 Amanda Braga Figueiredo1
Amanda Braga Figueiredo1 Míriam Conceição Souza-Testasicca1,2
Míriam Conceição Souza-Testasicca1,2 Tiago Wilson Patriarca Mineo3
Tiago Wilson Patriarca Mineo3 Luís Carlos Crocco Afonso1*
Luís Carlos Crocco Afonso1*
- 1Laboratório de Imunoparasitologia, ICEB/NUPEB, Universidade Federal de Ouro Preto, Ouro Preto, Brazil
- 2Coordenadoria da Área de Ciências Biológicas, Instituto Federal de Minas Gerais, Campus Ouro Preto, Ouro Preto, Brazil
- 3Laboratório de Imunoparasitologia “Dr. Mario Endsfeldz Camargo”, ICBIM, Universidade Federal de Uberlândia, Uberlândia, Brazil
Differently from others Leishmania species, infection by the protozoan parasite L. amazonensis is associated with a lack of antigen-specific T-cell responses. Dendritic cells (DC) are essential for the innate immune response and for directing the differentiation of T-helper lymphocytes. Previously, we showed that L. amazonensis infection impairs DC activation through the activation of adenosine A2B receptor, and here, we evaluated the intracellular events triggered by this receptor in infected cells. To this aim, bone marrow-derived DC from C57BL/6J mice were infected with metacyclic promastigotes of L. amazonensis. Our results show, for the first time, that L. amazonensis increases the production of cAMP and the phosphorylation of extracellular signal-regulated protein kinases 1/2 (ERK1/2) in infected DC by a mechanism dependent on the A2B receptor. Furthermore, L. amazonensis impairs CD40 expression and IL-12 production by DC, and the inhibition of adenylate cyclase, phosphoinositide 3-kinase (PI3K), and ERK1/2 prevent these effects. The increase of ERK1/2 phosphorylation and the inhibition of DC activation by L. amazonensis are independent of protein kinase A (PKA). In addition, C57BL/6J mice were inoculated in the ears with metacyclic promastigotes, in the presence of PSB1115, an A2B receptor antagonist. PSB1115 treatment increases the percentage of CD40+ DC on ears and draining lymph nodes. Furthermore, this treatment reduces lesion size and tissue parasitism. Lymph node cells from treated mice produce higher levels of IFN-γ than control mice, without altering the production of IL-10. In conclusion, we suggest a new pathway used by the parasite (A2B receptor → cAMP → PI3K → ERK1/2) to suppress DC activation, which may contribute to the decrease of IFN-γ production following by the deficiency in immune response characteristic of L. amazonensis infection.
Introduction
Leishmania parasites are protozoa transmitted between their hosts by female sand flies and cause in humans a group of diseases known as leishmaniasis. These diseases present a wide spectrum of clinical manifestations dependent on the parasite species and the host immune response. Leishmania amazonensis (L. amazonensis), in humans, causes diffuse leishmaniasis that is associated with diffuse non-ulcerative lesions with innumerous parasites. In this case, the parasite resistance to treatment is common (1, 2). The deficiency in immune responses associated with L. amazonensis infection, characterized by a lack of antigen-specific T-cell responses, contributes significantly to the failure of therapeutic approaches. Therefore, the understanding of evasion mechanisms used by L. amazonensis during infection has much to contribute to the development of new therapeutic strategies. In the murine model, this parasite causes non-healing chronic lesions in mouse strains otherwise resistant to other Leishmania species, such as Leishmania braziliensis and Leishmania major (3–5). The murine model has been extensively used to evaluate the mechanisms involved in the activation/evasion of the host immune response by the parasite.
Dendritic cells (DC) are essential players in the fight against infection where they link the innate and acquired immune responses. The role of these cells in infections induced by Leishmania has been clearly demonstrated. After contact with microorganisms, these cells initiate a maturation process characterized by increased expression of MHC class II and co-stimulators, such as CD80, CD86, and CD40 (6). Importantly, CD40–CD40L interaction is essential for antigen-specific T-helper lymphocyte priming (7, 8). Additionally, DC produce a wide array of cytokines and can direct T-helper cell differentiation (9). In this way, IL-12 production by DC induces the differentiation of IFN-γ-producing Th1 lymphocytes, which are critical to the control of Leishmania replication in the infected host (10–12). Several studies evaluated the interaction between Leishmania parasites and DC demonstrating that L. amazonensis can modulate several DC functions by modifying the expression of MHC class II, CD80 and CD86 and the production of IL-10 and IL-12 (13–18).
One important aspect of the infection by L. amazonensis in the murine model is the fact that, contrary to other Leishmania species, no mouse strain is completely resistant to the parasite [reviewed by Pereira and Alves (19)]. In addition, with the exception of BALB/c mice, the susceptibility to L. amazonensis infection is independent of disease-inducing cytokines such as IL-4 or IL-10, regardless of number or stage of development (purified metacyclic or stationary phase) of the promastigotes used for infection as well as the site of the infection (20–22). IL-10 only seems to play a relevant role, when its production is increased at the site of infection by the administration of sandfly saliva or adenosine and AMP (22, 23). Thus, finding an alternative immunomodulatory mechanism distinct from the participation of regulatory cytokines has been the aim of our laboratory for the last 15 years.
Extracellular ATP, released during infection or cellular injury, acts as a danger signal and a potent stimulator of inflammatory responses (24–26). Ectonucleotidases CD39 and CD73 hydrolyze ATP to adenosine, the latter of which presents immunomodulatory properties, such as inhibition of the production of inflammatory cytokines, such as TNF-α and IL-12, and stimulating the production of IL-10 (27, 28). Adenosine can act through A1, A2A, A2B, and A3 receptors. A2 receptors are able to stimulate adenylate cyclase, leading to the accumulation of cAMP (29–31), which impairs CD40 expression, the generation of inflammatory mediators, IL-12 production and microbicidal activity (32, 33).
Previously, we showed that L. amazonensis infection impairs DC activation (by decreasing the expression of MHC class II, CD86, and CD40) and, as a consequence, the triggering of an antigen-specific cellular response. This effect was dependent on the activation of A2B receptor (34), but the signaling pathways activated by this receptor remained unknown. Similarly, other studies demonstrated that inhibition of L. amazonensis-stimulated extracellular signal-regulated protein kinases 1/2 (ERK1/2) phosphorylation increases CD40 expression on DC (35) and decreases lesion size in mice infected by this parasite (36).
Given that L. amazonensis decreases DC activation, in particular CD40 expression, via A2B receptor, in this study, we evaluated the intracellular events triggered by this receptor in infected cells. Furthermore, we evaluated the role of A2B receptor on lesion development in mice infected by L. amazonensis. Our results show that L. amazonensis increases cAMP production by DC and stimulates the phosphorylation of ERK1/2 in these cells by mechanisms dependent on A2B receptor. Adenylate cyclase, phosphoinositide 3-kinase (PI3K), and ERK1/2 are involved in the decreased CD40 expression and IL-12 production in L. amazonensis-infected DC. In addition, A2B receptor blockade controls lesion development in mice infected by L. amazonensis, probably by increasing the percentage of CD40+ DC and the production of IFN-γ by lymph node cells.
Materials and Methods
Animals and Parasites
C57BL/6J (2–6 months old) mice were obtained from the Universidade Federal de Ouro Preto animal facility. Animals received water and food ad libitum. This study was carried out in accordance with the recommendations of the Brazilian Guidelines for animal experimentation. The protocols were approved by the University’s Ethical Committee on Animal Experimentation (CEUA 2012/02 and CEUA 2013/51). Leishmania amazonensis, PH8 strain (IFLA/BR/67/PH8) promastigotes were grown in Grace’s medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal calf serum (FCS, Cultilab, Campinas, SP, Brazil), 2 mM l-glutamine (Sigma-Aldrich) and 100 U/mL penicillin G potassium (Sigma-Aldrich), pH 6.5, at 25°C. Metacyclic promastigotes were purified by gradient centrifugation of parasites at the stationary phase of culture (day 5) over Ficoll 400 (Sigma-Aldrich), as previously described (5). In in vitro DC infection experiments, metacyclic promastigotes, suspended in PBS with 5% FCS, were incubated in the presence of 5 µM CFSE (Sigma-Aldrich) at 37°C for 10 min in the dark. The suspension was centrifuged and the parasites were washed in PBS, pH 7.2 (37). Alternatively, parasites were labeled with PKH26 (Sigma-Aldrich) according to the manufacturer’s instructions.
Differentiation of Bone Marrow-Derived DC
Bone-marrow-derived DC were obtained from C57BL/6J bone marrow as previously described (38). Briefly, bone marrow cells were isolated from the femur and tibia of C57BL/6J mice. Bone marrow cell suspensions were centrifuged and cells cultured in RPMI-1640 (Sigma-Aldrich) supplemented with 10% FCS, 2 mM l-glutamine, 100 U/mL penicillin G potassium, and 50 µM β-mercaptoetanol (Pharmacia Biotech AB, Uppsala, Sweden), pH 7.2. Cells were plated in Petri dishes at a concentration of 3 × 105 cells/mL and incubated at 37°C/5% CO2. GM-CSF (R&D Systems, Minneapolis, MN, USA) was added to each plate on the days 0, 3, and 6, at a concentration of 3 ng/mL (1,050 U/mL). Non-adherent DC were collected on the ninth day of culture. In regard to a recently published work (39), DC were extensively characterized. DC were CD11b+CD11c+F4/80−/lowMHCII+ cells and showed morphology characteristic of this population, with several and irregular dendrites. In addition, these cells were able to stimulate mixed leukocyte reaction and antigen-specific proliferation of CD4+ T lymphocyte (data not shown).
