 Namit Holay
Namit Holay Youra Kim1
Youra Kim1 Patrick Lee
Patrick Lee Shashi Gujar
Shashi Gujar- 1Department of Pathology, Dalhousie University, Halifax, NS, Canada
- 2Department of Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
- 3Department of Biology, Dalhousie University, Halifax, NS, Canada
- 4Centre for Innovative and Collaborative Health Sciences Research, Quality and System Performance, IWK Health Centre, Halifax, NS, Canada
Cancer immunotherapy represents a promising, modern-age option for treatment of cancers. Among the many immunotherapies being developed, oncolytic viruses (OVs) are slowly moving to the forefront of potential clinical therapeutic agents, especially considering the fact that the first oncolytic virus was recently approved by the Food and Drug Administration for the treatment of melanoma. OVs were originally discovered for their ability to kill cancer cells, but they have emerged as unconventional cancer immunotherapeutics due to their ability to activate a long-term antitumor immune response. This immune response not only eliminates cancer cells but also offers potential for preventing cancer recurrence. A fundamental requirement for the generation of such a strong antitumor T cell response is the recognition of an immunogenic tumor antigen by the antitumor T cell. Several tumor antigens capable of activating these antitumor T cells have been identified and are now being expressed through genetically engineered OVs to potentiate antitumor immunity. With the emergence of novel technologies for identifying tumor antigens and immunogenic epitopes in a myriad of cancers, design of “oncolytic vaccines” expressing highly specific tumor antigens provides a great strategy for targeting tumors. Here, we highlight the various OVs engineered to target tumor antigens and discuss multiple studies and strategies used to develop oncolytic vaccine regimens. We also contend how, going forward, a combination of technologies for identifying novel immunogenic tumor antigens and rational design of oncolytic vaccines will pave the way for the next generation of clinically efficacious cancer immunotherapies.
Introduction
Immunotherapies have steadily emerged as a powerful treatment option for patients with various types of cancers. While employing the immune system to fight cancer was first proposed in the late nineteenth century, it was only recently that the improved understanding and novel discoveries in classical and tumor immunology have led to the design of more targeted and efficacious immunotherapeutics (1–4). One such immunotherapy utilizes oncolytic viruses (OVs), which were originally discovered for their direct cancer-killing properties (5). Historically, case reports of cancer regression following infection with unrelated viruses started appearing around the early twentieth century (6). But it was not until the 1990s that concrete evidence demonstrated the ability of certain viruses to preferentially target cancer cells (7–9). In recent times, the advent of new technologies allowing for customization of viruses, combined with the urgent need for novel and effective therapies for cancer treatment, has led to a new impetus for OV research (10). OVs have a dual mechanism of action against tumors. First, they can preferentially replicate in and directly kill cancer cells, in a mechanism known as oncolysis (11). Second, the immunological events induced following the administration of OVs awaken the previously suppressed immune system to become activated and target tumor cells more effectively (12). This activation of the immune system is the most promising aspect of oncolytic virotherapy. One of the major players of the immune system responsible for targeting cancer cells are T cells and the effective activation of tumor-specific T cells can lead to long-term antitumor immunity and protection against cancer recurrence (13).
For the activation of antitumor T cells, the primary requirement is the presentation of a tumor antigen via major histocompatibility complex (MHC) molecules of antigen-presenting cells (APCs) (14). Antigens, usually identified as small peptide molecules of approximately 8–18 amino acids in length, are expressed via MHC class I and II molecules, and lead to the activation of antigen-specific CD8 and CD4 T cells, respectively (15). Tumor antigens can be derived from peptide fragments of mutated oncoproteins and tumor suppressors, aberrantly expressed cellular proteins, modified glycoproteins, oncofetal proteins, tissue-specific differentiation proteins, and proteins derived from oncogenic viruses (16, 17). Identification of such tumor antigens to activate antigen-specific T cell responses in tumors represent a highly attractive target for cancer immunotherapies today (16, 18). In addition to the antigenic peptide presented through the MHC molecule, the complete activation of T cells requires two other signals: costimulatory molecules on APCs and the presence of the appropriate cytokines in the immune milieu (19, 20). Thus, ongoing research to improve cancer immunotherapies aim to target one or more of these signals to effectively stimulate clinically relevant antitumor T cells.
In this mini-review, we highlight the studies that have incorporated tumor antigens in OVs to enhance antitumor immune responses and consequent therapeutic benefits in the context of cancer. We discuss recent studies completed using a variety of viral systems, as well as combinations of multiple strategies used to elicit the most efficacious immune response. We also throw light on some of the challenges in this area of research and emphasize the need for combining recent, cutting-edge technologies for tumor antigen discovery with oncolytic virus research for generating more efficacious cancer treatments.
Oncolytic Vaccine Therapy
The first generation of OVs primarily focused on direct killing of tumor cells. OVs can replicate preferentially in tumor cells due to deregulated signaling pathways (8, 9, 21, 22) resulting in increased susceptibility of tumor cells to viral infections (22–24). When it was observed that the direct killing of tumor cells led to the release of novel tumor antigens in the tumor microenvironment and the subsequent activation of immune responses (25–29), strategies began to be focused on the modulation and optimization of these immune responses to achieve maximum clinical benefit. The overexpression of tumor antigens via OVs represents one such strategy that makes OV-based cancer therapies more potent by driving immune responses to be directed specifically toward the tumor. OVs that are genetically modified to express tumor antigens are commonly known as “oncolytic vaccines.”
