 Evgeny Chichelnitskiy
Evgeny Chichelnitskiy Britta Himmelseher
Britta Himmelseher Malte Bachmann
Malte Bachmann Heiko Mühl
Heiko Mühl- Pharmazentrum Frankfurt/ZAFES, University Hospital Goethe-University Frankfurt, Frankfurt, Germany
Disturbed homeostasis as a result of tissue stress can provoke leukocyte responses enabling recovery. Since mild hypothermia displays specific clinically relevant tissue-protective properties and interleukin (IL)-22 promotes healing at host/environment interfaces, effects of lowered ambient temperature on IL-22 were studied. We demonstrate that a 5-h exposure of endotoxemic mice to 4°C reduces body temperature by 5.0° and enhances splenic and colonic il22 gene expression. In contrast, tumor necrosis factor-α and IL-17A were not increased. In vivo data on IL-22 were corroborated using murine splenocytes and human peripheral blood mononuclear cells (PBMC) cultured upon 33°C and polyclonal T cell activation. Upregulation by mild hypothermia of largely T-cell-derived IL-22 in PBMC required monocytes and associated with enhanced nuclear T-cell nuclear factor of activated T cells (NFAT)-c2. Notably, NFAT antagonism by cyclosporin A or FK506 impaired IL-22 upregulation at normothermia and entirely prevented its enhanced expression upon hypothermic culture conditions. Data suggest that intact NFAT signaling is required for efficient IL-22 induction upon normothermic and hypothermic conditions. Hypothermia furthermore boosted early signal transducer and activator of transcription 3 activation by IL-22 and shaped downstream gene expression in epithelial-like cells. Altogether, data indicate that hypothermia supports and fine-tunes IL-22 production/action, which may contribute to regulatory properties of low ambient temperature.
Introduction
Interleukin (IL)-22 is a member of the IL-10 cytokine family that, predominantly by engaging signal transducer and activator of transcription (STAT)-3 signaling, modulates gene expression foremost in epithelial (-like) cells (1). A hallmark of IL-22 activity is pro-proliferative and anti-apoptotic action that combines with antimicrobial properties (2–4). IL-22 is, due to restricted expression of the IL-22 receptor chain-1, generally unable to target leukocytes (5). Since IL-22 fails to efficiently activate nuclear factor-κB (6), this cytokine displays a unique tissue-protective quality in absence of a direct effect on immunoactivation, neither in proinflammatory nor in immunosuppressive sense. Those favorable properties concur with protection by IL-22 as detected in rodent models of infection- and/or tissue damage-driven diseases at host/environment interfaces (4), which include intestinal Citrobacter rodentium (7) as well as lung Klebsiella pneumonia (8) and influenza infections (9), ventilator-induced lung injury (10), experimental colitis (11, 12), and acute liver injury (13, 14). In addition, by strengthening the crucial parameter of intestinal barrier integrity (15), local IL-22 is supposed to prevent translocation of pathogenic bacteria that otherwise pose a risk for sepsis development (16). This function is also supported by superior intestinal wound healing under the influence of IL-22 (12).
Most relevant sources of IL-22 are lymphoid cells, in particular group 3 innate lymphoid cells, NKT cells, γδT cells as well as differentiated Th1, Th17, and Th22 cells (2). Research aiming at characterizing molecular mechanisms driving IL22 transcription has primarily focused on transcription factors that relate to lymphoid cell differentiation. For instance, STAT3, B-cell-activating transcription (Batf), retinoid orphan receptor γτ, and ligand-activated aryl hydrocarbon receptor all connect to Th17 differentiation, can bind the IL22 promoter, and accordingly promote IL-22 production (17). Notably, information on transcription factors enabling instant IL22 gene expression after T cell receptor activation is scarce. It is noteworthy that binding of NFATc2 to the IL22 promoter has been linked to rapid (within 1 h) cyclosporin A (CsA)-sensitive induction of IL-22 mRNA in activated human Jurkat T cells (18).
Therapeutic hypothermia, frequently associated with diminished inflammation, is employed or recommended for selected clinical conditions, among others, cardiac surgery and traumatic brain injury (19, 20). Interestingly, upregulation of tissue-protective IL-22 has been observed in experimental traumatic brain injury and in patients undergoing cardiac surgery (21, 22). Basic science revealed that, in similarity to IL-22 (10), hypothermia ameliorates tissue injury in rat ventilator-induced lung injury (23). Moreover, exposure of mice to low ambient temperature, like IL-22 (13, 14), reduces acute liver injury (24). Interestingly, upregulation of IL-22-related anti-inflammatory IL-10 (25) associates with hypothermia in the context of experimental ventilator-induced lung injury (26, 27), severe trauma by fracture and hemorrhage (28), cardiac surgery (29), and systemic inflammatory response syndrome/endotoxemia (30–35). Given the tissue-protective properties of IL-22 (4), it is an important topic of current research to understand and develop strategies that aim at controlled upregulation of IL-22, especially during acute injury. To assess a potential link between hypothermia, tissue-protective responses, and IL-22 during inflammation/immunoactivation, we set out to investigate IL-22 in the context of lowered ambient temperature.
Materials and Methods
Reagents
Endotoxin (lipopolysaccharide, LPS, O55:B5) and brefeldin A were from Sigma-Aldrich (Taufkirchen, Germany). 12-O-tetradecanoylphorbol-13-acetate (TPA) was from Enzo Life Sciences (LÖrrach, Germany) and A23187 from AppliChem (Karlsruhe, Germany). Cyclosporin A (CsA) and FK506 were purchased from Calbiochem-Novabiochem (Bad Soden, Germany). Human IL-22 and interferon (IFN)γ were obtained from Peprotech Inc. (Frankfurt, Germany). Murine (αCD3-#17A2, αCD28-#37.51) and human (αCD3-#OKT3, αCD28-#28.2) agonistic anti-CD3 and anti-CD28 antibodies were from BioLegend (San Diego, CA, USA).
In Vivo Mouse Experiments
All animal procedures were approved by local authorities (“Regierungspräsidium Darmstadt”) and are in accordance with National Institutes of Health guidelines. For experiments, 10- to 12-week-old C57Bl/6 male mice (MFD-Diagnostics GmbH, Wendelsheim, Germany) were transferred individually into cages early morning. The body weight and core temperature was determined using laboratory scales and a TH-5 + RET-3 mouse thermometer with rectal probes (Physitemp Instruments Inc., Clifton, NJ, USA). Mice were injected i.p. with LPS (1 µg/g body weight) or PBS and kept at either standard room temperature (RT, 23°C) or at 4°C with access to water only (36). After 5 h, mice underwent short isoflurane (Abbott, Wiesbaden, Germany) anesthesia and were sacrificed. Liver, lungs, spleen, colon, cecum, and blood plasma were snap frozen in liquid nitrogen and stored at −80°C.
Isolation of Human Peripheral Blood Mononuclear Cells (PBMC), CD3+ T-Cells, and Monocyte-Depleted PBMC
For isolation of PBMC, heparinized blood was taken from healthy donors. This procedure and the respective consent documents were approved by the “Ethik Kommission” of the University Hospital Goethe-University Frankfurt. PBMC were isolated from peripheral blood using Histopaque-1077 (Sigma-Aldrich) according to the manufacturer’s instructions. Untouched CD3+ T-cell were isolated from PBMC using the Pan-T-cell isolation kit according to the manufacturer’s instructions (Miltenyi, Bergisch Gladbach, Germany). Mean purity was 96.0 ± 0.7% (n = 37) determined by FACS analysis using anti-CD3-PerCP/Cy5.5-#UCHT1 (BioLegend). Monocyte-depleted PBMC were generated using anti-CD14 beads (Miltenyi) with a mean depletion efficiency of 98.1 ± 0.7% (n = 7) as assessed by FACS analysis using an anti-CD14eFluor450-#61D3 antibody (eBioscience, Frankfurt, Germany). Cells were resuspended in RPMI 1640 supplemented with 10 mM HEPES, 100 U/ml penicillin, 100 µg/ml streptomycin, and 1% human serum (Life Technologies, Darmstadt, Germany) and seeded at 3 × 106 cells/ml in round-bottom polypropylene tubes (Greiner, Frickenhausen, Germany).
Isolation of Murine Splenocytes
Spleens obtained from 8- to 12-week-old male C57Bl/6 mice (MFD-Diagnostics GmbH) were excised and transferred to 5 ml ice-cold RPMI 1640 medium without FCS. Tissue was destroyed over a nylon cell strainer (70 µm; BD Biosciences, Heidelberg, Germany). Cell suspensions were centrifuged at 500 g for 5 min at 4°C and resuspended in 2 ml 0.83% NH4Cl for 2 min at RT. Red blood cell lysis was stopped by adding 10 ml cold RPMI 1640 medium without FCS. Splenocytes were collected by centrifugation, washed once with RPMI and resuspended in RPMI 1640, supplemented with 10% heat-inactivated FCS and 100 U/ml penicillin, 100 µg/ml streptomycin. Cells were seeded on 24 well polystyrene plates (Greiner) with 0.5 ml media in a concentration 6 × 106 cells/ml.
Cultivation of Human Jurkat T Cells, DLD1 and Caco2 Colon Carcinoma Cells, and HepG2 Hepatoma Cells
Jurkat T cells (ATCC-TIB-152) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in RPMI 1640 (Life Technologies) supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% heat-inactivated FCS (Life Technologies). For experiments, cells were seeded on 6-well polystyrene plates (Greiner) at a density of 2.5 × 106 cells/ml. DLD1 colon epithelial/carcinoma cells (Center of Applied Microbiology and Research, Salisbury, UK), Caco2 colon epithelial/carcinoma cells, and HepG2 hepatoma cells (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) were maintained in DMEM supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% heat-inactivated FCS (Life Technologies). For experiments, cells were plated on 6-well polystyrene plates (Greiner) and used in subconfluent condition.