In Vitro DC Infection
CFSE-labeled metacyclic promastigotes and DC were co-incubated (1:3 cell to parasite ratio) in RPMI-1640 supplemented with 10% FCS, 2 mM l-glutamine, 100 U/mL penicillin G potassium, and 50 µM β-mercaptoetanol (Pharmacia Biotech AB, Uppsala, Sweden), pH 7.2, at 33°C/5% CO2 for 3 h and subsequently incubated at 37°C/5% CO2 for up to 17 h. In selected experiments A2B adenosine receptors antagonist, MRS1754 {N-(4-cyanophenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-acetamide, Tocris Bioscience, Park Ellisville, MO, USA}, or inhibitors of adenylate cyclase [SQ22536, 9-(tetrahydro-2-furanyl)-9H-purin-6-amine, Tocris Bioscience], protein kinase A (PKA) (KT5720, (9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3’,2’,1’-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester, Sigma-Aldrich), PI3K [LY294002, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride, Sigma-Aldrich] or ERK1/2 (U0126, 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene, Tocris Bioscience) were added at the moment of DC infection as described in figure legends. All drugs were diluted in DMSO (1% final concentration), which was added to control cultures.
cAMP Measurement
Briefly, metacyclic promastigotes and DC were co-incubated as already described, in the presence of 0,1 mM Ro 20-1724 (phosphodiesterase inhibitor, Sigma-Aldrich), at 33°C/5% CO2 for 15 min. In selected groups, MRS1754 was added at the moment of DC infection. In other groups, DC previously co-incubated with parasites and adenosine receptor antagonists were stimulated with 1 µM 5′-(N-ethylcarboxamido) adenosine (NECA, Sigma-Aldrich) at 33°C/5% CO2 for more 15 min. cAMP was measured by a bioluminescent assay according to the manufacturer’s instructions (Promega, Madison, WI, USA).
Infection of Mice
Female C57BL/6J mice were inoculated intradermally in the ears with 103 or 105 metacyclic promastigotes of L. amazonensis, in the presence or absence of 5 µM PSB1115 [1-propyl-8-(4-sulfophenyl) xanthine potassium salt hydrate]. Lesion size was measured weekly with a digital micrometer (Starrett, Athol, MA, USA). The lesion size was defined as the difference between the thickness of the infected and uninfected ears.
Culture of Lymph Node Cells
Single-cell suspensions were prepared from the auricular lymph nodes of mice infected for 12 weeks. Cell concentration was adjusted to 5 × 106 cells/mL in DMEM (Sigma-Aldrich) supplemented with 10% FCS, 2 mM l-glutamine, 100 U/mL penicillin G, 50 µM β-mercaptoethanol and 25 mM HEPES (Sigma-Aldrich), pH 7.2. Cell suspensions were distributed in culture plates and stimulated with 50 µg/mL of L. amazonensis particulate antigen. Supernatants were harvested after 48 h.
Parasite Load Estimation
The number of parasites in the ear lesion was estimated by the limiting dilution assay (3). After 12 weeks of infection, mice were euthanized and the ears removed and incubated in RPMI-1640/1 mg/mL collagenase A, pH 7.2, for 2 h at 37°C/5% CO2. The ears were ground in Grace’s medium, pH 6.5. Tissue debris were removed by centrifugation. Cells were resuspended in Grace’s medium supplemented with 10% FCS, 2 mM l-glutamine and 100 U/mL penicillin G, pH 6.5. The parasite suspension was serially diluted in 10-fold dilutions, pipette tips were replaced for each dilution. After 2 weeks of incubation at 25°C, plates were examined under an inverted microscope for the presence of parasites. Results were expressed as −log of the number of parasites corresponding to the last dilution in which they were observed.
Isolation of Cells from Ears and Draining Lymph Nodes
C57BL/6J mice were inoculated intradermally in both ears with 105 metacyclic promastigotes of L. amazonensis, in the presence or absence of 5 µM PSB1115. 7 days after infection, both ears and draining auricular lymph nodes were removed. Only ears were incubated in RPMI-1640/1 mg/mL collagenase A, pH 7.2, for 2 h at 37°C/5% CO2. The ears were ground in RPMI-1640, pH 7.2, using a BD Medimachine™ system and the suspension was filtered through a 30 µm Filcon (BD Biosciences, San Jose, CA, USA). The lymph nodes were ground in RPMI-1640, pH 7.2, using a tissue homogeneizer. Cells were stained and analyzed by flow cytometry as described below.
Cytokine Measurement
Supernatants from DC cultures were collected after 20 h and supernatants from lymph node cell cultures after 48 h and IL-12p70, IL-10, and IFN-γ cytokine levels were measured by ELISA using kits according to the manufacturer’s instructions (BD OptEIA, San Diego, CA, USA).
Flow Cytometry
For surface markers staining, cells in PBS with 1% BSA were submitted to FcγR blocking in the presence of anti-mouse CD16/CD32 (produced in our laboratory). Subsequently, cells were incubated with anti-mouse CD11c (HL3 clone), anti-mouse CD40 (3/23 clone—BD Pharmingen, San Diego, CA, USA), or their respective isotype controls, at 4°C for 30 min in the dark. The suspensions were centrifuged and the cells were washed in PBS, pH 7.2 and resuspended in a solution of 1% paraformaldehyde, 47.7 mM sodium cacodylate, and 113 mM NaCl, pH 7.2. Intracellular phospho-protein staining was performed according to the manufacturer’s instructions (BD Phosflow, San Diego, CA, USA). Briefly, DC previously co-incubated with parasites were stimulated with 1 µM NECA at 33°C/5% CO2 for 15 min, fixed in Lyse/fix buffer, permeabilized with Perm buffer III and incubated with anti-ERK1/2 pT202/pY204 (20 A clone), or its respective isotype control, at room temperature for 60 min in the dark. The suspensions were centrifuged and the cells were washed in PBS, pH 7.2 and resuspended in Stain buffer. The samples were analyzed in BD FACSCalibur™ flow cytometer. Cell acquisition was performed using BD CellQuest™ Pro software. Data analysis was performed using FlowJo software.
Statistical Analysis
Student’s t-test and one-way ANOVA were performed using Prism 5.0 software (GraphPad Software, La Jolla, CA, USA). p < 0.05 was considered statistically significant.
Results
L. amazonensis Infection Increases cAMP Production, Which Impairs DC Activation
As previously stated, our group showed that L. amazonensis impairs DC activation, especially CD40 expression, by a mechanism dependent on the A2B receptor (34). Here, we decided to investigate the signaling pathways involved in this process. In the figures, a schematic of the possible pathways triggered by the A2B receptor is shown, highlighting the pathways activated in L. amazonensis-infected DC.
Adenosine A2B receptors are G protein-coupled receptors that can be associated both to αs, leading to cAMP production, or to αq subunits, which stimulate phospholipase C and accumulation of intracellular calcium (29). Thus, to evaluate the accumulation of cAMP, DC were infected with L. amazonensis, in the presence or absence of MRS1754, a selective A2B receptor antagonist, and cAMP levels measured by a chemiluminescence assay before and after the addition of NECA, a non-selective adenosine receptor agonist. As shown in Figures 1A,B, L. amazonensis infection significantly increases cAMP production by DC. Interestingly, the blockade of A2B receptor reverses this effect. Addition of NECA to the culture does not substantially increase cAMP levels. The fact that cAMP production was inhibited by MRS1754 even in the absence of an exogenous stimulus (NECA) suggests that some level of extracellular adenosine production is present during the interaction between the parasite and the host cell.
