursors that encountered Acr1 during their maturation (AcrDCpre). We observed that AcrDCpost exhibited stimulatory function as evidenced by the DC’s ability to activate and differentiate T cells and restrict the growth of Mtb. The mechanism suggested was through the modulation of STAT-1, ERK, and NF-κB pathways. In contrast, AcrDCpre showed inhibitory function of DCs. This observation illustrates the diametric role of Acr1 in inhibiting the maturation of DCs precursor during early stage of differentiation, whereas the post-maturation encounter potentiates DCs development.
Materials and Methods
Animals
Female C3H/HeN, C3H/HeJ (Tlr4Lps-d), and C57BL/6 mice, 6–8 weeks, were procured from the Institute of Microbial Technology, Chandigarh, India. All the animal experiments were approved and complied by the Institutional Animal Ethics Committee (IAEC) and Regulatory Guidelines issued by the Committee for the Purpose of Supervision of Experiments on Animals (CPCSEA) (No. 55/1999), Ministry of Environment and Forest, Government of India.
Antibodies and Reagents
Recombinant GM-CSF, IL-4, and naïve CD4 T cell enrichment kit, Abs to IL-12p40/70, TNF-α, IL-6, IFN-γ, IL-17, PE-Cy7-CD11c, FITC-CD80, PE-CD86, efluor-MHCII, APC-CD40, STAT-1, pSTAT-1, STAT-4, pSTAT-4, JNK, and pJNK were purchased from BD Biosciences (San Diego, CA, USA). Biotinylated anti-CD83 and APC-conjugated CCR7 Abs were purchased from eBioscience (San Diego, CA, USA). Abs against actin were from Sigma (St. Louis, MO, USA), SOCS-3 from Abcam (Cambridge, MA, USA) and anti-pERK and ERK Abs from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The endotoxin removal column was purchased from Thermo Scientific (Rockford, IL, USA).
DC Culture
Bone marrow cells were collected from femurs and tibia. The BMCs (2 × 106/well) were cultured in media containing GM-CSF (2 ng/ml) + IL-4 (4 ng/ml) either with Acr1 (AcrDCpre) or without Acr1 (AcrDCpost). On the third day, both cultures were replenished with media containing GM-CSF and IL-4, with Acr1 added only to the AcrDCpre culture. On the sixth day, the AcrDCpost were stimulated with Acr1 (0–18 µg/ml) for 24 h, while the BMCs incubated with GM-CSF + IL-4 were used as controls (cDCs).
Western Blotting
AcrDCpre and AcrDCpost were harvested, washed, and lysed in lysis buffer (RIPA buffer, protease, and phosphatase inhibitor cocktail). The supernatants (SNs) of the lysates were estimated and equal amount of lysates were subjected to SDS-PAGE. After transfer to nitrocellulose membrane and subsequent blocking, the membranes were immunoblotted with their respective Abs against SOCS-3 or phosphorylated STAT-1, STAT-4, and ERK. Afterward, blot was stripped off to determine the expression of ERK and actin, and then developed using chemiluminescence kit (Lumigen, Inc., Southfield, MI, USA). Blots were scanned with the help of Image Quant LAS 4000 (GE Healthcare, Pittsburgh, PA, USA). Densitometric analysis was done using Scion image software.
Cytokine ELISA
The cytokines in the culture SNs of AcrDCpost and AcrDCpre were monitored by coating the ELISA plates overnight at 4°C with rat anti-mouse IFN-γ (2 µg/ml), IL-12 (1 µg/ml), TNF-α (2 µg/ml), IL-6 (1 µg/ml), and IL-10 (2 µg/ml) Abs in phosphate buffer (pH 9.6 and pH 6, 0.05 M). The unbound sites were blocked with BSA (1%) in PBS for 2 h at 37°C. The plates were washed with PBS-Tween-20 (0.05%) and culture SNs (50 µl) were added overnight at 4°C. Respective biotinylated Abs diluted in dilution buffer (1:1 solution of PBS-Tween-20 and blocking buffer) were added into plates and incubated for 2 h at 37°C. Thereafter, avidin-HRP (1:1,000) was added to plates and incubated at 37°C for 1 h. The usual procedures of washing and incubation were carried at each step. The color developed, due to H2O2-OPD substrate-chromogen, was stopped by H2SO4 (7%) and the plates were read at 492 nm. Serial dilutions of recombinant cytokines were used to prepare the standard curves to quantify the cytokines in the SNs.
Evaluation of NF-κB by EMSA
AcrDCpost and AcrDCpre were harvested on the sixth day, washed and treated with IFN-γ and LPS. After 30 min, nuclear extracts were prepared as mentioned elsewhere (6, 11). An equal amount of nuclear extract (10 µg) from each sample was incubated for 30 min/RT and labeled with P32-duplex oligonucleotides containing binding site for NF-κB in binding buffer. The DNA–protein complexes were resolved by electrophoresis on a native PAGE (7%). After electrophoresis, the gel was dried and exposed to screen at for 12 h/RT. The screen was scanned by phosphoimager (Fujifilm, Tokyo, Japan).
Syngeneic T Cell Differentiation
Naïve T cells were purified by MACS, as per manufacturer’s instructions (BD Biosciences, San Diego, CA, USA). Briefly, after preparing single cell suspension, splenocytes were treated with CD4 T cell enrichment cocktail supplemented with biotinylated anti-CD44 and CD25 Abs. Later, same volume of streptavidin-magnetic beads was added and cells were isolated using BD IMagnet. The purity of naïve T cells was >97%, as ascertained by flow cytometry. Naïve CD4 T cells (3 × 105/well, 24 well plate) were poured on anti-CD3 Ab (2 µg/ml) coated plate and cocultured with AcrDCpost and AcrDCpre under Th1 (IL-12: 10 ng/ml, anti-IL-4 Ab: 5 µg/ml, IL-2: 100 U/ml), Th2 (IL-4: 5 ng/ml, anti-IFN-γ Ab: 5 µg/ml, anti-IL-12 Ab: 5 µg/ml) and Th17 (IL-6: 30 ng/ml, TGF-β: 5 ng/ml, anti-IL-4 Ab: 5 µg/ml, anti-IFN-γ Ab: 5 µg/ml, anti-IL-2 Ab: 5 µg/ml) polarization conditions.
Allogeneic T Cell Differentiation
MACS purified naïve T cells (2 × 105/well) of C57BL/6 mice were cocultured with AcrDCpost and AcrDCpre of C3H/HeN in a 1:10 ratio (DC: T cells). After 3 days, the SNs were collected and estimated for IFN-γ and IL-17 by ELISA.
In Vivo Polarization of T Cells by AcrDCpost and AcrDCpre
Mice (n = 8) were injected subcutaneously (s.c.) with OVA (100 µg) in alum (2 mg) dissolved in PBS (0.2 ml). Ten days later, AcrDCpost and AcrDCpre incubated for 2 h with OVA (100 µg/ml) were adoptively transferred s.c. into OVA-primed mice. On the seventh day, the mice were sacrificed and LNs were isolated and single cell suspension was prepared. These cells were then cultured with OVA (100 µg/ml) for 48 h. Later, polarization of T cells was monitored by quantification of IFN-γ and IL-17 in the SNs by ELISA.
Differentiation of OVA-Specific T Cells by AcrDCpost and AcrDCpre
Mice (n = 8) were sensitized s.c. with OVA (100 µg). After 7 days, T cells were purified from LNs by MACS. T cells were cocultured with OVA-pulsed AcrDCpost and AcrDCpre at a ratio of 1:10 (DC: T cells) for 5 days. Later, IFN-γ and IL-17 were estimated in SNs by ELISA.
AcrDCpost and AcrDCpre-Mediated Proliferation of T Cells
Mice (n = 8) were s.c. immunized with OVA in alum. After 10 days, T cells were isolated from LNs by MACS. The T cells were labeled with CFSE (1 µM) by incubating in PBS for 8 min at 37°C. The cells were washed 3× and cocultured with OVA-pulsed AcrDCpost and AcrDCpre for 48 h. Proliferation was assayed by CFSE-dye dilution assay using a flowcytometer. The histogram data (stimulation index: SI) are represented as a bar diagram, SI = AcrDCpre/postcDCs. The flowcytometric analysis was done using BD FACS Diva software.
Migration Assay for AcrDCpost and AcrDCpre
AcrDCpost and AcrDCpre were labeled with either CFSE (2 µM) or efluor (2 µM) and adoptively transferred i.v. into mice (n = 8). After 2 days, animals were sacrificed. The presence of CFSE or efluor-positive cells in the spleen was monitored by FACSAria flowcytometer. The flowcytometric analysis was done using FACS Diva software.
