 Masaud Shah
Masaud Shah Sangdun Choi
Sangdun Choi- Department of Molecular Science and Technology, Ajou University, Suwon, South Korea
Our understanding of the pathophysiology of the pathological pain and the pharmacology of analgesic treatments has progressed tremendously over the past two decades. Among the well-documented pro-algesic factors, glia and other toll-like receptors (TLRs)-expressing cells in the neuroimmune interface have been recognized for their role in the development of neuropathic pain and for compromising the analgesic effects of opioids. Here, we comprehensively review the molecular mechanisms of pain initiation and progression, the role of TLRs in these processes, and the molecular mechanisms of morphine and morphine-3-glucuronide in TLR-dependent central immune signaling. The data reviewed here suggest that, while targeting glia to treat neuropathic pain, both analgesic and analgesia-opposing effects of opioids must be considered by acknowledging their role in TLR-mediated signaling.
Introduction
The interaction between the immunoprivileged nervous system and the immune system is not just limited to inflammatory conditions, as emphasized by studies reporting a two-way signaling between immunocompetent cells, including microglia, oligodendrocytes, astrocytes, and endothelial and neuronal cells present in the central nervous systems (CNS) and peripheral nervous systems (PNS) (1, 2). The interaction between immune and neuronal cells is crucial for the trauma-induced sensitization and pathophysiological changes that occur at the site of peripheral nerve injury. Physical or chemical incitements activate neuroimmune cells present at the site of injury, lead to the release of chemokines and cytokines, and enhance the neuroimmune response by enhanced expression of surface antigens on reacting cells (2).
Both the inflammatory and the neuropathic pain have been linked only to neuronal mechanisms until recently. However, advances in our understanding of the underlying pathophysiological mechanism and growing interest in the etiology of pain have highlighted the contribution of neuroimmune cells in the development and persistence of pain (3–5). Overproduction of cytokines and chemokines by reactive immune cells and expression of danger-associated molecular patterns (DAMPs)-recognizing receptors on neurons often enhance sensitivity to hyperalgesic stimuli and subsequently result in nociception, as reviewed previously (6).
The mechanism underlying the involvement of glia and other neuroimmune cells in nociception has been well documented. It has recently been reported that opioids, which are considered the gold standard for the treatment of pain, can also induce enhanced sensitization of neuronal and immune cells present in the neuroimmune interface and thereby lead to paradoxical hyperalgesia. The molecular mechanism of pain and its progression has been well elaborated previously; however, here, we will briefly review the role played by toll-like receptor (TLR)-expressing immune cells in hyperalgesia, the molecular mechanism of TLR-mediated signaling in pain sensitization, and the involvement of morphine and morphine-3-glucuronide (M3G) in hyperalgesia with respect to TLR-mediated signaling.
Neuroimmune Cells and Their Role in Pain Sensation
Glia, previously known to provide structural support to neuronal cells, is now recognized as playing a crucial role in the neuroimmune system, particularly in clearing cellular debris and providing immune surveillance (7). Nonetheless, glia is also endowed with the capacity to modulate pain and play a major role in sex-dependent pain sensitivity and the opioid response, as reviewed previously (7, 8).
Microglial cells cover approximately 15% of all cells in the CNS and are primarily derived from primitive myeloid precursors (9). Microglial cells play a crucial role in the tetrapartite synapse, relay post-injury plastic changes, and induce central sensitization. During neuronal injury or other pathological conditions, activated microglial cells release proinflammatory mediators that activate nearby glial and neuronal cells in the tetrapartite synapse. This potentiates the neuroinflammatory response, which can also lead to hyperalgesia (7, 10). The role of glia in normal pain progression is illustrated in Figure 1.