Vesicular Stomatitis Virus (VSV)
In recent times, many OVs have been found to be amenable for therapeutically desired genetic modifications. Among these, VSV has been the subject of extensive genetic manipulation and consequent investigation on antitumor immunity in the context of cancer treatment. For example, studies have shown that VSV-expressing tumor antigens human papilloma virus oncogene E7 (VSV-E7) and human dopachrome tautomerase (VSV-hDCT) can induce tumor antigen-specific CD8 cytotoxic T cell responses (30, 31). Therapeutic vaccination with VSV-E7 led to reduced TC-1 tumor volumes, and VSV-hDCT generated antigen-specific CD4 T cell responses in addition to CD8 T cells in murine melanoma (30, 31). Another study employed the popularly used ovalbumin (ova) as a surrogate tumor antigen expressed in murine melanoma cells to demonstrate that the administration of VSV engineered to express ova (VSV-ova) led to increased activation of naïve T cells, as well as increased number of ova-specific, antitumor T cells (32).
Furthermore, VSV-based oncolytic vaccines have also been indicated as promising candidates to be used in combination therapies. A recent study demonstrated that in combination with stereotactic ablative radiation therapy, VSV-ova helped control local and systemic disease in a murine oligometastatic melanoma model (33). In addition, oncolytic vaccines can be administered in combination with adoptive transfer of antigen-specific T cells for enhanced therapeutic benefits compared to either treatment alone. For instance, a melanoma-derived tumor antigen gp100-expressing VSV (VSV-gp100) combined with adoptive transfer of gp100-specific T cells resulted in increased survival of mice with established melanoma that was accompanied by the development of antitumor T cell responses (34, 35). Similar results were also observed by combining VSV-ova and adoptive transfer of ova-specific T cells (32). Taken together, these studies in preclinical models demonstrated that oncolytic vaccines may be combined with current clinical treatment options to achieve improved therapeutic and immune responses.
Another interesting approach to develop efficacious VSV-based oncolytic vaccines was to employ tumor-derived cDNA libraries. Specifically, cDNA libraries derived from cancer cell lines are expressed in VSV, followed by screening and administration of these library-based oncolytic vaccines in tumor-bearing mice. VSV expressing a cDNA library created from normal prostate cells has been shown to lead to the rejection of mouse prostate cancers with little autoimmunity as measured by whitening of whiskers and tail, hair depigmentation, abnormal immune cell infiltration, and tissue destruction (36). A subsequent study employed VSV to express a melanoma cDNA library, which was screened in vitro for immunogenic tumor antigens, and demonstrated that a combination of three specific VSV-cDNA viruses infected established melanoma tumors and induced tumor rejection via TH-17 responses (37).
These studies using cDNA library-expressing VSV also highlighted the importance of treating cancers according to their origins. First, it was observed that primary and recurring tumors must be targeted in a different manner and thus, “recurrence” libraries were developed. In one study employing a murine melanoma model, 14 out of 16 recurrences were found to have mutated BRAF, so VSV-BRAF was used to successfully target the recurring tumors (38). Second, the anatomical site of cancer development is another important consideration for oncolytic vaccine design. For example, a combination of VSV viruses derived from the melanoma cDNA library (VSV-N-RAS, VSV-CYTC-C, and VSV-TYRP-1) that was successful in treating subcutaneous melanoma could not treat intracranial melanoma tumors. Instead, a therapeutic combination targeting new tumor antigens in the context of intracranial tumors (VSV-HIF-2α, VSV-SOX-10, VSV-C-MYC, and VSV-TYRP-1) was shown to promote long-term survival of mice with intracranial melanoma (39). Building on this, another study showed that tumors of different histological origin shared immunological signatures based on their location and could be targeted specifically (40). The study used a glioma model in comparison to the intracranial melanoma model to establish that different tumors growing in the same location shared location-specific immunological signatures that could be targeted with the right combination of oncolytic vaccines. Of note, this study was among a few that combined oncolytic vaccines with immunological checkpoint inhibitors (ICIs) to reveal that ICIs enhanced the impact of oncolytic vaccines by reactivating TH-1 and TH-17 responses. Using ICIs like anti-PD1 and anti-CTLA4 antibodies in combination with OVs represents the next frontier in cancer immunotherapy as these complementary therapies are emerging as synergistic therapeutic partners of each other (41–44). Going forward, identification of novel tumor antigens (discussed in Section “Future directions and concluding remarks”) for expression in viral vectors combined with effective combination therapy via ICIs represents an emerging paradigm of cancer immunotherapy.
Vaccinia Virus (VV)
Another virus that has allowed for ease of genetic manipulation and, thus, lent itself to oncolytic vaccine research is VV. VV has been employed in multiple studies for prophylactic vaccine development. Studies using oncofetal tumor antigen animal models like carcinoembryonic antigen (CEA) and glycoprotein oncofetal tumor antigen (5T4) transgenic mice have been widely used to study the prophylactic capacity of VV vectors. One such study demonstrated that VV-expressing CEA administered in CEA transgenic mice led to the development of CEA-specific TH-1 responses and peptide-specific cytotoxicity. The authors noted that neither CEA antibodies nor CEA-specific T cell responses were elicited in CEA transgenic mice in response to endogenous or administered CEA in the absence of virus, indicating the usability of this animal model for more aggressive vaccinations (45). Upon virus infection, protection against CEA-specific cancer cells was observed without any effect on normal tissue-expressing CEA (45). Another study employing VV-expressing human and mouse 5T4 (VV-h5T4, VV-m5T4) demonstrated that mice vaccinated with these vectors showed retarded tumor growth upon challenge with syngeneic melanoma and colorectal cancer cells (46). No autoimmune toxicity in the form of wasting, respiratory problems, affected mobility or weight loss was seen in this study. Put together, these studies reaffirmed the potential of oncolytic vaccines as safe therapeutic options for treatment of cancers with low off-site toxicity.