Quantitative PCR for TGF-β
RNA was isolated using Trizol reagent; according to the manufacturer’s instruction (Invitrogen, Carisbad, CA). Briefly, RNA was quantified by NanoDrop spectrophotometer. A260/A280 ratio of samples was in the range of 1.90–2.00. Intactness of RNA samples was determined by formaldehyde denaturing agarose gel electrophoresis. DNA contamination of RNA samples was removed by amplification grade DNase. Later, DNase activity was terminated by adding stop solution. Further, the samples were heated to 70°C for 10 min to inactivate DNase. Analysis was done by comparative Ct method. RT-qPCR and data analysis were done by Realplex Mastercycler (Eppendorf, Hamburg, Germany). The following primers were used for the quantification.
Tgf-β
FV 5′ TGACGTCACTGGAGTTGTACGG 3′
RV 5′ GGTTCATGTCATGGATGGTGC 3′
β-Actin
FV 5′-AGAGGGAAATCGTGCGTGAC-3′
RV 5′-CAATAGTGATGACCTGGCCGT-3′
Results are represented in the form of relative expression to actin m-RNA (fold change).
Statistical Analysis
Statistical analysis was carried out using non-parametric Student’s t-test using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Results
Acr1 Exerts Distinct Effect on Encountering AcrDCpre and AcrDCpost
The optimum expression of MHC and costimulatory molecules is considered to be quite crucial in deciding the activation or anergy in T cells. Costimulatory molecules like CD80, CD86, CD40, and CD83 are regarded as maturation markers for DCs. Importantly, we noticed that Acr1 upregulates the CD80 molecule on AcrDCpost but downregulates CD80 on AcrDCpre. The change was noted in a dose-dependent manner (Figure 1A). Similarly, we observed the augmented expression of MHCII, CD86, CD40, and CD83 on AcrDCpost, while downregulation in the levels of these molecules on AcrDCpre (Figure 1B). Thus, substantiating the observed disparity in the expression of CD80 on AcrDCpre and AcrDCpost. Since, optimum expression of CD80 was induced at a dose of 9 µg/ml of Acr1, subsequently in all the experiments this concentration was used. In order to validate that the downregulation of costimulatory molecules in AcrDCpre is not due to cell death, we stained AcrDCpre and AcrDCpost with PI and no change in the viability of AcrDCpost and AcrDCpre was observed (Figure 1C).
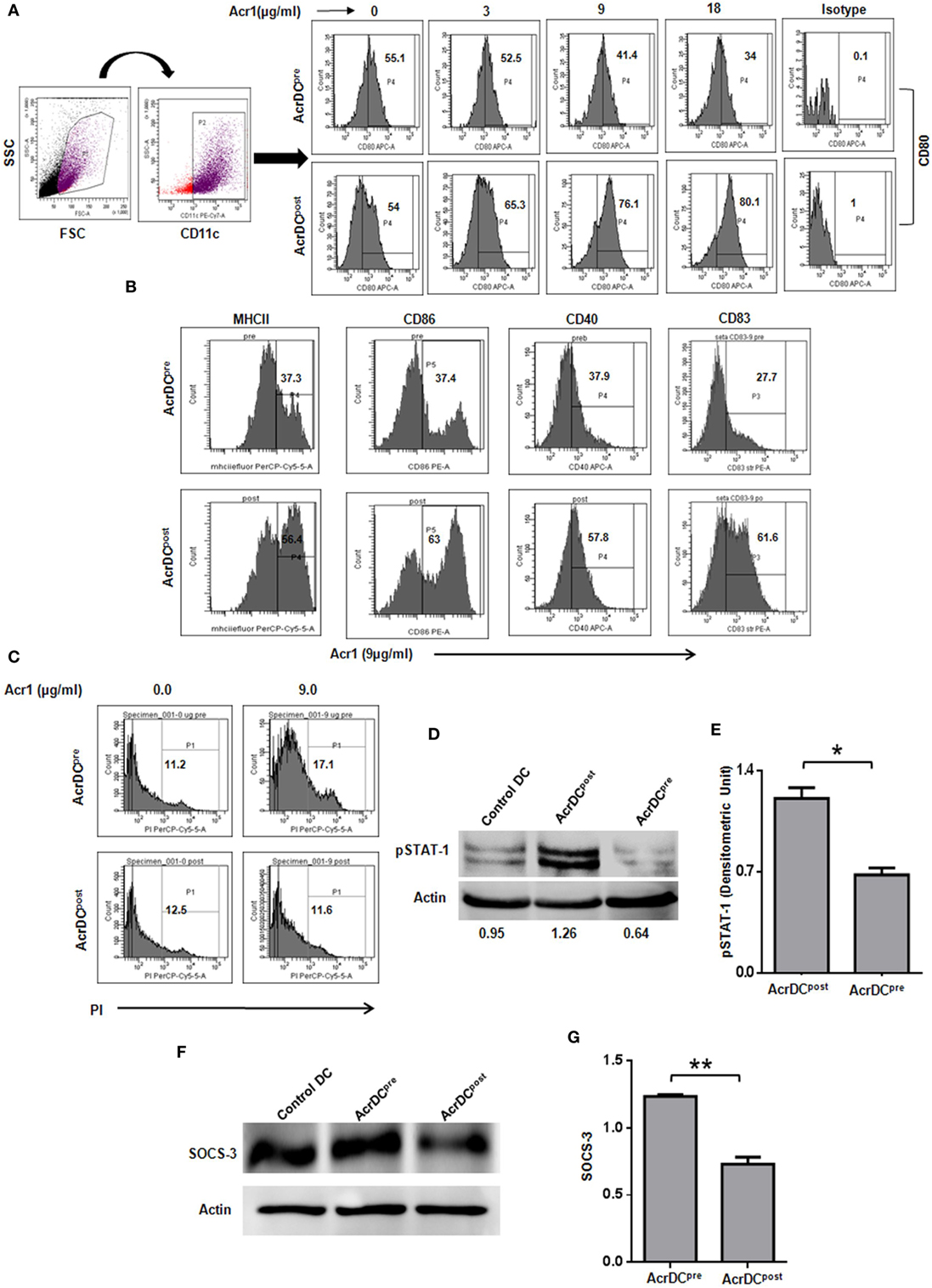
Figure 1. Distinct role of alpha-crystallin protein (Acr1) in the maturation of BMDCs. The BMDCs cultured with GM-CSF + IL-4 were exposed to Acr1 either during maturation (AcrDCpre) or after maturation (AcrDCpost) and the expression on CD11c gated population was monitored by flow cytometry for the expression of (A) CD80 (B); MHCII, CD86, CD40, CD83. (C) The AcrDCpre and AcrDCpost were checked for viability by PI staining. The maturation status of AcrDCpre and AcrDCpost was monitored by the phosphorylation of (D,E) STAT-1; (F,G) SOCS-3 by Western blotting and the densitometric analysis is expressed (mean ± SD) after normalizing with loading control actin. The values in the inset of flowcytometer histograms are percentage of cells. The results shown are representative of two to three independent experiments. *p < 0.05, **p < 0.01.
We next conducted experiments to identify the mechanism responsible for the distinct behavior of AcrDCpost and AcrDCpre. The STAT-1 is a critical transcription factor responsible for the activation and maturation of DCs (16). Therefore, we investigated the status of STAT-1 in AcrDCpost and AcrDCpre. Intriguingly, substantial enhancement in the level of STAT-1 phosphorylation in AcrDCpost but decline in AcrDCpre was observed (Figures 1D,E). The SOCS-3 is a suppressor of cytokine signaling that negatively regulates STAT-1 mediated maturation of DCs (17–19). SOCS-3 has also been linked with the inhibition of CD4 T cells activation and proliferation (20). Intriguingly, we observed an inverse correlation between STAT-1 and SOCS-3 (Figures 1D–G). AcrDCpre induced higher SOCS-3, whereas AcrDCpost displayed lower level. These results indicate that the mechanism responsible for the phenotypic and functional difference in AcrDCpost and AcrDCpre is mediated through SOCS-3, which in turn regulates the STAT-1 mediated maturation of DCs.
The cytokines released by DCs are indispensable for the differentiation of T cells. We observed significant (p < 0.001) increase in the secretion of IL-12 by C3H/HeN AcrDCpost, as compared to AcrDCpre (Figures 2A,B). A similar trend in the secretion of IL-12 was noted in C57BL/6 strain, thereby illustrating that the impact of Acr1 is independent of any genetic diversity in the mice. IL-12 is a key regulator for the differentiation of Th1 cells. Like IL-12, a similar trend was noted in the case of TNF-α (Figure 2C). TNF-α plays a vital role in restricting the growth of Mtb (21). The results of AcrDCpre are in consistent with our previous observation (6), we noticed substantial (p < 0.01) inhibition in the yield of IL-12 when BMCs were matured in the presence of Acr1. Further, as compared to AcrDCpost, we observed significantly (p < 0.05) higher levels of TGF-β in AcrDCpre (Figure 2D). Overall, these results indicate that Acr1 exposure inhibits the maturation of DC precursors during early stage of differentiation, whereas the post-maturation encounter of Acr1 potentiates DCs development.