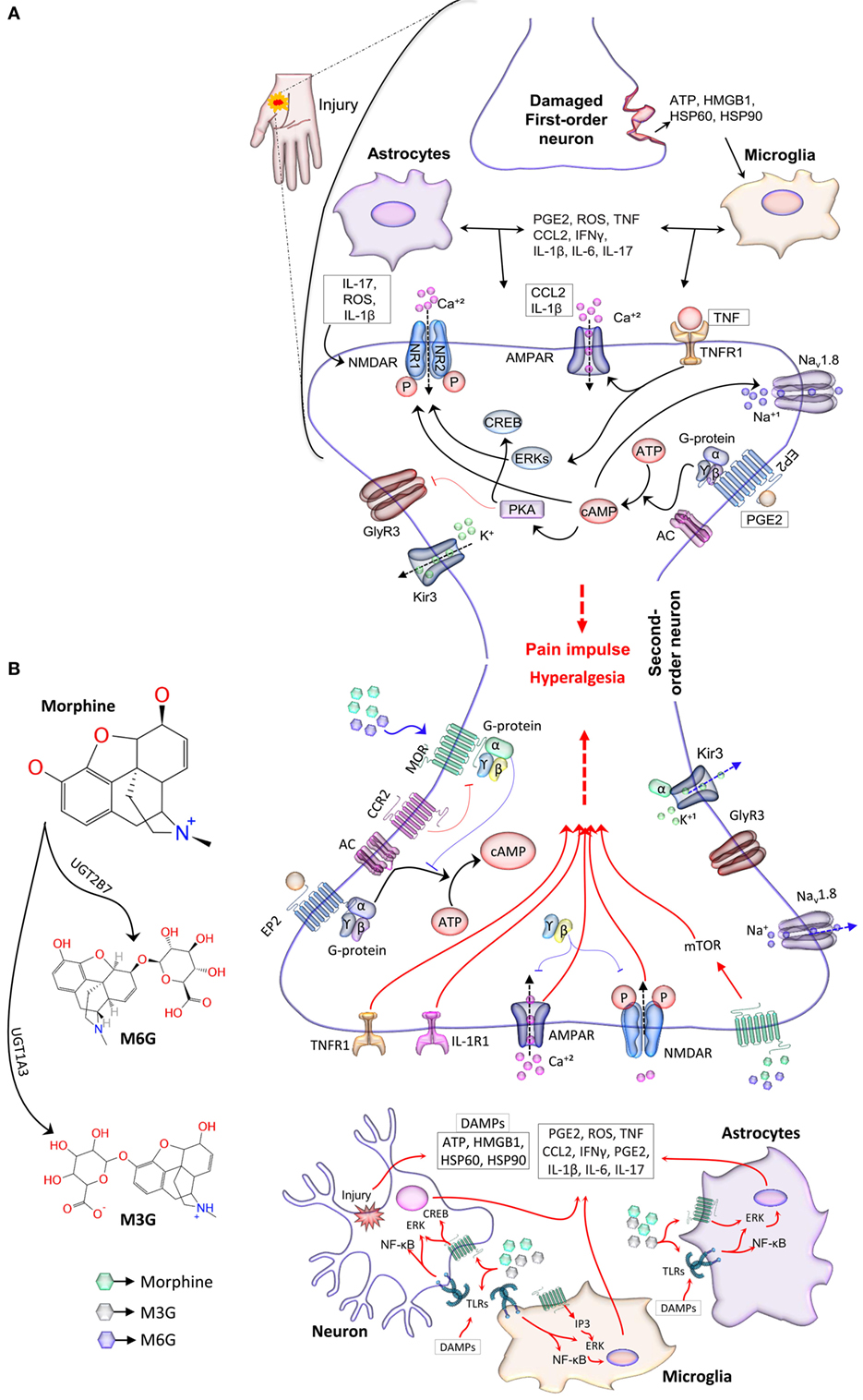
Figure 1. The neuroimmune interface and the role of glia in the pain response. (A) Reactive immunocompetent cells located at the site of injury release soluble mediators that diffuse into the neuroimmune interface and bind to synaptic terminals. These mediators modulate excitatory and inhibitory synaptic transmission and lead to nociceptive hypersensitivity. Mediators such as CCL2 and IL-1β elevate TNFR1 and AMPA signaling and the expression of Ca2+-permeable AMPARs. TNF also increases the phosphorylation of ERKs through the TNFR1 pathway, which then activates NMDAR activity. ROS and IL-17 induce the phosphorylation of the NR1 subunit of NMDAR. Other subunits, NR1, NR2A, and NR2B, are phosphorylated by IL-1β. Together, these mediators increase the influx of Ca2+ ions, thereby augmenting pain sensation. PGE2 activates AC through receptor stimulation of the G-protein (G), which then catalyzes the conversion of ATP into cAMP. The G-dependent rise in cAMP level is crucial for neuronal excitability. cAMP regulates the phosphorylation of ion channels, Nav1.8 and NMDAR, through PKA (11). PGE2-dependent EP2 signaling also leads to the PKA-dependent inhibition of glycinergic neurotransmission via GlyR3 receptors. Taken together, mediators released by glia, injured neurons, or other central immune cells promote pain sensation and result in pathological pain in severe conditions. (B) Opioid-induced analgesia and hyperalgesia. Morph is glucuronidated into M3G and M6G in hepatocytes. Morph and its MOR active metabolite, M6G, produce analgesic effects by modulating the Ca2+ and K+ ion channels through MOR mediated signal transduction (blue arrows). MORs are associated with G-proteins; after dissociation, the Gα subunit of the G-protein moves and directly interacts with the G-protein-gated inwardly rectifying K+ channel, Kir3 (12–14). The dissociated Gα subunit also decreases synaptic transmission partly by inhibiting ACs, thereby reducing the cAMP-dependent Ca2+ influx (15). Channel deactivation occurs after hydrolysis of GTP to GDP and Gα removal from the channel. This process causes cellular hyperpolarization and inhibits tonic neural activity. Opioid receptor-induced inhibition of Ca2+ conductance is mediated by binding of the dissociated Gβϒ subunit directly to the channel. This binding event is thought to reduce voltage activation of the channel pore opening and to enhance the analgesic effect of opioids. TLRs, expressed on neurons, glia, and other neuroimmune cells, are activated non-stereoselectively by both active and inactive isomers of Morph and its opioid-inactive metabolite M3G (8). Microglial activation and subsequent proinflammatory cytokine release sensitize neurons and diminish the analgesic effects of Morph and M6G (8, 16–18). This mechanism is thought to explain the negative effects of opioids (red arrows). Abbreviations: AC, adenylyl cyclase; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; CCL2, CC-chemokine