 Carina Gröschel1,2†
Carina Gröschel1,2† Daniela Hübscher2,3†Jessica Nolte4
Daniela Hübscher2,3†Jessica Nolte4 Sebastian Monecke1,2André Sasse1Leslie Elsner1
Sebastian Monecke1,2André Sasse1Leslie Elsner1 Walter Paulus5Claudia Trenkwalder6Bojan Polić7Ahmed Mansouri5,8
Walter Paulus5Claudia Trenkwalder6Bojan Polić7Ahmed Mansouri5,8 Kaomei Guan3,9‡
Kaomei Guan3,9‡ Ralf Dressel1,2*‡
Ralf Dressel1,2*‡
- 1Institute of Cellular and Molecular Immunology, University Medical Center Göttingen, Göttingen, Germany
- 2DZHK (German Center for Cardiovascular Research), Göttingen, Germany
- 3Department of Cardiology and Pneumology, University Medical Center Göttingen, Göttingen, Germany
- 4Institute of Human Genetics, University Medical Center Göttingen, Göttingen, Germany
- 5Department of Clinical Neurophysiology, University Medical Center Göttingen, Göttingen, Germany
- 6Department of Neurosurgery, University of Göttingen, Göttingen, Germany
- 7Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, Rijeka, Croatia
- 8Department of Molecular Cell Biology, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany
- 9Institute of Pharmacology and Toxicology, Technische Universität Dresden, Dresden, Germany
Natural killer (NK) cells play an important role as cytotoxic effector cells, which scan the organism for infected or tumorigenic cells. Conflicting data have been published whether NK cells can also kill allogeneic or even autologous pluripotent stem cells (PSCs) and which receptors are involved. A clarification of this question is relevant since an activity of NK cells against PSCs could reduce the risk of teratoma growth after transplantation of PSC-derived grafts. Therefore, the hypothesis has been tested that the activity of NK cells against PSCs depends on cytokine activation and specifically on the activating NK receptor NKG2D. It is shown that a subcutaneous injection of autologous iPSCs failed to activate NK cells against these iPSCs and can give rise to teratomas. In agreement with this result, several PSC lines, including two iPSC, two embryonic stem cell (ESC), and two so-called multipotent adult germline stem cell (maGSC) lines, were largely resistant against resting NK cells although differences in killing were found at low level. All PSC lines were killed by interleukin (IL)-2-activated NK cells, and maGSCs were better killed than the other PSC types. The PSCs expressed ligands of the activating NK receptor NKG2D and NKG2D-deficient NK cells from Klrk1−/− mice were impaired in their cytotoxic activity against PSCs. The low-cytotoxic activity of resting NK cells was almost completely dependent on NKG2D. The cytotoxic activity of IL-2-activated NKG2D-deficient NK cells against PSCs was reduced, indicating that also other activating receptors on cytokine-activated NK cells must be engaged by ligands on PSCs. Thus, NKG2D is an important activating receptor involved in killing of murine PSCs. However, NK cells need to be activated by cytokines before they efficiently target PSCs and then also other NK receptors become relevant. These features of NK cells might be relevant for transplantation of PSC-derived grafts since NK cells have the capability to kill undifferentiated cells, which might be present in grafts in trace amounts.
Introduction
Pluripotent stem cells (PSCs) hold a great potential for the therapy of diseases, which are currently incurable, including heart failure, Parkinson’s disease, and macular degeneration (1), since cellular grafts or even complex tissues can be generated in vitro by directed differentiation of these cells (2). Clinical trials to evaluate the therapeutic potential of PSCs have been initiated (3, 4), and first encouraging results of studies using human embryonic stem cells (ESCs) (5) as source of grafts to treat macular degeneration (6–8) and heart failure (9) have been published.
Embryonic stem cell-derived grafts have to be transplanted in an allogeneic setting and, therefore, the rejection of ESC-derived allografts is a challenge for these new therapies. Although immunosuppressive therapy can efficiently prevent the rejection of allogenic organs, the persistent use of immunosuppressive drugs is associated with problems of toxicity and increased risks of infections and cancer. Therefore, other PSC types, which can be obtained from adult cells including so-called multipotent adult germline stem cells (maGSCs) (10) and induced pluripotent stem cells (iPSCs) (11–14) gained attention as potential source of autologous grafts. Autologous grafts should be transplantable without the need of immunosuppression, and recently, first results on the transplantation of retinal epithelial cells derived from autologous iPSCs have been reported (15). However, it is still debated whether grafts derived from iPSCs can be immunogenic in syngeneic or autologous recipients (16, 17). While terminally differentiated grafts appear to be tolerated as expected (18–20), it has been reported that therapeutically relevant in vitro-differentiated human iPSC-derived grafts were at risk to be rejected in mice “humanized” with an “autologous immune system” depending on the direction of differentiation (21).
A further concern associated with the transplantation of PSC-derived grafts is the risk of tumorigenicity (22, 23). Notably, the risk of tumorigenicity might be even higher for iPSCs than ESCs due to mutations preexisting in the reprogrammed somatic cells, introduced during the reprogramming process or resulting from an increased genomic instability of iPSCs (24–26). However, all types of PSCs can give rise to teratomas. In immunodeficient mice, as few as 2 murine or 245 human ESCs were reported to elicit teratoma growth (27, 28). ESCs can also form teratomas in immunocompetent syngeneic recipients (29), although higher cell numbers appear to be required than in immunodeficient recipients (22, 30, 31). The risk of teratoma growth upon transplantation of autologous iPSCs into immunocompetent recipients has not been assessed to our knowledge. However, in allogeneic immunocompetent hosts, PSCs are usually rejected and do not form teratomas (22, 31) although exceptions from this rule have been reported (32).
Natural killer (NK) cells have important effector functions such as cytotoxicity and cytokine production. The activity of NK cells is regulated by a plethora of germ-line encoded receptors that recognize ligands on target cells (33). NK receptors include activating and inhibitory members. Inhibitory receptors of the Ly49 family in mice or the killer cell immunoglobulin-like receptor (KIR) family in humans interact with classical major histocompatibility complex (MHC) class Ia molecules, which are constitutively expressed on most cells (34, 35). In contrast, many activating receptors, including NKG2D (36), recognize ligands, which are mostly absent from healthy cells but become induced upon stress such as virus infection or malignant transformation (37). Classically, NK cells have been described as cells that, unlike cytotoxic T cells, do not require activation to exert cytotoxicity. However, stimulation with interleukin 2 (IL-2), IL-12, IL-15, IL-18, or combinations of these cytokines can markedly increase the activity of NK cells (38). While some targets such as the classical human NK cell target cell line K562 are efficiently killed also by resting NK cells, killing of others requires cytokine-activated NK cells (39).
Conflicting results have been published regarding the susceptibility of ESCs toward NK cells. Some studies have reported resistance or very low susceptibility (40, 41) while others found ESCs to be vulnerable to NK cells (42, 43), and this might be due to differences in the activation status of the NK cells in these studies. We have shown previously that murine PSCs, including ESCs, iPSCs, and maGSCs, are targets for IL-2-activated NK cells in vitro (31, 44) and that NK cells can impair or even suppress the teratoma growth upon transplantation of the PSCs in vivo (22, 44, 45). Murine PSCs are targets for allogeneic and syngeneic NK cells because they do not express MHC class I antigens, which serve as ligands for inhibitory NK receptors, at least at a level detectable by flow cytometry (31, 44, 46), but they do express ligands for activating NK receptors, such as NKG2D and DNAM-1 (31, 42, 44). Similar expression patterns of ligands for activating NK receptors as on murine PSCs were found on human iPSCs, which were also targets of IL-2-activated allogeneic and autologous NK cells (39). Notably, human ESCs and iPSCs, in contrast to the respective murine cells, do express MHC class I molecules although in lower amounts than most differentiated cell types (39, 47). We have previously shown by inhibition experiments that killing of murine PSCs by IL-2-activated NK cells was partly dependent on NKG2D while killing of human iPSCs was more dependent on DNAM-1 (31, 39, 44).