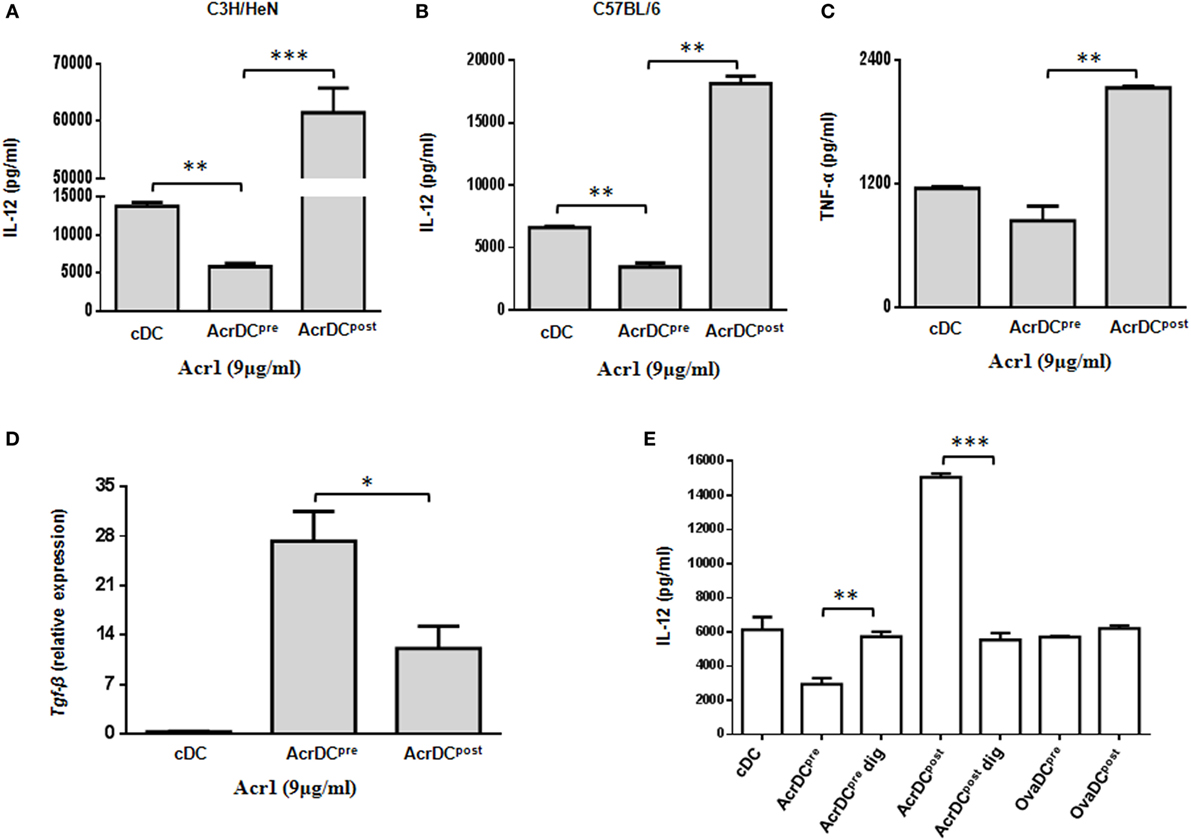
Figure 2. Discrete secretion of proinflammatory cytokines by AcrDCpre and AcrDCpost. The supernatants (SNs) collected from AcrDCpre and AcrDCpost derived from (A) C3H/HeN; (B) C57BL/6 mice were estimated for the secretion of IL-12 by ELISA. Similarly, the SNs obtained from C3H/HeN AcrDCpre and AcrDCpost were assessed for (C) TNF-α. (D) Tgf-β was monitored by RT-qPCR. The mRNA level was normalized with β-actin control and the data are depicted as relative expression. (E) The specificity of the activity of alpha-crystallin protein (Acr1) was established by culturing AcrDCpre and AcrDCpost with Acr1 digested with proteinase K and estimating the release of IL-12 in the SNs. OVA is used as a control protein. The results expressed as mean ± SD are the representative of two to three independent experiments. AcrDCpre dig and AcrDCpost dig: AcrDCpre and AcrDCpost cultured with proteinase K digested Acr; OvaDCpre and OvaDCpost: DCpre and DCpost cultured with OVA. *p < 0.05, **p < 0.01, ***p < 0.001.
To establish the specificity of Acr1 and its observed effect is not due to any artifact, we digested Acr1 with proteinase K and cultured it with AcrDCpre and AcrDCpost and studied the secretion of IL-12. Interestingly, no change in the release of IL-12 was observed by AcrDCpre and AcrDCpost cultured with proteinase K digested Acr1, but undigested Acr1 retained its modulatory function (Figure 2E), explicitly confirming the specificity of Acr1 in modulating DCs functionality. No change was noticed when AcrDCpre and AcrDCpost cultures were supplemented with OVA in lieu of Acr1, thereby further validating the specificity of Acr1.
Differential Production of IL-12 by AcrDCpre and AcrDCpost Is Mediated through ERK Signaling
Next, we elucidated the mechanism by which Acr1 elicited the IL-12 production. The MAPK pathway is critical in regulating the expression of IL-12. Therefore, we investigated the status of ERK and JNK activation. We observed augmented phosphorylation of ERK in AcrDCpre (Figure 3A). In contrast, suppression was noted in AcrDCpost. No change was discernible in JNK phosphorylation (Figure 3B), indicating that Acr1 mediates the regulation of IL-12 through the ERK pathway.
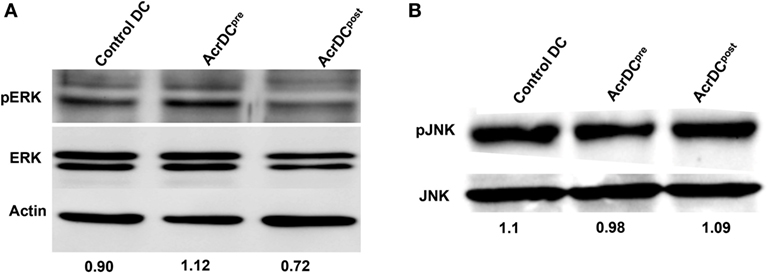
Figure 3. Difference in the yield of IL-12 by AcrDCpre and AcrDCpost is mediated through ERK pathway. (A,B) The variance in the levels of IL-12 by AcrDCpre and AcrDCpost is mediated through ERK but not JNK pathway, as evidenced by the modulation in the phosphorylation of ERK and JNK monitored by Western blotting. Western blots are the representative of two to three independent experiments.
The Upregulation in the Expression of CCR7 on AcrDCpost Correlates with Their Greater Migratory Potential and Ability to Help T cells
The trafficking of DCs is critical for the activation of T cells. To observe this phenomenon, we adoptively transferred CFSE-labeled AcrDCpost and AcrDCpre into mice and monitored their migration to the spleen. We noted a remarkable increase (p < 0.01) in the movement of AcrDCpost, as compared to AcrDCpre (Figure 4A). Interestingly, we observed higher expression of CCR7 on AcrDCpost that may explain their greater trafficking capability (Figure 4B). Furthermore, we investigated whether AcrDCpost exhibited enhancement in their potential to activate T cells. We observed that AcrDCpost showed greater ability to help T cells, as evidenced by improved (p < 0.01) proliferation of T cells (Figures 4C,D). In contrast, AcrDCpre significantly (p < 0.01) inhibited the proliferation of T cells, as compared to control DCs (cDCs).