ligand 2; CREB, cAMP response element-binding protein; EP2, prostaglandin E receptor 2; ERK, extracellular signal-regulated kinase; GlyR3, glycine receptor 3; IFN, interferon; IL, interleukin; IP3, inositol triphosphate; Morph, morphine; MOR: μ-opioid receptor; mTOR, mammalian target of rapamycin; M3G, morphine-3-glucuronide; M6G, morphine-6-glucuronide; NMDAR, N-methyl-d-aspartic acid receptors; PGE2, prostaglandin E2; PKA, protein kinase A; HSP, heat shock protein; HMGB1, high mobility group box 1 protein; ROS, reactive oxygen species; TNF, tumor necrosis factor; TNFRs, tumor necrosis factor receptors.
Astrocytes also play a key role in neuroimmune signaling; they are involved in the modulation of glutamate uptake by altering glutamate transporters and releasing proinflammatory cytokines that heighten pain sensation (19–21). On activation of astrocytes by neuronal injury or activated microglial cells, the expression of plasma membrane-localized glutamate (glutamate transporter 1) and glutamate-aspartate transporters is downregulated in the astrocytes, thereby resulting in decreased glutamate uptake and thus increased nociception (22). Moreover, these cells recognize DAMPs and induce the receptor-dependent activation of cellular kinases, c-jun N-terminal kinase (JNK), and extracellular signal-regulated kinase, leading to the release of proinflammatory cytokines (10) and thus pathological and inflammatory pain progression (Figure 1A).
Molecular Mechanism of Pain Sensation and Progression
Injury caused by physical or chemical stressors leads to high-threshold biochemical activity and release of elevated inflammatory mediators from damaged neurons, which initiate neuroimmune signaling at the innervation level (23). Astrocytes and T cells are reported to receive and convey neurochemical signals; however, microglial cells initiate the response to the mediators released by damaged neurons. These mediators include chemokines, adenosine triphosphate, and DAMPs, in particular high mobility group box 1 protein, heat shock protein (HSP) 60, and HSP90 (2, 24). A transition into reactive gliosis takes place when microglia detect these signals. Reactive gliosis leads to astrogliosis and the infiltration of peripheral immune cells owing to the release of chemokines, cytokines, and DAMPs (Figure 1A). Beside other cytokines and chemokine-recognizing receptors, recent studies have found that glia also express TLRs, which respond to the DAMPs released by damaged neurons or other central immune cells (25, 26). Heightened activation of TLR-expressing glial cells has been reported to play a crucial role in neuropathic pain. The finding that TLRs play a critical role in the nervous system-related pathologies and are capable of interacting with ligands other than those associated with pathogens has further emphasized that TLR-inhibiting molecules could be useful in alleviating glial-mediated allodynia (27).