According to the protocols indicated in the figure legends, cells (PBMC, epithelial-like cell lines) were cultivated in parallel at 30, 33, or 37°C incubator temperature. Incubators used for different temperatures were switched occasionally in order to exclude incubator effects on cell behavior different from incubator temperature.
Cytokine Analysis by Enzyme-Linked Immunosorbent Assay (ELISA)
Murine and human IL-22 (R&D-Systems, Wiesbaden, Germany) and human IL-8 (BD Biosciences) secretion were determined by ELISA according to the manufacturer’s instructions.
Intracellular Cytokine Staining and Flow Cytometry
Peripheral blood mononuclear cells were kept as unstimulated control or stimulated for 7 h with agonistic anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C. Thereafter, brefeldin A (2 µg/ml) was added for another 4 h, followed by intracellular staining and flow cytometry. After harvesting, PBMC were reconstituted in FACS buffer (1 × PBS + 1% FCS) and stained with surface marker antibody (anti-CD4-PE-Cy7-#SK3, eBioscience) for 30 min on ice. Thereafter, PBMC were fixed and permeabilized [BD Cytofix/Cytoperm Kit (BD Biosciences)], followed by resuspension in FACS buffer, and intracellular staining (2 h on ice) using IL-22-PE-#22URTI or IFNγ-FITC-B27 (both eBioscience) and flow cytometry with gates set to exclude cell debris.
Analysis of mRNA Expression by Real-time Polymerase Chain Reaction (PCR)
Total RNA was extracted from homogenized mouse tissue or cultured cells using Tri-Reagent according to the manufacturer’s instructions (Sigma-Aldrich). Tissues were homogenized using OMNI TIP Homogenizing KIT (Kennesaw, GA, USA). 0.5 µg RNA was transcribed using random hexameric primers and Moloney Murine Leukemia Virus Reverse Transcriptase (Thermo Scientific, Darmstadt, Germany) according to the manufacturer’s instructions. cDNA was amplified using assay-on-demand kits (Taqman probes/assay kit from Thermo Scientific) and an AbiPrism 7500 Fast Sequence Detector (Thermo Scientific). During real-time PCR, changes in fluorescence are caused by the Taq polymerase degrading a probe containing a fluorescent dye [glyceraldehyde 3-phosphate dehydrogenase (GAPDH): VIC; all others: FAM]. Two initial steps at 50°C for 2 min and 95°C for 20 s were followed by 40 cycles at 95°C for 3 s and 60°C for 30 s. Target mRNA was normalized to that of GAPDH and quantified by the 2–ΔCT method (raw data, Figures 1, 2, 4 and 5) or the 2–ΔΔCT method (fold-induction, Figure 6). The following probes were used: hs-GAPDH (4310884E), hs-IL-22 (Hs01574152_g1), hs-IL-10 (Hs99999035_m1), hs-IFNγ (Hs00174143_m1), α1ACT (Hs00153674_m1), hs-IL-8 (Hs00174103_m1), hs-IL-2 (Hs00174114_m1), mm-GAPDH (4352339E), mm-IL-22 (Mm00444241_m1), mm-MIP2 (Mm00436450_m1), mm-IL-10 (Mm00439614_m1), mm-TNF-α (Mm00443285_m1), mm-IFNγ (Mm01168134_m1), and mm-IL17A (Mm00439618_m1). Primers and probe for IL-18BPa were designed using Primer Express (Applied Biosystems) according to AF110798: forward, 5′-ACCTCCCAGGCCGACTG-3′; reverse, 5′-CCTTGCACAGCTGCGTACC-3′; probe 5′-CACCAGCCGGGAACGTGGGA-3′. GAPDH was not a target of regulation by hypothermia under all conditions investigated (data not shown).
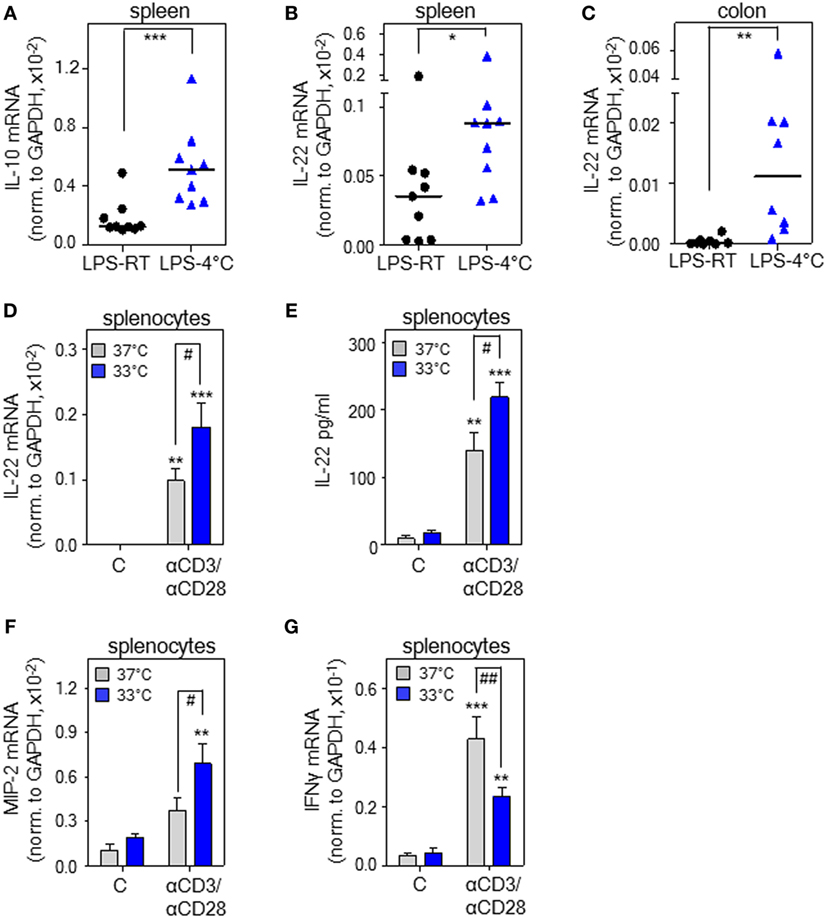
Figure 1. Cold stress and hypothermia promote interleukin (IL)-22 expression as detected in endotoxemic mice and cultured murine splenocytes. (A–C) Mice received i.p. either PBS or lipopolysaccharide (LPS, 1 µg/g) under conditions of room temperature (RT) [LPS-RT, n = 9 for (A,B) and n = 8 for (C)] or at 4°C [LPS-4°C, n = 9 for (A,B) and n = 8 for (C)]. After 5 h, splenic IL-10 (A) and IL-22 (B) as well as colonic (C) IL-22 mRNA expression were determined by real-time polymerase chain reaction (PCR) using GAPDH for normalization. Depicted are raw data for each tissue sample together with the group median. Expression of IL-22 mRNA in PBS-treated control mice (spleen: RT or 4°C, n = 8; colon: RT or 4°C, n = 6) was undetectable (spleen) or below 0.07 × 10–5 (colon) (raw data, IL-22 normalized to GAPDH). (A–C) *P < 0.05, **P < 0.01, ***P < 0.001; statistical analysis, Mann–Whitney U-test. (D,F,G) Ex vivo cultured murine splenocytes were either kept as unstimulated control or stimulated with agonistic anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C. After 8 h, mRNA expression of IL-22 (n = 10) (D), macrophage inflammatory protein (MIP)-2 (n = 5) (F), and interferon (IFN)γ (n = 10) (G) was determined by real-time PCR using GAPDH for normalization. Depicted are raw data (means ± SEM). (E) Murine splenocytes were either kept as unstimulated control or stimulated with agonistic anti-CD3 (5 µg/ml)/-CD28 (0.5 µg/ml) antibodies at either 37 or 33°C. After 72 h, IL-22 secretion was determined by enzyme-linked immunosorbent assay. Data are shown as means ± SEM (unstimulated control at either temperature, n = 4; anti-CD3/-CD28 at either temperature, n = 5). (D–G) **P < 0.01, ***P < 0.001 compared to unstimulated control; #P < 0.05, ##P < 0.01; statistical analysis, one-way ANOVA with post hoc Bonferroni correction.