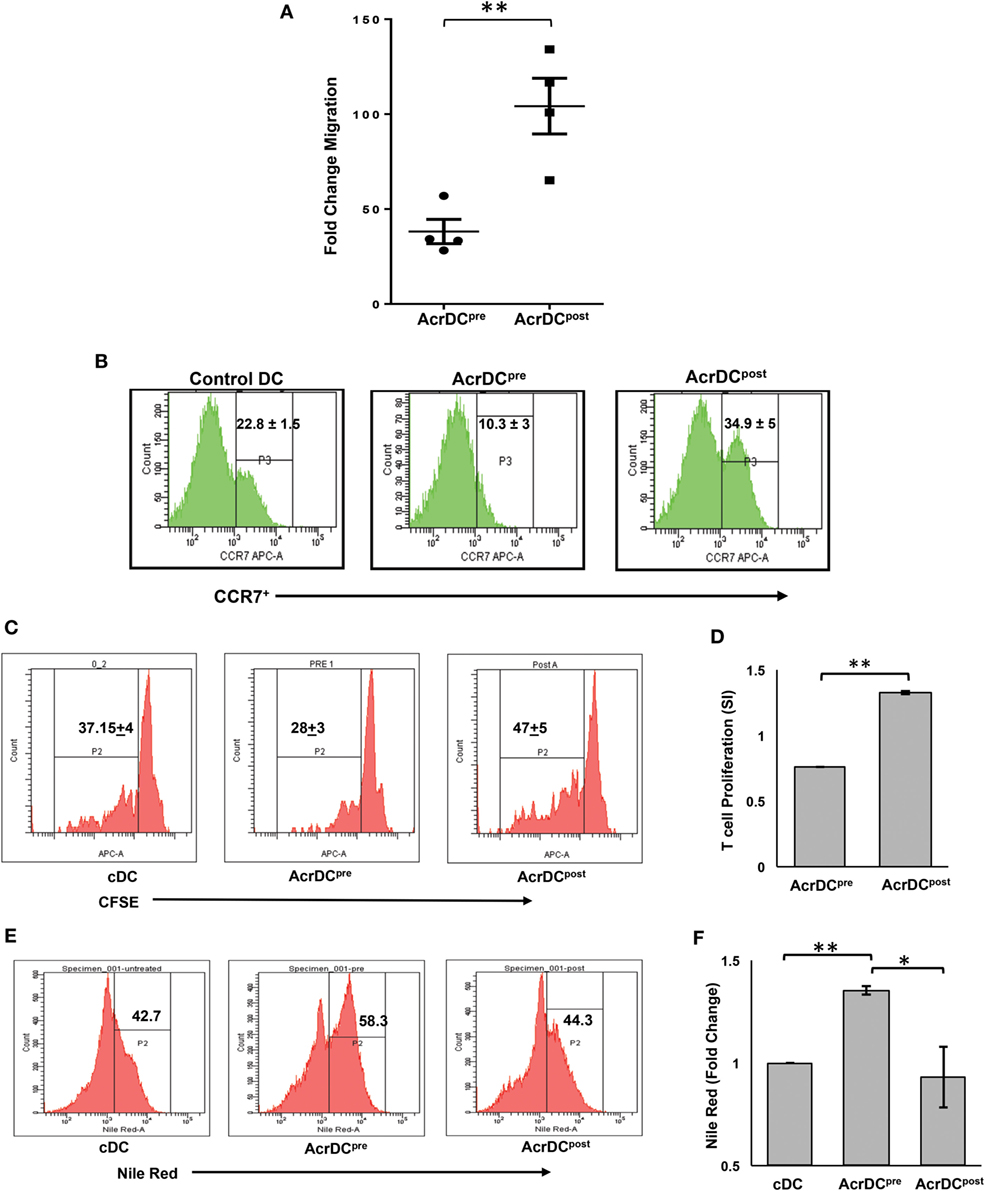
Figure 4. AcrDCpre and AcrDCpost exhibited disparity in their migratory preferences and activation of T cells. (A) The adoptive transfer of CFSE-labeled AcrDCpre and AcrDCpost in the mice established that AcrDCpre migration to spleen was significantly lesser than AcrDCpost. Each dot indicates single animal. The fold change in the migration was calculated by normalizing with untreated dendritic cells (DCs). (B) AcrDCpre showed downregulation in the expression of migratory chemokine CCR7. In contrast, AcrDCpost displayed upregulation of CCR7, as monitored by flow cytometry. (C,D) T cells isolated from OVA immunized mice labeled with CFSE were cocultured with AcrDCpre and AcrDCpost for 48 h. The proliferation was enumerated by flow cytometry and the data are expressed as histograms. The histogram data (stimulation index: SI) are represented as bar diagram, SI = AcrDCpre/post/cDCs. (E,F) The AcrDCpre and AcrDCpost were stained with Nile red dye to estimate the accumulation of lipids and data are expressed as flow cytometry histograms and bar diagram. The figures in the inset of histograms signify percent positive cells. The data represented as mean ± SD are of two to three independent experiments (n = 4/group). *p < 0.05, **p < 0.01.
We next examined why AcrDCpre expressed inhibitory activity. Recently, it has been reported that DCs with accumulated lipids exhibit dysfunction in their activity (22). This prompted us to check whether the malfunctioning of AcrDCpre can be attributed due to their lipid content. We observed that AcrDCpre accumulated a substantially larger amount of lipids than AcrDCpost (p < 0.05) or cDCs (p < 0.01) (Figures 4E,F; Figure S1 in Supplementary Material). Consequently, higher lipid content may be one of the reasons for the suppressive nature of AcrDCpre.
AcrDCpost Promotes but AcrDCpre Inhibits the Differentiation of Th1 Cells and Th17 Cells
We noticed the discrete role of AcrDCpost and AcrDCpre in modulating the secretion of IL-12 (Figure 2). DCs play a cardinal role in the activation and differentiation of naïve CD4 T cells. IL-12 is a differentiation factor for Th1 cells. Therefore, it was worth to monitor the influence of AcrDCpost and AcrDCpre in the generation of Th1 cells. AcrDCpost and AcrDCpre were cultured with either anti-CD3 Ab stimulated naïve CD4 T cells under Th1 differentiation conditions (Figure 5A) or allogeneic naïve CD4 T cells (Figure 5B) or antigen specific T cells (Figure 5C). Intriguingly, AcrDCpost promoted but AcrDCpre retarded the differentiation of Th1 cells, as evidenced by higher expression of IFN-γ by AcrDCpost. This change was detected in both antigen-specific and allogeneic CD4 T cells (Figures 5A–C). Furthermore, we also adoptively transferred OVA-pulsed AcrDCpost and AcrDCpre into the mice that were primed with OVA. Like the in vitro results, it was noticed in vivo, as well that AcrDCpost potentiated while AcrDCpre inhibited the differentiation of Th1 cells (Figure 5D).
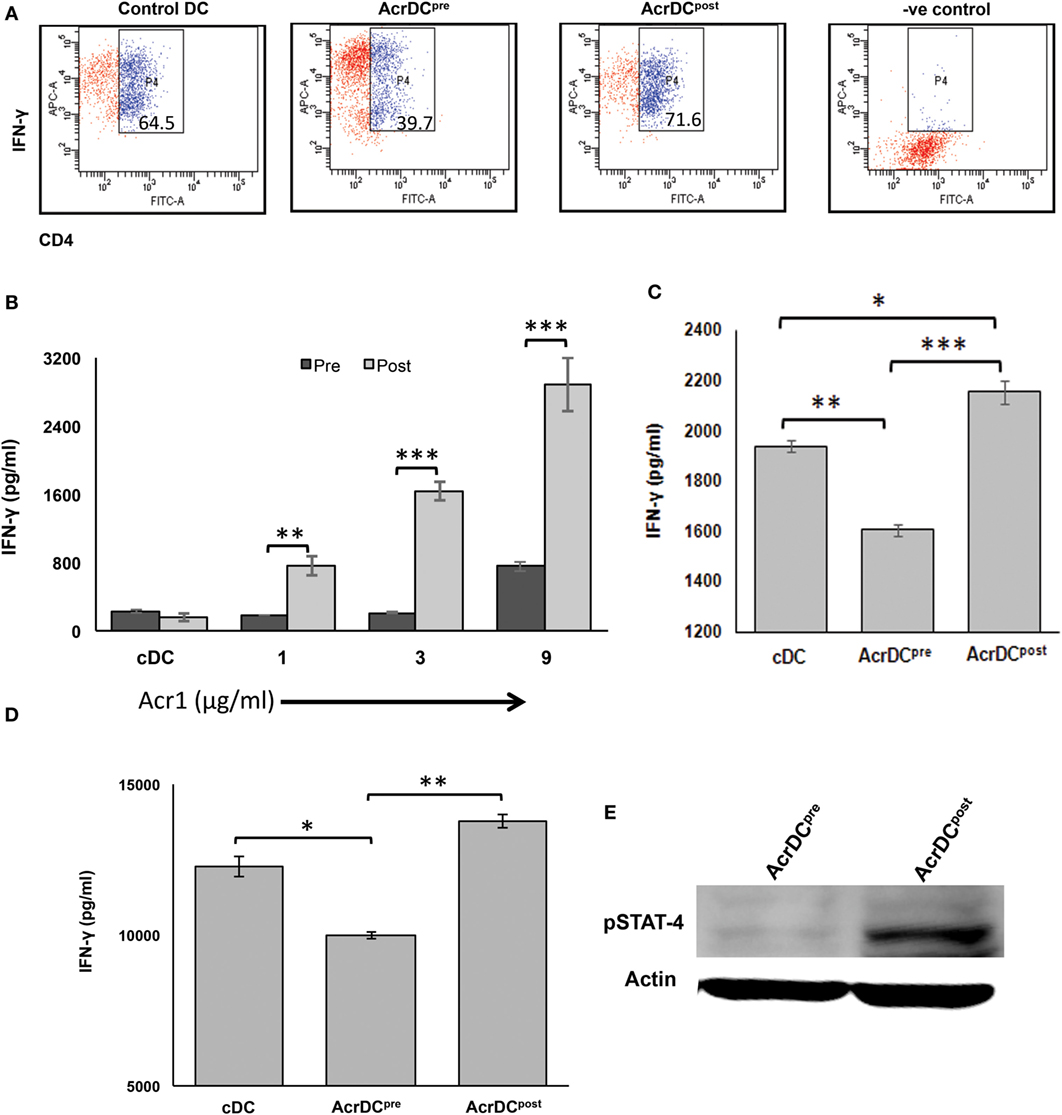
Figure 5. AcrDCpre inhibits, while AcrDCpost supports the differentiation of naïve T cells to Th1 cells by modulating the expression of STAT-4. (A) Anti-CD3 Ab-stimulated naïve CD4 T cells were cocultured with AcrDCpre and AcrDCpost under Th1 cell differentiation conditions (IL-12 + anti-IL4 Ab), and monitored for the intracellular expression of IFN-γ by flow cytometry. AcrDCpost facilitates, in contrast AcrDCpre suppresses the secretion of IFN-γ when cocultured with (B) allogeneic (C57BL/6) naïve T cells; (C) CD4 T cells isolated from OVA immunized animals were cocultured with AcrDCpre and AcrDCpost for 5 days. Later, IFN-γ was estimated in the supernatants (SNs) by ELISA. (D) OVA-pulsed AcrDCpre and AcrDCpost were adoptively transferred in the OVA-primed mice. After 7 days, LN cells were isolated and cultured with OVA for 48 h. The IFN-γ was estimated in the culture SNs by ELISA. The flow cytometry data in the inset illustrate percent positive cells and bar diagram of ELISA (mean ± SD) are representative of two to three independent experiments. *p < 0.05, **p < 0.01; ***p < 0.001. (E) Phosphorylation status of STAT-4 in AcrDCpre and AcrDCpost was monitored by Western blotting. Actin was used as a loading control. The blot shown is an illustrative of three individual experiments.