Engineering genetic elements other than tumor antigens for immunomodulation is a common strategy for developing viral vectors. VV has provided an excellent platform for engineering costimulatory elements in conjunction with tumor antigens to incorporate other important factors for T cell activation. One such study demonstrated the expression of a T cell engager (TCE) element along with tumor antigen EphA2 in a VV vector (47). The TCE is a special secretory element used to specifically bind and activate T cells via CD3. The study noted that the virus killed tumor cells and induced a bystander killing of non-infected tumor cells in vitro. The modified VV also had potent antitumor activity in vivo in a lung cancer xenograft model (47). Another study employed VV to express a triad of costimulatory molecules (B7-1, ICAM-1, and LFA-3) along with oncofetal tumor antigen CEA, and the administration of the modified virus increased survival of colon adenocarcinoma tumor-bearing mice due to induced CD8 and CD4 T cell responses. Clinical serum and urine assays combined with histopathology showed no classical indicators of autoimmune responses, a typical complication when targeting oncofetal antigens like CEA (48).
The final factor that determines the quality of the generated T cell responses is the presence of cytokines in the immune environment. Expressing cytokines via viral vectors represents a promising strategy for the development of a robust antitumor immune response, and thus far, many cytokines including IL-2, TNF, IFN, and GM-CSF have been expressed through OVs (49–52). In 2015, the U.S. Food and Drug Administration approved the first oncolytic virus T-VEC for use in clinics. T-VEC employs modified herpes simplex virus (HSV)-expressing GM-CSF to enhance the generation of APCs (53). In the context of oncolytic vaccines and cytokines, a study using MB49 cancer cells expressing male tumor antigen HY was used to evaluate the efficacy of VV-overexpressing HY (VV-HY) and GM-CSF (VV-GM-CSF) to overcome tumor-associated immune tolerance. The study noted that the administration of both viruses together led to the generation of splenic HY-specific CD8 T cells, indicating development of systemic immunity (54). Overall, consideration of all the signals required for an effective T cell response is an important parameter for future studies employing oncolytic vaccines to activate holistic antitumor responses that are qualitatively superior to current therapeutic regimens.
Other Viruses
Several other viruses have been employed as oncolytic vaccines to target tumor antigens. A unique study worth highlighting employed Sindbis virus engineered with β-galactosidase (SV-β-gal) to demonstrate memory T cell responses that conferred protection against tumor re-challenge with antigen-specific and non-specific colon cancer cells in mice (55). The authors demonstrated that the influx of NKG2D-expressing antigen-specific CD8 T cells in the tumor was important for the development of long-lasting memory responses (55). Another study compared homologous vaccination strategies with Semliki Forest virus, adenovirus, and pox virus, and found that Semliki Forest virus provided potent protection in P185 tumors and showed increased levels of systemic antitumor-specific central memory T cells (56). However, the role of memory T cell development upon oncolytic vaccine administration remains largely unknown and poorly characterized. Considering the importance of antitumor T cell memory responses in long-term protection, the focus of future studies aimed at dissecting the immunological implications of oncolytic vaccines should consider memory T cells.
Other viruses that have been employed to target tumor antigens include Newcastle disease virus (NDV) and HSV. A study using NDV-expressing β-galactosidase-derived antigenic peptide (NDV-β-gal) found enhanced antitumor immune responses in a murine colon cancer model (57). The importance of cytokines in enhancing antitumor T cell responses has been discussed earlier. This study also demonstrated that coadministration of NDV-β-gal with NDV-expressing IL-2 led to increased frequency of tumor-infiltrating antigen-specific T cells and enhanced tumor regression (57). In another study, HSV was employed to express xenoantigen prostatic acid phosphatase (HSV-hPAP). This study demonstrated that HSV-hPAP caused reduced tumor growth and increased survival in mice bearing prostate tumors (58). A complete list of studies employing OVs to target tumor antigens has been summarized in Table 1.