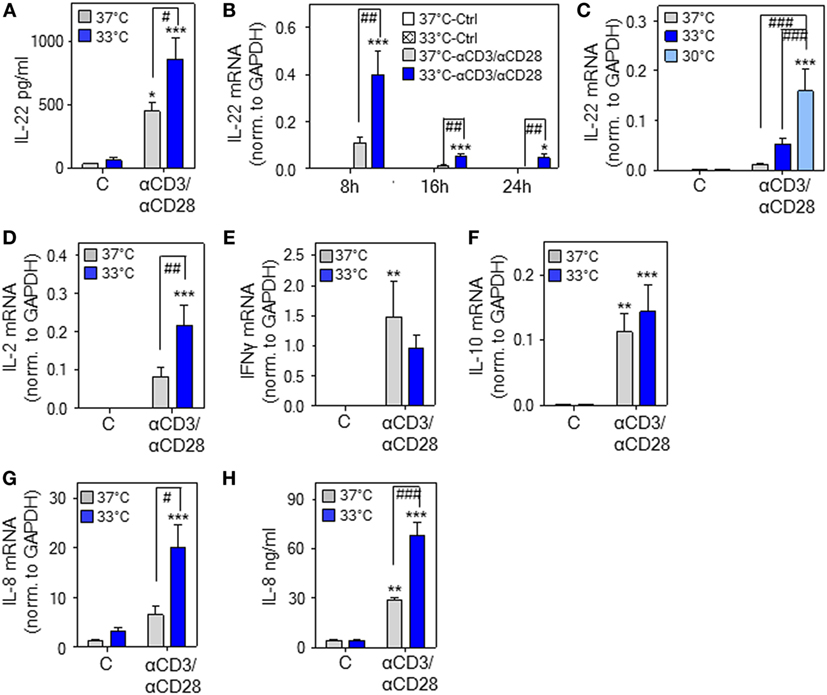
Figure 2. Hypothermia enhances interleukin (IL)-22 expression and release by human peripheral blood mononuclear cells (PBMC). (A–H) PBMC were kept as unstimulated control or stimulated with agonistic anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C (A–H) or 30°C (C). (A) After 48 h, IL-22 secretion was determined by enzyme-linked immunosorbent assay. Data are shown as means ± SEM (n = 6). *P < 0.05, ***P < 0.001 compared to unstimulated control at the respective temperature; #P < 0.05; statistical analysis, one-way ANOVA with post hoc Bonferroni correction. (B) After 8 h (n = 13), 16 h (unstimulated control at either temperature, n = 8; anti-CD3/-CD28 at either temperature, n = 14), or 24 h (unstimulated control at either temperature, n = 9; anti-CD3/-CD28 at either temperature, n = 14), IL-22 mRNA expression was determined by real-time polymerase chain reaction (PCR) using GAPDH for normalization. Depicted are raw data (means ± SEM). *P < 0.05, ***P < 0.001 compared to unstimulated control at the respective temperature and time point; ##P < 0.01; statistical analysis for individual time points, one-way ANOVA with post hoc Bonferroni correction. (C) After 16 h [unstimulated at all temperatures, n = 8; anti-CD3/-CD28 at 37°C (n = 14), at 33°C (n = 14), or at 30°C (n = 10)], IL-22 mRNA expression was determined by real-time PCR using GAPDH for normalization. Depicted are raw data (means ± SEM). ***P < 0.001 compared to unstimulated control at the respective temperature; ###P < 0.001; statistical analysis, one-way ANOVA with post hoc Bonferroni correction. (D–G) After 8 h, IL-2 [(D), n = 12], interferon (IFN)γ [(E), n = 12], IL-10 [(F), n = 14], and IL-8 [(G), n = 14] mRNA expression was determined by real-time PCR using GAPDH for normalization. Depicted are raw data (means ± SEM). **P < 0.01, ***P < 0.001 compared to unstimulated control at the respective temperature; #P < 0.05, ##P < 0.01. (H) After 48 h, IL-8 secretion was determined by enzyme-linked immunosorbent assay. Data are shown as means ± SEM (n = 6). **P < 0.01, ***P < 0.001 compared to unstimulated control at the respective temperature; ###P < 0.001. (D–H) Statistical analysis, one-way ANOVA with post hoc Bonferroni correction.
Immunoblot Analysis
Immunoblot analysis for cellular STAT1/3 in DLD1, Caco2, and HepG2 cells was performed as previously described (37) using total cell lysis buffer [150 mM NaCl, 1 mM CaCl2, 25 mM Tris–Cl (pH 7.4), 1% Triton X-100] supplemented with protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), DTT, Na3VO4, PMSF (each 1 mM), and NaF (20 mM). Antibodies: total STAT3-#124H6 (mouse monoclonal antibody); total STAT1, pSTAT1-Y701-#D4A7 (both rabbit polyclonal antibodies); pSTAT3-Y705-#D3A7 (rabbit monoclonal antibody); all from Cell Signaling, Frankfurt, Germany. For detection of total STAT1 or total STAT3 blots were stripped and reprobed.
Isolation of nuclei (38) for immunoblot analysis of nuclear NFAT-c2 in human T-cells was performed by lysis using nuclear extraction buffer A (10 mM HEPES at pH 7.9, 10 mM KCL, 0.1 mM EDTA, 0.1 mM EGTA) supplemented with protease inhibitor cocktail (Roche Diagnostics). After 10 min on ice and addition of Triton X-100, nuclei were collected by centrifugation at 12,000 g for 1 min at 4°C. Pellets containing nuclei were resuspended in nucleic-lysis buffer C (20 mM HEPES at pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 25% glycerin) supplemented with protease inhibitor cocktail (Roche Diagnostics). Antibodies: NFAT-c2-#4G6-G5, mouse monoclonal antibody (Santa Cruz Biotechnology, Heidelberg, Germany); β-actin-#AC15, mouse monoclonal antibody (Sigma-Aldrich). For detection of NFAT-c2 and β-actin on the same blot, the blot was cut. Data quantifications were performed by Quantity-One analysis software (Bio-Rad, Munich, Germany).
Statistical Analysis
Data are shown as group median, means ± SD, or means ± SEM and presented as [raw data], [fold-induction], [percent], [pg/ml], [ng/ml], and [Adj.Vol. INT*mm2]. The D’Agostino–Pearson normality test was used to assess data distribution. Statistical analysis was performed on raw data as indicated in the legends by one-way ANOVA with post hoc Bonferroni correction, unpaired Student’s t-test, or Mann–Whitney U-test. Differences were considered significant in case of P values below 0.05 (Prism 5.0, GraphPad, La Jolla, CA, USA).
Results
Cold Stress and Hypothermia Promote il22 Gene Expression As Detected in Endotoxemic Mice and Cultured Splenocytes
Experimental endotoxemia is regarded a standard model for the hyper-inflammatory phase of sepsis. Both, rodent endotoxemia (39) and sepsis (40) associate with enhanced IL-22 production. To investigate il22 gene expression under the influence of cold stress, PBS-treated control mice and mice undergoing endotoxemia were exposed to either an ambient temperature of 4°C or to RT (23°C) for 5 h. Notably, a low endotoxin dosage of 1 µg/g, by itself unable to induce hypothermia, was administered for induction of systemic inflammation. Analysis of rectal temperature after 5 h at 4°C revealed that only endotoxemic mice (n = 9) but not PBS-treated control mice (n = 8) developed mild-to-moderate hypothermia with a significant drop from 35.9± 0.3°C to a core temperature of 30.9 ± 0.9°C (P < 0.001, Student’s t-test). Under conditions of RT, core body temperature of PBS-treated control (n = 8) or endotoxemic mice (n = 9) was indistinguishable at 35.8 ± 0.4 or 35.5 ± 0.4°C, respectively. In accord with previous in vivo data on IL-10 and anti-inflammatory properties of hypothermia (26–35), enhanced splenic il10 gene expression was observed in endotoxemic mice exposed to 4°C (Figure 1A). To verify the hypothesis that IL-22 is affected by hypothermia, these same specimens were analyzed for expression of this cytokine. As shown in Figure 1B, splenic il22 gene expression was significantly upregulated in endotoxemic mice exposed to 4°C ambient temperature. On the contrary, in endotoxemic mice, upregulation of splenic IL-17A mRNA [0.40 × 10–4 versus 0.42 × 10–4 (median of target gene expression normalized to GAPDH) for RT versus 4°C ambient temperature (n = 9), not significantly different by Mann–Whitney U-test; IL-17A mRNA, undetectable in PBS-treated control mice—irrespective of ambient temperature] and tumor necrosis factor (TNF)-α mRNA [6.7 ± 0.9- versus 6.4 ± 1.7-fold-induction for RT (n = 8) versus 4°C ambient temperature (n = 9)] was unaffected by changes in ambient temperature. Since local IL-22 is crucial for maintenance of intestinal barrier function (41) and sepsis pathogenesis (16), colonic IL-22 expression was determined and found likewise upregulated upon cold stress (Figure 1C). Induction of colonic TNF-α mRNA by endotoxemia was, in accord with observations in spleen, not further increased by changes in ambient temperature (data not shown). Notably, as compared to RT, an ambient temperature of 4°C without endotoxemia failed to significantly affect splenic and colonic il22 expression. Specifically, using the current protocol, IL-22 mRNA was neither detectable in total colonic RNA obtained from PBS-treated control mice exposed to RT (n = 6) nor in splenic specimens irrespective of the tested ambient temperature (n = 8). In colonic tissues obtained from PBS-treated control mice exposed to 4°C, il22 expression was very low and barely detectable with a median of 0.01 × 10–5 (n = 6) for IL-22 mRNA normalized to that of GAPDH. Notably, very low il22 gene expression in colonic tissue of healthy untreated mice concurs with previous observations (42).
Exposure of mice to cold stress engages a complex systemic response that involves, among others, activation of the β-adrenergic/cAMP-axis (20) with its documented potential for immunomodulation (43), possibly by upregulation of IL-10 (43, 44). In order to evaluate whether hypothermia is able to directly upregulate il22 expression on the level of cultured murine leukocytes, freshly isolated splenocytes were stimulated using agonistic anti-CD3/-CD28 antibodies in the context of an ambient temperature of either 37 or 33°C—the latter condition resembling mild hypothermia. Figure 1D demonstrates that mild hypothermia amplifies IL-22 mRNA expression by activated splenocytes. Moreover, we also verified the potential of hypothermia to upregulate IL-22 protein release (Figure 1E). Whereas the murine functional IL-8 homolog and stress-responsive parameter macrophage inflammatory protein (MIP)-2 displayed similar upregulation by hypothermia (Figure 1F), expression of IFNγ mRNA was retarded under these same conditions (Figure 1G). In light of the proinflammatory pathological functions of IFNγ (45), this latter observation agrees with immunosuppressive properties of hypothermia (46). Taken together, data suggest that cold stress and hypothermia can serve as a cofactor enhancing murine il22 gene expression. Since stimulatory effects of hypothermia on IL-22 are detectable on cell culture level (Figures 1D,E), upregulation of the cytokine in vivo may also be independent from activation of the β-adrenergic/cAMP-axis.