STAT-4 activation is responsible for the IFN-γ-inducible genes (23). Therefore, we further validated AcrDCpost mediated differentiation of Th1 cells by examining the activation of STAT-4. It was noticed that AcrDCpost showed higher phosphorylation of STAT-4 (Figure 5E). Contrary to this, AcrDCpre exhibited substantial decline in STAT-4. This is, therefore, indicative of the role of STAT-4 pathway in promoting the differentiation of Th1 cells by AcrDCpost.
Th17 cells play a dual role not only in protection but also in the pathogenesis of the diseases, and TGF-β and IL-6 cytokines are necessary for their differentiation (24). We observed disparate production of TGF-β (Figure 2D) and IL-6 (Figure 6A) by AcrDCpost and AcrDCpre. Consequently, we were curious to know whether AcrDCpost and AcrDCpre can distinctly regulate Th17 cells. AcrDCpost and AcrDCpre were cultured with anti-CD3 Ab stimulated naïve CD4 T cells under Th17 differentiation conditions (Figure 6B), allogeneic naïve CD4 T cells (Figure 6C), and antigen-specific T cells (Figure 6D). Like Th1 cells, AcrDCpost facilitated, whereas AcrDCpre impeded the differentiation of Th17 cells (Figures 6B–D). No change was observed in Th2 cells differentiation by AcrDCpost and AcrDCpre (Figure 6E).
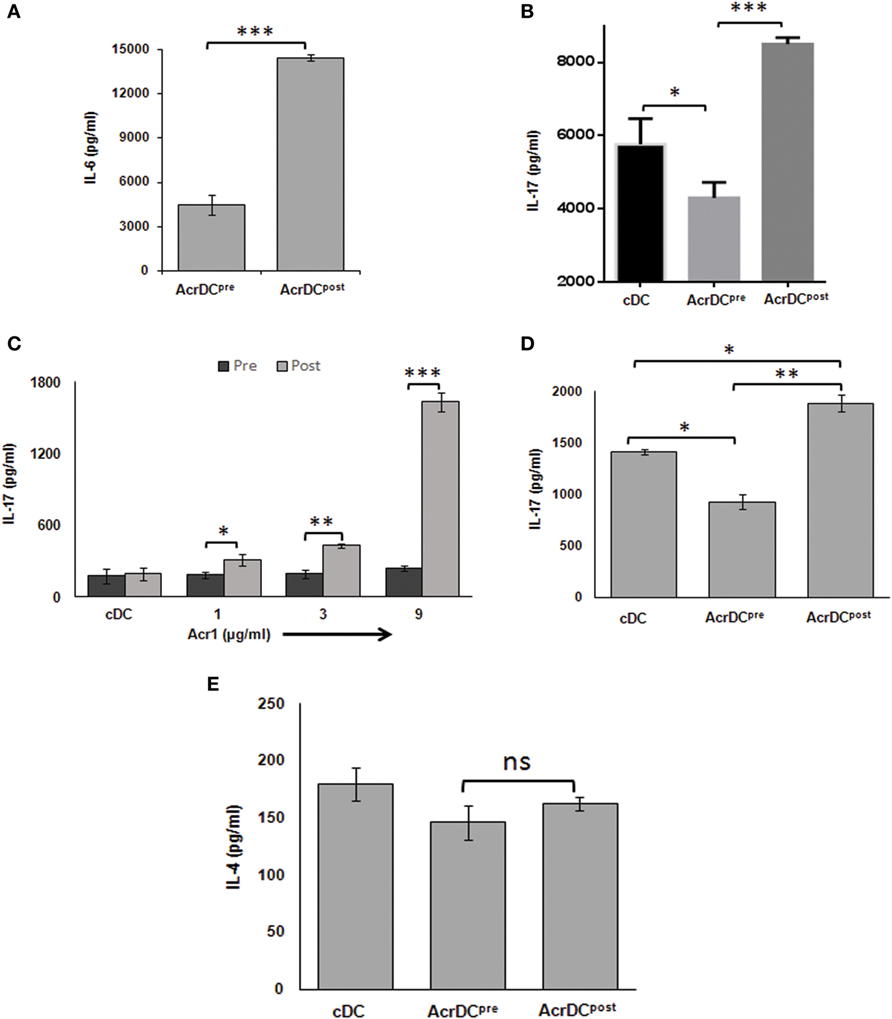
Figure 6. AcrDCpre impedes but AcrDCpost promotes the differentiation of Th17 cells. (A) IL-6 was estimated in the supernatants (SNs) of AcrDCpost and AcrDCpre. IL-17 was monitored in SNs of AcrDCpost and AcrDCpre cultured with (B) anti-CD3 Ab stimulated naïve T cells under Th17 differentiation conditions (IL-6 ± TGF-β); (C) allogeneic naïve T cells (C57BL/6); (D) OVA-specific T cells. (E) AcrDCpre and AcrDCpost showed no difference in the yield of IL-4 when cocultured with OVA-specific T cells. The cytokines were measured by ELISA. The data (mean ± SD) expressed as bar diagram are from two to three independent experiments. *p < 0.05, **p < 0.01; ***p < 0.001.
AcrDCpost Controls but AcrDCpre Fails to Restrict the Intracellular Growth of Mtb
Mycobacterium tuberculosis is an extremely successful pathogen because it can thwart its host’s immune system and thrives in dormant state in a hostile environment of one-third of world’s population. Acr1 is predominantly expressed by Mtb during latency (8, 25). The current study illustrates that Acr1 alters the function of AcrDCpost and AcrDCpre, so we wanted to investigate their abilities to restrict the growth of Mtb. Importantly, as compared to AcrDCpre, AcrDCpost notably (p < 0.01) constrained the survival of Mtb (Figure 7A). However, Mtb showed unobstructed growth in AcrDCpre; which also supports our earlier study (6). These results suggest the dual role of Acr1 in controlling the stage-specific activity of DCs.