Differentiation of PSCs usually increases the expression of MHC class I molecules (48, 49) and decreases the expression of NKG2D ligands (31, 44) and consequently in vitro-differentiated cells acquire resistance against NK cells (44). Thus, NK cells have the capability to target undifferentiated PSCs while ignoring differentiated cells. NK cells might, therefore, increase the safety of transplantations of PSC-derived grafts because they potentially increase the number of undifferentiated cells that can be tolerated in a graft. However, it needs to be clarified whether NK cells need activation by cytokines to target PSCs and which receptor–ligand interactions between NK cells and PSCs are important.
In this study, we set out to demonstrate that murine iPSCs can indeed form teratomas upon truly autologous transplantation and we tested whether NK cells become activated in vivo upon transplantation. Moreover, to resolve previous conflicting results, we directly compared the cytotoxic activity of resting and IL-2-activated NK cells against PSCs, including ESCs, iPSCs, and maGSCs and analyzed the role of the activating NK receptor NKG2D by comparing wild-type and NKG2D-deficient NK cells.
Materials and Methods
Mouse Strains
C57BL/6, 129Sv, and NKG2D-deficient Klrk1−/− mice (50) as well as immunodeficient Rag2−/− and SCID/beige mice (C.B-17/IcrHsd-scid-bg) were bred in the central facility for animal experimentation at the University Medical Center Göttingen under specific pathogen-free conditions in individually ventilated cages and in a 12 h light–dark cycle. The Klrk1−/− and Rag2−/− mice were on a C57BL/6 background. All animal experiments had been approved by the local government (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit) and were carried out in compliance with German and EU legislation (Directive 2010/63/EU).
Stem Cell Lines and Cell Culture
Generation of New iPSC Lines
Tail biopsies were taken aseptically, cut into pieces, and incubated in dispase solution (2 mg/ml, Thermo Fisher Scientific, Waltham, MA, USA) for 20 min at 37°C, washed with phosphate-buffered saline (PBS), and incubated again for 20 min at 37°C in 0.05% trypsin with 0.005% EDTA. After being washed, the isolated cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 plus GlutaMAX™ (Thermo Fisher Scientific) supplemented with 10% fetal calf serum (FCS; selected batch, Lonza, Basel, Switzerland), 1× N2 supplement (Thermo Fisher Scientific), 1× non-essential amino acids (NEAA; Thermo Fisher Scientific), 50 µM β-mercaptoethanol (β-ME; Promega, Mannheim, Germany), 0.3 µg/ml hydrocortisone (Sigma-Aldrich, Munich, Germany), 10 ng/ml human epidermal growth factor (Peprotech, Rocky Hill, NJ, USA), 10 ng/ml human basic fibroblast growth factor (hbFGF, Peprotech), and 100 U/ml penicillin plus 100 µg/ml streptomycin (Thermo Fisher Scientific). Outgrowing fibroblasts were transduced with the STEMCCA virus (pHAGE2-EF1αFull-hOct4-F2A-hKlf4-IHRES-hSox2-P2A-hc-Myc-W-loxP), which was kindly provided by Dr. Darrell Kotton, University of Boston. Cells transduced by the STEMCCA virus express the human pluripotency genes OCT4, KLF4, SOX2, and cMYC. Fibroblasts were transduced at a multiplicity of infection of 1 in 24-well-plates with 700 µl FIII medium [minimum essential medium α (MEMα; Thermo Fisher Scientific) with 4.5% FCS, 4.5% knockout serum replacement (Thermo Fisher Scientific), 50 µM β-ME, 0.35% d-(+)-glucose (Sigma-Aldrich), and 1,000 U/ml leukemia inhibitory factor (LIF; Merck Millipore, Darmstadt, Germany)] and 5 µg/ml polybrene (hexadimethrine bromide, Sigma-Aldrich). After culture for 24 h at 37°C, the virus was washed away and the cells were cultured for 5–7 days. Then, the cells were seeded on mitomycin C-inactivated mouse embryonic feeder cells in DMEM (Thermo Fisher Scientific) supplemented with 15% FCS, 2 mM glutamine (Thermo Fisher Scientific), 1× NEAA, 50 µM β-ME, and 1,000 U/ml LIF as described previously (10). Single colonies of reprogrammed cells were picked to obtain stable cell lines for further analysis.
Culture of Stem Cell Lines
The ESC line MPI-II derived from a 129Sv mouse at the Max Planck Institute for Biophysical Chemistry (Göttingen, Germany) has been extensively characterized previously including the demonstration of pluripotency by teratoma assays (31). After transfer of breeders of that colony to the University Medical Center Göttingen, a new ESC line (ESC BTL1) has been generated from a 129Sv embryo as described (51). In parallel, two iPSC lines were generated as described above, one from the same 129Sv colony (iPSC129Sv) and one from a C57BL/6J mouse (Janvier Labs, Le Genest Saint Isle, France). The maGSC lines, maGSC 129Sv, and maGSC C57BL/6 have been described earlier (10) and their pluripotency has also been demonstrated in teratoma assays (44). The well-characterized ESC R1 cell line (52) has been used as positive control for expression of pluripotency genes in quantitative PCR (qPCR) experiments.
Before the PSC lines were used as target cells for NK cells, they were cultured on Geltrex-coated cell culture plates (333 µg/T25 flask), prepared according to manufacturer’s instructions (Thermo Fisher Scientific) in DMEM supplemented with 10% FCS, 1× NEAA, 50 µM β-ME, and 1,000 U/ml LIF. For harvesting at 80% confluence, the cells were incubated in stem cell trypsin (0.25% trypsin, 0.02% EDTA, 0.1% glucose, 0.3% Tris–HCl in PBS) for several minutes at 37°C until dissociation of stem cell colonies occurred. A single cell suspension was prepared by diluting cells in 10 volumes of stem cell medium. Stem cells were tested by PCR assays prior to experiments to exclude fungal, bacterial, and mycoplasmal infections.
Culture of YAC-1 Cells
The murine T-lymphoma cell line YAC-1 (H2a), which was used as positive control for the cytotoxic activity of NK cells, was maintained in DMEM supplemented with 10% FCS, 2 mM glutamine, 1 mM sodium pyruvate, 50 µM β-ME, 100 U/ml penicillin, and 100 µg/ml streptomycin.
Pluripotency Assays
Alkaline Phosphatase Staining and Immunofluorescence Staining of Pluripotency Marker Proteins
The alkaline phosphatase activity of PSCs was detected using a kit according to the manufacturer’s instructions (Sigma-Aldrich). Expression of marker proteins of PSCs (LIN28, NANOG, and SSEA1) was analyzed by immunofluorescence staining. The stem cells were grown on cover-slips coated by 0.1% gelatin. The cells were fixed with 4% paraformaldehyde in PBS before blocking in 1% bovine serum albumin solution. For detection of NANOG, the cells were permeabilized (0.1% Triton X-100, Sigma-Aldrich) for 10 min before incubation with the primary antibody (Ab). The primary Abs (Table S1 in Supplementary Material) were added in 100 µl PBS to the cover-slips and incubated for 1 h at 37°C in a humidified chamber. Afterward, the cover-slips were washed three times with PBS before the secondary Ab was added. After incubation for 1 h at 37°C, the cover-slips were washed again before nuclei were stained with 4′,6-diamidino-2-phenylindol (DAPI, 0.4 µg/ml, Sigma-Aldrich) for 10 min. The samples were mounted in Vectashield mounting medium (Vector Laboratories, Cambridgeshire, UK) and analyzed with the fluorescence microscope Axio Observer Z1 (Zeiss, Jena, Germany) and Axio Vision 4.6 software.
Immunoblotting for Detection of Pluripotency Marker Proteins
In ESC BTL1 cells, the pluripotency marker proteins OCT4, SALL4, SOX2, KLF4, and ZPF206 were detected by immunoblotting and α-tubulin served as loading control. The antibodies used are given in Table S1 in Supplementary Material, and the immunoblotting was done as described previously (53).