Hypothermia Enhances IL-22 Expression and Release by Human PBMC
To broaden aforementioned murine in vitro and in vivo data, experiments were performed using human PBMC. We confirm previous observations (47) on IL-22 secretion by PBMC in response to stimulatory anti-CD3/-CD28 antibodies at physiological 37°C. Herein, we demonstrate that cultivation of PBMC at 33°C significantly amplifies IL-22 protein secretion (Figure 2A). Enhanced expression of IL-22 under the influence of mild hypothermia and anti-CD3/-CD28-stimulation was likewise detectable on mRNA level (Figure 2B). Upregulation of IL-22 by hypothermia was even more pronounced by further reducing the cultivation temperature to 30°C (Figure 2C) indicating a concurrent response upon more severe hypothermic culture conditions. IL2 gene expression was, like IL22, enhanced upon cultivation at 33°C (Figure 2D). Notably, regulation by mild hypothermia displayed specificity since neither expression of IFNG (Figure 2E) nor that of IL10 (Figure 2F) was affected by cultivation of anti-CD3/-CD28-stimulated PBMC at 33°C. Unresponsiveness of IL-10 concerning hypothermia in vitro contrasts with robust IL-10 upregulation in vivo [(28–35) and Figure 1A]. IL-10 production in vivo is, however, induced by the β-adrenergic/cAMP-axis (44), which is activated by acute hypothermia (20). This likely explains the observed differences compared to in vitro cultured PBMC. In contrast to IFNγ and IL-10, mRNA expression (Figure 2G) and release (Figure 2H) of IL-8 was increased by mild hypothermia. This latter observation concurs with the notion that IL8 is a stress-responsive gene (48).
Stimulation of human PBMC by treatment with anti-CD3/-CD28 antibodies and associated IL-22 secretion has been linked to the CD4+ memory T-cell compartment being the chief IL-22 PBMC-source under conditions of polyclonal activation (47). Herein, intracellular cytokine staining demonstrated that upregulation of IL-22+ cells by anti-CD3/-CD28 in total PBMC (Figure 3A) and the CD4+ PBMC fraction (Figure 3B) is further amplified by mild hypothermia. In contrast, exposure to 33°C did not significantly affect numbers of IFNγ+ cells in total PBMC (Figure 3C) thus confirming aforementioned mRNA data (Figure 2E).
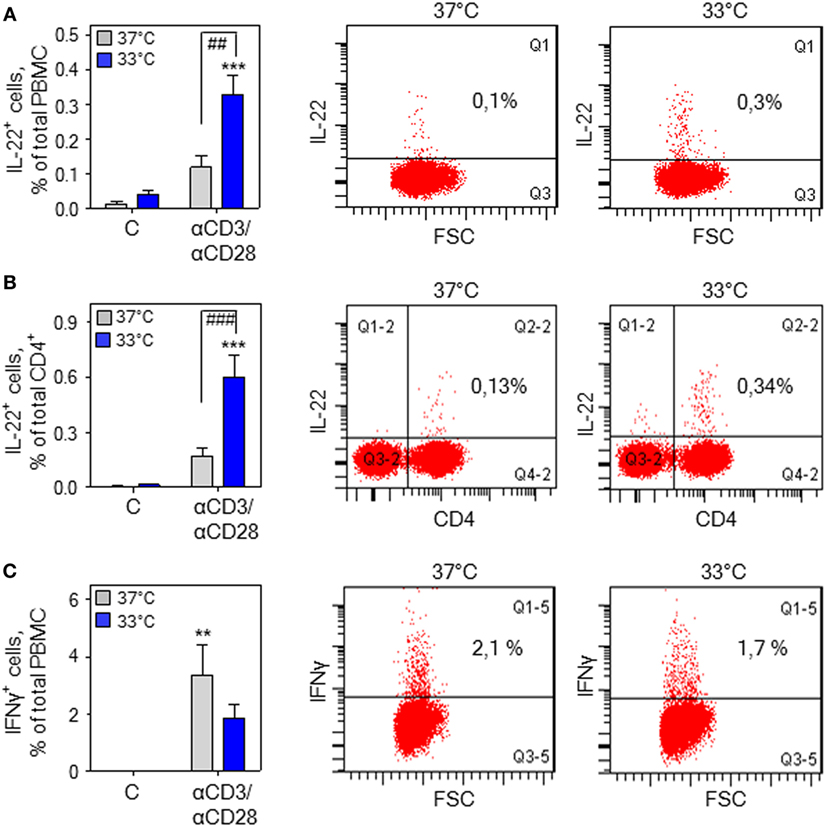
Figure 3. Hypothermia increases interleukin (IL)-22+ cells in total peripheral blood mononuclear cells (PBMC). (A) PBMC were kept as unstimulated control or stimulated for 7 h with agonistic anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C. Thereafter, brefeldin A (2 µg/ml) was added for another 4 h, followed by intracellular staining and flow cytometry for IL-22 (A,B) and interferon (IFN)γ (C) detection. Left panel (A–C): depicted are percentages of IL-22+ cells in total PBMC (n = 6) (A) and the CD4+ PBMC fraction (n = 7) (B) as well as IFNγ+ cells in total PBMC (n = 6) (C). Data are shown as means ± SEM. **P < 0.01, ***P < 0.001 compared to unstimulated control at the respective temperature; ##P < 0.01, ###P < 0.001; statistical analysis, one-way ANOVA with post hoc Bonferroni correction. Right panel: depicted are representative dot blots displaying IL-22+ cells in total PBMC (A) and the CD4+ PBMC fraction (B) as well as IFNγ+ cells in total PBMC (C).
Hypothermia Amplifying IL22 Expression in PBMC Demands the Presence of Monocytes
Monocytes have previously been identified as crucial cellular component supporting T-cell-derived IL-22 production by PBMC in response to diverse stimuli (49, 50). To specify their role in mild hypothermia enhancing IL-22 production, PBMC were depleted from monocytes. As shown in Figure 4A, depletion of monocytes entirely prevented upregulation of IL22 expression under the influence of hypothermia. To further investigate this matter, whole T-cells were isolated from PBMC and cultivated thereafter at either 37 or 33°C. Analysis after stimulation of isolated T-cells by anti-CD3/-CD28 antibodies actually revealed no significant difference in IL22 expression between both ambient temperatures (Figure 4B, left panel). Notably, whole PBMC from these same donors simultaneously cultivated displayed amplified IL-22 mRNA expression at 33°C (Figure 4B, right panel). Human leukemic Jurkat T-cells activate IL22 gene expression in response to polyclonal stimulation (18). In accord with aforementioned data on isolated primary T-cells, mild hypothermia left IL22 expression by TPA/A23187-stimulated Jurkat T-cells unaffected (Figure 4C). Taken together, observations indicate that upregulation of IL-22 by mild hypothermia is not a T-cell autonomous process but requires interactions between T cells and additional PBMC subsets, especially monocytes.
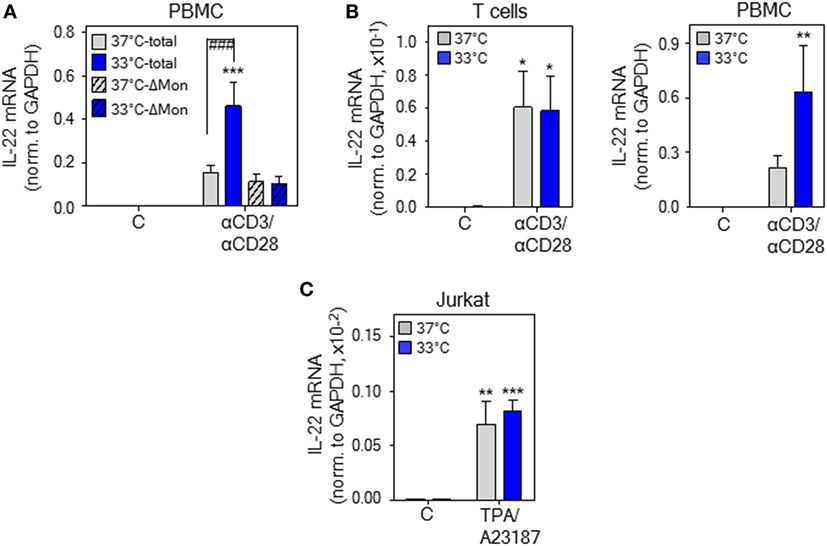
Figure 4. Upregulation of interleukin (IL)-22 expression by hypothermia demands monocyte presence. (A) Peripheral blood mononuclear cells (PBMC) were used in parallel as whole population (total) or depleted for monocytes (ΔMon). Total and monocyte-depleted PBMC were kept as unstimulated control or stimulated with anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C. After 8 h, IL-22 mRNA expression was determined by real-time polymerase chain reaction (PCR) using GAPDH for normalization. Data are shown as means ± SEM (n = 7). (B) PBMC were used in parallel as whole population or as starting point for isolation of CD3+ T-cells. CD3+ T-cells (left panel) or total PBMC (right panel) were kept as unstimulated control or stimulated with anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C. After 8 h, IL-22 mRNA expression was determined by real-time PCR using GAPDH for normalization. Data are shown as means ± SEM (n = 9). (C) Jurkat T cells were kept as unstimulated control or stimulated with TPA (20 ng/ml) plus A23187 (2 µM) at 37 or 33°C. After 6 h, IL-22 mRNA expression was assessed by real-time PCR using GAPDH for normalization. Data are shown as means ± SD (n = 5). (A–C), *P < 0.05, **P < 0.01, ***P < 0.001 compared to unstimulated control at the respective temperature; ###P < 0.001; statistical analysis, one-way ANOVA with post hoc Bonferroni correction.