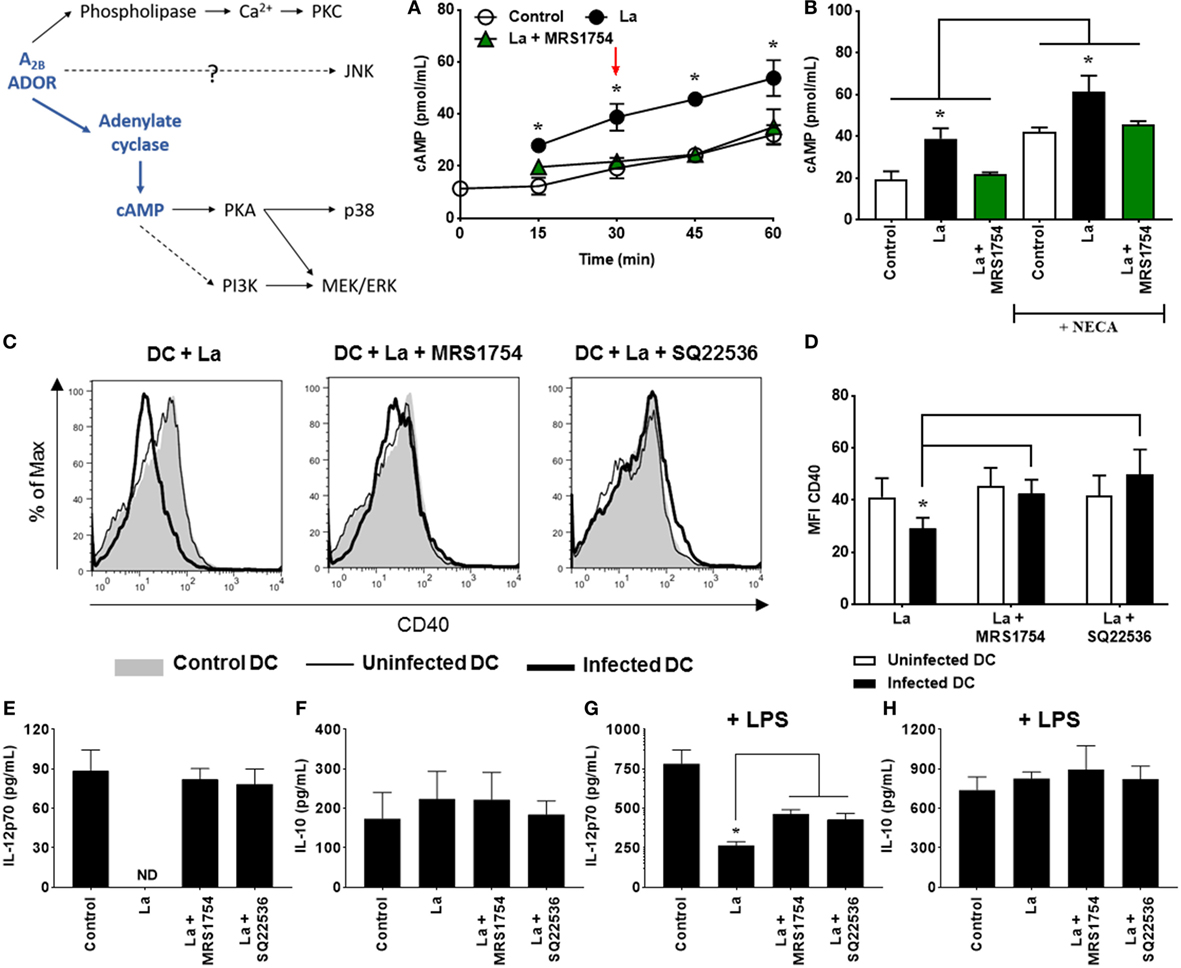
Figure 1. Leishmania amazonensis infection increases cAMP production by dendritic cells (DC), that is involved in the decrease of CD40 expression and IL-12p70 production. (A) DC obtained after 9 days of culture with GM-CSF were infected with metacyclic promastigotes (1:3 cell to parasite ratio) in the presence of 0.1 mM Ro 20-1724 and 5 µM MRS1754 (adenosine A2B receptor antagonist) and the cAMP production evaluated after 15–60 min. (B) After 30 min of infection (as shown by arrow in graph A), DC were stimulated with 1 µM NECA (non-selective adenosine receptor agonist) for more 15 min. The results represent the mean + SD from three independent experiments. *p < 0.05 between La and Control or La + MRS1754 groups, or between linked groups, two-tailed Student’s t-test. (C–H) DC were infected with CFSE-labeled metacyclic promastigotes in the presence of 5 µM MRS1754 or 100 µM SQ22536 (adenylate cyclase inhibitor) and CD40 expression and IL-12p70 and IL-10 production evaluated after 20 h. (C,D) CD11c+ DC were gated into populations of uninfected (CFSE− cells, white bars) and infected (CFSE+ cells, black bars) cells and the MFI of CD40 analyzed in both populations. Control is DC that had not contact with parasites. (C) Histograms are representative of at least three independent experiments. IL-12p70 (E,G) and IL-10 (F,H) cytokine levels were measured in the supernatants using an ELISA. (G,H) 2 µg/mL LPS was added after 3 h of infection. ND, not detected. The results represent the mean + SD from five independent experiments. *p < 0.05 between uninfected and infected DC or between linked groups, two-way ANOVA and Tukey’s post-test (A,B,D) two-tailed Student’s t-test (G).
Previous studies have shown that cAMP plays a critical role in the inhibition of immune cells (32), including DC (40, 41). To confirm that cAMP production induced by A2B receptor activation is important for the inhibition of DC activation by L. amazonensis, we evaluated the expression of CD40 and the production of IL-12p70 and IL-10 in cells treated with SQ22536, an inhibitor of adenylate cyclase. As previously shown (34), L. amazonensis inhibits CD40 expression on DC and this effect is abolished in the presence of MRS1754 (Figures 1C,D). In addition, we showed that inhibition of adenylate cyclase by SQ22536 treatment also restores CD40 expression on infected DC (Figures 1C,D). Moreover, cells infected with L. amazonensis are unable to produce basal levels of IL-12p70, but this ability is restored after the blockade of A2B receptor or inhibition of adenylate cyclase (Figure 1E). The same effect is observed when we stimulated infected cells with LPS (Figure 1G). Finally, we find no changes in IL-10 production by infected DC as compared to uninfected DC and MRS1754 or SQ22536 treatments do not interfere with IL-10 production by these cells (Figures 1F,H). Taken together, our first set of results show that L. amazonensis infection increases cAMP production by DC, and that the production of this intracellular messenger is critical for the decrease of CD40 expression and IL-12p70 production by infected cells.
In addition to Gs proteins, the adenosine A2B receptor has been shown to also engage Gq protein capable of stimulating intracellular calcium accumulation (29). To exclude the role of calcium in DC inhibition by L. amazonensis, L. amazonensis metacyclic promastigotes were labeled with PKH26 and used to infected DC cells loaded with Oregon Green 488. Our results show that although L. amazonensis infection increases the amount of intracellular calcium in DC, this increase is independent of A2B receptor activation, since treatment with MRS1754 does not reverse calcium accumulation (Figure S1A in Supplementary Material). Furthermore, we observe no change in intracellular calcium levels after addition of NECA, a non-selective adenosine receptor agonist, both in uninfected cells and infected cells (Figure S1A in Supplementary Material). These results show that L. amazonensis infection leads to the accumulation of calcium in DC, but adenosine is not responsible for this effect. Moreover, since calcium accumulation triggers protein kinase C (PKC) activation, DC were infected in the presence of staurosporine, a PKC inhibitor, and this treatment was unable to reverse the inhibition of CD40 expression and IL-12p70 production in infected cells (Figures S1B–D in Supplementary Material) confirming that calcium accumulation was not related to inhibition of CD40 expression.
Decrease of CD40 Expression and IL-12p70 Production by L. amazonensis-Infected DC Is Dependent on PI3K
cAMP can binds PKA or Epac (exchange protein activated by cyclic AMP), which is able to phosphorylate residues on several target proteins (32). cAMP may also lead to the activation of another kinase, PI3K (29, 42). In order to verify whether these proteins are involved in cAMP-mediated DC inhibition, cells were infected by L. amazonensis in the presence of KT5720 or LY294002, known inhibitors of PKA and PI3K, respectively, and after 20 h of infection, CD40 expression and cytokine production were evaluated. As shown in Figure 2, inhibition of PKA does not modify CD40 expression (Figure 2A) and IL-12p70 production (Figure 2B). In addition, it also does not alter IL-10 production (Figure 2C) by L. amazonensis-infected cells. On the other hand, treatment with LY294002 significantly increases CD40 expression and IL-12p70 production by infected cells, showing that PI3K activation takes part in the inhibition of DC activation by L. amazonensis.
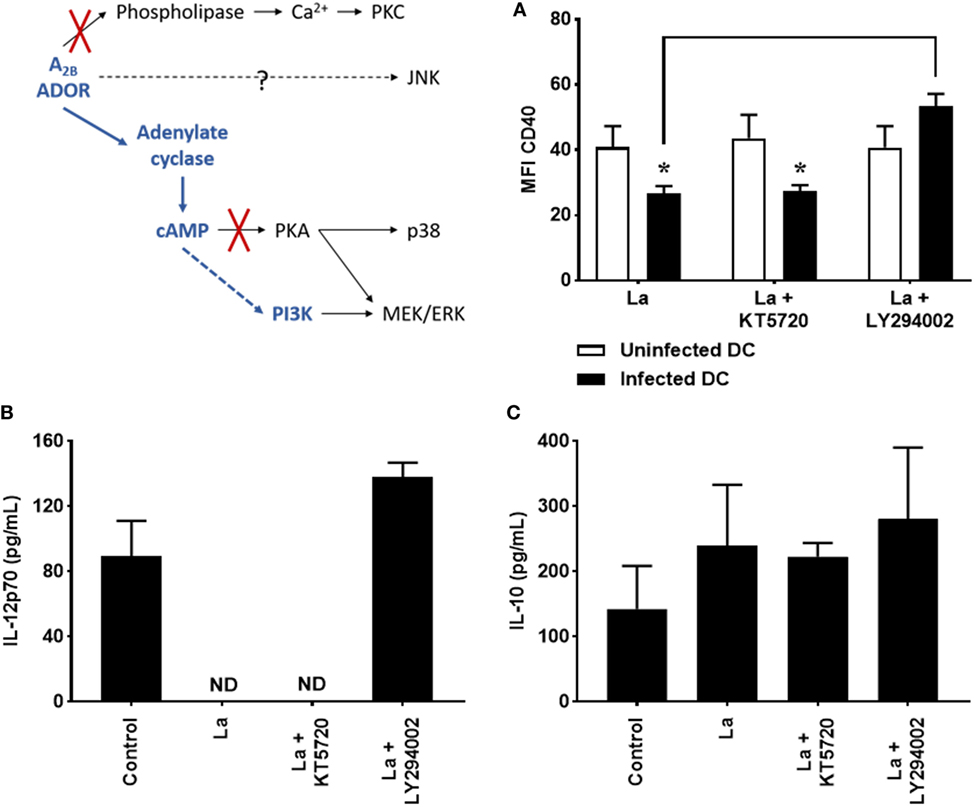
Figure 2. Decrease of CD40 expression and IL-12p70 production by Leishmania amazonensis-infected dendritic cell (DC) is dependent on phosphoinositide 3-kinase (PI3K). DC were infected as described in Figure 1 in the presence of 5 µM MRS1754 (adenosine A2B receptor antagonist) or 10 µM KT5720 [protein kinase A (PKA) inhibitor] or 1 µM LY294002 (PI3K inhibitor) and CD40 expression and IL-12p70 and IL-10 production evaluated after 20 h. (A) CD11c+ DC were gated into populations of uninfected (CFSE− cells, white bars) and infected (CFSE+ cells, black bars) cells and the MFI of CD40 analyzed in both populations. IL-12p70 (B) and IL-10 (C) cytokine levels were measured in the supernatants using an ELISA. Control is uninfected DC. ND, not detected. The results represent the mean + SD from three independent experiments. *p < 0.05 between uninfected and infected DC or between linked groups, two-way ANOVA and Tukey’s post-test.