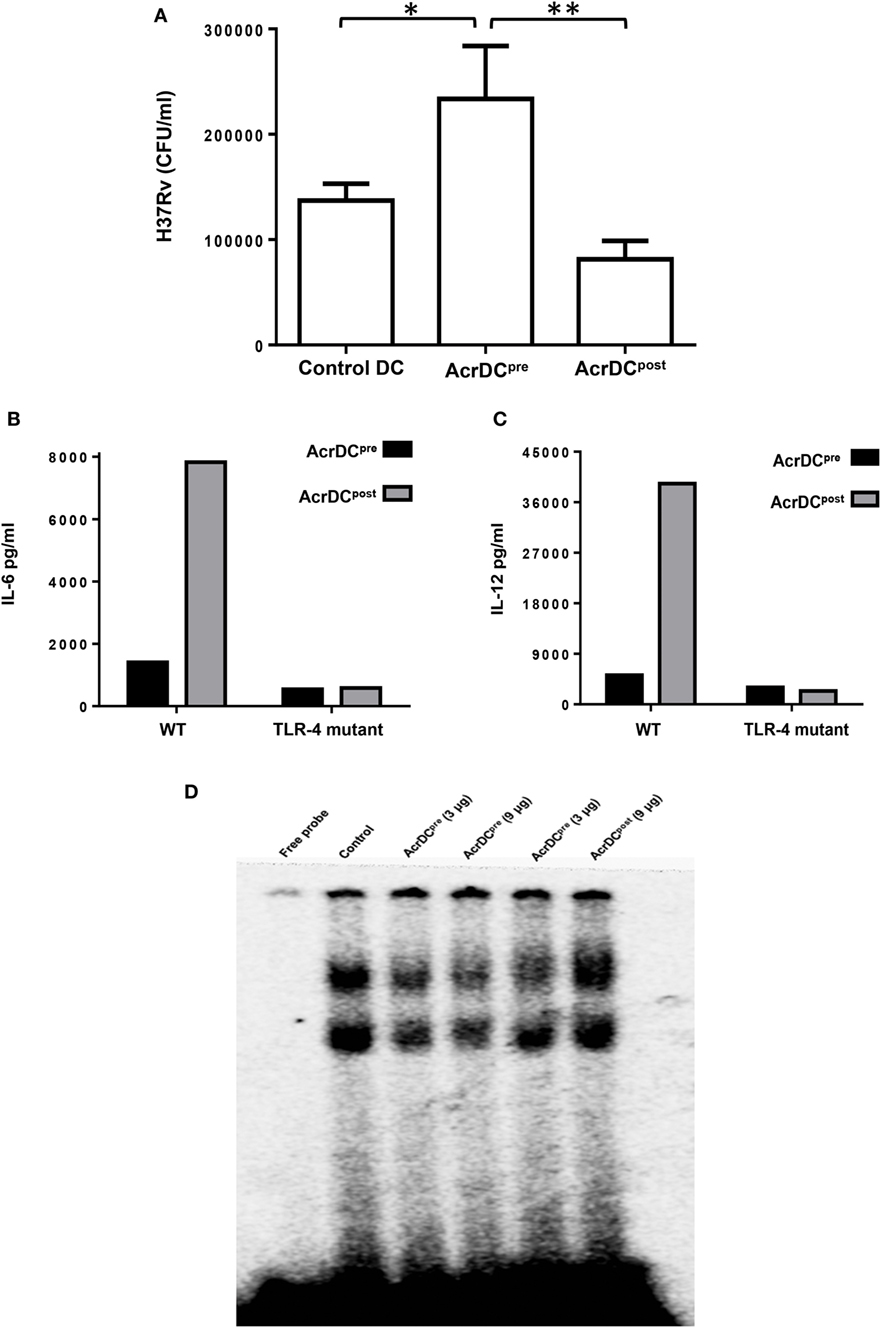
Figure 7. AcrDCpost but not AcrDCpre restricted the intracellular growth of Mycobacterium tuberculosis (Mtb). (A) AcrDCpre and Acr1DCpost were infected with Mtb H37Rv for 24 h. Later, the infected dendritic cells (DCs) were lysed and plated on 7H11 agar plates and bacterial load was counted after 20 days and expressed as CFU/ml. (B) Dissimilar response in AcrDCpre and AcrDCpost is mediated by TLR-4. AcrDCpre and AcrDCpost were generated from WT and TLR-4 mutant mice and estimated the secretion of (B) IL-6; (C) IL-12 in the supernatants by ELISA. (D) AcrDCpre prevented but AcrDCpost promoted the nuclear translocation of NF-κB. Nuclear extracts of AcrDCpre and AcrDCpost cultured with the indicated concentrations of alpha-crystallin protein (Acr1) were prepared and checked for NF-κB binding to DNA by EMSA. Data (mean ± SD) are representative of two to three independent experiments. *p < 0.05, **p < 0.01; ***p < 0.001.
Differential Regulation of AcrDCpre and AcrDCpost by Acr1 Is Mediated through TLR-4
It is reported that Acr1 binds to TLR-4 (15). Therefore, we thought it would be prudent to evaluate the role of TLR-4 in Acr1 mediated modulation of DCs. Interestingly, AcrDCpost derived from TLR-4 mutant mice (C3H/HeJ) showed abrogation in the secretion of IL-6 and IL-12 (Figures 7B,C). Wild-type mice (C3H/HeN), on the other hand, exhibited augmented production of these cytokines. These results indicate that the modulatory activity of Acr1 on DCs may be dependent on the extent of TLR-4 signaling. Triggering through TLR-4 is reported to induce both activation and inhibition in the function of DCs (19). Consequently, this study validates our results.
The Modulatory Function of AcrDCpost and AcrDCpre Is Regulated through NF-κB Activation
NF-κB plays a key role in orchestrating the immune response upon microbial infection. NF-κB translocation to the nucleus leads to upregulation of various cytokines, chemokines, and inflammatory mediators, which are required for the migration of immune cells to the site of infection. This study suggests the stimulatory phenotype of AcrDCpost (CD80hi, CD86hi, CD40hi, CD83hi, MHCIIhi, IL-6hi, IL-12hi, TNF-αhi, TGF-βlo) and suppressive phenotype of AcrDCpre(CD80lo, CD86lo, CD40lo, CD83lo, MHCIIlo, IL-6lo, IL-12lo, TNF-αlo, TGF-βhi). Therefore, we investigated the NF-κB activation in AcrDCpost and AcrDCpre. AcrDCpost exhibited significant enhancement but AcrDCpre showed impediment in the nuclear translocation of NF-κB (Figure 7D), indicating that the differential responses detected in AcrDCpre and AcrDCpost related to costimulatory molecules, cytokines, and proliferation are coordinated through NF-κB signaling.
Discussion
One-third of world’s population is estimated to have latent tuberculosis infection. These individual serves as a reservoir for the pathogen and therefore pose a serious threat of spreading tuberculosis and providing a major impediment in the complete eradication of the disease. Although the host employs its immuno-artillery to combat Mtb, the pathogen not only diffuses the attack and survives but also releases factors to overpower the immunity. Acr1 is one of the predominantly expressed proteins during the dormancy. Its role as a virulence factor that impairs the differentiation and function of DCs has been surfaced (6, 26). In contrast, due to its immunodominant nature, it can be a potential vaccine candidate (27).
We have demonstrated that during differentiation if DCs encounter Acr1, it leads to the impairment in their maturation and functionality, resulting in the generation of tolerogenic phenotype (6). The Mtb showed better survival in such DCs. In contrast, a recent study suggested an adjuvant property of Acr1 treated DCs, in protection against tumor. Consequently, we thought to evaluate the impact of Acr1 on the differentiated DCs. To do so, we stimulated differentiated DCs with Acr1 (AcrDCpost) and following major findings have emerged in context with the promotion of the maturation of DCs as evidenced by augmented (1) expression of MHCII and costimulatory molecules; (2) release of proinflammatory cytokines; (3) CCR7 display and migratory capacity; (4) potential to restrict the growth of Mtb; (5) ability to proliferate and differentiate T cells; (6) mechanism involved in this phenomenon was through TLR-4-mediated activation of STAT-1, ERK, and NF-κB; and inhibition in the SOCS-3 pathways (Figure S2 in Supplementary Material).
The delivery of signals through costimulatory molecules like CD80, CD86, CD83, and CD40 by DCs in conjunction with MHC–peptide complex is crucial for the activation of T cells. Contrary to this, downregulation of costimulatory molecules can lead to anergy in T cells. Costimulatory molecules are widely considered as maturation and differentiation markers for DCs and other APCs (28, 29). CD83 is an established maturation marker for DCs (30). DCs deficient in CD83 resulted in lesser T cell proliferation with impaired IFN-γ secretion (30). CD83 induces the expression of CD80, CD86, and MHCII molecules (31–33), thus boosting the overall functionality of DCs. Recruitment of CD86 and CD80 to lipid rafts on DCs is essential for optimal T cell activation (34). Similarly, we noted that AcrDCpost but not AcrDCpre could optimally activate T cells. We postulate that the diminished activity of AcrDCpre to activate T cells may be due to their reduced migratory capacity, as evidenced by the decreased display of CCR7. Furthermore, downregulation of the expression of costimulatory molecules and increased lipid content may be one of the reasons for the impairment of the function of AcrDCpre to activate T cells to proliferate and secrete cytokines (22).
AcrDCpost chiefly promoted the proliferation and activation of naïve CD4 T cells toward Th1 cells and Th17 cells. In contrast, inhibition in the differentiation capacity of AcrDCpre was observed. This may be due to the fact that AcrDCpost showed enhanced production of IL-12, IL-6, and TGF-β, which are the important factors for the differentiation of Th1 cells and Th17 cells, respectively (35, 36). Th1 cells and Th17 cells play a fundamental role in controlling the survival of intracellular pathogens like Mtb, leishmania, malaria, HIV, etc. (5, 37, 38). We were interested to know whether the modulation in the function of AcrDCpost and AcrDCpre was explicitly due to Acr1. To establish this, we digested Acr1 with proteinase K and observed abrogation in the function of AcrDCpost, as evidenced by inhibition in the secretion of IL-12. In addition, we used OVA, a non-Acr1 antigen and detected no discernible change in the yield of IL-12 by either AcrDCpost or AcrDCpre. Consequently, we established that the observed results are specifically due to Acr1.