Hypothermia Promotes NFAT-c2 in Human PBMC
The calcineurin/NFAT inhibitors cyclosporin A (CsA) and FK506 (tacrolimus) (51) potently suppress IL22 expression by activated human Jurkat T-cells, primary T-cells, and PBMC (18, 52). This observation concurs with inhibition of T-cell-derived IL-22 in psoriatic skin of tacrolimus-treated mice (53) and downregulation of IL-22 in patients undergoing CsA therapy (54, 55). Moreover, NFAT-c2 (51) binding to a specific site within the human IL22 promoter contributes to gene activation (18), conceivably by cooperation with an adjacent binding site for ATF2-jun heterodimers (56). Since aforementioned data suggested IL-22 as NFAT-inducible cytokine, CsA was tested in the current experimental protocol. In fact, coincubation with CsA not only impaired IL22 expression by activated PBMC upon normothermia but entirely prevented potentiation of gene induction at 33°C (Figure 5A). In contrast, CsA failed to suppress anti-CD3/-CD28-induced IL-8, a monocyte-derived inflammatory chemokine that was determined to control for unspecific inhibitory effects of the agent on PBMC cytokine expression. Interestingly, CsA actually amplified IL8 expression upon hypothermia (Figure 5B), which corresponds to observations on human smooth muscle cells where stimulation of activator protein-1 by CsA enforces IL-8 (57). Notably, the alternate NFAT antagonist FK506 (tacrolimus) displayed very similar inhibitory action on IL-22 (Figure 5C). As expected, we confirm previous observations (58) on potent suppression of IFNγ production by PBMC under the influence of CsA (Figure S1 in Supplementary Material).
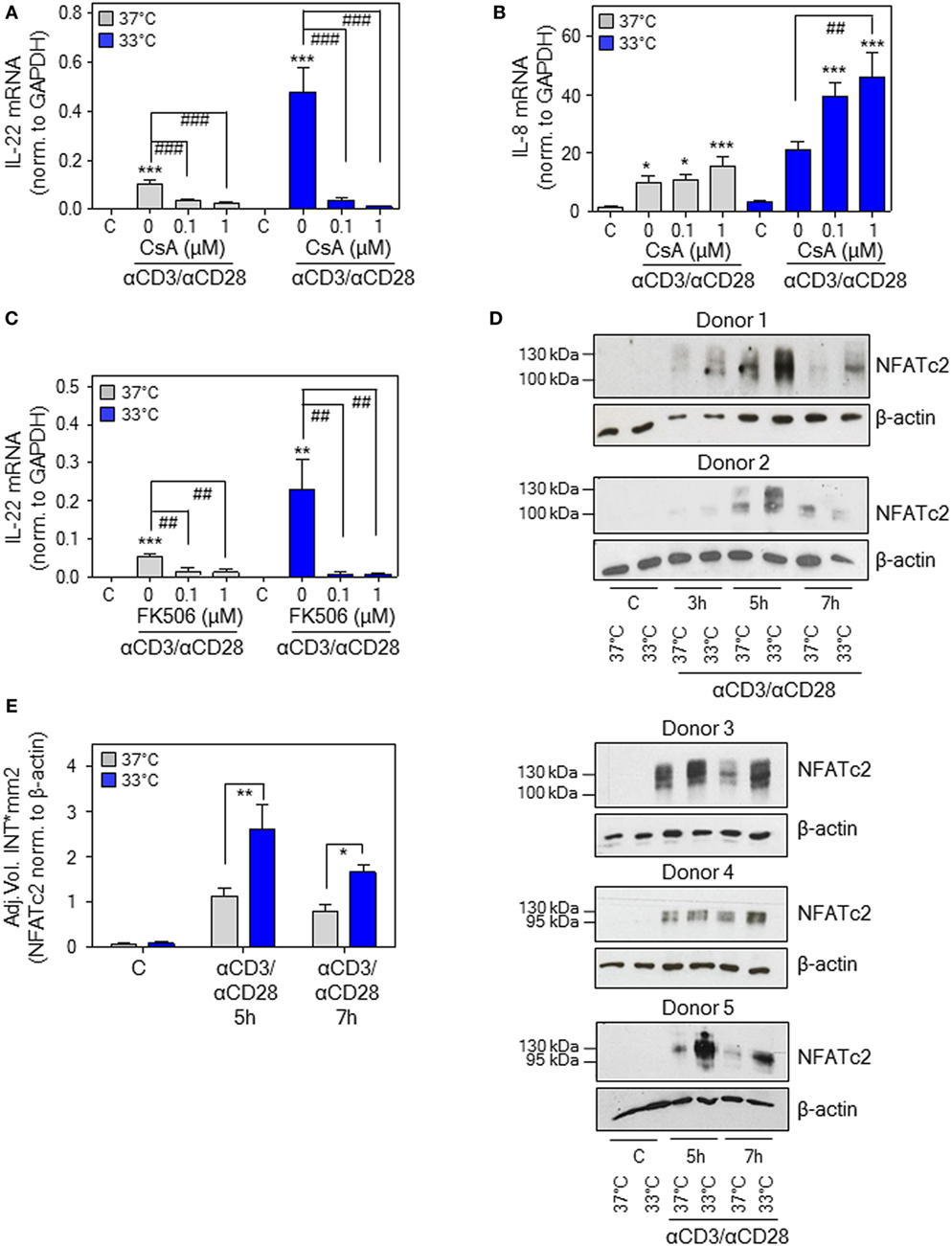
Figure 5. Nuclear factor of activated T cells (NFAT) signaling in peripheral blood mononuclear cells (PBMC) is mandatory for interleukin (IL)-22 upregulation by anti-CD3/-CD28 upon normothermic or hypothermic culture conditions. (A–C) Where indicated, PBMC were pretreated for 30 min with CsA (A,B) or with FK506 (C) at the specified concentrations. Cells were further kept as unstimulated control or stimulated with anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C. All cultures were adjusted to a final concentration of 0.002% (A,B) or 0.008% DMSO (C) (vehicle for CsA and FK506). After 8 h, IL-22 [(A), n = 10; (C), n = 6] or IL-8 [(B), n = 10] mRNA expression was assessed by real-time polymerase chain reaction using GAPDH for normalization. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 compared to unstimulated control; ##P < 0.01, ###P < 0.001; statistical analysis for either 37 or 33°C, one-way ANOVA with post hoc Bonferroni correction. (D) PBMC from five different donors were kept unstimulated or stimulated with anti-CD3 (0.2 µg/ml)/-CD28 (0.02 µg/ml) antibodies at 37 or 33°C for the indicated time periods. Thereafter, CD3+ T-cell isolation was performed followed by immunoblot analysis for detection of nucleic NFAT-c2. (E) Densitometric quantification of experiments shown in Figure 5D. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01; statistical analysis, unpaired Student’s t-test.
In order to more directly investigate the role of NFAT-c2 in this context, immunoblot analysis was performed using T-cells isolated instantly after stimulation of PBMC at 33 or 37°C. Herein, we demonstrate that hypothermia increases nuclear NFAT-c2 accumulation, thus transcription factor activation, in primary human T-cells after activation by anti-CD3/-CD28 (as part of whole PBMC). Notably, results obtained from five different donors indicated the most prominent effect of low ambient temperature at 5–7 h after onset of polyclonal T-cell stimulation (Figure 5D). Densitometric quantification of these experiments is shown in Figure 5E. Data altogether suggest that exposure of PBMC to mild hypothermia in CsA/FK506-sensitive manner supports IL22 expression, which associates with and is likely mediated by enhanced nuclear NFAT-c2.
Hypothermia Supports Activation of Epithelial-Like Cells by IL-22
To investigate effects of hypothermia on IL-22 biological activity, IL-22-responsive epithelial-like human Caco2 and DLD1 colon carcinoma cells (59) as well as HepG2 hepatoma cells (13) were adjusted for 6 h to an ambient temperature of either 30 or 37°C followed by stimulation with IL-22. As detected by analysis of pSTAT3 after 1 h, early IL-22 signal transduction was significantly enhanced upon hypothermia in all three cell types investigated. Notably, this effect vanished at the later 3-h time point. Figure 6 displays densitometric quantification of results (three independently performed experiments per cell line) obtained from DLD1 (Figure 6A), Caco2 (Figure 6B), and HepG2 cells (Figure 6C). Representative immunoblots for each cell line are shown in Figure 6D. To determine consequences of temperature-sensitive STAT3 activation for downstream gene regulation, prototypic IL-22-inducible α1-antichymotrypsin (α1ACT) (1, 14) was investigated in DLD1 cells. Time course analysis revealed enhanced expression of α1ACT mRNA at 30°C, a phenomenon that started at 4 h and persisted thereafter (Figure 6E). The specificity of hypothermia supporting STAT3 was assessed by analysis of IFNγ-mediated activation of STAT1 using the same experimental protocol. As shown by immunoblot analysis (Figure 6F) and respective densitometric quantification of three independently performed experiments (Figure 6G), STAT1 activation was, in stark contrast to STAT3, not enhanced but moderately inhibited by exposure of DLD1 cells to hypothermia. Moreover, expression of prototypic STAT1-dependent IL-18 binding protein (IL-18BP) mRNA expression (60, 61) was curbed by cultivation at 30°C (Figure 6H).