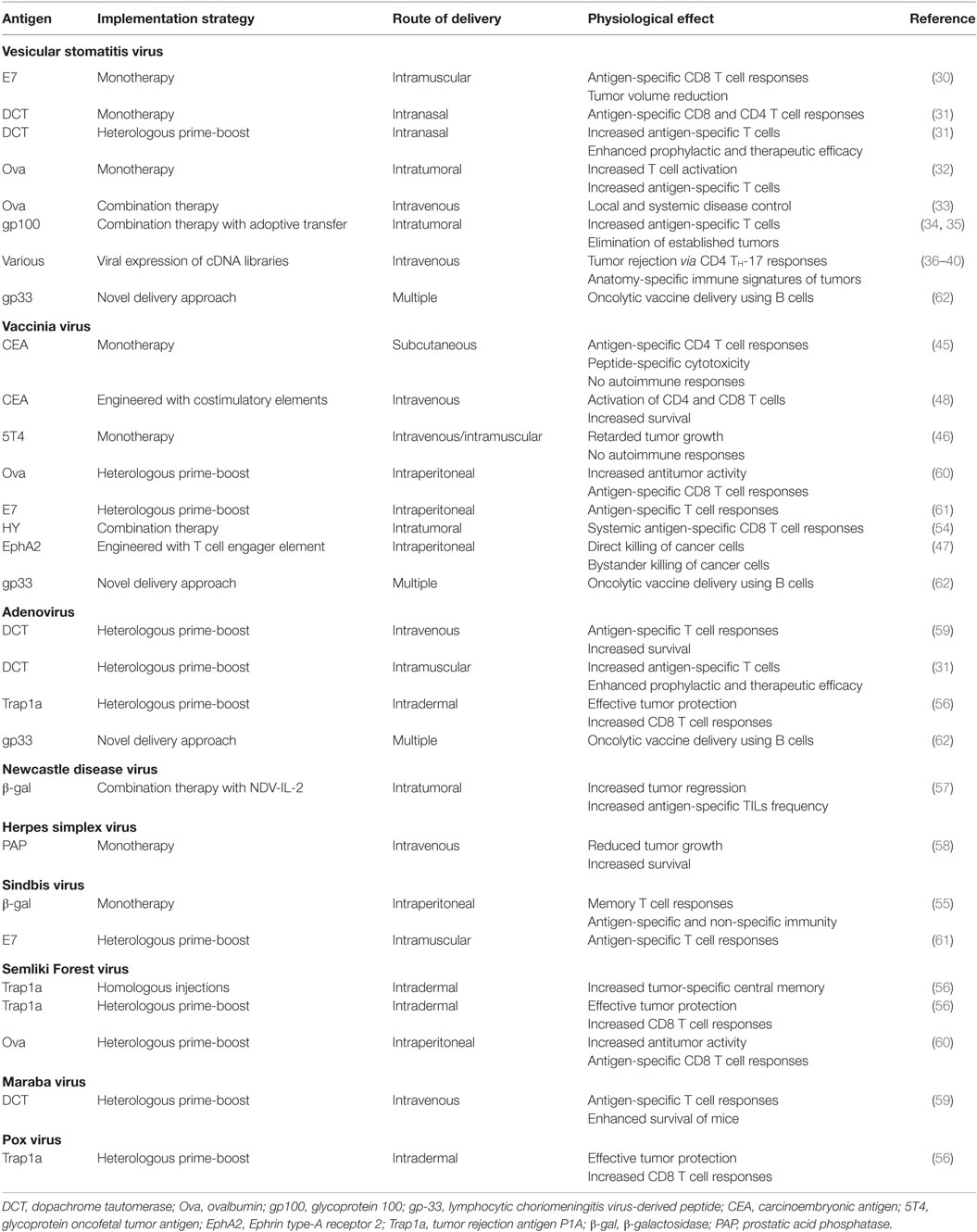
Table 1. Summary of oncolytic vaccine studies using a variety of viral vectors targeting respective tumor antigens.
Heterologous Virus Prime-Boost Strategies
One of the major challenges that oncolytic vaccines face is the induction of an undesired antiviral immune response against the viral vector. This antiviral immunity reduces the efficacy of the OV treatment by clearing the virus prematurely. In the context of oncolytic vaccines, immune responses against the viral vector may dominate the response against the tumor antigen, generating a much more subdued antitumor response. To overcome this obstacle, strategies that effectively redirect the immune responses against the tumor must be employed. One such strategy that we shall discuss is the heterologous virus prime-boost strategy.
The heterologous virus prime-boost strategy exploits the quick and effective immunological recall responses to redirect the maximal potential of immunity toward the tumor antigen. In the first priming step, a tumor antigen is expressed using one oncolytic virus vector, whereby the immune system “sees” and responds to the tumor and viral antigen for the first time. In the second boost step, a different viral vector is chosen to express the same tumor antigen, resulting in a primary immune response against the second viral vector while a stronger memory immune response is induced against the tumor antigen. Thus, this clever manipulation allows for the skewing of immunity toward antitumor responses over antiviral responses.
In preclinical studies, adenoviruses (Ad), VSV, and VV have emerged as common viral vectors that can be adapted for evaluation of priming and boosting strategies. Using the previously discussed tumor antigen hDCT, studies have shown that priming with Ad-hDCT and boosting with Maraba virus-expressing hDCT led to increased hDCT-specific T cell responses and survival of melanoma tumor-bearing mice (59). Another study using the same tumor antigen in a murine melanoma model demonstrated that priming with VSV-hDCT followed by a booster with Ad-hDCT also greatly enhanced antigen-specific T cell responses and led to enhanced efficacy in a prophylactic as well as a therapeutic setting (31). This study also demonstrated that VSV could boost hDCT-specific T cell responses generated by Ad-hDCT priming, indicating that VSV could successfully be used to either prime or boost tumor T cell responses (31). Another study demonstrating that the prime-boost strategy is effective irrespective of the order in which certain versatile viral vectors are administered used VV and Semliki Forest virus both overexpressing ova, resulting in enhanced antitumor activity and increased levels of ova-specific CD8 T cells in a murine ovarian surface epithelial carcinoma animal model (60). VV has also been used in combination with Sindbis virus in TC-1 tumor models for the prime-boost strategy, where priming with Sindbis-E7 and boosting with VV-E7 resulted in effective antitumor immune responses and the generation of increased numbers of E7-specific CD8 T cells (61).
Unfortunately, one of the major challenges with the heterologous virus prime-boost strategy involves the rapid killing of APCs by effector T cells before the memory T cells are engaged to produce a strong antitumor T cell response. A unique study with oncolytic vaccines addressed this issue by delivering oncolytic vaccines with B cells. Here, the immunodominant gp33 peptide derived from lymphocytic choriomeningitis virus was expressed in three different viruses VSV, Ad, and VV to demonstrate that B cells loaded with the booster virus can elicit better antigen-specific secondary T cell responses, which were dependent on antigen presentation by dendritic cells (62).
Altogether, the heterologous virus prime-boost strategy represents an innovative approach to redirecting immune responses in oncolytic vaccine therapy and warrants further research in the areas of viral vector selection and delivery of vaccines. These strategies combined with effective oncolytic vaccine design and vector development can help maximize antitumor immune responses and the development of efficacious clinical regimens.