In Vitro Differentiation of iPSCs
For in vitro differentiation of iPSCs, the hanging drop method was applied. Hanging drops containing 300 cells in 20 µl Iscove’s modified Dulbecco’s medium plus GlutaMAX™ (Thermo Fisher Scientific) supplemented with 20% FCS, 1× NEAA, 450 µM α-Monothioglycerol (Sigma-Aldrich) were cultured for 2 days. In this time, embryoid bodies had formed and were subsequently transferred into Petri dishes for a 3-day suspension culture period before transfer to gelatinized (0.1%) cell culture dishes for further 5, 15, or 25 days. Medium was exchanged every second or third day. The cells were harvested at the indicated time points, and expression of genes that indicate differentiation into the three germ layers was analyzed.
Reverse Transcription Polymerase Chain Reaction (RT-PCR) and qPCR for Expression Analysis of Pluripotency and Differentiation Genes
Total RNA was extracted from PSCs or in vitro-differentiated cells, treated with DNase I to avoid contamination with genomic DNA, and used for cDNA synthesis as described previously (46). The analyzed genes and the primer pairs used are given in Table S2 in Supplementary Material. The iPSC lines and their differentiation products were analyzed by qualitative RT-PCR as described previously (10, 54). The expression of pluripotency genes in ESC BTL1 cells was determined by qPCR as described before (53).
Teratoma Assays
The PSCs were injected in 100 µl PBS subcutaneously into the flank of the mice. Tumor growth was monitored by palpation and size was recorded using linear calipers. Animals were sacrificed after 3 months or when a tumor volume of 1 cm3 was reached. The tumor volume was calculated by the formula V = πabc/2, where a, b, and c are the orthogonal diameters. Autopsies of all animals were performed and tumor tissues were immediately frozen in liquid nitrogen or directly placed in phosphate-buffered formalin (30 mM NaH2PO4, 40 mM Na2HPO4, 4% formalin) for 16 h before being embedded in paraffin. Tissue sections (2.5 µm) were stained with hematoxylin and eosin for histological examination.
NK Cells and Cytotoxicity Assay
Natural killer cells were isolated from spleens of C57BL/6 wild-type and NKG2D-deficient Klrk1−/− mice by magnetic-activated cell sorting using a kit for negative selection (mouse NK Cell Isolation Kit II; Miltenyi Biotec; Bergisch-Gladbach, Germany) according to the manufacturer’s protocol. The proportion of NK cells in the spleens and the purity of the isolated CD49b+CD3− NK cells were always determined by flow cytometry and were similar in wild-type and NKG2D-deficient mice (Figure S1 in Supplementary Material). In parallel, we always confirmed the expression of NKG2D on CD49b+ wild-type NK cells and the NKG2D-deficiency on Klrk1−/− CD49b+ NK cells (data not shown). The purified NK cells were either directly used as cytotoxic effector cells (day 0) or after stimulation for 4 days with 10 ng/ml mouse IL-2 (Immunotools, Friesoythe, Germany). The cytotoxic activity of the NK cells against the PSCs and YAC-1 control cells was measured in 51Cr-release assays as described previously (45). The 51Cr-labeled target cells were exposed to the NK cells in triplicates at several effector to target (E:T) ratios for 4 h. The E:T ratios always indicate the ratio of CD49b+CD3− effector cells to target cells. Spontaneous release of 51Cr was determined by incubation of target cells in the absence of effector cells and specific lysis was calculated by subtracting the spontaneous 51Cr-release.
Flow Cytometry
Flow cytometry was performed on a FACS Calibur flow cytometer (BD Biosciences, Heidelberg, Germany) using CellQuestPro data acquisition and analysis software. The Abs used for flow cytometry are described in Table S3 in Supplementary Material. Isotype controls were used for directly labeled monoclonal antibodies (mAbs). For staining, 5 × 105 cells were incubated in 100 µl PBS with 1 µg of the respective primary mAb for 30 min at 4°C before washing with PBS. To detect the unlabeled mAbs, the cells were incubated subsequently in 100 µl PBS with 1 µl of a fluorescein isothiocyanate-labeled goat anti-rat IgG Ab (for anti-RAE-1, anti-MULT-1, and anti-H60). In these experiments, cells stained with the secondary reagent only served as control. The percentage of positive cells expressing the analyzed molecule and their mean fluorescence intensity (MFI) was determined by subtracting the values of the appropriate controls.
Statistics
Results are shown as mean with SD or as mean with SEM for cytotoxicity assays. The data were evaluated with the SPSS software (IBM, Armonk, NY, USA). Analyses of variance (ANOVA) adjusted for effector to target (E:T) ratios were used to evaluate the cytotoxicity data and Bonferroni tests were selected as post hoc tests. A P-value of ≤ 0.05 in two-sided tests was considered significant.
Results
No NK Cell Activation against Autologous iPSCs In Vivo
We generated fibroblasts from the tail tips of five C57BL/6J mice at the age of 11 days. They were transduced with the STEMCCA virus for reprogramming into iPSCs. Colonies of cells with PSC morphology, which were positive for alkaline phosphatase, were obtained from all transductions as exemplified in Figure 1A. Single colonies were picked and iPSC lines were established from the five mice. These lines expressed the endogenous pluripotency genes Oct4, Sox2, Nanog, and Lin28 (Figure S2A in Supplementary Material). They were positive for the pluripotency markers SSEA1, LIN28, and NANOG at the protein level as shown by immunofluorescence staining (Figure 1B). After in vitro differentiation via the hanging drop method, marker genes for differentiated cells were expressed, including Afp for the endodermal lineage, Mash1 for the ectodermal linage, and Flk1 for the mesodermal lineage. Moreover, Myh6 expression was detected, which is expressed in cardiomyocytes (Figure S2B in Supplementary Material).
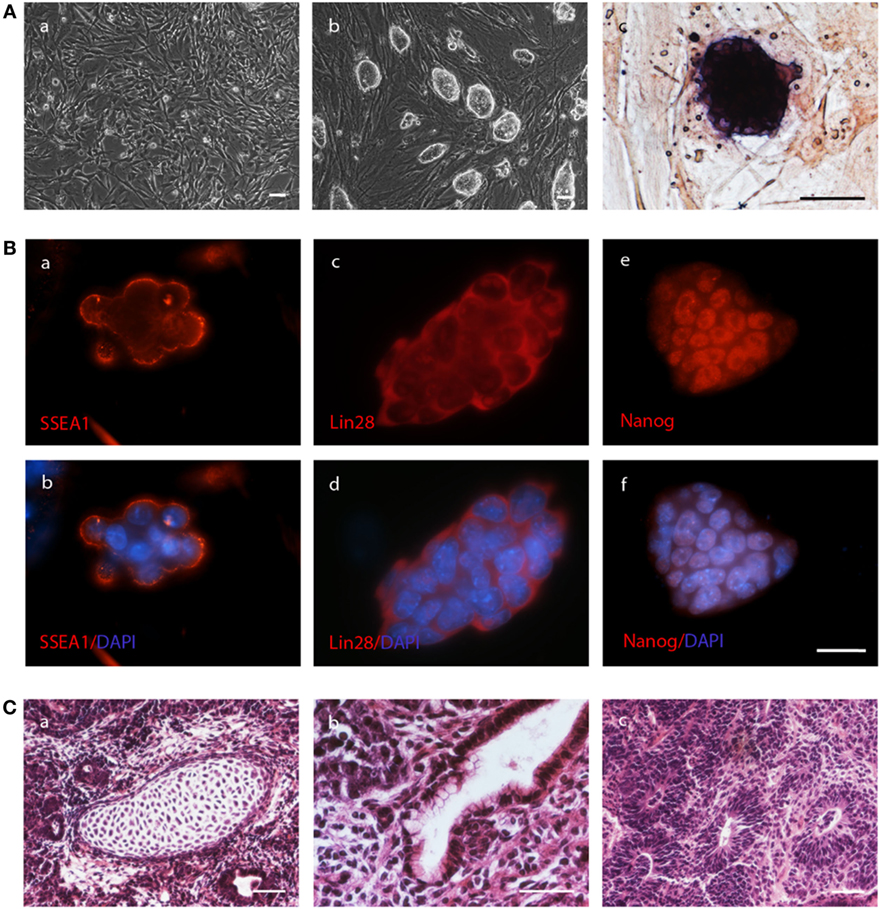
Figure 1. Autologous induced pluripotent stem cell (iPSC) lines are pluripotent and can give rise to teratomas. (A) Fibroblasts (a) isolated from tail tip biopsies of C57BL/6J mice gave rise to colonies of iPSCs (b) upon transduction with the reprogramming vector. The colonies were positive for alkaline phosphatase (c). The scale bars indicate 50 µm. (B) Colonies of the iPSCs also expressed the pluripotency marker proteins SSEA1 (a, b), LIN28 (c, d), and NANOG (e, f) as demonstrated by immunofluorescence staining alone (a, c, e) or in combination with DAPI to counterstain nuclei (b, d, f). The scale bar indicates 25 µm. (C) Cells of the iPSC line 1–2 were subcutaneously injected into the mouse that donated the fibroblast and a teratoma was obtained after 35 days. The mesodermal differentiation into cartilage (a), endodermal differentiation into intestinal epithelium (b), and ectodermal differentiation into neural rosettes (c) is shown. The scale bars indicate 100 µm.