The IL-12-mediated expression of CCR7 is critical for DCs migration to secondary lymphoid organs during Mtb infection (39). Similarly, we observed augmented production of IL-12 and higher expression of CCR7 in the case of AcrDCpost, which explains the greater migratory potential of AcrDCpost. These results corroborated with earlier findings (39).
One of the important features of APCs such as macrophages and DCs is to phagocytose and eliminate the pathogen. If the APCs fail to kill the phagocytosed bacteria, it will lead to bacterial survival and disease progression. Therefore, it was important to monitor whether the AcrDCpost were able to confine the intracellular growth and persistence of Mtb. AcrDCpost revealed significantly greater restriction of Mtb, as compared to AcrDCpre, indicating that Acr1 enhanced the killing potential of AcrDCpost.
This study suggests the stimulatory as well as inhibitory role of Acr1, depending on the different stages of maturation of DCs. Hence, we propose here a mechanism responsible for this discrepancy. STAT-1 phosphorylation is an important indicator for the maturation and activation of DCs (16). Accordingly, we observed that AcrDCpost exhibited remarkably higher expression of pSTAT-1 but AcrDCpre showed sizable decline in the activation of STAT-1. Further, we validated the data by examining the expression of the SOCS-3, which is a negative regulator of DCs maturation (16). We observed sufficient decrease in the SOCS-3 level, which may be responsible for the activation of AcrDCpost. Furthermore, the stimulatory function of AcrDCpost was confirmed by the enhancement in the nuclear translocation of NF-κB. NF-κB contributes indispensably to the maturation of DCs and its blockage abrogates the process (40). ERK is responsible for regulating the secretion of IL-12 (41). DCs obtained from Erk1 knockout mice showed profound production of IL-12 (42), illustrating that IL-12 production is negatively regulated by the ERK pathway (41). We observed that the phosphorylation of ERK was significantly higher in AcrDCpre, as compared to AcrDCpost. Like ERK, JNK activation is also known to regulate IL-12 expression; however, no change in the phosphorylation of JNK was seen. This suggests that Acr1 regulates the production of IL-12 through the ERK pathway.
Recently, it has been demonstrated that Acr1 binds to TLR-4 and triggers the immuno-adjuvanting effect of DCs against tumors. Furthermore, Acr1 was shown to be a potent TLR-4 agonist that can enhance both DC activation and Th1 polarization through the MyD88 and TRIF signaling pathways (15). Acr1 activates the tyrosine phosphorylation of TLR-4, thus enhancing the MyD88 and TRIF-mediated signaling cascade. MyD88-dependent pathway is critical for the production of proinflammatory cytokines, while TRIF pathway is required for the expression of costimulatory molecules. Therefore, we thought it would be judicious to monitor the influence of Acr1 on DCs derived from TLR-4 mutant mice. Consequently, we noted that Acr1 failed to evoke the release of IL-12 and IL-6 by AcrDCpost and AcrDCpre derived from TLR-4 mutant mice. In contrast, AcrDCpost from wild-type animals revealed substantially higher yield of IL-12 and IL-6. Thus, it may be inferred that Mtb employs its Acr1 to deliver signals through TLR-4 in order to modulate the function and phenotype of DCs. The inhibitory activity of Acr1 on AcrDCpre through TLR-4 is in accordance with the study depicting that the engagement of DC precursors by pathogens or their TLRs ligand lead to the obstruction in the differentiation of DCs (43, 44). In contrast, Acr1 stimulation of AcrDCpost through TLR-4 promotes their maturation and activation capabilities (19, 40, 45). We suggest that this may be one of the reasons for the dual role of Acr1 on DCs.
Dendritic cell lineage comprises of cells at various stages of maturation and differentiation. Consequently, when either of these DCs encounter Acr1, it may lead to their stimulation or tolerization, which ultimately influences the activation or suppression of T cells. Earlier, it has been suggested that Mtb can reach and hide within bone marrow. It implies that Mtb might utilize its Acr1 to interfere with the proper differentiation of DCs precursor, which may be considered as one of its immune evasion strategy (46). On the other hand, at the site of infection, the host overpowers the bug by priming the differentiated DCs with Acr1 to activate the cells of the immune system and foil Mtb attack. This is also supported by the previously published study demonstrating immunoprotective role of recombinant BCG expressing Acr1. Thus, this study suggests that besides Acr1 utility in TB, its role as an immunotherapeutic agent may also be of immense importance in potentiating the activity of DCs in cancer therapy. In contrast, inhibition of the function of DCs can be a viable strategy for treating autoimmune diseases.
Ethics Statement
Mice were procured from the animal house facility of Institute of Microbial Technology, Chandigarh, India. Institutional Animal Ethics Committee (IAEC) and regulatory guideline issued by the Committee for the Purpose of Supervision of Experiments on Animals (No. 55/1999/CPCSEA), Ministry of Environment and forest, Government of India.
Author Contributions
The origin of concept was done by JA and designing of experiments by JA and MA. The experimental procedures and data analysis were conducted by MA, M Aqdas, SN, KS, NK, and JS. The manuscript was written by MA and JA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are grateful to the Council of Scientific and Industrial Research (CSIR) and Department of Biotechnology (DBT), India for financial support. MA is a recipient of fellowship of the University Grant Commission, M Aqdas of Inspire Fellowship of the Department of Science and Technology, SN, KS, NK of DBT, and JS of CSIR. We are grateful to Kate O’Brien of Scripps, Florida for English editing of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00624/full#supplementary-material.
Figure S1. The AcrDCpre and AcrDCpost display distinct lipid levels. The AcrDCpre and AcrDCpost were stained with Nile red dye and the expression of lipids was observed under confocal microscopy (magnification: 60×). The data are the representative of two independent experiments.
Figure S2. Alpha-crystallin protein (Acr1) differentially regulates the functionality of DCs. (A) During the differentiation, Acr1 abrogates the activation and maturation of DCs. In AcrDCpre, Acr1 suppresses the maturation of DCs by upregulating SOCS-3 which in turn inhibits activation of STAT-1. Moreover in AcrDCpre the phosphorylation of ERK is promoted while the translocation of NF-κB is hampered, leading to suppressed expression of proinflmmatory cytokines. (B) In contrast, Acr1 interaction with TLR-4 after maturation leads to SOCS-3 downregulation and enhanced activation of STAT-1. Furthermore, it also leads to downregulation of phosphorylation of ERK and facilitates the nuclear translocation of NF-κB. The activation of NF-κB leads to the upregulation of proinflammatory cytokines.