Challenges for Oncolytic Vaccine Therapy
Oncolytic vaccine development has come a long way from its initial stages but still has ways to go. As alluded to earlier, implementation of strategies like the heterologous virus prime-boost warrants more research. An important factor when extending such strategies to clinics is a consideration of pre-existing immunity to common viral vectors, which may lead to reduced efficacy in humans. Understanding pre-existing immunity combined with choosing the most effective routes of delivery, which depends on the type of tumor being targeted, will help achieve maximum efficacy of oncolytic vaccines in clinics. Routes of delivery employed in the reviewed studies, along with other parameters and findings, have been summarized in Table 1. More research on developing the criteria for matching tumor type with routes of delivery is much needed for effective translation of preclinical models to bedside treatments. Moreover as discussed above, development of autoimmunity upon implementation of oncolytic vaccines has been a long-standing scientific concern. Minimizing autoimmune responses and maximizing tumor-specific T cell responses needs to be a major focus of future studies aimed at developing oncolytic vaccines. In this context, choice of tumor antigen can play an important role in not only maximizing antitumor immunity but also reducing off-site toxicity if carefully selected. In light of ongoing burgeoning developments in the areas of cancer genomics and proteomics, it is believed that novel antigens that are highly tumor-specific, and thus are non-autoreactive, will be available for such targeting (63, 64). In the following section, we discuss how, going forward, novel tumor antigens discovered via a combination of computational and high throughput approaches provide the best chance for selecting antigens to be targeted in tumors and represent an emerging frontier in cancer immunotherapy.
Future Directions and Concluding Remarks
Novel tumor antigen discovery is a fundamental cornerstone in cancer immunotherapy and provides an enormous knowledge base for oncolytic vaccine research to draw from. Cutting-edge technological advances in tumor antigen discovery have identified a wide range of potential targets that can be incorporated in the next generation of oncolytic vaccines for designing highly targeted and efficacious therapies. One of these major advances is in the field of sequencing; new and improved sequencing technologies are now available to identify and categorize mutations at the level of genes, exons, and RNA from many different cancer types (65–67). In addition, recent developments in computational techniques allow better in silico prediction and identification of immunogenic tumor antigens (68–74). Finally, advances in peptide isolation and identification by immunoprecipitation and mass spectrometry-based proteomics, respectively, have led to the development of novel approaches to unearth new tumor antigens with greater precision (75–77). One such result of modern research combining these advances in different fields has led to the discovery of the high-confidence tumor MHC ligandome, which represents an array of tumor-derived peptide antigens that are bound to MHC molecules. These tumor antigens are identified using immunoprecipitation of peptide-MHC molecules followed by mass spectrometry analysis of the peptides eluted from the MHC molecules. Such antigens have been collectively called as ligandome-derived tumor-associated antigens (LiTAAs) (78–81). A recent breakthrough study used a combination of exome sequencing, proteomics, and computational modeling to identify novel tumor antigens that could be targeted via peptide-based vaccines (82). Going forward, the tumor MHC ligandome will serve as a unique and effective library for identifying novel tumor antigens to be targeted using oncolytic vaccines. More importantly, as opposed to the traditional peptide-based vaccines to deliver LiTAAs, oncolytic vaccines provide the advantage of administering LiTAAs in the presence of all the necessary immune signals for the proper activation of long-term antitumor immunity.
Combining this unique capability of oncolytic vaccines with novel tumor antigen discovery paradigms and effective, timely immunological intervention via strategies like the heterologous virus prime-boost will aid the design of the most clinically efficacious cancer immunotherapeutics (summarized in Figure 1). With the ever-increasing knowledge bases that have not yet been effectively translated to the bedside, concerted efforts to adapt these uniquely exciting strategies to develop therapeutic models of high clinical relevance are more important today than ever before and mark the next frontier in precision cancer immunotherapy research.
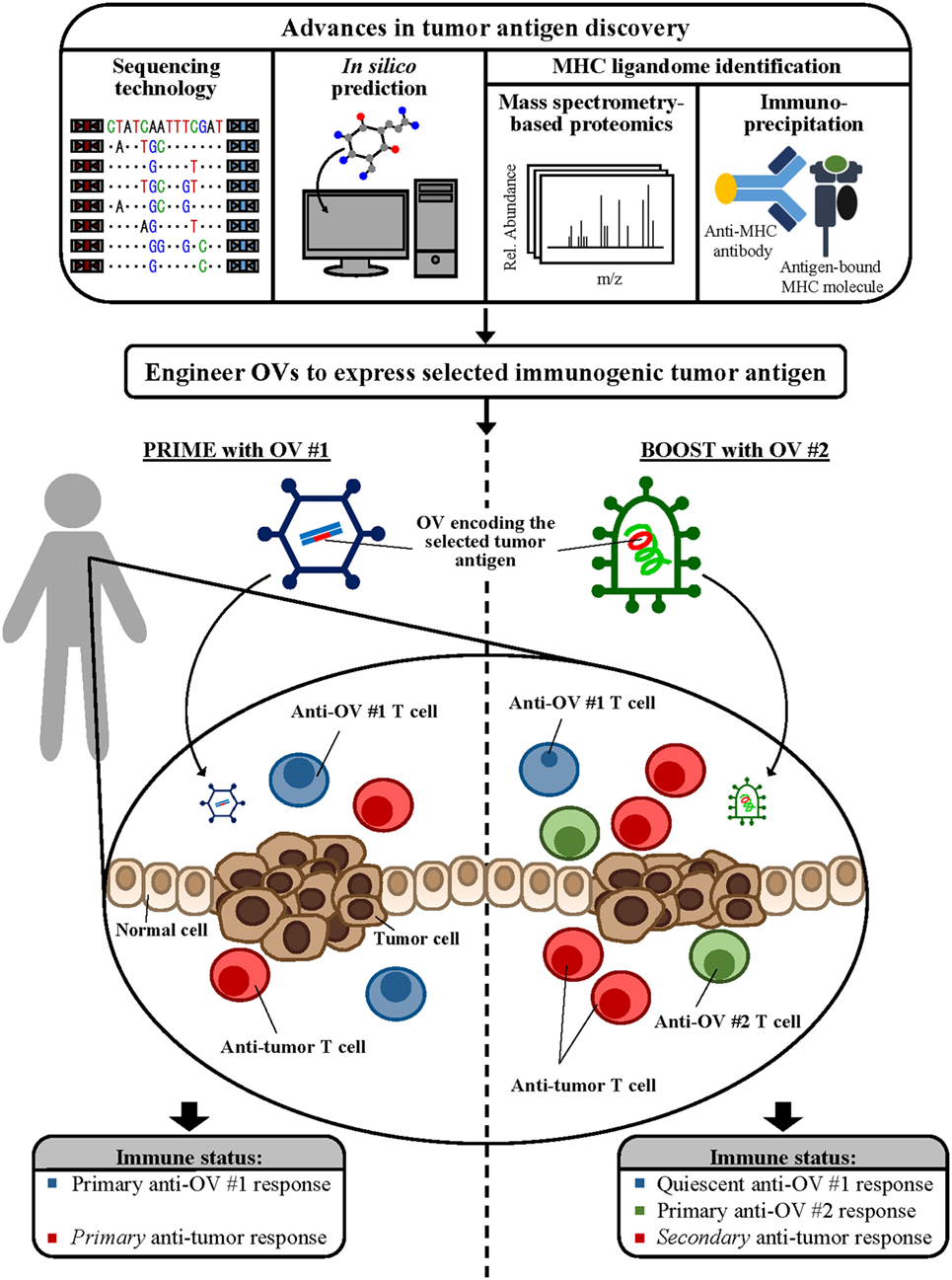