ERK1/2 Is Involved in the Decrease of CD40 Expression and IL-12p70 Production by L. amazonensis-Infected DC
Mitogen-activated protein kinases (MAPK) is a diverse protein family that consists of three main groups: the c-Jun N-terminal kinases (JNK), the stress-activated protein kinase (SAPK) p38, and the extracellular signal-regulated protein kinases (ERK). These kinases are involved in intracellular signaling events triggered by adenosine (29). Although L. amazonensis infection increased the phosphorylation of JNK and p38, these effects are independent of the A2B receptor; furthermore, the inhibition of both proteins is unable to alter the levels of CD40 expression and the production of IL-12p70 and IL-10 by infected cells (Figure S2 in Supplementary Material).
As previously mentioned, it has been shown that L. amazonensis is able to stimulate the phosphorylation of ERK1/2, which is involved in the inhibition of CD40 on L. amazonensis-infected DC (35, 43); however, the mechanisms that lead to the activation of ERK1/2 in infected DC remain unknown. To investigate whether the activation of the A2B receptor is involved in ERK1/2 phosphorylation, DC were infected with L. amazonensis in the presence or absence of MRS1754. As shown in Figure 3A, phosphorylation of ERK1/2 is significantly higher in infected cells if compared to uninfected controls, after 1 h of infection. Interestingly, the blockade of the A2B receptor by MRS1754 treatment completely abrogates this effect (Figure 3B), demonstrating that the A2B receptor is critical for the phosphorylation of ERK1/2 induced by L. amazonensis infection.
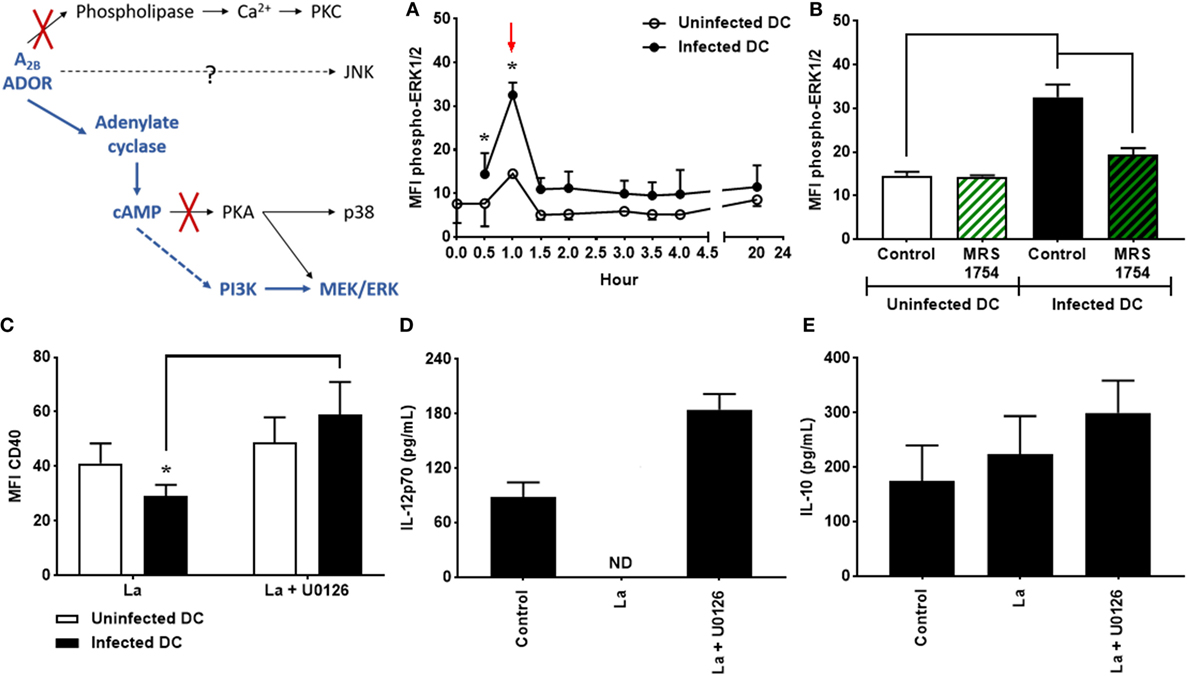
Figure 3. Extracellular signal-regulated protein kinases 1/2 (ERK1/2) is involved in the decrease of CD40 expression and IL-12p70 production by Leishmania amazonensis-infected dendritic cells (DC). (A) DC were infected as described in Figure 1 and the MFI of phospho-ERK1/2 analyzed in populations of uninfected and infected DC after multiple periods of infection. After infection, cells were stimulated with 1 µM NECA for 15 min, fixed and then analyzed by flow cytometry. (B) MFI of phospho-ERK1/2 in uninfected and infected CD11c+ DC in the absence (Control) or presence of 5 µM MRS1754 (adenosine A2B receptor antagonist), after 1 h of infection (as shown by arrow in graph A). The results represent the mean + SD from three independent experiments. *p < 0.05 between uninfected and infected DC or between linked groups, two-way ANOVA and Tukey’s post-test. (C–E) DC were infected in the presence of 5 µM MRS1754 or 10 µM U0126 (MEK inhibitor) and CD40 expression and IL-12p70 and IL-10 production evaluated after 20 h. (C) CD11c+ DC were gated into populations of uninfected (CFSE− cells, white bars) and infected (CFSE+ cells, black bars) cells and the MFI of CD40 analyzed in both populations. IL-12p70 (D) and IL-10 (E) cytokine levels were measured in the supernatants using an ELISA. Control is uninfected DC. ND, not detected. The results represent the mean + SD from five independent experiments. *p < 0.05 between uninfected and infected DC or between linked groups, two-way ANOVA and Tukey’s post-test.
In addition, DC infection in the presence of U0126, a potent inhibitor of MEK, results in a considerable increase in the expression levels of CD40 in infected cells (Figure 3C). Furthermore, L. amazonensis-infected DC treated with U0126 recover their ability to produce IL-12p70 (Figure 3D). Again, IL-10 production by DC is not affected (Figure 3E). The same effects are observed in DC infected in the presence of PD98059, another inhibitor of ERK1/2 (data not shown). Taken together, our results show that ERK1/2 phosphorylation driven by A2B receptor activation plays a relevant role in the inhibition of CD40 expression and IL-12p70 production by L. amazonensis-infected DC.
Our results demonstrate that inhibition of CD40 expression and IL-12p70 production by L. amazonensis-infected DC is dependent on A2B receptor activation, cAMP production, PI3K activation, and ERK1/2 phosphorylation. To assess whether ERK1/2 phosphorylation, as previously found for A2B receptor, is also dependent on the cAMP production and PI3K activation, DC were infected in the presence of SQ22536 or LY294002. U0126 was used as control. Interestingly, both adenylate cyclase and PI3K inhibition are able to reverse the phosphorylation of ERK1/2 stimulated by L. amazonensis (Figure 4), suggesting that A2B receptor, adenylate cyclase, PI3K, and ERK1/2 act in a direct pathway, instead of these proteins act in parallel and independent pathways.
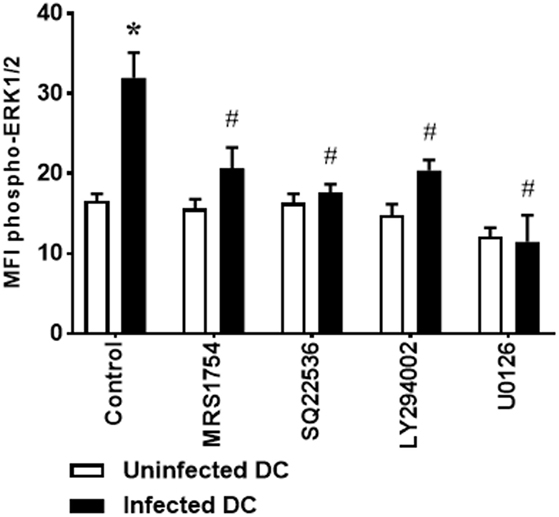
Figure 4. Extracellular signal-regulated protein kinases 1/2 (ERK1/2) phosphorylation in Leishmania amazonensis-infected dendritic cells (DC) is dependent on A2B receptor, AMPc production, and phosphoinositide 3-kinase (PI3K) activation. DC were infected as described in Figure 1, in the absence (Control) or presence of 5 µM MRS1754 (adenosine A2B receptor antagonist), 100 µM SQ22536 (adenylate cyclase inhibitor), 1 µM LY294002 (PI3K inhibitor), or 10 µM U0126 (MEK inhibitor), and the MFI of phospho-ERK1/2 analyzed in populations of uninfected and infected CD11c+ DC after 1 h of infection. After infection, cells were stimulated with 1 µM NECA for 15 min, fixed and then analyzed by flow cytometry. The results represent the mean + SD from three independent experiments. *p < 0.05 between uninfected and infected DC, #p < 0,05 between Control and treated DC, two-way ANOVA and Tukey’s post-test.
In our previous study, we observed that infection by L. amazonensis in addition to decrease CD40 expression, also inhibited the expression of MHCII and CD86. We also demonstrated that the inhibition of the expression of these molecules was reversed by the A2B receptor antagonist, MRS1754. To verify if the pathway involved in the inhibition of CD40 expression is also associated with the inhibition of MHCII and CD86 expression, we analyzed the expression of these molecules in the presence of the inhibitors of the key enzymes studied here. As shown in Figure S3 in Supplementary Material, although the blockade of the A2B receptor is able to reverse the inhibition of MHCII and CD86, inhibition of adenylate cyclase, PI3K and of ERK phosphorylation has no effect on these parameters suggesting the activation of a different pathway starting at the A2B receptor.