The Role of Glial TLRs in Pain
The association between pain and TLRs can be traced back to the era before the discovery of lipopolysaccharides as the ligand of TLR4. TLRs are receptors expressed on various types of cells, including those present in the CNS and constitute a vital link between the immune system and the CNS. In addition to their expression on immunocompetent cells, TLRs have been reported to be expressed on endothelial and neuronal cells that are originally considered non-immunocompetent cells (25). TLRs expressed on neuroimmune cells have mainly been linked to hyperalgesia; however, accrued evidences now confirm that glial TLRs are also involved in suppressing inflammation and neuronal repair (7, 28). The role of TLRs in pathological pain has been well documented in the literature (8, 18, 25). TLRs expressed by central immune cells are mainly activated by DAMPs, released from damaged neurons at the site of injury, which further enhance the proinflammatory cascade and leads to the augmentation of the pain impulse ultimately resulting in pathological pain (29, 30). The involvement of TLRs in nociception makes them a critical component of analgesics and other pain-relieving substances (Figure 2).
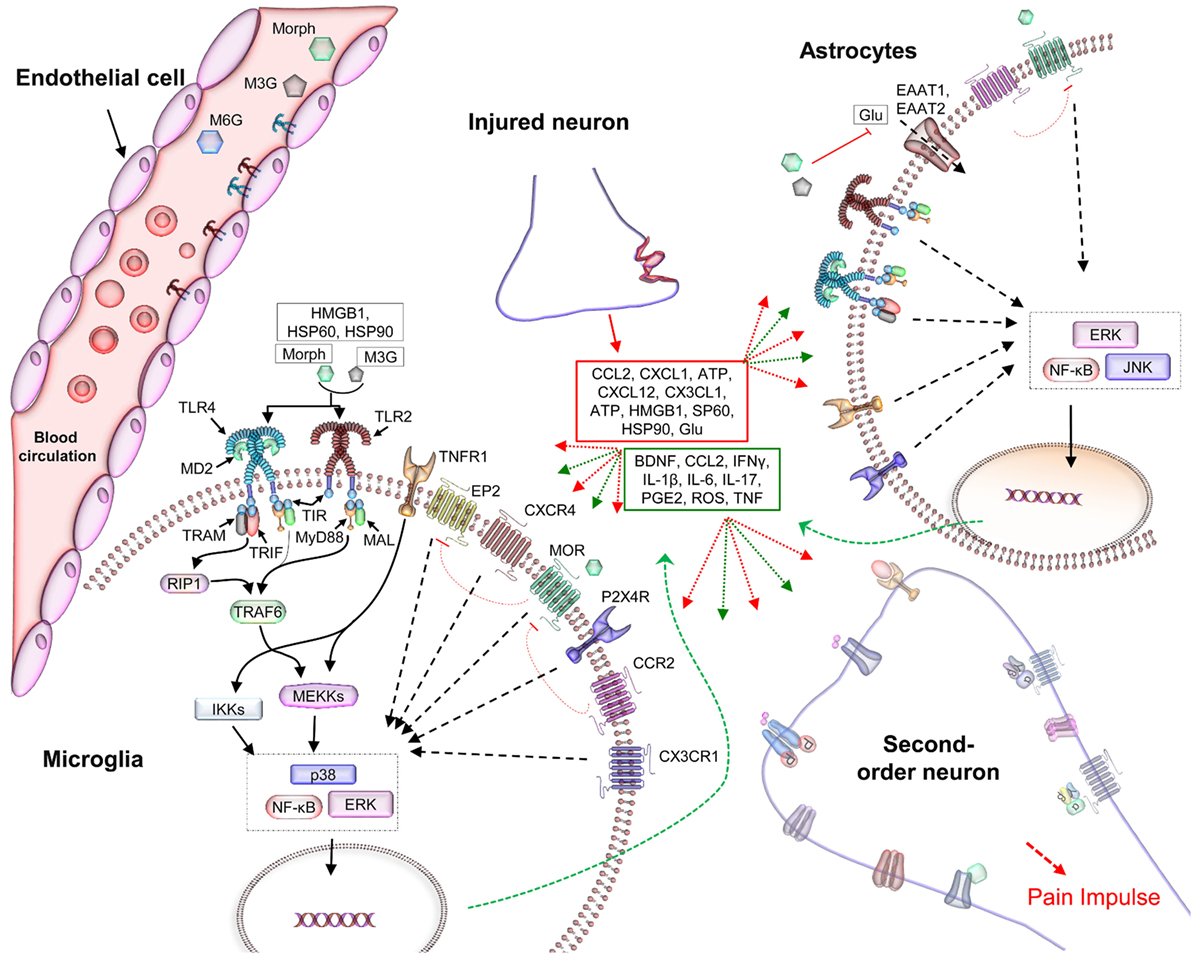
Figure 2. The role of TLRs in hyperalgesia and the negative effects of opioids in analgesia. Soluble mediators, released by injured tissues and immune cells in the neuroimmune interface, interact with the synaptic terminals and TLRs on glial and endothelial cells (25, 26). This activates complex signaling processes that intersect at multiple points, modulating inhibitory and excitatory synaptic processes and thereby resulting in nociceptive hypersensitivity. Among the TLRs, TLR2 and TLR4 have been widely reported to be associated with gliosis in studies of the neuroimmune interface (2, 31, 32). Microglia, astrocytes, and endothelial cells, located in the CNS, express TLR4 (32) and TLR2 (17), which can recognize potentially harmful substances such as DAMPs released by the damaged tissues and neurons (red box) and activate an immune-like response (18, 33). This immune cascade activates second-order neurons that transmit the pain impulse to the CNS (Figure 1). Activation of TLRs in CNS cells provokes immune-like responses via the production of proinflammatory cytokines (green box). These cytokines are then released into the extracellular environment, where they activate other receptors within the synaptic cleft (red and green dotted arrows) (34). Together, these activated