Figure 1. Combining novel antigen discovery platforms to design oncolytic vaccines. Advances in sequencing technology, in silico prediction methods, and mass spectrometry-based proteomics and immunoprecipitation for major histocompatibility complex (MHC) ligandome elucidation allow identification of novel tumor antigen targets. Engineering these novel antigens into powerful viral vectors will provide a platform for the development of the next generation of oncolytic vaccines. Incorporating immunomodulatory strategies, such as the heterologous virus prime-boost as shown, during oncolytic vaccine administration can maximize antitumor immune responses, leading to the development of complete and clinically efficacious antitumor treatment options.
Author Contributions
NH, YK, and SG developed the concept. NH prepared the figures and wrote the manuscript. YK participated in writing and technical editing of the manuscript. PL and SG supervised and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by research grants from the Canadian Cancer Society Research Institute (CCSRI), Canadian Institutes of Health Research (CIHR), and Terry Fox Research Institute (TFRI) to PL and SG. NH is currently funded by the Nova Scotia Graduate Scholarship (NSGS). YK is currently funded by the CIHR Doctoral Award.
References
1. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi:10.1016/j.cell.2017.01.017
2. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov (2017) 7(3):264–76. doi:10.1158/2159-8290.CD-16-0828
3. Dustin ML. Cancer immunotherapy: killers on sterols. Nature (2016) 531(7596):583–4. doi:10.1038/nature17310
4. Shipman L. Tumour immunology: interrogating intratumoral Treg cells. Nat Rev Immunol (2017) 17(1):4–5. doi:10.1038/nri.2016.137
5. Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun (2016) 8:14340. doi:10.1038/ncomms14340
6. Kuruppu D, Tanabe KK. Viral oncolysis by herpes simplex virus and other viruses. Cancer Biol Ther (2005) 4(5):524–31. doi:10.4161/cbt.4.5.1820
7. McKie EA, MacLean AR, Lewis AD, Cruickshank G, Rampling R, Barnett SC, et al. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours – evaluation of a potentially effective clinical therapy. Br J Cancer (1996) 74(5):745–52. doi:10.1038/bjc.1996.431
8. Coffey MC, Strong JE, Forsyth PA, Lee PWK. Reovirus therapy of tumors with activated Ras pathway. Science (1998) 282(5392):1332. doi:10.1126/science.282.5392.1332
9. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PWK. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J (1998) 17(12):3351. doi:10.1093/emboj/17.12.3351
10. Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther (2017) 15(4):651–9. doi:10.1038/sj.mt.6300108
11. Parato KA, Senger D, Forsyth PAJ, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer (2005) 5(12):965–76. doi:10.1038/nrc1750
12. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov (2015) 14(9):642–62. doi:10.1038/nrd4663
13. Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther (2011) 19(6):1008–16. doi:10.1038/mt.2011.65
14. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285(5425):221. doi:10.1126/science.285.5425.221
15. Neefjes J, Ovaa H. A peptide’s perspective on antigen presentation to the immune system. Nat Chem Biol (2013) 9(12):769–75. doi:10.1038/nchembio.1391
16. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14(2):135–46. doi:10.1038/nrc3670
17. Srinivasan R, Wolchok JD. Tumor antigens for cancer immunotherapy: therapeutic potential of xenogeneic DNA vaccines. J Transl Med (2004) 2(1):12. doi:10.1186/1479-5876-2-12
18. Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol (2011) 18(1):23–34. doi:10.1128/CVI.00286-10
19. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol (2010) 22(3):333–40. doi:10.1016/j.coi.2010.02.013
20. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi:10.1038/nri3405
21. Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res (2016) 76(22):6747. doi:10.1158/0008-5472.CAN-16-1404
22. Cuddington BP, Mossman KL. Permissiveness of human cancer cells to oncolytic bovine herpesvirus 1 is mediated in part by KRAS activity. J Virol (2014) 88(12):6885–95. doi:10.1128/JVI.00849-14
23. Dold C, Rodriguez Urbiola C, Wollmann G, Egerer L, Muik A, Bellmann L, et al. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol Ther Oncolytics (2016) 3:16021. doi:10.1038/mto.2016.21
24. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med (2000) 6(7):821–5. doi:10.1038/77558
25. Prestwich RJ, Errington F, Ilett EJ, Morgan RSM, Scott KJ, Kottke T, et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res (2008) 14(22):7358. doi:10.1158/1078-0432.CCR-08-0831
26. Li H, Dutuor A, Fu X, Zhang X. Induction of strong antitumor immunity by an HSV-2-based oncolytic virus in a murine mammary tumor model. J Gene Med (2007) 9(3):161–9. doi:10.1002/jgm.1005
27. Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science (2006) 311(5768):1780. doi:10.1126/science.1121411
28. Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB, et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther (1999) 10(17):2741–55. doi:10.1089/10430349950016483
29. Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther (1999) 10(3):385–93. doi:10.1089/10430349950018832
30. Liao JB, Publicover J, Rose JK, DiMaio D. Single-dose, therapeutic vaccination of mice with vesicular stomatitis virus expressing human papillomavirus type 16 E7 protein. Clin Vaccine Immunol (2008) 15(5):817–24. doi:10.1128/CVI.00343-07
31. Bridle BW, Boudreau JE, Lichty BD, Brunellière J, Stephenson K, Koshy S, et al. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol Ther (2009) 17(10):1814–21. doi:10.1038/mt.2009.154
32. Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res (2007) 67(6):2840. doi:10.1158/0008-5472.CAN-06-3974
33. Blanchard M, Shim KG, Grams MP, Rajani K, Diaz RM, Furutani KM, et al. Definitive management of oligometastatic melanoma in a murine model using combined ablative radiation therapy and viral immunotherapy. Int J Radiat Oncol Biol Phys (2015) 93(3):577–87. doi:10.1016/j.ijrobp.2015.07.2274
34. Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, et al. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum Gene Ther (2011) 22(11):1343–53. doi:10.1089/hum.2010.216
35. Rommelfanger DM, Wongthida P, Diaz RM, Kaluza KM, Thompson JM, Kottke TJ, et al. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res (2012) 72(18):4753–64. doi:10.1158/0008-5472.CAN-12-0600
36. Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P, et al. Broad antigenic coverage induced by viral cDNA library-based vaccination cures established tumors. Nat Med (2011) 17(7):854–9. doi:10.1038/nm.2390
37. Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol (2012) 30(4):337–43. doi:10.1038/nbt.2157
38. Zaidi S, Blanchard M, Shim K, Ilett E, Rajani K, Parrish C, et al. Mutated BRAF emerges as a major effector of recurrence in a murine melanoma model after treatment with immunomodulatory agents. Mol Ther (2014) 23(5):845–56. doi:10.1038/mt.2014.253
39. Alonso-Camino V, Rajani K, Kottke T, Rommelfanger-Konkol D, Zaidi S, Thompson J, et al. The profile of tumor antigens which can be targeted by immunotherapy depends upon the tumor’s anatomical site. Mol Ther (2014) 22(11):1936–48. doi:10.1038/mt.2014.134
40. Cockle JV, Rajani K, Zaidi S, Kottke T, Thompson J, Diaz RM, et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol (2016) 18(4):518–27. doi:10.1093/neuonc/nov173
41. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol (2017) 34(22):2619–26. doi:10.1200/JCO.2016.67.1529
42. Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res (2015) 21(24):5543. doi:10.1158/1078-0432.CCR-14-2009
43. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med (2014) 6(226):226ra32. doi:10.1126/scitranslmed.3008095
44. Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther (2014) 22(11):1949–59. doi:10.1038/mt.2014.160
45. Kass E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW. Induction of protective host immunity to carcinoembryonic antigen (CEA), a self-antigen in CEA transgenic mice, by immunizing with a recombinant vaccinia-CEA virus. Cancer Res (1999) 59(3):676.
46. Mulryan K, Ryan MG, Myers KA, Shaw D, Wang W, Kingsman SM, et al. Attenuated recombinant vaccinia virus expressing oncofetal antigen (tumor-associated antigen) 5T4 induces active therapy of established tumors. Mol Cancer Ther (2002) 1(12):1129.
47. Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song X. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther (2013) 22(1):102–11. doi:10.1038/mt.2013.240
48. Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res (2003) 9(5):1837.
49. Chen H, Sampath P, Hou W, Thorne SH. Regulating cytokine function enhances safety and activity of genetic cancer therapies. Mol Ther (2013) 21(1):167–74. doi:10.1038/mt.2012.225
50. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19(3):329–36. doi:10.1038/nm.3089
51. Wang LS, Lynn RC, Cheng G, Alexander E, Kapoor V, Moon EK, et al. Treating tumors with a vaccinia virus expressing IFNÎ2 illustrates the complex relationships between oncolytic ability and immunogenicity. Mol Ther (2012) 20(4):736–48. doi:10.1038/mt.2011.228
52. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQM, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature (2011) 477(7362):99–102. doi:10.1038/nature10358
53. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res (2016) 22(5):1048. doi:10.1158/1078-0432.CCR-15-2667
54. Yang AS, Monken CE, Lattime EC. Intratumoral vaccination with vaccinia-expressed tumor antigen and granulocyte macrophage colony-stimulating factor overcomes immunological ignorance to tumor antigen. Cancer Res (2003) 63(20):6956.