Inhibition of the Adenosine A2B Receptor Increases CD40 Expression by DC in Mice Infected by L. amazonensis
Considering that cAMP production triggered by A2B receptor is important to the inhibition of CD40 in L. amazonensis-infected DC, that this effect is not present in infection by other species of Leishmania (34) and that CD40 plays a central role in the activation of T lymphocytes by DC (8), we evaluated the expression of CD40 on DC from ears and draining lymph nodes of mice infected by L. amazonensis, in the presence or absence PSB1115, an A2B receptor antagonist. Due to its higher solubility in water, PSB1115 is more appropriate for in vivo studies (44) and hence was used in the following experiments. Administration of PSB1115 in the infective inoculum increases the percentage of CD40+ DC in both the ear and draining lymph nodes (Figure 5). The percentage of DC, evaluated by CD11c expression, on injection sites and draining lymph nodes is not modify by infection or PSB1115 treatment nor is the expression of CD40 in DC from uninfected mice (Figure 5). PSB1115 has no direct effect on the viability or proliferation of promastigotes (data not shown). These results corroborate in vivo our findings of the previous experiments.
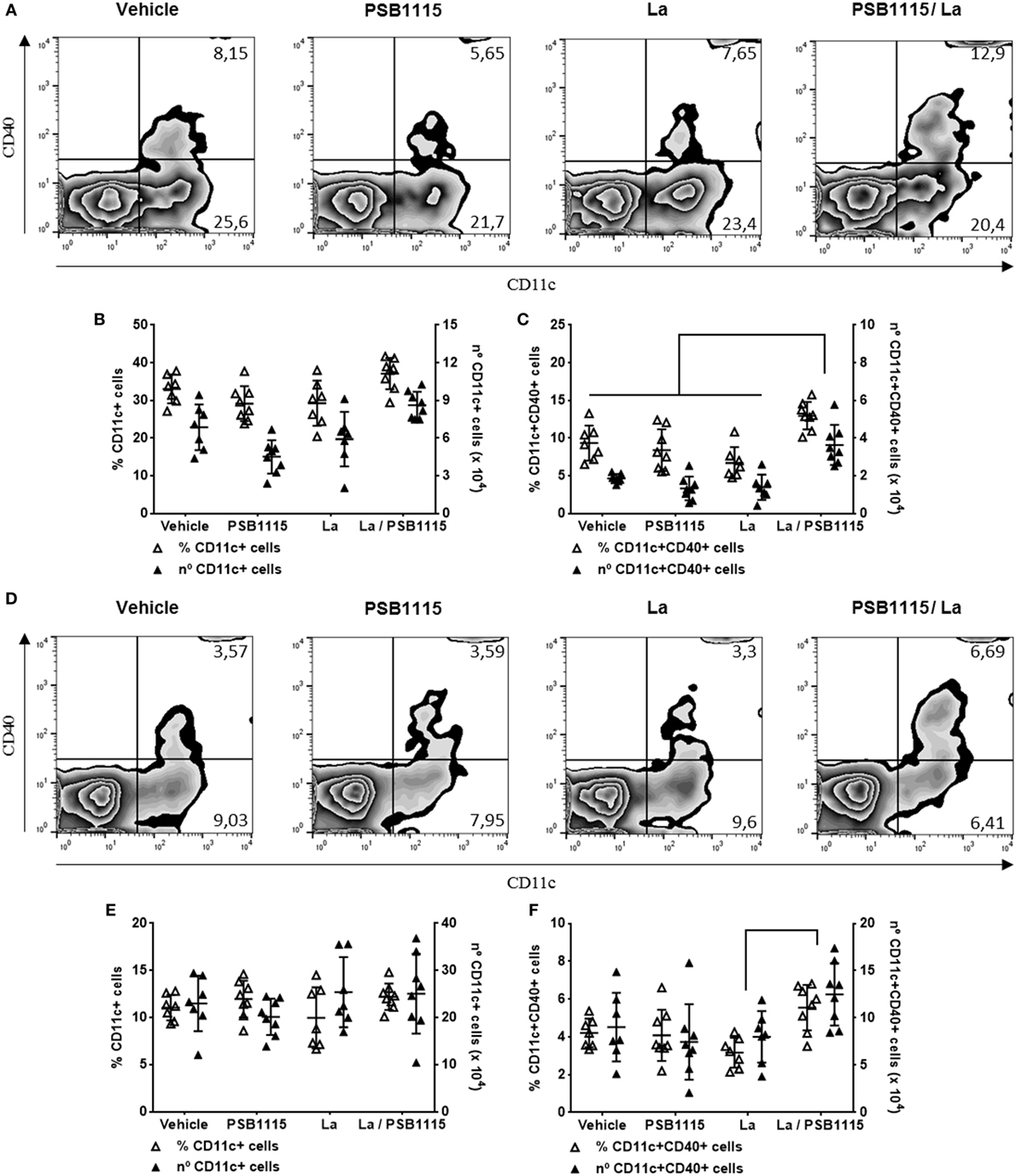
Figure 5. A2B receptor blockade increases the percentage of CD40+ dendritic cells on ears and draining lymph nodes of L. amazonensis-infected mice. C57BL/6J mice were inoculated intradermally in both ears with 105 metacyclic promastigotes of L. amazonensis, in the presence or absence of 5 µM PSB1115 (adenosine A2B receptor antagonist). 7 days after infection, both ears and draining auricular lymph nodes were removed. The percentage or the total number of cells in both tissues of CD11c+ (B,E) or CD11c+CD40+ (C,F) cells in the ears (B,C) or lymph nodes (E,F) were analyzed using flow cytometry. Representative dot plots of the expression of CD11c and CD40 in the ears (A) or lymph nodes (D). The results represent the mean + SD from two independent experiments with three or four mice per group. *p < 0.05 between linked groups, one-way ANOVA and Tukey’s post-test.
Adenosine A2B Receptor Blockade Controls Lesion Development in Mice Infected by L. amazonensis
To investigate the role of the A2B receptor activation on lesion development during L. amazonensis infection, C57BL/6J mice were inoculated in the ears with metacyclic promastigotes and lesion size measured weekly. Tissue parasitism and cytokine production were evaluated at the 12th week of infection. Interestingly, our results show that the blockade of A2B receptor by PSB1115 reduces lesion size (Figure 6A) starting at the eighth week of infection. The reduction in lesion size was accompanied by a decrease in tissue parasitism (90%) after 12 weeks of infection (Figure 6B). Evaluation of cytokine production by antigen stimulated lymph node cells from treated mice demonstrated that the reduction in lesion size and tissue parasitism is associated with higher levels of IFN-γ in culture supernatants when compared with cells from control mice (Figure 6C). However, no alteration was detected on the production of IL-10 by these cells also demonstrating in vivo the apparent lack of role of this cytokine in L. amazonensis infection even with the inhibition of A2B receptor at the beginning of the infection (Figure 6D).
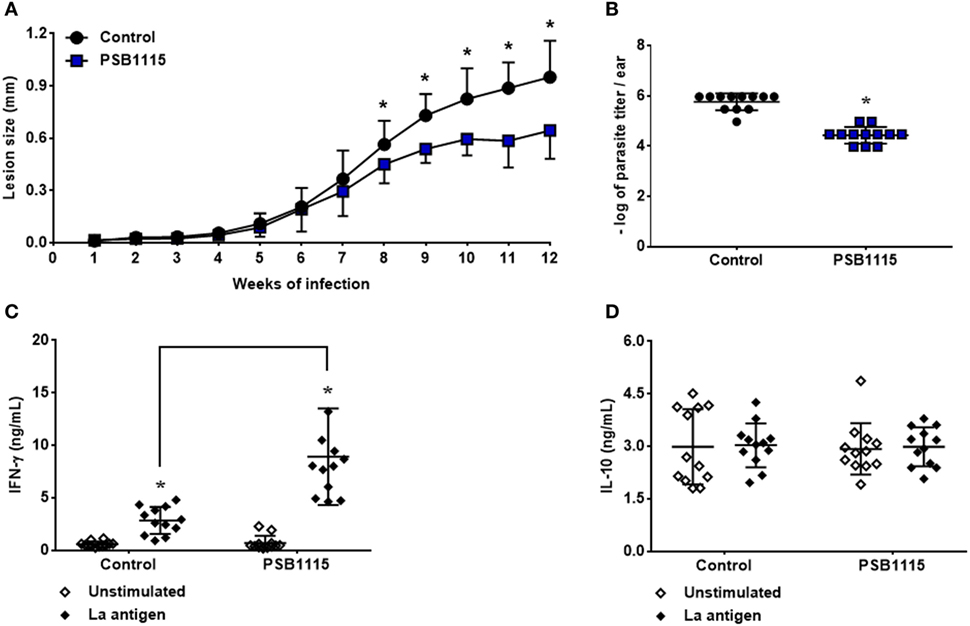
Figure 6. A2B receptor blockade controls lesion development in mice infected by L. amazonensis. C57BL/6J mice were inoculated intradermally in the ears with 103 metacyclic promastigotes of L. amazonensis, in the presence or absence of 5 µM PSB1115 (adenosine A2B receptor antagonist). (A) Lesion sizes were measured weekly. The lesion size was defined as the difference between the infected and uninfected contralateral ear. (B) Tissue parasitism at the 12th week of infection. Lesions from infected mice were excised and parasitism evaluated by limiting dilution. IFN-γ (C) or IL-10 (D) production by draining lymph node cells from mice infected for 12 weeks. Cells were stimulated for 48 h with particulate antigen of L. amazonensis. The results represent the mean + SD from three independent experiments with four mice per group. *p < 0.05 between Control and PSB1115 groups (A,B) or between unstimulated and stimulated cells or linked groups (C), two-tailed Student’s t-test (A,B), two-way ANOVA and Tukey’s post-test (C).