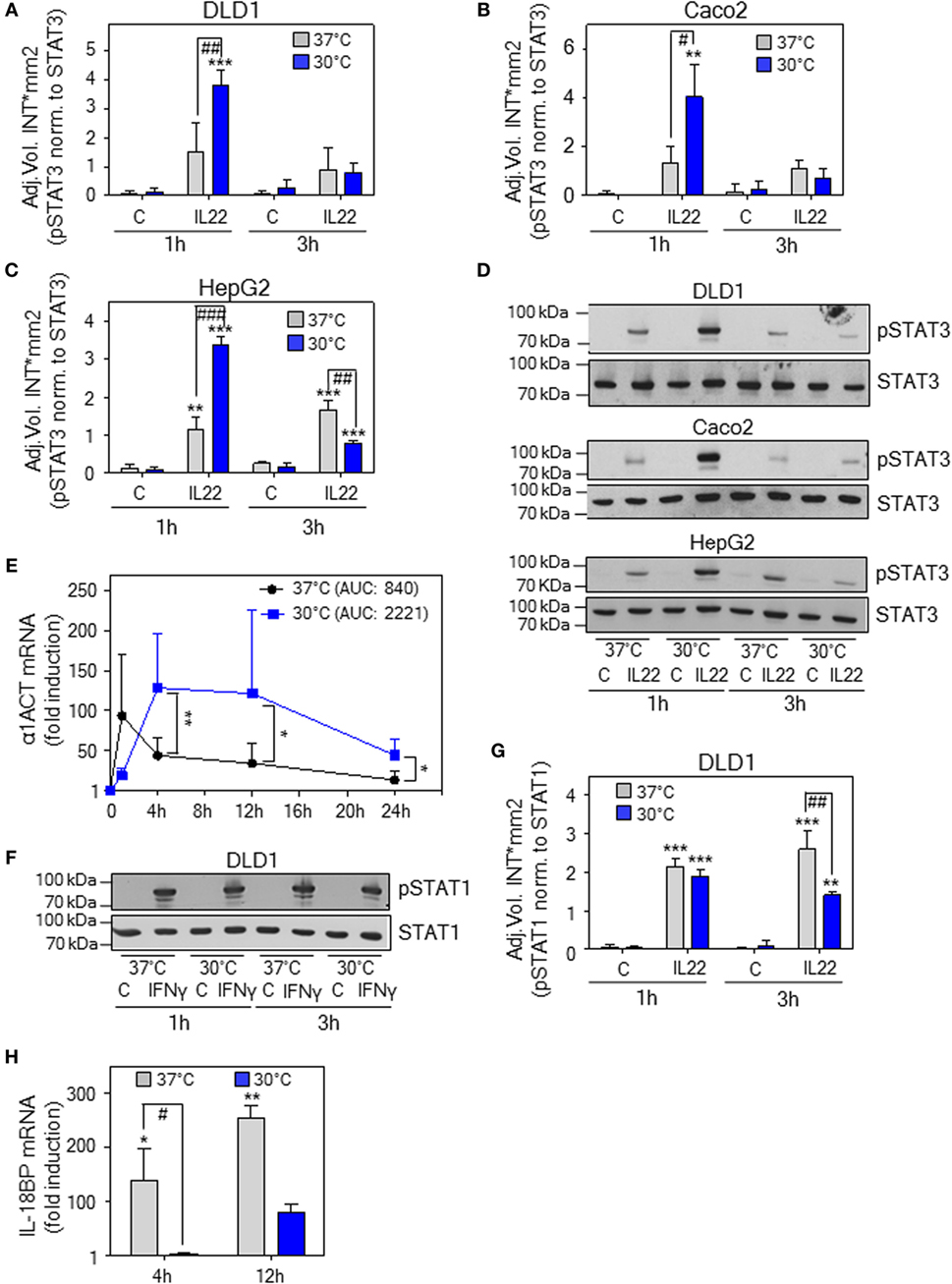
Figure 6. Effects of hypothermic preconditioning on interleukin (IL)-22 bioactivity. Epithelial (-like) IL-22-responsive human DLD1 (A) and Caco2 colon (B) carcinoma cells as well as HepG2 hepatoma cells (C) were preincubated for 6 h without additional stimulation at 30 or 37°C. Thereafter, cells maintained under the respective ambient temperatures were further kept as unstimulated control or stimulated with IL-22 (20 ng/ml). After 1 or 3 h, cells were harvested and IL-22-induced signal transducer and activator of transcription (STAT)-3 activation was determined by immunoblot analysis for pSTAT3. (A–C) Densitometric quantification of experiments (n = 3) is depicted as raw data (means ± SD). **P < 0.01, ***P < 0.001 compared to unstimulated control at the respective temperature and time point; #P < 0.05, ##P < 0.01, ###P < 0.001; statistical analysis for individual time points, one-way ANOVA with post hoc Bonferroni correction. (D) One representative of the three independently performed experiments (for each cell type) is shown. (E–H) DLD1 cells were preincubated for 6 h without additional stimulation at 30 or 37°C. (E) Thereafter, cells were further kept under the respective ambient temperatures as unstimulated control or were stimulated with IL-22 (20 ng/ml). After the indicated time periods, α1ACT mRNA expression was determined by real-time polymerase chain reaction (PCR) using GAPDH for normalization. Data are shown as means ± SD [1 h, n = 5 (30 and 37°C); 4 h, n = 8 (30°C) and n = 7 (37°C); 12 h, n = 8 (30 and 37°C); 24 h, n = 6 (30°C), and n = 4 (37°C)]; *P < 0.05, **P < 0.01; statistical analysis on fold-induction at each time point, unpaired Student’s t-test. AUC, area under the curve. (F) Thereafter, cells were further kept under the respective ambient temperatures as unstimulated control or stimulated with interferon (IFN)γ (20 ng/ml). After 1 or 3 h, cells were harvested and IFNγ-induced STAT1 activation was determined by immunoblot analysis for pSTAT1. One representative of three independently performed experiments is shown. Densitometric quantification of these experiments (n = 3) is depicted in (G) as raw data (means ± SD). **P < 0.01, ***P < 0.001 compared to unstimulated control at the respective temperature and time point; ##P < 0.01; statistical analysis for individual time points, one-way ANOVA with post hoc Bonferroni correction. (H) Thereafter, cells were further kept under the respective ambient temperatures as unstimulated control or stimulated with IFNγ (20 ng/ml). After 4 or 12 h, IL-18 binding protein (IL-18BP) mRNA expression was determined by real-time PCR using GAPDH for normalization. Data are shown as means ± SD (4 h, n = 4; 12 h, n = 5); *P < 0.05, **P < 0.01 compared to unstimulated control of the respective temperature and time point; #P < 0.05; statistical analysis on raw data for individual time points, one-way ANOVA with post hoc Bonferroni correction.
Discussion
Herein, we identify in vitro and in vivo hypothermia as novel parameter augmenting IL-22 expression in the context of immunoactivation. Hypothermia as single stimulus, however, failed to significantly upregulate IL-22 under all conditions investigated. Fine-tuning by hypothermia of largely T-cell-derived IL-22 in activated PBMC was dependent on monocytes, sensitive to inhibition by CsA or FK506, and associated with increased nuclear NFAT-c2. The notion of enhanced NFAT-c2 function under the influence of hypothermia is furthermore supported by the current observation of similarly regulated IL-2 expression, a well-defined prototypic NFAT-c2-inducible gene (62). Notably, early data already indicated the capability of monocytes to amplify Ca2+-signaling, thus NFAT function, in adjacent T-cells (63). Since NFAT in T-cells can be engaged by T-cell receptor-independent mechanisms connecting to innate immunity (64, 65), enhanced NFAT-c2 may also contribute to the current observation of hypothermia-associated IL-22 upregulation during murine endotoxemia.
Hypothermia may support NFAT function by action on T cell receptor/Ca2+-signaling and calcineurin activation or by inducing a state of NFAT hypophosphorylation via suppression of deactivating kinases. Interestingly, inhibition of glycogen synthase kinase-3β, regarded key to NFAT inactivation (51), has been associated with hypothermia in rat lung injury (66). In light of increased nuclear NFAT-c2 and enhanced IL-2 as well as IL-22 expression, lack of IFNγ upregulation, likewise an NFAT target (67), indicates complexity of hypothermia-regulated cytokine production. Hypothermia may thus be able to inhibit additional signals that are required for IFNγ production but leave IL-22 unaffected. Whereas inhibition of IFNγ agrees with tissue-protective properties of hypothermia, future studies need to shed light on mechanisms of differential regulation of IFNγ and IL-22 by low ambient temperature.
In order to relate effects of lowered ambient temperature to IL-22 biological activity, STAT3 activation was investigated. Previous reports indicated that activation of hepatic STAT3 is enforced by hypothermia during piglet cardiac surgery (29) and murine liver regeneration (68). Both studies connect hypothermia and hepatic STAT3 activation to liver protection, though upregulation of either IL-10 (29) or IL-6 (68) was associated with those observations—leaving open the possibility that hypothermia directly affects signal transduction. Herein, we report that cultivation of human DLD1 and Caco2 colon carcinoma as well as HepG2 hepatoma cells under the influence of hypothermia amplifies initial IL-22 signal transduction. Data concur with increased STAT3 activation in murine brain endothelial cells exposed to hypothermia (69). Notably, tissue-protective IL-22/STAT3 (4) but decisively not IFNγ/STAT1 signaling, the latter known to support pathological inflammation (45), was enhanced by hypothermia in the current study.
Tissue stress unbalancing homeostasis interconnects in a regulatory network with a fine-tuned inflammatory program, recently coined para-inflammation, which has the potential to enable adaptation and possibly protective preconditioning (70, 71). In that broader context, upregulation of IL-22 by hypothermia in endotoxemic mice suggests this cytokine to be part of a protective agenda not only directly serving preservation of stressed/injured tissues. By its capability to enforce insulin action (72, 73), enhanced IL-22 biological activity may, moreover, support insulin-dependent glucose uptake by brown fat and muscle tissue, which likely contributes to or supports heat generation of the hypothermic organism (74–76). Notably, when studied individually, endotoxemia and hypothermia are generally associated with reduced insulin function (77–80).
Rodent models of severe systemic inflammation/sepsis display endogenous hypothermia (81, 82) that can serve defined protective functions (19, 30–35). Current data suggest upregulation of IL-10 (30–35) and IL-22 as part of a hypothermia-associated cytokine profile counteracting overt pathological inflammation and strengthening biological barriers during severe systemic inflammation and infection. Observations presented likewise imply that IL-22 may contribute to specific tissue-protective properties of elective hypothermia.