receptors exacerbate the inflammatory response, leading to allodynia (35). In addition, the signaling complex modulates ion channels and the electrical potential of the CNS, and reduces the analgesic effect of morphine and its opioid receptors’ active metabolite, M6G. Abbreviations: ATP, adenosine triphosphate; BNDF, brain-derived neurotrophic factor; CCL2, CC-chemokine ligand 2; DAMPs, danger-associated molecular patterns; EP2, prostaglandin E receptor 2; ERK, extracellular signal-regulated kinase; HSP, heat shock protein; IFNγ, interferon γ; IKK, inhibitor of κB-kinase; IL-1β, interleukin-1β; JNK, c-jun N-terminal kinase; MAL, MyD88-adapter-like; MyD88, myeloid differentiation protein 88; Morph, morphine; MOR, μ-opioid receptor; M6G, morphine-6-glucuronide; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; PGE2, prostaglandin E2; RIP1, receptor-interacting protein-1; TAB, TAK1-binding protein; TIR, toll/IL-1 receptor homology domain; TNF, tumor necrosis factor; TRAF6, TNFR-associated factor 6; TLRs, toll-like receptors; TRAM, TRIF-related adaptor molecule; TRIF, TIR-domain-containing adapter-inducing interferon-β.
The involvement of TLRs in pathological pain has recently been highlighted in studies on animal models, wherein TLR4 is determined as a pain initiator (32, 36). Furthermore, the inhibition of the TLR2 and TLR4 pathways has also been reported to prevent and relieve neuropathic pain in animal models (37, 38). In addition to the well-studied role of TLR4 in neuropathic pain, other TLRs, such as TLR2 and TLR3, have also been reported to be involved in the potentiation of pain in preclinical pain models (39–41). Kim et al. suggested that glial cells, activated by neuronal injury, participate in processes leading to pain hypersensitivity through the direct activation of TLR2; this was further confirmed in TLR2-knockout mice, wherein the induced allodynia vanished (42). The association between TLRs, glia, and neuropathic pain has been extensively investigated in vitro. The discovery that glia and some neurons in the CNS and PNS express TLRs has highlighted their substantial role in the modulation of neuropathic and inflammatory pain. It should be noted that, while resolving the problem of hyperalgesia, using analgesics that can activate both μ-opioid receptor (MOR) and TLRs would worsen the general scenario; alternatively, pain killers, such as psychotropic agents, including ketamine and clonidine, COX-inhibitors, and non-steroidal anti-inflammatory drugs should therefore be used for pain relief.