Discussion
Leishmania amazonensis infection is characterized by a deficiency in antigen-specific T cell response, which contributes to disease progression and failure in therapy (1, 2). It is, thus, relevant to study the mechanisms responsible for this anergy and DC would be a preferential target for intervention, given the essential role of these cells in the differentiation of effector T lymphocytes. Having previously demonstrated that L. amazonensis inhibit DC response by a mechanism dependent on A2B adenosine receptor (34), here we decided to evaluate the intracellular events triggered by this receptor in infected cells.
Of the four adenosine receptors (A1, A2A, A2B, and A3), A2A and A2B are responsible for the main immunosuppressive effects of this nucleoside. In a previous work, we showed that A2A receptor is not involved in the inhibition of L. amazonensis-infected DC (34); therefore, we focused this work only on the A2B receptor.
The adenosine A2B receptor has been shown to engage Gs or Gq proteins, capable of stimulating adenylate cyclase and phospholipase C, respectively (29). The increase in intracellular cAMP concentration by adenylate cyclase activity has been strongly associated with inhibition of immune cells, including monocytes/macrophages (32, 45), DC (40, 41, 46) and T lymphocytes (47). Here, we show, for the first time, that L. amazonensis infection, via activation of the A2B receptor, increases cAMP production by DC and uses this mechanism to inhibit the co-stimulatory activity of infected cells. Furthermore, we showed that this messenger is essential for the inhibition of infected DC, since the inhibition of adenylate cyclase by SQ22536 treatment restores the ability of L. amazonensis-infected DC to express CD40 and produce IL-12p70.
Phospholipase C leads to the accumulation of intracellular calcium (29) that is involved in the maturation of human monocyte-derived DC (48, 49) and may be involved in the generation of a population of DC that produces low amounts of IL-12 and drives the differentiation of Th2 lymphocytes (48). Our results show that, although L. amazonensis infection increases the intracellular levels of calcium, this effect was independent of A2B receptor triggering. In addition, NECA, a non-specific adenosine receptor agonist, did not alter the levels of calcium in infected cells, showing that although calcium may be important in L. amazonensis-infected DC response, the increase in intracellular calcium concentration does not seem to be dependent on the activation of adenosine receptors.
Impairment of DC activation was independent of PKA activity but triggered by a PI3K-dependent pathway. Considering that the inhibition of adenylate cyclase and the inhibition of PI3K have the same effect on DC response, and that cAMP can lead to the PI3K activation (29, 42), we suggest that the inhibition of CD40 expression and IL-12p70 production by infected DC is mediated by a cAMP-PI3K pathway.
The triggering of adenosine receptors, especially A2B receptor, can lead to the activation of any of three major MAPK cascades, known as JNK, p38 and ERK1/2 (29, 30). As previously shown for macrophages infected with Leishmania mexicana (50), we also found that DC infection by L. amazonensis increases the phosphorylation of these three kinases. However, the increase of phosphorylation of JNK and p38 in infected cells was independent of A2B receptor. Moreover, the inhibition of these proteins by SP600125 or SB203580 was unable to reverse the inhibition of DC activation as measured by CD40 expression and IL-12 production. It has been previously described that L. amazonensis stimulates the expression of phospho-ERK1/2 in infected DC (35). In another study, Schulte and Fredholm (51) demonstrated that the phosphorylation of ERK1/2 induced by the activation of the A2B receptor is dependent on PI3K and independent of PKA. Our results link these previous observations by showing that ERK1/2 phosphorylation in L. amazonensis-infected DC is dependent on A2B receptor activation and cAMP production. Our data also strongly suggest that the previously unknown G protein-coupled receptor associated with ERK1/2 phosphorylation (43) is, in fact, the adenosine A2B receptor. In addition, by showing that the inhibition of CD40 expression and IL-12p70 production by DC caused by L. amazonensis was dependent on ERK1/2, we provide further evidence that ERK1/2 phosphorylation is associated with the pathogenesis of L. amazonensis infection.
Several pathogens are also able to modulate MAPK signaling (reviewed by Ref. (52)). In Toxoplasma gondii infection, the blockade of ERK phosphorylation decreases parasite proliferation (53) and increase IL-12 production by host cells (54), corroborating our results with L. amazonensis infection. Interestingly, Trypanosoma cruzi, another protozoan parasite, triggers ERK phosphorylation which stimulates the production of IL-10 and decreases lymphocyte proliferation by regulatory DC (55). Also, ERK phosphorylation is associated to cardiac damage induced by TGF-β in T. cruzi-infected mice (56). Thus, ERK phosphorylation seems to be a common observation in situations where immune modulation by pathogens occurs. However, the participation of purinergic signaling in these settings has not yet been addressed. It would be interesting to investigate the participation of adenosine mediated ERK phosphorylation in infections by these parasites.
Differently from the infection with other Leishmania species (57–59) IL-10 does not seem to play a relevant role in L. amazonensis infection (20, 60). As shown before by our group (34) and in the present study, IL-10 production by DC was not altered by L. amazonensis infection or by any of the treatments used. These findings contrast with the observation that ERK1/2 phosphorylation is important for IL-10 production by L. amazonensis-infected macrophages (36). Possible explanations for the discrepancy observed are the cell type (macrophages versus DC), the parasite stage (amastigotes versus metacyclic promastigotes), the strain of mice used (BALB/c—highly susceptible to Leishmania infection versus C57BL/6—resistant to most Leishmania species) and the fact that low molecular weight hyaluronic acid was necessary for IL-10 production. In our studies, cells were not further stimulated (present work) or were stimulated with LPS (34). Interestingly, however, the study by Yang and colleagues (36) shows that treatment of infected mice with U0126, to inhibit ERK1/2 phosphorylation, restrains parasite growth and lesion development. The finding that cAMP is involved in the regulation of DC activation is relevant in the context of L. amazonensis infection since it provides an explanation for the lack of role of IL-10 in this infection.
Our results confirm, to some extent, a very recent work published with human macrophages demonstrating an association between A2B receptor activation and ERK1/2 phosphorylation (61). However, contrary to our results, the study reports an association between IL-10 production and A2B receptor activation, although this observation was not directly tested. We believe the discrepancy between the two studies could be related to the model used and/or the high concentration of MRS1754 used in the human study which may have an overlapping action on other adenosine receptors. The inhibition of other adenosine receptors, particularly, the A2A may interfere with IL-10 production Nevertheless, the study by Vijayamahantesh and colleagues reinforces our observation and extends it to the human system, thus proving the validity of our findings.
The results described in this report link several previous “unrelated” observations regarding the mechanism by which L. amazonensis inhibits the establishment of an adequate immune response. Our data implicate the activation of the adenosine A2B receptor during the infection by this parasite species to the inhibition of CD40 expression (14, 35), the lack of IL-12 production (16, 35), the phosphorylation of ERK1/2 (35, 36) (Figure 7) and also provide an explanation (increased cAMP) for the inhibition of the immune response by this parasite in the absence of a Th2 response (3) and IL-10 production (20, 60). The impairment of CD40 expression and IL-12p70 production by DC caused by L. amazonensis is an important immune response evasion mechanism, since both molecules are essential for the development of a Th1 response necessary to control the parasite infection (7, 10). Moreover, the lower expression of these molecules could explain the lack of T cell activation observed in patients with diffuse cutaneous leishmaniasis caused by L. amazonensis infection (62, 63).
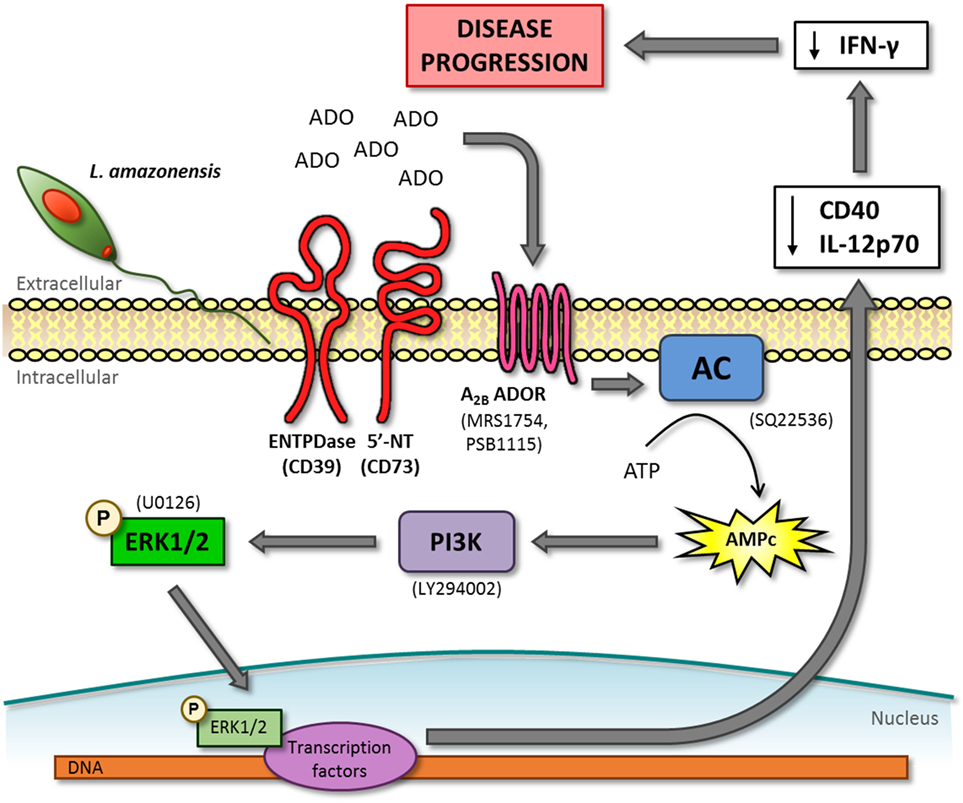
Figure 7. cAMP-phosphoinositide 3-kinase (PI3K)–extracellular signal-regulated protein kinases 1/2 (ERK1/2) pathway activated by the adenosine A2B receptor is important to dendritic cell inhibition and lesion development in mice infected by L. amazonensis. L. amazonensis infection leads to the accumulation of adenosine (ADO) in the extracellular environment that activates adenosine A2B receptor (A2B ADOR) on dendritic cells (DC). The following steps of this pathway are the activation of adenylate cyclase (AC) and consequent production of cAMP, activation of PI3K and, finally, the phosphorylation of ERK1/2. In order to identify this pathway, we used inhibitors that are showed in parenthesis. phospho-ERK1/2 is able to translocate to the nucleus to interacts with transcription factors that leads to the decrease of CD40 expression and IL-12p70 production. DC inhibited by L. amazonensis can decrease IFN-γ production by lymph node cells, resulting in the suppression of immune response and lesion development in mice.