Ethics Statement
For isolation of PBMC, heparinized blood was taken from healthy donors. This procedure was carried out in accordance with the recommendations of “Ethik Kommission of the University Hospital Goethe-University Frankfurt” with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the “Ethik Kommission of the University Hospital Goethe-University Frankfurt.” In vivo mouse experiments were carried out in accordance with the recommendations of ‘Regierungspräsidium Darmstadt’. The protocol was approved by the ‘Regierungspräsidium Darmstadt’.
Author Contributions
EC, BH, and MB performed experiments, analyzed the data, and performed manuscript editing. JP analyzed the data and contributed to manuscript editing. HM analyzed the data, designed the study, wrote the paper, and performed manuscript editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work is supported by a grant from the DFG (MU 1284/6-1) to HM. BH was supported by the “Frankfurter Promotionsförderung,” Medical Faculty, Goethe-University Frankfurt am Main.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00742/full#supplementary-material.
References
1. Dumoutier L, Van Roost E, Colau D, Renauld JC. Human interleukin-10-related T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc Natl Acad Sci U S A (2000) 97:10144–9. doi:10.1073/pnas.170291697
2. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol (2015) 33:747–85. doi:10.1146/annurev-immunol-032414-112123
3. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov (2014) 13:21–38. doi:10.1038/nrd4176
4. Mühl H, Scheiermann P, Bachmann M, Härdle L, Heinrichs A, Pfeilschifter J. IL-22 in tissue-protective therapy. Br J Pharmacol (2013) 169:761–71. doi:10.1111/bph.12196
5. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity (2004) 21:241–54. doi:10.1016/j.immuni.2004.07.007
6. Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol (2010) 31:354–61. doi:10.1016/j.it.2010.06.004
7. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14:282–9. doi:10.1038/nm1720
8. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14:275–81. doi:10.1038/nm1710
9. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem (2012) 287:8816–29. doi:10.1074/jbc.M111.304758
10. Hoegl S, Bachmann M, Scheiermann P, Goren I, Hofstetter C, Pfeilschifter J, et al. Protective properties of inhaled IL-22 in a model of ventilator-induced lung injury. Am J Respir Cell Mol Biol (2011) 44:369–76. doi:10.1165/rcmb.2009-0440OC
11. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest (2008) 118:534–44. doi:10.1172/JCI33194
12. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med (2009) 206:1465–72. doi:10.1084/jem.20082683
13. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology (2004) 39:1332–42. doi:10.1002/hep.20184
14. Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Mühl H. Application of interleukin-22 mediates protection in experimental acetaminophen-induced acute liver injury. Am J Pathol (2013) 182:1107–13. doi:10.1016/j.ajpath.2012.12.010
15. Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM II, Buchman TG, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA (2002) 287:1716–21. doi:10.1001/jama.287.13.1716
16. Abt MC, Buffie CG, Sušac B, Becattini S, Carter RA, Leiner I, et al. TLR-7 activation enhances IL-22-mediated colonization resistance against vancomycin-resistant enterococcus. Sci Transl Med (2016) 8:327ra25. doi:10.1126/scitranslmed.aad6663
17. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev (2013) 252:116–32. doi:10.1111/imr.12027
18. Rudloff I, Bachmann M, Pfeilschifter J, Mühl H. Mechanisms of rapid induction of interleukin-22 in activated T cells and its modulation by cyclosporin A. J Biol Chem (2012) 287:4531–43. doi:10.1074/jbc.M111.286492
19. Frink M, Flohé S, van Griensven M, Mommsen P, Hildebrand F. Facts and fiction: the impact of hypothermia on molecular mechanisms following major challenge. Mediators Inflamm (2012) 2012:762840. doi:10.1155/2012/762840
20. Karp CL. Unstressing intemperate models: how cold stress undermines mouse modeling. J Exp Med (2012) 209:1069–74. doi:10.1084/jem.20120988
21. Chio CC, Lin HJ, Tian YF, Chen YC, Lin MT, Lin CH, et al. Exercise attenuates neurological deficits by stimulating a critical HSP70/NF-κB/IL-6/synapsin I axis in traumatic brain injury rats. J Neuroinflammation (2017) 14:90. doi:10.1186/s12974-017-0867-9
22. Hsing CH, Hsieh MY, Chen WY, Cheung So E, Cheng BC, Chang MS. Induction of interleukin-19 and interleukin-22 after cardiac surgery with cardiopulmonary bypass. Ann Thorac Surg (2006) 81:2196–201. doi:10.1016/j.athoracsur.2006.01.092
23. Aslami H, Kuipers MT, Beurskens CJ, Roelofs JJ, Schultz MJ, Juffermans NP. Mild hypothermia reduces ventilator-induced lung injury, irrespective of reducing respiratory rate. Transl Res (2012) 159:110–7. doi:10.1016/j.trsl.2011.10.005
24. Sakurai T, Kudo M, Watanabe T, Itoh K, Higashitsuji H, Arizumi T, et al. Hypothermia protects against fulminant hepatitis in mice by reducing reactive oxygen species production. Dig Dis (2013) 31:440–6. doi:10.1159/000355242
25. Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev (2010) 21:331–44. doi:10.1016/j.cytogfr.2010.09.002
26. Morita Y, Oda S, Sadahiro T, Nakamura M, Oshima T, Otani S, et al. The effects of body temperature control on cytokine production in a rat model of ventilator-induced lung injury. Cytokine (2009) 47:48–55. doi:10.1016/j.cyto.2009.04.004
27. Cruces P, Erranz B, Donoso A, Carvajal C, Salomón T, Torres MF, et al. Mild hypothermia increases pulmonary anti-inflammatory response during protective mechanical ventilation in a piglet model of acute lung injury. Paediatr Anaesth (2013) 23:1069–77. doi:10.1111/pan.12209
28. Hildebrand F, van Griensven M, Giannoudis P, Luerig A, Harwood P, Harms O, et al. Effects of hypothermia and re-warming on the inflammatory response in a murine multiple hit model of trauma. Cytokine (2005) 31:382–93. doi:10.1016/j.cyto.2005.06.008
29. Qing M, Nimmesgern A, Heinrich PC, Schumacher K, Vazquez-Jimenez JF, Hess J, et al. Intrahepatic synthesis of tumor necrosis factor-alpha related to cardiac surgery is inhibited by interleukin-10 via the Janus kinase (Jak)/signal transducers and activator of transcription (STAT) pathway. Crit Care Med (2003) 31:2769–75. doi:10.1097/01.CCM.0000098858.64868.9C
30. Lim CM, Kim MS, Ahn JJ, Kim MJ, Kwon Y, Lee I, et al. Hypothermia protects against endotoxin-induced acute lung injury in rats. Intensive Care Med (2003) 29:453–9. doi:10.1007/s00134-002-1529-6
31. Hofstetter C, Boost KA, Flondor M, Basagan-Mogol E, Betz C, Homann M, et al. Anti-inflammatory effects of sevoflurane and mild hypothermia in endotoxemic rats. Acta Anaesthesiol Scand (2007) 51:893–9. doi:10.1111/j.1399-6576.2007.01353.x
32. Stewart CR, Landseadel JP, Gurka MJ, Fairchild KD. Hypothermia increases interleukin-6 and interleukin-10 in juvenile endotoxemic mice. Pediatr Crit Care Med (2010) 11:109–16. doi:10.1097/PCC.0b013e3181b01042
33. Sarcia PJ, Scumpia PO, Moldawer LL, DeMarco VG, Skimming JW. Hypothermia induces interleukin-10 and attenuates injury in the lungs of endotoxemic rats. Shock (2003) 20:41–5. doi:10.1097/01.shk.0000071080.50028.f2
34. Scumpia PO, Sarcia PJ, Kelly KM, DeMarco VG, Skimming JW. Hypothermia induces anti-inflammatory cytokines and inhibits nitric oxide and myeloperoxidase-mediated damage in the hearts of endotoxemic rats. Chest (2004) 125:1483–91. doi:10.1378/chest.125.4.1483
35. Huet O, Kinirons B, Dupic L, Lajeunie E, Mazoit JX, Benhamou D, et al. Induced mild hypothermia reduces mortality during acute inflammation in rats. Acta Anaesthesiol Scand (2007) 51:1211–6. doi:10.1111/j.1399-6576.2007.01419.x
36. Chichelnitskiy E, Vegiopoulos A, Berriel Diaz M, Ziegler A, Kleiman A, Rauch A, et al. In vivo phosphoenolpyruvate carboxykinase promoter mapping identifies disrupted hormonal synergism as a target of inflammation during sepsis in mice. Hepatology (2009) 50:1963–71. doi:10.1002/hep.23194
37. Bachmann M, Ulziibat S, Härdle L, Pfeilschifter J, Mühl H. IFNα converts IL-22 into a cytokine efficiently activating STAT1 and its downstream targets. Biochem Pharmacol (2013) 85:396–403. doi:10.1016/j.bcp.2012.11.004
38. Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res (1989) 17:6419. doi:10.1093/nar/17.15.6419
39. Wolk K, Witte E, Reineke U, Witte K, Friedrich M, Sterry W, et al. Is there an interaction between interleukin-10 and interleukin-22? Genes Immun (2005) 6:8–18. doi:10.1038/sj.gene.6364144
40. Ziesché E, Scheiermann P, Bachmann M, Sadik CD, Hofstetter C, Zwissler B, et al. Dexamethasone suppresses interleukin-22 associated with bacterial infection in vitro and in vivo. Clin Exp Immunol (2009) 157:370–6. doi:10.1111/j.1365-2249.2009.03969.x