Opioid-Induced TLR-Dependent Hyperalgesia
Opioids, considered the benchmark therapy for both chronic and acute pain, are also associated with paradoxical hyperplasia. Even though opioids are continuously used in relieving pain, their immune signaling mechanisms in the CNS are not well documented. It has been reported that, apart from their direct interaction with opioid-receptors expressed on glial and neuronal cells, opioids can interact with other receptors and activate exacerbated immune-like signaling in the CNS (8). Neuroinflammatory cells, including glia, have been associated with opioid-induced hyperalgesia, and this association has been well studied over the past decade (1).
Generally, opioids including morphine produce analgesic effects by modulating Ca2+ and K+ ion channels through MOR-mediated signal transduction. MORs are associated with G-proteins; after dissociation, the Gα subunit moves and directly interacts with the G-protein-gated inwardly rectifying K+ channel, Kir3 (12–14). The dissociated Gα subunit also decreases synaptic transmission partly by inhibiting adenylyl cyclase, thereby reducing the cyclic adenosine monophosphate-dependent Ca2+ influx (15). Channel deactivation occurs after hydrolysis of GTP to GDP and Gα removal from the channel. This process causes cellular hyperpolarization and inhibits tonic neural activity. Opioid receptor-induced inhibition of Ca2+ conductance is mediated by binding of the dissociated Gβϒ subunit directly to the channel. This binding event is thought to reduce voltage activation of the channel pore opening and enhance the analgesic effect of opioids (Figure 1B).
The off-target exacerbated signaling at the neuroimmune interface and the classical analgesic mechanism have been linked to the stereoselective interaction of morphine and its metabolites, M3G and morphine-6-glucuronide (M6G), with MOR and TLRs. Morphine, when administered into the body, is glucuronidated into M3G and M6G in the hepatocytes. M6G stereoselectively binds to the MOR (43, 44), while morphine, with and without MOR activity, and M3G have been reported to oppose analgesia and boost nociception (8, 45, 46). Morphine, a classical opioid, has been shown to bind non-stereoselectively to TLR4/MD2 and to activate the TLR4 pathway (47, 48). Besides morphine and its opioid-inactive metabolite, M3G, other clinically significant opioids have also been reported to bind to the TLR4/MD2 complex (1, 47, 49). After binding to the TLRs expressed on glia and dorsal root ganglion neuron, opioids, including morphine and M3G, activate the downstream signaling pathways. Activation of TLRs in these cells has been linked to the release of proinflammatory mediators, including but not limited to nitric oxide, reactive oxygen species, interleukins, interferons, monocyte chemotactic protein-1, CC-chemokine ligand 5, CC-chemokine ligand 2, CXCL10, inducible nitric oxide synthase, and prostaglandin E2. Excluding TLR3, all TLRs are known to convey their signaling via myeloid differentiation protein 88 (MyD88). After activation, MyD88 initiates a signaling cascade, which ultimately results in the release of proinflammatory mediators. These mediators exacerbate the pain sensation and progression leading to nociception and hyperalgesia. The molecular mechanism of opioid-induced TLR-dependent hyperalgesia has been illustrated in Figure 2. Ligands that can selectively block the signaling of TLRs have therefore been used to restore the analgesic effect of opioids in experimental animal models and in vitro experiments. Accordingly, the involvement of TLRs in the pharmacodynamics of opioids has become obvious.
Conclusion
After understanding the role of individual cells at the tetrapartite synapse and the neuroimmune interface in general, one could suggest that pain initiated by a stimulus or pathologically, is the result of a complex neuroimmune-signaling cascade. Furthermore, TLRs expressed on the cells present in the nervous system are pivotal in the maintenance and sustention of neuropathic pain and counter the analgesic effect of opioids. To avoid negative side effects of opioids, the development of non-opioid therapies for nociceptive pain needs serious consideration; however, some alternative therapies are already being investigated in preclinical trials. This will not only resolve the problem of heightened pain sensitivity in pathological conditions but also reduce the chances of opioid-related drug abuse in patients with depression, stress, or acquired immunodeficiency syndrome.
Author Contributions
MS and SC designed the study, wrote the manuscript, proofread, and approved this work. MS generated the figures and SC approved them.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Mid-Career Researcher Program through the National Research Foundation of Korea, which is funded by the Ministry of Education, Science, and Technology (NRF-2015R1A2A2A09001059), and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (HI14C1992). This work was also partially supported by a grant from the Priority Research Centers Program (NRF 2012-0006687).