55. Granot T, Yamanashi Y, Meruelo D. Sindbis viral vectors transiently deliver tumor-associated antigens to lymph nodes and elicit diversified antitumor CD8(+) T-cell immunity. Mol Ther (2013) 22(1):112–22. doi:10.1038/mt.2013.215
56. Näslund TI, Uyttenhove C, Nordström EK, Colau D, Warnier G, Jondal M, et al. Comparative prime-boost vaccinations using Semliki forest virus, adenovirus, and ALVAC vectors demonstrate differences in the generation of a protective central memory CTL response against the P815 tumor. J Immunol (2007) 178(11):6761. doi:10.4049/jimmunol.178.11.6761
57. Vigil A, Martinez O, Chua MA, GarcÃa-Sastre A. Recombinant Newcastle disease virus as a vaccine vector for cancer therapy. Mol Ther (2008) 16(11):1883–90. doi:10.1038/mt.2008.181
58. Castelo-Branco P, Passer BJ, Buhrman JS, Antoszczyk S, Marinelli M, Zaupa C, et al. Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances anti-tumor efficacy in prostate cancer. Gene Ther (2010) 17(6):805–10. doi:10.1038/gt.2010.20
59. Pol JG, Zhang L, Bridle BW, Stephenson KB, Rességuier J, Hanson S, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther (2013) 22(2):420–9. doi:10.1038/mt.2013.249
60. Zhang Y, Tsai Y, Monie A, Wu T, Hung C. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol Ther (2009) 18(4):692–9. doi:10.1038/mt.2009.318
61. Lin C, Hung C, Juang J, He L, Lin K, WooKim T, et al. Boosting with recombinant vaccinia increases HPV-16 E7-specific T cell precursor frequencies and antitumor effects of HPV-16 E7-expressing Sindbis virus replicon particles. Mol Ther (2003) 8(4):559–66. doi:10.1016/S1525-0016(03)00238-7
62. Zhang L, Bridle BW, Chen L, Pol J, Spaner D, Boudreau JE, et al. Delivery of viral-vectored vaccines by B cells represents a novel strategy to accelerate CD8+ T-cell recall responses. Blood (2013) 121(13):2432. doi:10.1182/blood-2012-06-438481
63. Dersh D, Yewdell JW. I’ve got algorithm: predicting tumor and autoimmune peptide targets for CD8+ T cells. J Clin Invest (2016) 126(12):4399–401. doi:10.1172/JCI91302
64. Iranzo J, Villoslada P. Autoimmunity and tumor immunology: two facets of a probabilistic immune system. BMC Syst Biol (2014) 8(1):120. doi:10.1186/s12918-014-0120-4
65. Robbins PF, Lu Y, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi:10.1038/nm.3161
66. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest (2015) 125(9):3413–21. doi:10.1172/JCI80008
67. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res (2014) 24(5):743–50. doi:10.1101/gr.165985.113
68. Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res (2008) 36:W509–12. doi:10.1093/nar/gkn202
69. Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics (2013) 65:711–24. doi:10.1007/s00251-013-0720-y
70. Erup Larsen M, Kloverpris H, Stryhn A, Koofhethile CK, Sims S, Ndung’u T, et al. HLArestrictor – a tool for patient-specific predictions of HLA restriction elements and optimal epitopes within peptides. Immunogenetics (2011) 63:43–55. doi:10.1007/s00251-010-0493-5
71. Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci (2003) 12:1007–17. doi:10.1110/ps.0239403
72. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics (1999) 50:213–9. doi:10.1007/s002510050595
73. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR. The immune epitope database (IEDB) 3.0. Nucleic Acids Res (2015) 43:D405–12. doi:10.1093/nar/gku938
74. Murphy JP, Konda P, Kowalewski DJ, Schuster H, Clements D, Kim Y, et al. MHC-I ligand discovery using targeted database searches of mass spectrometry data: implications for T-cell immunotherapies. J Proteome Res (2017) 16(4):1806–16. doi:10.1021/acs.jproteome.6b00971
75. Klade CS, Voss T, Krystek E, Ahorn H, Zatloukal K, Pummer K, et al. Identification of tumor antigens in renal cell carcinoma by serological proteome analysis. Proteomics (2001) 1(7):890–8. doi:10.1002/1615-9861(200107)1:7<890::AID-PROT890>3.3.CO;2-Q
76. Stevanovic S. Identification of tumour-associated t-cell epitopes for vaccine development. Nat Rev Cancer (2002) 2(7):514–514. doi:10.1038/nrc841
77. Berlin C, Kowalewski DJ, Schuster H, Mirza N, Walz S, Handel M, et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia (2015) 29(3):647–59. doi:10.1038/leu.2014.233
78. Kowalewski DJ, Schemionek M, Kanz L, Brümmendorf TH, Salih HR, Rammensee H, et al. HLA ligandome analysis of chronic myeloid leukemia (CML), revealed novel tumor associated antigens for peptide based immunotherapy. Blood (2013) 122(21):3975.
79. Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, et al. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci U S A (2014) 112(2):E166–75. doi:10.1073/pnas.1416389112
80. Rammensee H, Singh-Jasuja H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev Vaccines (2013) 12(10):1211–7. doi:10.1586/14760584.2013.836911
81. Antwi K, Hanavan PD, Myers CE, Ruiz YW, Thompson EJ, Lake DF. Proteomic identification of an MHC-binding peptidome from pancreas and breast cancer cell lines. Mol Immunol (2009) 46(15):2931–7. doi:10.1016/j.molimm.2009.06.021
Keywords: cancer immunotherapy, oncolytic vaccines, tumor antigens, antitumor immunity, T cells, tumor major histocompatibility complex ligandome
Citation: Holay N, Kim Y, Lee P and Gujar S (2017) Sharpening the Edge for Precision Cancer Immunotherapy: Targeting Tumor Antigens through Oncolytic Vaccines. Front. Immunol. 8:800. doi: 10.3389/fimmu.2017.00800
Received: 30 April 2017; Accepted: 26 June 2017;
Published: 13 July 2017
Edited by:
Joshua Pesach Rosenzweig, Cell-El Ltd., IsraelReviewed by:
Raymond B. Birge, Rutgers University, The State University of New Jersey, United StatesGraham Robert Leggatt, The University of Queensland, Australia
Copyright: © 2017 Holay, Kim, Lee and Gujar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shashi Gujar, c2hhc2hpLmd1amFyQGRhbC5jYQ==