41. Parks OB, Pociask DA, Hodzic Z, Kolls JK, Good M. Interleukin-22 signaling in the regulation of intestinal health and disease. Front Cell Dev Biol (2016) 3:85. doi:10.3389/fcell.2015.00085
42. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491:259–63. doi:10.1038/nature11535
43. Lorton D, Bellinger DL. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int J Mol Sci (2015) 16:5635–65. doi:10.3390/ijms16035635
44. Platzer C, Meisel C, Vogt K, Platzer M, Volk HD. Up-regulation of monocytic IL-10 by tumor necrosis factor-alpha and cAMP elevating drugs. Int Immunol (1995) 7:517–23. doi:10.1093/intimm/7.4.517
45. Mühl H. Pro-inflammatory signaling by IL-10 and IL-22: bad habit stirred up by interferons? Front Immunol (2013) 4:18. doi:10.3389/fimmu.2013.00018
46. Du G, Liu Y, Li J, Liu W, Wang Y, Li H. Hypothermic microenvironment plays a key role in tumor immune subversion. Int Immunopharmacol (2013) 17:245–53. doi:10.1016/j.intimp.2013.06.018
47. Liu Y, Yang B, Zhou M, Li L, Zhou H, Zhang J, et al. Memory IL-22-producing CD4+ T cells specific for Candida albicans are present in humans. Eur J Immunol (2009) 39:1472–9. doi:10.1002/eji.200838811
48. Shapiro L, Dinarello CA. Osmotic regulation of cytokine synthesis in vitro. Proc Natl Acad Sci U S A (1995) 92:12230–4. doi:10.1073/pnas.92.26.12230
49. Bachmann M, Horn K, Rudloff I, Goren I, Holdener M, Christen U, et al. Early production of IL-22 but not IL-17 by peripheral blood mononuclear cells exposed to live Borrelia burgdorferi: the role of monocytes and interleukin-1. PLoS Pathog (2010) 6:e1001144. doi:10.1371/journal.ppat.1001144
50. Truchetet ME, Allanore Y, Montanari E, Chizzolini C, Brembilla NC. Prostaglandin I(2) analogues enhance already exuberant Th17 cell responses in systemic sclerosis. Ann Rheum Dis (2012) 71:2044–50. doi:10.1136/annrheumdis-2012-201400
51. Müller MR, Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol (2010) 10:645–56. doi:10.1038/nri2818
52. Hoffmann U, Neudörfl C, Daemen K, Keil J, Stevanovic-Meyer M, Lehner F, et al. NK cells of kidney transplant recipients display an activated phenotype that is influenced by immunosuppression and pathological staging. PLoS One (2015) 10:e0132484. doi:10.1371/journal.pone.0132484
53. Takenaka N, Edamatsu H, Suzuki N, Saito H, Inoue Y, Oka M, et al. Overexpression of phospholipase Cε in keratinocytes upregulates cytokine expression and causes dermatitis with acanthosis and T-cell infiltration. Eur J Immunol (2011) 41:202–13. doi:10.1002/eji.201040675
54. Haider AS, Lowes MA, Suárez-Fariñas M, Zaba LC, Cardinale I, Khatcherian A, et al. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol (2008) 180(3):1913–20. doi:10.4049/jimmunol.180.3.1913
55. Khattri S, Shemer A, Rozenblit M, Dhingra N, Czarnowicki T, Finney R, et al. Cyclosporine in patients with atopic dermatitis modulates activated inflammatory pathways and reverses epidermal pathology. J Allergy Clin Immunol (2014) 133:1626–34. doi:10.1016/j.jaci.2014.03.003
56. Macián F, López-Rodríguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene (2001) 20:2476–89. doi:10.1038/sj.onc.1204386
57. Murakami R, Kambe F, Mitsuyama H, Okumura K, Murohara T, Niwata S, et al. Cyclosporin A enhances interleukin-8 expression by inducing activator protein-1 in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol (2003) 23:2034–40. doi:10.1161/01.ATV.0000094234.60166.78
58. Espevik T, Figari IS, Shalaby MR, Lackides GA, Lewis GD, Shepard HM, et al. Inhibition of cytokine production by cyclosporin A and transforming growth factor beta. J Exp Med (1987) 166:571–6. doi:10.1084/jem.166.2.571
59. Ziesché E, Bachmann M, Kleinert H, Pfeilschifter J, Mühl H. The interleukin-22/STAT3 pathway potentiates expression of inducible nitric-oxide synthase in human colon carcinoma cells. J Biol Chem (2007) 282:16006–15. doi:10.1074/jbc.M611040200
60. Paulukat J, Bosmann M, Nold M, Garkisch S, Kämpfer H, Frank S, et al. Expression and release of IL-18 binding protein in response to IFN-gamma. J Immunol (2001) 167:7038–43. doi:10.4049/jimmunol.167.12.7038
61. Bachmann M, Paulukat J, Pfeilschifter J, Mühl H. Molecular mechanisms of IL-18BP regulation in DLD-1 cells: pivotal direct action of the STAT1/GAS axis on the promoter level. J Cell Mol Med (2009) 13:1987–94. doi:10.1111/j.1582-4934.2008.00604.x
62. Podtschaske M, Benary U, Zwinger S, Höfer T, Radbruch A, Baumgrass R. Digital NFATc2 activation per cell transforms graded T cell receptor activation into an all-or-none IL-2 expression. PLoS One (2007) 2:e935. doi:10.1371/journal.pone.0000935
63. Lederman HM, Lee JW, Cheung RK, Grinstein S, Gelfand EW. Monocytes are required to trigger Ca2+ uptake in the proliferative response of human t lymphocytes to Staphylococcus aureus protein A. Proc Natl Acad Sci U S A (1984) 81:6827–30. doi:10.1073/pnas.81.21.6827
64. Raab M, Pfister S, Rudd CE. CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity (2001) 15:921–33. doi:10.1016/S1074-7613(01)00248-5
65. Martelli MP, Lin H, Zhang W, Samelson LE, Bierer BE. Signaling via LAT (linker for T-cell activation) and Syk/ZAP70 is required for ERK activation and NFAT transcriptional activation following CD2 stimulation. Blood (2000) 96:2181–90.
66. Kim K, Kim W, Rhee JE, Jo YH, Lee JH, Kim KS, et al. Induced hypothermia attenuates the acute lung injury in hemorrhagic shock. J Trauma (2010) 68:373–81. doi:10.1097/TA.0b013e3181a73eea
67. Sica A, Dorman L, Viggiano V, Cippitelli M, Ghosh P, Rice N, et al. Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. J Biol Chem (1997) 272:30412–20. doi:10.1074/jbc.272.48.30412
68. Debonera F, Aldeguer X, Shen X, Gelman AE, Gao F, Que X, et al. Activation of interleukin-6/STAT3 and liver regeneration following transplantation. J Surg Res (2001) 96:289–95. doi:10.1006/jsre.2001.6086
69. Park YH, Lee YM, Kim DS, Park J, Suk K, Kim JK, et al. Hypothermia enhances induction of protective protein metallothionein under ischemia. J Neuroinflammation (2013) 10:21. doi:10.1186/1742-2094-10-21
70. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi:10.1038/nature07201
71. Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell (2014) 54:281–8. doi:10.1016/j.molcel.2014.03.030
72. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature (2014) 514:237–41. doi:10.1038/nature13564
73. Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med (2014) 20:1417–26. doi:10.1038/nm.3705
74. Gumbiner B, Thorburn AW, Henry RR. Reduced glucose-induced thermogenesis is present in noninsulin-dependent diabetes mellitus without obesity. J Clin Endocrinol Metab (1991) 72:801–7. doi:10.1210/jcem-72-4-801
75. Orava J, Nuutila P, Lidell ME, Oikonen V, Noponen T, Viljanen T, et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab (2011) 14:272–9. doi:10.1016/j.cmet.2011.06.012
76. Inokuma K, Ogura-Okamatsu Y, Toda C, Kimura K, Yamashita H, Saito M. Uncoupling protein 1 is necessary for norepinephrine-induced glucose utilization in brown adipose tissue. Diabetes (2005) 54:1385–91. doi:10.2337/diabetes.54.5.1385
77. Vila IK, Badin PM, Marques MA, Monbrun L, Lefort C, Mir L, et al. Immune cell toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep (2014) 7:1116–29. doi:10.1016/j.celrep.2014.03.062
78. Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes (2010) 59:172–81. doi:10.2337/db09-0367
79. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
80. Sah Pri A, Chase JG, Pretty CG, Shaw GM, Preiser JC, Vincent JL, et al. Evolution of insulin sensitivity and its variability in out-of-hospital cardiac arrest (OHCA) patients treated with hypothermia. Crit Care (2014) 18:586. doi:10.1186/s13054-014-0586-x
81. Souza LL, Duchene J, Todiras M, Azevedo LC, Costa-Neto CM, Alenina N, et al. Receptor MAS protects mice against hypothermia and mortality induced by endotoxemia. Shock (2014) 41:331–6. doi:10.1097/SHK.0000000000000115
Keywords: interleukin-22, signal transducer and activator of transcription 3, endotoxemia, hypothermia, peripheral blood mononuclear cells
Citation: Chichelnitskiy E, Himmelseher B, Bachmann M, Pfeilschifter J and Mühl H (2017) Hypothermia Promotes Interleukin-22 Expression and Fine-Tunes Its Biological Activity. Front. Immunol. 8:742. doi: 10.3389/fimmu.2017.00742
Received: 28 March 2017; Accepted: 12 June 2017;
Published: 29 June 2017
Edited by:
Sofie Struyf, KU Leuven, BelgiumReviewed by:
Jung Ho Lee, Yonsei University, South KoreaLauren A. Zenewicz, University of Oklahoma Health Sciences Center, United States
Timothy Frankel, University of Michigan, United States
Copyright: © 2017 Chichelnitskiy, Himmelseher, Bachmann, Pfeilschifter and Mühl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heiko Mühl, aC5tdWVobEBlbS51bmktZnJhbmtmdXJ0LmRl