References
1. Roeckel LA, Le Coz GM, Gaveriaux-Ruff C, Simonin F. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience (2016) 338:160–82. doi: 10.1016/j.neuroscience.2016.06.029
2. Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol (2014) 14:217–31. doi:10.1038/nri3621
3. Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci (2005) 28:661–9. doi:10.1016/j.tins.2005.10.001
4. Waxman AR, Arout C, Caldwell M, Dahan A, Kest B. Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett (2009) 462:68–72. doi:10.1016/j.neulet.2009.06.061
5. Grace PM, Rolan PE, Hutchinson MR. Peripheral immune contributions to the maintenance of central glial activation underlying neuropathic pain. Brain Behav Immun (2011) 25:1322–32. doi:10.1016/j.bbi.2011.04.003
6. Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron (2006) 52:77–92. doi:10.1016/j.neuron.2006.09.021
7. Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci (2009) 10:23–36. doi:10.1038/nrn2533
8. Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev (2011) 63:772–810. doi:10.1124/pr.110.004135
9. Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol (2010) 119:89–105. doi:10.1007/s00401-009-0622-0
10. Ben Achour S, Pascual O. Glia: the many ways to modulate synaptic plasticity. Neurochem Int (2010) 57:440–5. doi:10.1016/j.neuint.2010.02.013
11. Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron (2007) 55:365–76. doi:10.1016/j.neuron.2007.07.008
12. Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest (2010) 120:3779–87. doi:10.1172/JCI43766
13. Pizzo PA, Clark NM. Alleviating suffering 101 – pain relief in the United States. N Engl J Med (2012) 366:197–209. doi:10.1056/NEJMp1109084
14. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain (2009) 10:895–926. doi:10.1016/j.jpain.2009.06.012
15. Tegeder I, Geisslinger G. Opioids as modulators of cell death and survival – unraveling mechanisms and revealing new indications. Pharmacol Rev (2004) 56:351–69. doi:10.1124/pr.56.3.2
16. Jou I, Lee JH, Park SY, Yoon HJ, Joe EH, Park EJ. Gangliosides trigger inflammatory responses via TLR4 in brain glia. Am J Pathol (2006) 168:1619–30. doi:10.2353/ajpath.2006.050924
17. Zhang Y, Li H, Li Y, Sun X, Zhu M, Hanley G, et al. Essential role of toll-like receptor 2 in morphine-induced microglia activation in mice. Neurosci Lett (2011) 489:43–57. doi:10.1016/j.neulet.2010.11.063
18. Wang W, Deng M, Liu X, Ai W, Tang Q, Hu J. TLR4 activation induces nontolerant inflammatory response in endothelial cells. Inflammation (2011) 34:509–18. doi:10.1007/s10753-010-9258-4
19. Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science (2002) 295:2282–5. doi:10.1126/science.1067859
20. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci (2003) 23:8692–700.
21. Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res (2005) 1034:172–9. doi:10.1016/j.brainres.2004.11.018
22. Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci (2003) 23:2899–910.