Four iPSC lines from four different mice formed teratomas in immunodeficient recipients indicating successful reprogramming of the fibroblasts into iPSCs (Figure S2C in Supplementary Material). These features are summarized in Table 1. The iPSCs were also injected subcutaneously into the respective donors of the fibroblasts. In three of the five mice, the autologous iPSCs formed teratomas (Figure 1C). The iPSC lines were also injected in syngeneic (not autologous) C57BL/6J recipients (H2b) as well as into MHC-matched but otherwise allogeneic 129Sv mice (H2b). Teratomas were observed in about 40% of the syngeneic but none of the allogeneic recipients (Table 1).

Table 1. Features of induced pluripotent stem cell (iPSC) lines generated for testing teratoma growth in autologous recipients.
From three of these mice injected with autologous iPSCs [two without (8–7 and 9–2) and one with a teratoma (0–3)], we obtained NK cells at the end of the experiment and tested their activity against the autologous iPSCs and the control cell line YAC-1 directly ex vivo without any further in vitro stimulation. The NK cells of all mice killed YAC-1 targets but failed to lyse the respective iPSC lines (Figures 2A–C) despite expressing ligands of activating NK receptors, including the NKG2D ligand RAE-1 and the DNAM-1 ligand CD155 and despite being largely negative for MHC class I molecules, which serve as ligands for inhibitory NK receptors of the Ly49 family (Figures 2D,E).
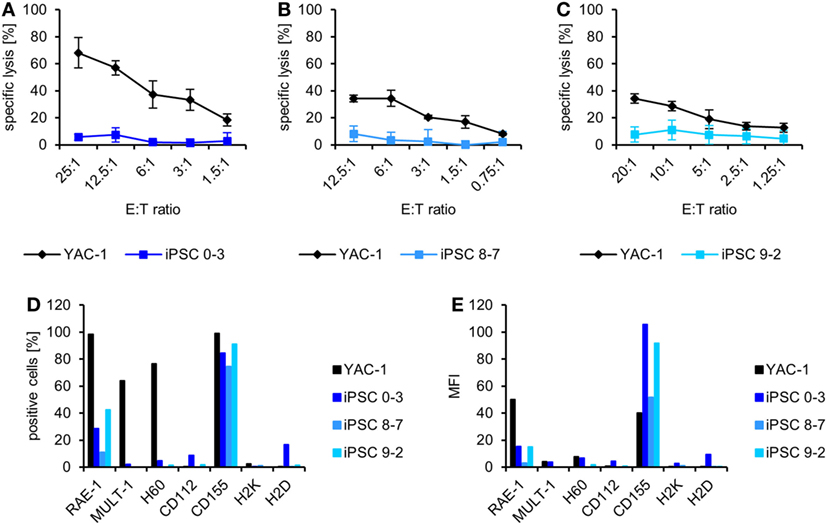
Figure 2. Natural killer (NK) cells of mice injected with autologous induced pluripotent stem cell (iPSCs) do not kill the respective iPSC line ex vivo. (A) Means and SD of triplets are displayed of the specific lysis of YAC-1 and iPSC 0–3 cells by NK cells derived from the donor mouse of this iPSC line, which had been injected with these cells and developed a teratoma. (B) Means and SD of triplets are shown of the specific lysis of YAC-1 and iPSC 8–7 cells by NK cells derived from the donor mouse of this iPSC line, which had been injected with these cells and developed no teratoma. (C) Means and SD of triplets are displayed of the specific lysis of YAC-1 and iPSC 9–2 cells by NK cells derived from the donor mouse of this iPSC line, which had been injected with these cells but developed no teratoma. (D) In parallel to the 51Cr-release assays, shown in panels (A–C), the expression of ligands of activating and inhibitory NK receptors was determined by flow cytometry and the percentage of cells expressing the indicated ligands is shown. (E) The expression intensity of the ligands is displayed as mean fluorescence intensity.
Efficient Killing of PSCs Requires Cytokine-Activated NK Cells
We have previously shown that IL-2-activated NK cells efficiently kill PSCs, including iPSCs, ESCs, and maGSCs (44). However, the NK cells from the C57BL/6 mice, which had been injected with autologous iPSCs in this study, failed to kill the iPSCs, irrespective of whether they developed a teratoma or not. Therefore, we next compared the efficacy of resting and IL-2-activated NK cells against several PSC lines. These included an iPSC line from a C57BL/6 mouse and an iPSC line from a 129Sv mouse, which were generated similarly by transduction of fibroblasts with the STEMCCA virus. We also included a newly established ESC line from a 129Sv embryo (ESC BTL1) (Figure S3 in Supplementary Material) and the long-established ESC line MPI-II also derived from a 129Sv embryo (31). Two maGSC lines, one from a C57BL/6 and one from a 129Sv mouse, which have been previously described (10, 44), were also included in the experiments. The pluripotency of the new stem cell lines iPSC 129Sv, iPSC C57BL/6, and ESC BTL1 cells was demonstrated by their ability to differentiate in vivo into derivatives of the three germ layers in teratoma assays (Figure S4 in Supplementary Material).
We purified NK cells from C57BL/6 mice and either stimulated them for 4 days in vitro with IL-2 or used them directly at day 0 as effector cells in 51Cr-release assays against the PSCs and YAC-1 targets. Resting NK cells (day 0) killed the target cells with different efficacy (P = 0.0001, ANOVA) (Figure 3A). The YAC-1 control target cells were more susceptible to resting NK cells than iPSCs (P = 9.47 × 10−6) and maGSCs (P = 0.0096) and there was a similar trend for ESCs (P = 0.0766, Bonferroni post hoc test). In a comparison of the PSC types only, a trend for variation in their susceptibility to resting NK cells was found (P = 0.0575). Similarly, the four target types varied in killing by IL-2-activated NK cells at day 4 (P = 0.0003) (Figure 3B). The YAC-1 cells were less susceptible to activated NK cells than maGSCs (P = 0.0010, Bonferroni post hoc test). The susceptibility of the PSC types only also varied (P = 0.0007), and the post hoc test indicated a higher killing of maGSCs than iPSCs (P = 0.0006) and ESCs (P = 0.0389). Thus, maGSCs were more susceptible to resting and activated NK cells than the other PSC types.