References
1. Wayne LG, Sohaskey CD. Nonreplicating persistence of Mycobacterium tuberculosis. Annu Rev Microbiol (2001) 55:139–63. doi:10.1146/annurev.micro.55.1.139
2. Das G, Vohra H, Saha B, Agrewala JN, Mishra GC. Apoptosis of Th1-like cells in experimental tuberculosis (TB). Clin Exp Immunol (1999) 115(2):324–8. doi:10.1046/j.1365-2249.1999.00755.x
3. Khan N, Vidyarthi A, Javed S, Agrewala JN. Innate immunity holding the flanks until reinforced by adaptive immunity against Mycobacterium tuberculosis infection. Front Microbiol (2016) 7:328. doi:10.3389/fmicb.2016.00328
4. Khan N, Vidyarthi A, Pahari S, Agrewala JN. Distinct strategies employed by dendritic cells and macrophages in restricting Mycobacterium tuberculosis infection: different philosophies but same desire. Int Rev Immunol (2015) 35(5):386–98. doi:10.3109/08830185.2015.1015718
5. Khan N, Gowthaman U, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici!! PLoS Pathog (2012) 8(6):e1002676. doi:10.1371/journal.ppat.1002676
6. Siddiqui KF, Amir M, Gurram RK, Khan N, Arora A, Rajagopal K, et al. Latency-associated protein Acr1 impairs dendritic cell maturation and functionality: a possible mechanism of immune evasion by Mycobacterium tuberculosis. J Infect Dis (2014) 209(9):1436–45. doi:10.1093/infdis/jit595
7. Chodisetti SB, Gowthaman U, Rai PK, Vidyarthi A, Khan N, Agrewala JN. Triggering through Toll-like receptor 2 limits chronically stimulated T-helper type 1 cells from undergoing exhaustion. J Infect Dis (2015) 211(3):486–96. doi:10.1093/infdis/jiu472
8. Siddiqui KF, Amir M, Agrewala JN. Understanding the biology of 16 kDa antigen of Mycobacterium tuberculosis: scope in diagnosis, vaccine design and therapy. Crit Rev Microbiol (2011) 37(4):349–57. doi:10.3109/1040841X.2011.606425
9. Yuan Y, Crane DD, Simpson RM, Zhu YQ, Hickey MJ, Sherman DR, et al. The 16-kDa alpha-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc Natl Acad Sci U S A (1998) 95(16):9578–83. doi:10.1073/pnas.95.16.9578
10. Reis e Sousa C. Dendritic cells as sensors of infection. Immunity (2001) 14(5):495–8. doi:10.1016/S1074-7613(01)00136-4
11. Natarajan K, Latchumanan VK, Singh B, Singh S, Sharma P. Down-regulation of T helper 1 responses to mycobacterial antigens due to maturation of dendritic cells by 10-kDa Mycobacterium tuberculosis secretory antigen. J Infect Dis (2003) 187(6):914–28. doi:10.1086/368173
12. Latchumanan VK, Singh B, Sharma P, Natarajan K. Mycobacterium tuberculosis antigens induce the differentiation of dendritic cells from bone marrow. J Immunol (2002) 169(12):6856–64. doi:10.4049/jimmunol.169.12.6856
13. Tian T, Woodworth J, Skold M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J Immunol (2005) 175(5):3268–72. doi:10.4049/jimmunol.175.5.3268
14. Sinha A, Singh A, Satchidanandam V, Natarajan K. Impaired generation of reactive oxygen species during differentiation of dendritic cells (DCs) by Mycobacterium tuberculosis secretory antigen (MTSA) and subsequent activation of MTSA-DCs by mycobacteria results in increased intracellular survival. J Immunol (2006) 177(1):468–78. doi:10.4049/jimmunol.177.1.468
15. Jung ID, Shin SJ, Lee MG, Kang TH, Han HD, Lee SJ, et al. Enhancement of tumor-specific T cell-mediated immunity in dendritic cell-based vaccines by Mycobacterium tuberculosis heat shock protein X. J Immunol (2014) 193(3):1233–45. doi:10.4049/jimmunol.1400656
16. Jackson SH, Yu CR, Mahdi RM, Ebong S, Egwuagu CE. Dendritic cell maturation requires STAT1 and is under feedback regulation by suppressors of cytokine signaling. J Immunol (2004) 172(4):2307–15. doi:10.4049/jimmunol.172.4.2307
17. Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem (1998) 273(52):35056–62. doi:10.1074/jbc.273.52.35056
18. Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol (2003) 4(6):540–5. doi:10.1038/ni931
19. Bartz H, Avalos NM, Baetz A, Heeg K, Dalpke AH. Involvement of suppressors of cytokine signaling in toll-like receptor-mediated block of dendritic cell differentiation. Blood (2006) 108(13):4102–8. doi:10.1182/blood-2006-03-008946
20. Yu CR, Mahdi RM, Ebong S, Vistica BP, Gery I, Egwuagu CE. Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J Biol Chem (2003) 278(32):29752–9. doi:10.1074/jbc.M300489200
21. Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol (2010) 161(1):1–9. doi:10.1111/j.1365-2249.2010.04146.x
22. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi:10.1038/nm.2172
23. Lawless VA, Zhang S, Ozes ON, Bruns HA, Oldham I, Hoey T, et al. Stat4 regulates multiple components of IFN-gamma-inducing signaling pathways. J Immunol (2000) 165(12):6803–8. doi:10.4049/jimmunol.165.12.6803
24. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
25. Belay M, Legesse M, Mihret A, Bekele Y, Ottenhoff TH, Franken KL, et al. Pro- and anti-inflammatory cytokines against Rv2031 are elevated during latent tuberculosis: a study in cohorts of tuberculosis patients, household contacts and community controls in an endemic setting. PLoS One (2015) 10(4):e0124134. doi:10.1371/journal.pone.0124134
26. Wieczorek AE, Troudt JL, Knabenbauer P, Taylor J, Pavlicek RL, Karls R, et al. HspX vaccination and role in virulence in the guinea pig model of tuberculosis. Pathog Dis (2014) 71(3):315–25. doi:10.1111/2049-632X.12147
27. Siddiqui KF, Amir M, Khan N, Rama Krishna G, Sheikh JA, Rajagopal K, et al. Prime-boost vaccination strategy with bacillus Calmette-Guerin (BCG) and liposomized alpha-crystalline protein 1 reinvigorates BCG potency. Clin Exp Immunol (2015) 181(2):286–96. doi:10.1111/cei.12634
28. Suvas S, Singh V, Sahdev S, Vohra H, Agrewala JN. Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J Biol Chem (2002) 277(10):7766–75. doi:10.1074/jbc.M105902200
29. Tan JK, O’Neill HC. Maturation requirements for dendritic cells in T cell stimulation leading to tolerance versus immunity. J Leukoc Biol (2005) 78(2):319–24. doi:10.1189/jlb.1104664
30. Aerts-Toegaert C, Heirman C, Tuyaerts S, Corthals J, Aerts JL, Bonehill A, et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur J Immunol (2007) 37(3):686–95. doi:10.1002/eji.200636535
31. Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, et al. Activation of human dendritic cells through CD40 cross-linking. J Exp Med (1994) 180(4):1263–72. doi:10.1084/jem.180.4.1263
32. Hancock WW, Sayegh MH, Zheng XG, Peach R, Linsley PS, Turka LA. Costimulatory function and expression of CD40 ligand, CD80, and CD86 in vascularized murine cardiac allograft rejection. Proc Natl Acad Sci U S A (1996) 93(24):13967–72. doi:10.1073/pnas.93.24.13967
33. Tze LE, Horikawa K, Domaschenz H, Howard DR, Roots CM, Rigby RJ, et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J Exp Med (2011) 208(1):149–65. doi:10.1084/jem.20092203
34. Meyer zum Bueschenfelde CO, Unternaehrer J, Mellman I, Bottomly K. Regulated recruitment of MHC class II and costimulatory molecules to lipid rafts in dendritic cells. J Immunol (2004) 173(10):6119–24. doi:10.4049/jimmunol.173.10.6119
35. Bullens DM, Kasran A, Thielemans K, Bakkus M, Ceuppens JL. CD40L-induced IL-12 production is further enhanced by the Th2 cytokines IL-4 and IL-13. Scand J Immunol (2001) 53(5):455–63. doi:10.1046/j.1365-3083.2001.00900.x
36. Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med (1996) 184(2):747–52. doi:10.1084/jem.184.2.747
37. Li J, Liu D, Mou Z, Ihedioha OC, Blanchard A, Jia P, et al. Deficiency of prolactin-inducible protein leads to impaired Th1 immune response and susceptibility to Leishmania major in mice. Eur J Immunol (2015) 45(4):1082–91. doi:10.1002/eji.201445078
38. Sacramento LA, Cunha FQ, de Almeida RP, da Silva JS, Carregaro V. Protective role of 5-lipoxigenase during Leishmania infantum infection is associated with Th17 subset. Biomed Res Int (2014) 2014:264270. doi:10.1155/2014/264270
39. Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med (2006) 203(7):1805–15. doi:10.1084/jem.20052545
40. Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med (1998) 188(11):2175–80. doi:10.1084/jem.188.11.2175
41. Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol (2010) 11(8):647–55. doi:10.1038/ni.1894
42. Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, et al. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol (2004) 172(8):4733–43. doi:10.4049/jimmunol.172.8.4733
43. Palucka K, Banchereau J. Dendritic cells: a link between innate and adaptive immunity. J Clin Immunol (1999) 19(1):12–25. doi:10.1023/A:1020558317162
44. Rotta G, Edwards EW, Sangaletti S, Bennett C, Ronzoni S, Colombo MP, et al. Lipopolysaccharide or whole bacteria block the conversion of inflammatory monocytes into dendritic cells in vivo. J Exp Med (2003) 198(8):1253–63. doi:10.1084/jem.20030335
45. Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect (2004) 6(15):1382–7. doi:10.1016/j.micinf.2004.08.018
Keywords: Acr1, Mycobacterium tuberculosis, immunomodulation, dendritic cells, Th1 cells, Th17 cells
Citation: Amir M, Aqdas M, Nadeem S, Siddiqui KF, Khan N, Sheikh JA and Agrewala JN (2017) Diametric Role of the Latency-Associated Protein Acr1 of Mycobacterium tuberculosis in Modulating the Functionality of Pre- and Post-maturational Stages of Dendritic Cells. Front. Immunol. 8:624. doi: 10.3389/fimmu.2017.00624
Received: 24 March 2017; Accepted: 10 May 2017;
Published: 30 May 2017
Edited by:
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Utpal Sengupta, The Leprosy Mission, IndiaSumana Sanyal, University of Hong Kong, Hong Kong
Copyright: © 2017 Amir, Aqdas, Nadeem, Siddiqui, Khan, Sheikh and Agrewala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javed N. Agrewala, amF2ZWRAaW10ZWNoLnJlcy5pbg==