Corroborating this hypothesis, we demonstrated, in vivo, that inhibition of the A2B receptor at the moment of infection not only increases CD40 expression by DC present not only in the injection site but also at the draining lymph nodes but also increases the Leishmania-specific Th1 response resulting in decreased lesion size and tissue parasitism. Furthermore, this enhanced Th1 response was not associated with changes in the production of IL-10 reinforcing the apparent lack of role of this cytokine in the control of the infection by L. amazonensis in C57BL/6 mice. Our data point to a new mechanism of control of immune response by the parasite that is associated with autocrine production of cAMP by the infected cell rather than the secretion of immunomodulatory cytokines. The fact that the treatment used in this study is not able to completely control parasite development indicates that other factors may control the enhanced Th1 response. The role of purinergic signaling on macrophages and other cells involved in the immune response against L. amazonensis is currently under investigation.
Finally, the recent advances of the role of purinergic signaling in the establishment and control of the immune response has triggered a series of clinical studies designed to evaluate the use of agonists, as well as antagonists, of purine receptors in different diseases with emphasis in cancer treatment (64). The confirmation of the pathway used by the L. amazonensis to suppress the immune response (A2B receptor → cAMP → PI3K → ERK1/2) in humans may suggest possible targets for new therapeutic approaches to control L. amazonensis infection specially in the case of diffuse cutaneous leishmaniasis which is, as mentioned earlier, usually refractory to treatment.
Ethics Statement
This study was carried out in accordance with the recommendations of the Brazilian Guidelines for animal experimentation. The protocols were approved by the University’s Ethical Committee on Animal Experimentation (CEUA 2012/02 and CEUA 2013/51).
Author Contributions
Conceived and designed the experiments: AF, TM, and LA. Performed the experiments: AF and MS-T. Analyzed the data: AF, MS-T, TM, and LA. Wrote the paper: AF and LA. All authors reviewed the results and the manuscript and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewers, RS and AF, and handling editor declared their shared affiliations, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors wish to thank Leandro H. Santos and Marcorelio D. Souza for technical assistance and Dr. Leda Q. Vieira and Dr. Ieso M. Castro for providing essential reagents. The authors are also indebted to the helpful discussions of Dr. Helton Santiago, Dr. Rogelio Brandão, Dr. Cláudio Salgado, and Dr. Tiago D. Serafim.
Funding
This work was supported by grants from FAPEMIG, CNPq, CAPES, UFOP, Rede de Pesquisa em Doenças Infecciosas Humanas e Animais no Estado de Minas Gerais/FAPEMIG, and Rede Mineira de Bioterismo/FAPEMIG. Luís Afonso and Tiago Mineo are fellow researchers from CNPq.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00849/full#supplementary-material.
Abbreviations
DC, dendritic cell; ERK1/2, extracellular signal-regulated protein kinases 1/2; JNK, c-Jun N-terminal kinase; La, Leishmania amazonensis; MAPK, mitogen-activated protein kinase; NECA, 5’-(N-ethylcarboxamido) adenosine; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; PKC, protein kinase C; SAPK/p38, stress-activated protein kinases/p38.
References
1. Silveira FT, Lainson R, Corbett CE. Clinical and immunopathological spectrum of American cutaneous leishmaniasis with special reference to the disease in Amazonian Brazil: a review. Mem Inst Oswaldo Cruz (2004) 99(3):239–51. doi:10.1590/S0074-02762004000300001
2. Silveira FT, Lainson R, De Castro Gomes CM, Laurenti MD, Corbett CE. Immunopathogenic competences of Leishmania (V.) braziliensis and L. (L.) amazonensis in American cutaneous leishmaniasis. Parasite Immunol (2009) 31(8):423–31. doi:10.1111/j.1365-3024.2009.01116.x
3. Afonso LCC, Scott P. Immune responses associated with susceptibility of C57BL/10 mice to Leishmania amazonensis. Infect Immun (1993) 61(7):2952–9.
4. Maioli TU, Takane E, Arantes RME, Fietto JLR, Afonso LCC. Immune response induced by New World Leishmania species in C57BL/6 mice. Parasitol Res (2004) 94(3):207–12. doi:10.1007/s00436-004-1193-6
5. Marques-da-Silva EA, Oliveira JC, Figueiredo AB, Lima-Junior DS, Carneiro CM, Fietto JLR, et al. Extracellular nucleotide metabolism in Leishmania: influence of adenosine in the establishment of infection. Microbes Infect (2008) 10(8):850–7. doi:10.1016/j.micinf.2008.04.016
6. Brandonisio O, Spinelli R, Pepe M. Dendritic cells in Leishmania infection. Microbes Infect (2004) 6(15):1402–9. doi:10.1016/j.micinf.2004.10.004
7. Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature (1995) 378(6557):617–20. doi:10.1038/378617a0
8. Grewal IS, Flavell RA. The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev (1996) 153:85–106. doi:10.1111/j.1600-065X.1996.tb00921.x
9. Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol (2007) 37(Suppl 1):S53–60. doi:10.1002/eji.200737400
10. Afonso LCC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science (1994) 263(5144):235–7. doi:10.1126/science.7904381
11. Sacks DL, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol (2002) 2(11):845–58. doi:10.1038/nri933
12. Scott P, Hunter CA. Dendritic cells and immunity to leishmaniasis and toxoplasmosis. Curr Opin Immunol (2002) 14(4):466–70. doi:10.1016/S0952-7915(02)00353-9
13. Qi H, Popov V, Soong L. Leishmania amazonensis-dendritic cell interactions in vitro and the priming of parasite-specific CD4+ T cells in vivo. J Immunol (2001) 167(8):4534–42. doi:10.4049/jimmunol.167.8.4534
14. Prina E, Abdi SZ, Lebastard M, Perret E, Winter N, Antoine JC. Dendritic cells as host cells for the promastigote and amastigote stages of Leishmania amazonensis: the role of opsonins in parasite uptake and dendritic cell maturation. J Cell Sci (2004) 117(Pt 2):315–25. doi:10.1242/jcs.00860
15. Favali C, Tavares N, Clarencio J, Barral A, Barral-Netto M, Brodskyn CI. Leishmania amazonensis infection impairs differentiation and function of human dendritic cells. J Leukoc Biol (2007) 82(6):1401–6. doi:10.1189/jlb.0307187
16. Vasquez RE, Xin L, Soong L. Effects of CXCL10 on dendritic cell and CD4+ T-cell functions during Leishmania amazonensis infection. Infect Immun (2008) 76(1):161–9. doi:10.1128/IAI.00825-07
17. Xin L, Li K, Soong L. Down-regulation of dendritic cell signaling pathways by Leishmania amazonensis amastigotes. Mol Immunol (2008) 45(12):3371–82. doi:10.1016/j.molimm.2008.04.018
18. Hermida MD, Doria PG, Taguchi AM, Mengel JO, Santos W. Leishmania amazonensis infection impairs dendritic cell migration from the inflammatory site to the draining lymph node. BMC Infect Dis (2014) 14:450. doi:10.1186/1471-2334-14-450
19. Pereira BA, Alves CR. Immunological characteristics of experimental murine infection with Leishmania (Leishmania) amazonensis. Vet Parasitol (2008) 158(4):239–55. doi:10.1016/j.vetpar.2008.09.015
20. Jones DE, Ackermann MR, Wille U, Hunter CA, Scott P. Early enhanced Th1 response after Leishmania amazonensis infection of C57BL/6 interleukin-10-deficient mice does not lead to resolution of infection. Infect Immun (2002) 70(4):2151–8. doi:10.1128/IAI.70.4.2151-2158.2002
21. Cortes DF, Carneiro MB, Santos LM, Souza TC, Maioli TU, Duz AL, et al. Low and high-dose intradermal infection with Leishmania major and Leishmania amazonensis in C57BL/6 mice. Mem Inst Oswaldo Cruz (2010) 105(6):736–45. doi:10.1590/S0074-02762010000600002
22. Carregaro V, Sa-Nunes A, Cunha TM, Grespan R, Oliveira CJ, Lima-Junior DS, et al. Nucleosides from Phlebotomus papatasi salivary gland ameliorate murine collagen-induced arthritis by impairing dendritic cell functions. J Immunol (2011) 187(8):4347–59. doi:10.4049/jimmunol.1003404
23. Norsworthy NB, Sun J, Elnaiem D, Lanzaro G, Soong L. Sand fly saliva enhances Leishmania amazonensis infection by modulating interleukin-10 production. Infect Immun (2004) 72(3):1240–7. doi:10.1128/IAI.72.3.1240-1247.2004
24. Di Virgilio F. Purinergic mechanism in the immune system: a signal of danger for dendritic cells. Purinergic Signal (2005) 1(3):205–9. doi:10.1007/s11302-005-6312-z
25. Chaves SP, Torres-Santos EC, Marques C, Figliuolo VR, Persechini PM, Coutinho-Silva R, et al. Modulation of P2X7 purinergic receptor in macrophages by Leishmania amazonensis and its role in parasite elimination. Microbes Infect (2009) 11(10–11):842–9. doi:10.1016/j.micinf.2009.05.001