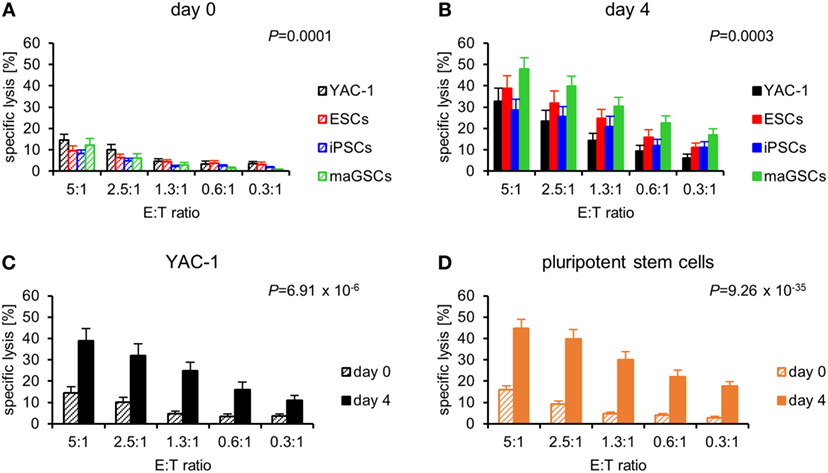
Figure 3. Pluripotent stem cells (PSCs) are more susceptible to interleukin (IL)-2-activated natural killer (NK) cells than resting NK cells. (A) A summary of means and SEM of specific lysis of YAC-1 cells (n = 16), embryonic stem cells (ESCs) (n = 14), induced pluripotent stem cell (iPSCs) (n = 18), and multipotent adult germline stem cell (maGSCs) (n = 16) by freshly purified NK cells (day 0) is shown as determined by 51Cr-release assays. (B) A summary of means and SEM of specific lysis of YAC-1 cells (n = 12), ESCs (n = 12), iPSCs (n = 12), and maGSCs (n = 12) by IL-2-activated NK cells (day 4) is shown as determined by 51Cr-release assays. The cytotoxic activity of resting (day 0) and IL-2-activated NK cells (day 4) has been compared for (C) YAC-1 cells and (D) all PSCs together. P-values for the comparisons (2-way-analyses of variance adjusted for E:T ratios) are indicated.
The comparison of the susceptibility of the targets to resting and cytokine-activated NK cells indicated that the YAC-1 cells were more susceptible to IL-2-activated NK cells (P = 6.91 × 10−6) (Figure 3C), similarly also the PSCs were overall significantly better lysed by IL-2-activated than by resting NK cells (P = 9.26 × 10−35) (Figure 3D). This difference was confirmed for all three types of PSCs in separate analyses (Figure S5 in Supplementary Material).
The Cytotoxic Activity of Resting NK Cells against PSCs Depends on the Activating NK Receptor NKG2D
In inhibition experiments with recombinant NKG2D-Fc proteins, we have found previously that the killing of PSCs by IL-2-activated NK cells partly depends on the activating NK receptor NKG2D (31, 44). To verify this result and to compare resting and IL-2-activated NK cells, we used NKG2D-deficient mice (Klrk1−/−) as NK cell donors in addition to the wild-type mice. Resting NKG2D-deficient NK cells were significantly impaired in killing of YAC-1 target cells (Figure 4A). The PSC lines were hardly killed and overall more resistant to NKG2D-deficient than to wild-type NK cells (P = 4.47 × 10−19) (Figure 4B). The separate analysis of the three PSC types revealed that all types were significantly more resistant to NKG2D-deficient than to wild-type NK cells (Figures 4C–E).
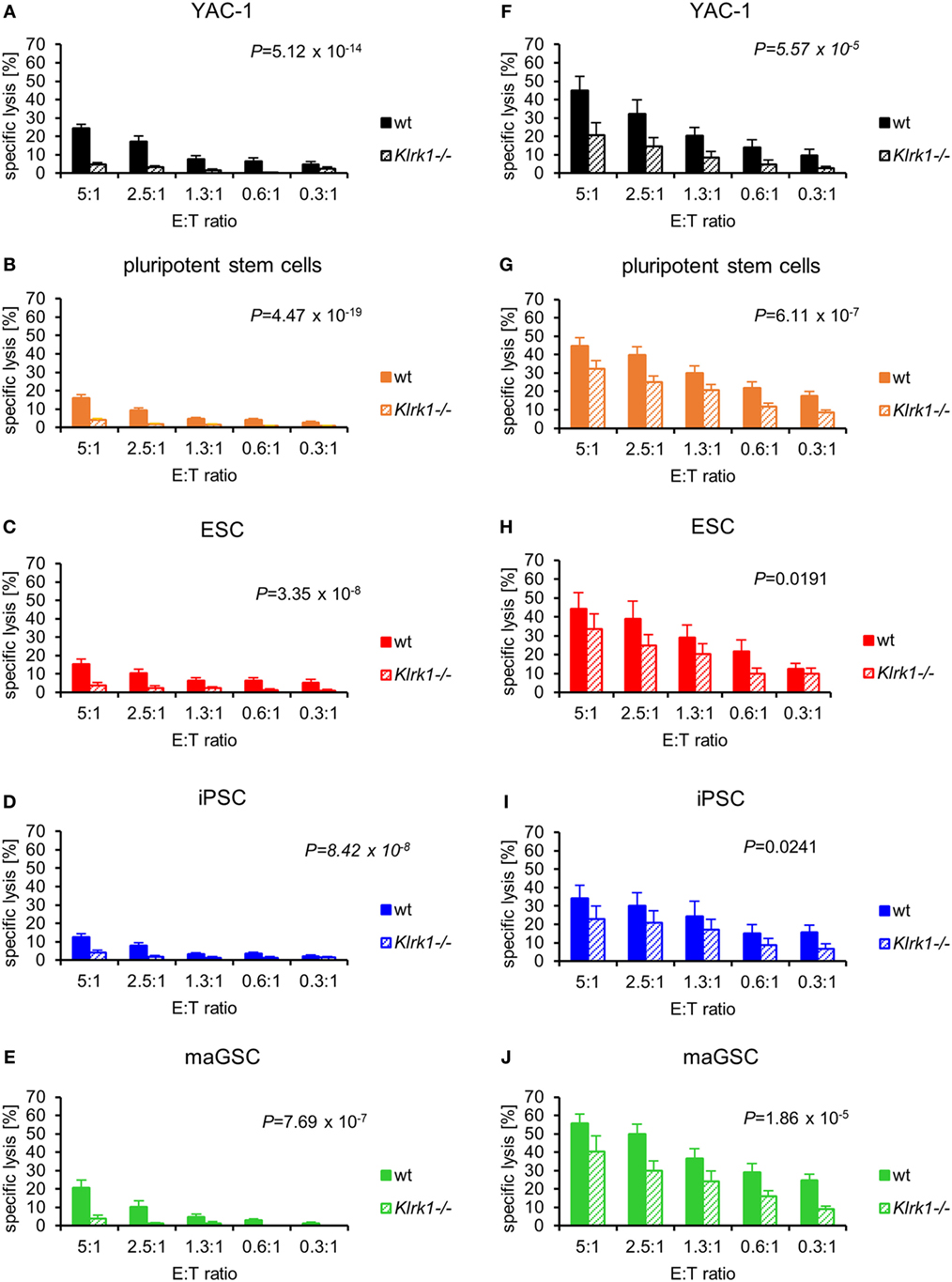
Figure 4. Pluripotent stem cells (PSCs) are largely resistant against resting NKG2D-deficient natural killer (NK) cells, whereas the cytotoxic activity of interleukin-2-activated NK cells depends only partly on NKG2D. The cytotoxic activity of resting wild-type (wt) and NKG2D-deficient (Klrk1−/−) NK cells has been compared for (A) YAC-1 cells (wt: n = 8, Klrk1−/−: n = 8), (B) all PSCs together (wt: n = 24, Klrk1−/−: n = 24), (C) embryonic stem cells (ESCs) (wt: n = 7, Klrk1−/−: n = 7), (D) induced pluripotent stem cells (iPSCs) (wt: n = 9, Klrk1−/−: n = 9), and (E) multipotent adult germline stem cell (maGSCs) (wt: n = 8, Klrk1−/−: n = 8). The cytotoxic activity of IL-2-activated wt and Klrk1−/− NK cells has been compared for (F) YAC-1 cells (wt: n = 6, Klrk1−/−: n = 6), (G) all PSCs together (wt: n = 18, Klrk1−/−: n = 18), (H) ESCs (wt: n = 6, Klrk1−/−: n = 6), (I) iPSCs (wt: n = 6, Klrk1−/−: n = 6), and (J) maGSCs (wt: n = 6, Klrk1−/−: n = 6). P-values for the comparisons (2-way-analyses of variance adjusted for E:T ratios) are indicated.