23. Suter MR, Berta T, Gao YJ, Decosterd I, Ji RR. Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Mol Pain (2009) 5:53. doi:10.1186/1744-8069-5-53
24. Calvo M, Zhu N, Tsantoulas C, Ma Z, Grist J, Loeb JA, et al. Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J Neurosci (2010) 30:5437–50. doi:10.1523/JNEUROSCI.5169-09.2010
25. Nicotra L, Loram LC, Watkins LR, Hutchinson MR. Toll-like receptors in chronic pain. Exp Neurol (2012) 234:316–29. doi:10.1016/j.expneurol.2011.09.038
26. Saijo K, Crotti A, Glass CK. Regulation of microglia activation and deactivation by nuclear receptors. Glia (2013) 61:104–11. doi:10.1002/glia.22423
27. Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci (2011) 34:269–81. doi:10.1016/j.tins.2011.02.005
28. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol (2009) 9:429–39. doi:10.1038/nri2565
29. Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem (2010) 114:13–27. doi:10.1111/j.1471-4159.2010.06736.x
30. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci (2007) 10:1361–8. doi:10.1038/nn1992
31. Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in toll-like receptor-mediated neuronal injury. Glia (2010) 58:253–63. doi:10.1002/glia.20928
32. Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A (2005) 102:5856–61. doi:10.1073/pnas.0501634102
33. Faure E, Thomas L, Xu H, Medvedev A, Equils O, Arditi M. Bacterial lipopolysaccharide and IFN-gamma induce toll-like receptor 2 and toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol (2001) 166:2018–24. doi:10.4049/jimmunol.166.3.2018
34. Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci (2009) 30:581–91. doi:10.1016/j.tips.2009.08.002
35. DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist (2004) 10:40–52. doi:10.1177/1073858403259950
36. Hutchinson MR, Ramos KM, Loram LC, Wieseler J, Sholar PW, Kearney JJ, et al. Evidence for a role of heat shock protein-90 in toll like receptor 4 mediated pain enhancement in rats. Neuroscience (2009) 164:1821–32. doi:10.1016/j.neuroscience.2009.09.046
37. Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur J Neurosci (2008) 28:20–9. doi:10.1111/j.1460-9568.2008.06321.x
38. Jurga AM, Rojewska E, Piotrowska A, Makuch W, Pilat D, Przewlocka B, et al. Blockade of toll-like receptors (TLR2, TLR4) attenuates pain and potentiates buprenorphine analgesia in a rat neuropathic pain model. Neural Plast (2016) 2016:5238730. doi:10.1155/2016/5238730
39. Kim D, You B, Lim H, Lee SJ. Toll-like receptor 2 contributes to chemokine gene expression and macrophage infiltration in the dorsal root ganglia after peripheral nerve injury. Mol Pain (2011) 7:74. doi:10.1186/1744-8069-7-74
40. Shi XQ, Zekki H, Zhang J. The role of TLR2 in nerve injury-induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia (2011) 59:231–41. doi:10.1002/glia.21093
41. Obata K, Katsura H, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, et al. Toll-like receptor 3 contributes to spinal glial activation and tactile allodynia after nerve injury. J Neurochem (2008) 105:2249–59. doi:10.1111/j.1471-4159.2008.05353.x
42. Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, et al. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem (2007) 282:14975–83. doi:10.1074/jbc.M607277200
43. Lotsch J, Stockmann A, Kobal G, Brune K, Waibel R, Schmidt N, et al. Pharmacokinetics of morphine and its glucuronides after intravenous infusion of morphine and morphine-6-glucuronide in healthy volunteers. Clin Pharmacol Ther (1996) 60:316–25. doi:10.1016/S0009-9236(96)90058-2
44. Ohno S, Kawana K, Nakajin S. Contribution of UDP-glucuronosyltransferase 1A1 and 1A8 to morphine-6-glucuronidation and its kinetic properties. Drug Metab Dispos (2008) 36:688–94. doi:10.1124/dmd.107.019281
45. Komatsu T, Sakurada S, Katsuyama S, Sanai K, Sakurada T. Mechanism of allodynia evoked by intrathecal morphine-3-glucuronide in mice. Int Rev Neurobiol (2009) 85:207–19. doi:10.1016/S0074-7742(09)85016-2
46. Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, et al. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience (2010) 165:569–83. doi:10.1016/j.neuroscience.2009.10.011
47. Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A (2012) 109:6325–30. doi:10.1073/pnas.1200130109
48. Shah M, Anwar MA, Yesudhas D, Krishnan J, Choi S. A structural insight into the negative effects of opioids in analgesia by modulating the TLR4 signaling: an in silico approach. Sci Rep (2016) 6:39271. doi:10.1038/srep39271
Keywords: hyperalgesia, morphine, opioid, pain, toll-like receptor
Citation: Shah M and Choi S (2017) Toll-like Receptor-Dependent Negative Effects of Opioids: A Battle between Analgesia and Hyperalgesia. Front. Immunol. 8:642. doi: 10.3389/fimmu.2017.00642
Received: 13 April 2017; Accepted: 17 May 2017;
Published: 31 May 2017
Edited by:
Alexandre Corthay, Oslo University Hospital, NorwayReviewed by:
Kushagra Bansal, Harvard Medical School, United StatesJohn P. Vasilakos, 3M Company, United States
Copyright: © 2017 Shah and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangdun Choi, c2FuZ2R1bmNob2lAYWpvdS5hYy5rcg==