26. Ren H, Teng Y, Tan B, Zhang X, Jiang W, Liu M, et al. Toll-like receptor-triggered calcium mobilization protects mice against bacterial infection through extracellular ATP release. Infect Immun (2014) 82(12):5076–85. doi:10.1128/IAI.02546-14
27. Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112(2):358–404. doi:10.1016/j.pharmthera.2005.04.013
28. Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal (2014) 10:529–64. doi:10.1007/s11302-014-9427-2
29. Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal (2003) 15(9):813–27. doi:10.1016/S0898-6568(03)00058-5
30. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13(12):842–57. doi:10.1038/nrc3613
31. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16(3):177–92. doi:10.1038/nri.2016.4
32. Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol (2008) 39(2):127–32. doi:10.1165/rcmb.2008-0091TR
33. Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta (2011) 1808(5):1329–39. doi:10.1016/j.bbamem.2010.05.016
34. Figueiredo AB, Serafim TD, Marques-da-Silva EA, Meyer-Fernandes JR, Afonso LCC. Leishmania amazonensis impairs DC function by inhibiting CD40 expression via A2B adenosine receptor activation. Eur J Immunol (2012) 42(5):1203–15. doi:10.1002/eji.201141926
35. Boggiatto PM, Jie F, Ghosh M, Gibson-Corley KN, Ramer-Tait AE, Jones DE, et al. Altered dendritic cell phenotype in response to Leishmania amazonensis amastigote infection is mediated by MAP kinase, ERK. Am J Pathol (2009) 174(5):1818–26. doi:10.2353/ajpath.2009.080905
36. Yang Z, Mosser DM, Zhang X. Activation of the MAPK, ERK, following Leishmania amazonensis infection of macrophages. J Immunol (2007) 178(2):1077–85. doi:10.4049/jimmunol.178.2.1077
37. Goncalves R, Vieira ER, Melo MN, Gollob KJ, Mosser DM, Tafuri WL. A sensitive flow cytometric methodology for studying the binding of L. chagasi to canine peritoneal macrophages. BMC Infect Dis (2005) 5(1):39. doi:10.1186/1471-2334-5-39
38. Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods (1999) 223(1):77–92. doi:10.1016/S0022-1759(98)00204-X
39. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity (2015) 42(6):1197–211. doi:10.1016/j.immuni.2015.05.018
40. Li K, Anderson KJ, Peng Q, Noble A, Lu B, Kelly AP, et al. Cyclic AMP plays a critical role in C3a-receptor-mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood (2008) 112(13):5084–94. doi:10.1182/blood-2008-05-156646
41. Challier J, Bruniquel D, Sewell AK, Laugel B. Adenosine and cAMP signaling skew human dendritic cell differentiation towards a tolerogenic phenotype with defective CD8+ T-cell priming capacity. Immunology (2012) 138:402–10. doi:10.1111/imm.12053
42. Yen JH, Kocieda VP, Jing H, Ganea D. Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. J Biol Chem (2011) 286(45):38913–23. doi:10.1074/jbc.M111.252932
43. Boggiatto PM, Martinez PA, Pullikuth A, Jones DE, Bellaire B, Catling A, et al. Targeted extracellular signal-regulated kinase activation mediated by Leishmania amazonensis requires MP1 scaffold. Microbes Infect (2014) 16:328–36. doi:10.1016/j.micinf.2013.12.006
44. Muller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta (2011) 1808(5):1290–308. doi:10.1016/j.bbamem.2010.12.017
45. Sciaraffia E, Riccomi A, Lindstedt R, Gesa V, Cirelli E, Patrizio M, et al. Human monocytes respond to extracellular cAMP through A2A and A2B adenosine receptors. J Leukoc Biol (2014) 96:113–22. doi:10.1189/jlb.3A0513-302RR
46. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol (2010) 265(2):91–6. doi:10.1016/j.cellimm.2010.07.007
47. Mosenden R, Tasken K. Cyclic AMP-mediated immune regulation – overview of mechanisms of action in T cells. Cell Signal (2011) 23(6):1009–16. doi:10.1016/j.cellsig.2010.11.018
48. Faries MB, Bedrosian I, Xu S, Koski G, Roros JG, Moise MA, et al. Calcium signaling inhibits interleukin-12 production and activates CD83(+) dendritic cells that induce Th2 cell development. Blood (2001) 98(8):2489–97. doi:10.1182/blood.V98.8.2489
49. Bagley KC, Abdelwahab SF, Tuskan RG, Lewis GK. Calcium signaling through phospholipase C activates dendritic cells to mature and is necessary for the activation and maturation of dendritic cells induced by diverse agonists. Clin Diagn Lab Immunol (2004) 11(1):77–82. doi:10.1128/CDLI.11.1.77-82.2004
50. Shweash M, Adrienne McGachy H, Schroeder J, Neamatallah T, Bryant CE, Millington O, et al. Leishmania mexicana promastigotes inhibit macrophage IL-12 production via TLR-4 dependent COX-2, iNOS and arginase-1 expression. Mol Immunol (2011) 48(15–16):1800–8. doi:10.1016/j.molimm.2011.05.013
51. Schulte G, Fredholm BB. The G(s)-coupled adenosine A(2B) receptor recruits divergent pathways to regulate ERK1/2 and p38. Exp Cell Res (2003) 290(1):168–76. doi:10.1016/S0014-4827(03)00324-0
52. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol (2013) 13(9):679–92. doi:10.1038/nri3495
53. Barbosa BF, Paulesu L, Ietta F, Bechi N, Romagnoli R, Gomes AO, et al. Susceptibility to Toxoplasma gondii proliferation in BeWo human trophoblast cells is dose-dependent of macrophage migration inhibitory factor (MIF), via ERK1/2 phosphorylation and prostaglandin E2 production. Placenta (2014) 35(3):152–62. doi:10.1016/j.placenta.2013.12.013
54. Quan JH, Chu JQ, Kwon J, Choi IW, Ismail HA, Zhou W, et al. Intracellular networks of the PI3K/AKT and MAPK pathways for regulating Toxoplasma gondii-induced IL-23 and IL-12 production in human THP-1 cells. PLoS One (2015) 10(11):e0141550. doi:10.1371/journal.pone.0141550
55. Poncini CV, Gimenez G, Pontillo CA, Alba-Soto CD, Isola EL, Piazzon I, et al. Central role of extracellular signal-regulated kinase and toll-like receptor 4 in IL-10 production in regulatory dendritic cells induced by Trypanosoma cruzi. Mol Immunol (2010) 47(11–12):1981–8. doi:10.1016/j.molimm.2010.04.016
56. Ferreira RR, Souza EM, Oliveira FL, Ferrao PM, Gomes LH, Mendonca-Lima L, et al. Proteins involved on TGF-beta pathway are up-regulated during the acute phase of experimental Chagas disease. Immunobiology (2016) 221(5):587–94. doi:10.1016/j.imbio.2016.01.009
57. Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med (2001) 194(10):1497–506. doi:10.1084/jem.194.10.1497
58. Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol (2001) 166(2):1141–7. doi:10.4049/jimmunol.166.2.1141
59. Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 mediates susceptibility to Leishmania donovani infection. Eur J Immunol (2001) 31(10):2848–56. doi:10.1002/1521-4141(2001010)31:10<2848:AID-IMMU2848>3.0.CO;2-T
60. Ji J, Sun J, Soong L. Impaired expression of inflammatory cytokines and chemokines at early stages of infection with Leishmania amazonensis. Infect Immun (2003) 71(8):4278–88. doi:10.1128/IAI.71.8.4278-4288.2003
61. Vijayamahantesh, Amit A, Kumar S, Dikhit MR, Jha PK, Singh AK, et al. Up regulation of A2B adenosine receptor on monocytes are crucially required for immune pathogenicity in Indian patients exposed to Leishmania donovani. Cytokine (2016) 79:38–44. doi:10.1016/j.cyto.2015.12.016
62. Convit J, Pinardi ME, Rondon AJ. Diffuse cutaneous leishmaniasis: a disease due to an immunological defect of the host. Trans R Soc Trop Med Hyg (1972) 66(4):603–10. doi:10.1016/0035-9203(72)90306-9
63. Castes M, Agnelli A, Verde O, Rondon AJ. Characterization of the cellular immune response in American cutaneous leishmaniasis. Clin Immunol Immunopathol (1983) 27(2):176–86. doi:10.1016/0090-1229(83)90068-5
Keywords: A2B adenosine receptor, cAMP, extracellular signal-regulated protein kinases, CD40, IL-12p70, dendritic cell, Leishmania amazonensis
Citation: Figueiredo AB, Souza-Testasicca MC, Mineo TWP and Afonso LCC (2017) Leishmania amazonensis-Induced cAMP Triggered by Adenosine A2B Receptor Is Important to Inhibit Dendritic Cell Activation and Evade Immune Response in Infected Mice. Front. Immunol. 8:849. doi: 10.3389/fimmu.2017.00849
Received: 22 April 2017; Accepted: 05 July 2017;
Published: 25 July 2017
Edited by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilReviewed by:
Robson Coutinho-Silva, Federal University of Rio de Janeiro, BrazilAlessandra D’Almeida Filardy, Federal University of Rio de Janeiro, Brazil
Carlos Roberto Alves, Oswaldo Cruz Foundation, Brazil
Copyright: © 2017 Figueiredo, Souza-Testasicca, Mineo and Afonso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luís Carlos Crocco Afonso, YWZvbnNvQG51cGViLnVmb3AuYnI=