The Cytotoxic Activity of Cytokine-Activated NK Cells against PSCs Depends Partly on NKG2D
The cytotoxicity of IL-2-activated NK cells against YAC-1 cells was largely dependent on NKG2D (P = 5.57 × 10−5) (Figure 4F). Overall, also the lysis of PSCs by NKG2D-deficient NK cells was reduced (P = 6.11 × 10−7) (Figure 4G). Although the IL-2-activated NK cells were less dependent on NKG2D than resting NK cells, the killing of ESCs, iPSCs, and maGSCs by NKG2D-deficient NK cells was lower than the killing by wild-type IL-2-activated NK cells (Figures 4H–J).
Individual PSC Lines Vary in Their Susceptibility to NK Cells Independent of the Stem Cell Type
Separate analyses of the single PSC lines for their susceptibility to resting and IL-2-activated wild-type and NKG2D-deficient NK cells are shown in the Figure S6 in Supplementary Material. The six PSC lines included in the analysis varied in their susceptibility to IL-2-activated NK cells (P = 0.0010, ANOVA), but to resting NK cells only with borderline significance (P = 0.0634, ANOVA). The maGSC 129Sv cells were killed better by IL-2-activated NK cells than ESC MPI-II (P = 0.0078) and iPSC 129Sv cells (P = 0.0092, Bonferroni post hoc test). All cell lines were more susceptible to IL-2-activated NK cells than resting NK cells and the cytotoxic activity of resting NK cells toward all PSC lines was NKG2D-dependent. However, only for ESC BTL1, maGSC 129Sv, and maGSC C57BL/6 cells the killing by IL-2-activated NK cells was significantly impaired by NKG2D deficiency (Figure S6 in Supplementary Material).
The PSC Lines Express NKG2D Ligands
The PSC lines expressed the NKG2D ligands RAE-1, MULT-1, and H60 in variable amounts as determined by flow cytometry in parallel to the cytotoxicity assays (Figure 5). On average more than 50% of the ESC BTL1, ESC MPI-II, and maGSC 129Sv cells expressed RAE-1 (Figure 5A) and the MFI of RAE-1 (21.6, 36.6, and 36.6) was higher than for some other PSC lines, such as iPSC 129Sv (4.9) (Figure 5B). Taking into account the proportion of positive cells and the MFI, these data are summarized in Table 2. The expression of MHC class I molecules (H2Db and H2Kb) has also been determined and the newly established PSC lines were negative similarly as the old lines (Table 2), which had been tested also in previous studies (31, 44, 46). This expression pattern fits well to the observation that the killing of these cell lines by IL-2-activated NK cells was in part dependent on NKG2D. However, the lowest expression of NKG2D ligands was observed on the maGSC C57BL/6 cells but their killing by IL-2-activated NKG2D-deficient NK cells was also clearly reduced. Thus, further factors appear to contribute to these variations and lack of inhibition by inhibitory NK receptors, which bind to MHC class I molecules is presumably one of them.

Figure 5. Analysis of the expression of NKG2D ligands on target cells. (A) The percentage of cells expressing the indicated NKG2D receptor ligands (mean plus SEM) is shown as determined by flow cytometry in 3–13 individual experiments [YAC-1: n = 13, embryonic stem cell (ESC) BTL1: n = 3, ESC MPI-II: n = 7, induced pluripotent stem cell (iPSC) 129Sv: n = 5, iPSC C57BL/6: n = 7, multipotent adult germline stem cell (maGSC) 129Sv: n = 6, maGSC C57BL/6: n = 7]. (B) The mean fluorescence intensity has been determined in parallel and is displayed plus SEM.

Table 2. Summary of the expression of NKG2D ligands and major histocompatibility complex class I molecules on pluripotent stem cells and YAC-1 cells.
Discussion
It is well-known that ESCs can form teratomas not only in immunodeficient but also in immunocompetent syngeneic hosts (29, 31). Therefore, it is reasonable to expect that iPSCs will form teratomas upon transplantation into autologous recipients, although this has previously not been demonstrated to our knowledge. In this study, we have shown that murine iPSCs derived from fibroblasts indeed can form teratomas in the individual mice from which the fibroblasts have been taken. Notably, teratomas are regularly formed in syngeneic immunocompetent recipients although syngeneic NK cells can attack PSCs (31, 44). When we compared the teratoma growth upon subcutaneous injections of ESC MPI-II cells into immunocompetent syngeneic 129Sv mice and immunodeficient SCID mice, which have no B and T cells, and SCID/beige mice, which in addition lack also functional NK cells, we found that the teratoma growth was similarly impaired in 129Sv and SCID mice compared to SCID/beige mice (22). Moreover, depletion of NK cells in SCID mice supported the early outgrowth of teratomas after injection of PSCs (44). These results demonstrated that NK cells indeed impair teratoma growth but are not sufficient to completely prevent tumors if high enough numbers of PSCs are injected. Activation of NK cells in vivo by poly I:C further reduced the teratoma frequency and size in SCID but not in SCID/beige recipients (44), suggesting that the activation status of NK cells is important for the outcome. Importantly, in vitro-differentiated cells have been shown to acquire resistance to NK cells (44), and therefore, PSCs are expected to escape finally the NK cell-mediated rejection by differentiation into other cell types which then form the teratoma.
The autologous transplantations of iPSCs performed in this study did not lead to a significant cytotoxic activity of NK cells against the autologous iPSCs. This was observed similarly in one mouse that developed a teratoma and in two recipients without a teratoma. We have recently shown that NK cells impair the outgrowth of teratomas from maGSCs not only in the subcutaneous tissue but also after transplantation into a therapeutically relevant tissue such as the myocardium (45). In these experiments, it turned out that the operation procedure, which included an open chests surgery, was substantially increasing the cytotoxic activity of NK cells up to 4 weeks after transplantation (45). The subcutaneous injections of the autologous iPSCs performed in the experiments reported here, produced presumably less inflammatory responses than a major surgery and consequently did not activate NK cells against the iPSCs at least at the time point when the mice were sacrificed. However, individual differences might exist since NK cells from mouse 0–3 showed a higher cytotoxic activity against YAC-1 control target cells than the NK cells of the other two recipients of autologous iPSCs (8–7, 9–2). NK cells of these two mice killed YAC-1 cells to a similar extent as NK cells from untreated control C57BL/6 mice that we have analyzed previously (45).
Taking the importance of the activation status of NK cells into account, we now directly compared the cytotoxic activity of resting and IL-2-activated NK cells against PSCs. As in our previous studies (31, 44), all PSC lines, including ESCs, iPSCs, and maGSCs, were targets for IL-2-activated NK cells. However, they were much less attacked by resting than activated NK cells. The maGSCs appeared to be killed even better than ESCs and iPSCs by activated NK cells but differences of individual PSC lines might be more important for this finding than principal differences between the PSC types. The finding that murine PSCs are good targets for cytokine-activated NK cells but are killed less by resting NK cells likely explains why some early studies reported resistance of ESCs to NK cells (40, 41) while we and others found them to be susceptible to cytokine-activated NK cells (31, 42, 44). The result is also in agreement with our findings on human iPSCs, which were killed by allogeneic and syngeneic IL-2-activated NK cells but much less by resting NK cells (39), and with results reported by others for human cardiac-derived stem/progenitor cells (55).
Murine PSCs do not express MHC class I molecules at least at a level that is readily detectable by flow cytometry (31, 44, 46). MHC class I molecules function as ligands mainly for inhibitory NK receptors of the Ly49 family in mice (35). Cells that lack MHC class I molecules cannot inhibit NK cell activity and can, therefore, become targets of NK cells (56). Importantly, the activity of NK cells is regulated by the balance of inhibitory and activating signals received via inhibitory and activating NK receptors (37). One of the most important activating NK receptor is NKG2D that interacts with a group of stress-inducible ligands including the RAE-1 and H60 families and MULT-1 in the mouse (36, 57). We and others have shown that murine ESCs are among the few cell types that constitutively express NKG2D ligands (31, 42). In addition to ESCs, also iPSCs and maGSCs express variable patterns of NKG2D ligands mainly of the RAE-1 family and ligands of further activating NK receptors including the DNAM-1 ligands CD112 and CD155 (44). The system of NKG2D and NKG2D ligands has been shown to be highly important for tumor immunity (58). It also affects the outcome of hematopoietic stem cell transplantation (59–62) and solid organ transplantation (63, 64). NKG2D has also been reported to be involved in the rejection of neural progenitor cells by NK cells (65). In inhibition experiments with a recombinant NKG2D protein, we have previously shown that the killing of murine ESCs (31) and other PSCs (44) by IL-2-activated NK cells is partly dependent on NKG2D. Here, we have confirmed this finding by comparing the activity of wild-type and genetically NKG2D-deficient NK cells. Notably, the killing of some individual PSC lines appeared to depend little on NKG2D while killing of others was clearly affected by NKG2D-deficiency. Thus, the only partial inhibition of killing by recombinant NKG2D, which we observed in previous experiments, was not due to an incomplete inhibition of NKG2D activity and other activating NK receptors than NKG2D must contribute to the recognition of PSCs by activated NK cells. DNAM-1 is an obvious candidate among others because especially the ligand CD155 is prominently expressed on PSCs including the autologous iPSCs used in this study. Moreover, we have recently shown that the killing of human iPSCs by NK cells is mediated in part by DNAM-1 (39). The killing of human iPSCs was not dependent on NKG2D with exception of one combination of a specific iPSC line and an individual NK cell donor (39). Thus, individual variations in NK receptor–ligand interaction need to be considered for their implications on human transplantations.
This potential individual variation in recognitions of PSCs by NK cells is further emphasized by results obtained with human ESCs that have been engineered not to express β2-microglobulin (66). In contrast to murine PSCs, human ESCs as well as iPSCs do express significant amounts of MHC class I molecules (39, 47). The β2-microglobulin and, therefore, also MHC class I-deficient human ESCs were killed more efficiently by NK cells than wild-type ESCs (66). This demonstrates the relevance of NK cell inhibition via human MHC class I molecules recognized by inhibitory KIRs on human NK cells. Recognition of human MHC class I molecules by KIRs shows a considerable variability (34) and the activity of NK cells toward allogeneic PSCs and their differentiation products can, therefore, be expected to vary depending on the MHC polymorphism of the PSCs and the KIR polymorphism of the recipients.
Notably, the low cytotoxic activity of resting NK cells against PSCs, which we observed, was almost completely depend on NKG2D in all investigated PSC lines. Therefore, any in vivo activity of NK cells against PSCs under non-inflammatory conditions must be expected to depend largely on the recognition of NKG2D ligands by NKG2D at least in murine transplantation models. Subcutaneous injections of PSCs presumably cause little inflammatory responses compared to, e.g., intramyocardial injections by open chest surgery. Nonetheless, we observed measurable effects of NK cells on the teratoma growth kinetics after injection of PSCs in both models (44, 45). Thus, also the low largely NKG2D-dependent cytotoxic activity of resting NK cells against PSCs appears to be relevant in vivo. It might be highly important if PSCs would be present in trace amounts among in vitro-differentiated cells intended for grafting. If NK cells contribute to a fail-safe system against the tumorigenicity of PSCs after transplantation (67), this system might largely rely on NKG2D.
As a caveat, it should be mentioned that the initial step or even steps of differentiation of PSCs in vivo must not immediately remove the risk of tumorigenicity associated with PSCs but it could alter the recognition of the differentiated cells by the immune system and particularly NK cells. When we transplanted the murine ESC line MPI-II into rats immunosuppressed with cyclosporine A, no teratomas occurred likely due to the rejection of the ESCs by NK cells (31) which are not suppressed by cyclosporine A in their activity against PSCs (45). However, when the same numbers of neuronal cells with a purity of more than 95% that were obtained from these ESCs by a directed in vitro differentiation protocol were injected, we observed teratomas in 61% of the recipients (31). Thus, a cell type in the differentiation culture that differed from the original ESCs by resistance to NK cells but that retained pluripotency presumably caused the tumors in these experiments. Therefore, it is important to carefully investigate the individual immunological properties of any graft that is evaluated in preclinical animal models or in clinical studies. Differentiation of PSCs into progenitor cells with a limited differentiation potential, e.g., neural progenitor cells, might be a strategy to significantly reduce the risk of tumorigenicity (68).
Conclusion
Transplantation of autologous iPSCs can lead to teratomas as formally demonstrated here in a murine model to our knowledge for the first time. Transplantation of grafts derived from autologous iPSCs into immunocompetent recipients as well as grafts derived from allogeneic PSCs into immunosuppressed or immunomodulated recipients has the unavoidable risk of tumorigenicity if undifferentiated or not sufficiently differentiated cells are present in the grafts. The cytotoxic activity of NK cells against PSCs might reduce this risk although NK cells need to be activated by cytokines to exhibit a high cytotoxicity against PSCs. The IL-2-activated NK cells killed PSCs in a partly NKG2D-dependent manner while the low killing of PSCs by resting NK cells was almost completely dependent on NKG2D. Thus, any activity of NK cells against PSCs present in grafts transplanted under non-inflammatory conditions, which fail to produce NK cell-activating cytokines, will presumably depend largely on NKG2D at least in murine models. The analysis of the activity of cytokine-activated NK cells against PSCs and of the receptors involved might allow for the development of immunomodulatory protocols, which stimulate NK cells to function efficiently as safeguard against undifferentiated PSCs in PSC-derived grafts.
Ethics Statement
All animal experiments had been approved by the local government (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit) and were carried out in compliance with German and EU legislation (Directive 2010/63/EU).
Author Contributions
RD and KG designed the study; CG, DH, JN, SM, LE, AS, and RD acquired data; JN, CT, WP, BP, AM, and KG provided important reagents, CG, DH, JN, SM, AS, AM, KG, and RD analyzed and interpreted data; RD wrote the manuscript; all authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Yvonne Hintz and Anke Bode for excellent technical assistance. Moreover, we thank Professor Dirk Busch, Technische Universität München, for sending a breeding pair of the Klrk1−/− mice to the University Medical Center Göttingen.
Funding
The study was supported by the Bundesministerium für Bildung und Forschung (01GN018, 01GN0819) and the Deutsche Forschungsgemeinschaft (SFB 1002 TP C05, GU 595/2-1). We acknowledge support by the Open Access Publication Funds of the Göttingen University.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00870/full#supplementary-material.
References
1. Neofytou E, O’brien CG, Couture LA, Wu JC. Hurdles to clinical translation of human induced pluripotent stem cells. J Clin Invest (2015) 125:2551–7. doi:10.1172/JCI80575
2. Tiburcy M, Hudson JE, Balfanz P, Schlick SF, Meyer T, Chang Liao ML, et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modelling and repair. Circulation (2017) 135:1832–47. doi:10.1161/CIRCULATIONAHA.116.024145
3. Ilic D, Devito L, Miere C, Codognotto S. Human embryonic and induced pluripotent stem cells in clinical trials. Br Med Bull (2015) 116:19–27. doi:10.1093/bmb/ldv045
4. Ilic D, Ogilvie C. Concise review: human embryonic stem cells – what have we done? What are we doing? Where are we going? Stem Cells (2017) 35:17–25. doi:10.1002/stem.2450
5. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science (1998) 282:1145–7. doi:10.1126/science.282.5391.1145
6. Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet (2012) 379:713–20. doi:10.1016/S0140-6736(12)60028-2
7. Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet (2015) 385:509–16. doi:10.1016/S0140-6736(14)61376-3
8. Song WK, Park KM, Kim HJ, Lee JH, Choi J, Chong SY, et al. Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: preliminary results in Asian patients. Stem Cell Reports (2015) 4:860–72. doi:10.1016/j.stemcr.2015.04.005
9. Menasche P, Vanneaux V, Hagege A, Bel A, Cholley B, Cacciapuoti I, et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: first clinical case report. Eur Heart J (2015) 36:2011–7. doi:10.1093/eurheartj/ehv189
10. Guan K, Nayernia K, Maier LS, Wagner S, Dressel R, Lee JH, et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature (2006) 440:1199–203. doi:10.1038/nature04697
11. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126:663–76. doi:10.1016/j.cell.2006.07.024
12. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131:861–72. doi:10.1016/j.cell.2007.11.019
13. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (2007) 318:1917–20. doi:10.1126/science.1151526
14. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature (2008) 451:141–6. doi:10.1038/nature06534
15. Mandai M, Watanabe A, Kurimoto Y, Hirami Y, Morinaga C, Daimon T, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med (2017) 376:1038–46. doi:10.1056/NEJMoa1608368
16. Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature (2011) 474:212–5. doi:10.1038/nature10135
17. Kaneko S, Yamanaka S. To be immunogenic, or not to be: that’s the iPSC question. Cell Stem Cell (2013) 12:385–6. doi:10.1016/j.stem.2013.03.008
18. Araki R, Uda M, Hoki Y, Sunayama M, Nakamura M, Ando S, et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature (2013) 494:100–4. doi:10.1038/nature11807
19. Guha P, Morgan JW, Mostoslavsky G, Rodrigues NP, Boyd AS. Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell (2013) 12:407–12. doi:10.1016/j.stem.2013.01.006
20. De Almeida PE, Meyer EH, Kooreman NG, Diecke S, Dey D, Sanchez-Freire V, et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat Commun (2014) 5:3903. doi:10.1038/ncomms4903
21. Zhao T, Zhang ZN, Westenskow PD, Todorova D, Hu Z, Lin T, et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell (2015) 17:353–9. doi:10.1016/j.stem.2015.07.021
22. Dressel R. Effects of histocompatibility and host immune responses on the tumorigenicity of pluripotent stem cells. Semin Immunopathol (2011) 33:573–91. doi:10.1007/s00281-011-0266-8
23. Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med (2013) 19:998–1004. doi:10.1038/nm.3267
24. Tapia N, Schöler HR. Molecular obstacles to clinical translation of iPSCs. Cell Stem Cell (2016) 19:298–309. doi:10.1016/j.stem.2016.06.017
25. Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature (2017) 545:229–33. doi:10.1038/nature22312
26. Yoshihara M, Hayashizaki Y, Murakawa Y. Genomic instability of iPSCs: challenges towards their clinical applications. Stem Cell Rev (2017) 13:7–16. doi:10.1007/s12015-016-9680-6
27. Lawrenz B, Schiller H, Willbold E, Ruediger M, Muhs A, Esser S. Highly sensitive biosafety model for stem-cell-derived grafts. Cytotherapy (2004) 6:212–22. doi:10.1080/14653240410006031
28. Hentze H, Soong PL, Wang ST, Phillips BW, Putti TC, Dunn NR. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res (2009) 2:198–210. doi:10.1016/j.scr.2009.02.002
29. Wobus AM, Holzhausen H, Jakel P, Schöneich J. Characterization of a pluripotent stem cell line derived from a mouse embryo. Exp Cell Res (1984) 152:212–9. doi:10.1016/0014-4827(84)90246-5
30. Nussbaum J, Minami E, Laflamme MA, Virag JA, Ware CB, Masino A, et al. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J (2007) 21:1345–57. doi:10.1096/fj.06-6769com
31. Dressel R, Schindehütte J, Kuhlmann T, Elsner L, Novota P, Baier PC, et al. The tumorigenicity of mouse embryonic stem cells and in vitro differentiated neuronal cells is controlled by the recipients’ immune response. PLoS One (2008) 3:e2622. doi:10.1371/journal.pone.0002622
32. Didie M, Christalla P, Rubart M, Muppala V, Doker S, Unsöld B, et al. Parthenogenetic stem cells for tissue-engineered heart repair. J Clin Invest (2013) 123:1285–98. doi:10.1172/JCI66854
33. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol (2013) 132:536–44. doi:10.1016/j.jaci.2013.07.006
34. Manser AR, Weinhold S, Uhrberg M. Human KIR repertoires: shaped by genetic diversity and evolution. Immunol Rev (2015) 267:178–96. doi:10.1111/imr.12316
35. Rahim MM, Makrigiannis AP. Ly49 receptors: evolution, genetic diversity, and impact on immunity. Immunol Rev (2015) 267:137–47. doi:10.1111/imr.12318
36. Carapito R, Bahram S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol Rev (2015) 267:88–116. doi:10.1111/imr.12328
37. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12:239–52. doi:10.1038/nri3174
38. Granzin M, Wagner J, Köhl U, Cerwenka A, Huppert V, Ullrich E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol (2017) 8:458. doi:10.3389/fimmu.2017.00458
39. Kruse V, Hamann C, Monecke S, Cyganek L, Elsner L, Hübscher D, et al. Human induced pluripotent stem cells are targets for allogeneic and autologous natural killer (NK) cells and killing is partly mediated by the activating NK receptor DNAM-1. PLoS One (2015) 10:e0125544. doi:10.1371/journal.pone.0125544
40. Bonde S, Zavazava N. Immunogenicity and engraftment of mouse embryonic stem cells in allogeneic recipients. Stem Cells (2006) 24:2192–201. doi:10.1634/stemcells.2006-0022
41. Koch CA, Jordan CE, Platt JL. Complement-dependent control of teratoma formation by embryonic stem cells. J Immunol (2006) 177:4803–9. doi:10.4049/jimmunol.177.7.4803
42. Frenzel LP, Abdullah Z, Kriegeskorte AK, Dieterich R, Lange N, Busch DH, et al. Role of natural-killer group 2 member D ligands and intercellular adhesion molecule 1 in natural killer cell-mediated lysis of murine embryonic stem cells and embryonic stem cell-derived cardiomyocytes. Stem Cells (2009) 27:307–16. doi:10.1634/stemcells.2008-0528
43. Perez-Cunningham J, Ames E, Smith RC, Peter AK, Naidu R, Nolta JA, et al. Natural killer cell subsets differentially reject embryonic stem cells based on licensing. Transplantation (2014) 97:992–8. doi:10.1097/TP.0000000000000063
44. Dressel R, Nolte J, Elsner L, Novota P, Guan K, Streckfuss-Bömeke K, et al. Pluripotent stem cells are highly susceptible targets for syngeneic, allogeneic, and xenogeneic natural killer cells. FASEB J (2010) 24:2164–77. doi:10.1096/fj.09-134957
45. Hübscher D, Kaiser D, Elsner L, Monecke S, Dressel R, Guan K. The tumorigenicity of multipotent adult germline stem cells transplanted into the heart is affected by natural killer cells and by cyclosporine A independent of its immunosuppressive effects. Front Immunol (2017) 8:67. doi:10.3389/fimmu.2017.00067
46. Dressel R, Guan K, Nolte J, Elsner L, Monecke S, Nayernia K, et al. Multipotent adult germ-line stem cells, like other pluripotent stem cells, can be killed by cytotoxic T lymphocytes despite low expression of major histocompatibility complex class I molecules. Biol Direct (2009) 4:31. doi:10.1186/1745-6150-4-31
47. Drukker M, Katz G, Urbach A, Schuldiner M, Markel G, Itskovitz-Eldor J, et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci U S A (2002) 99:9864–9. doi:10.1073/pnas.142298299
48. Swijnenburg RJ, Tanaka M, Vogel H, Baker J, Kofidis T, Gunawan F, et al. Embryonic stem cell immunogenicity increases upon differentiation after transplantation into ischemic myocardium. Circulation (2005) 112:I166–72. doi:10.1161/CIRCULATIONAHA.104.525824
49. Robertson NJ, Brook FA, Gardner RL, Cobbold SP, Waldmann H, Fairchild PJ. Embryonic stem cell-derived tissues are immunogenic but their inherent immune privilege promotes the induction of tolerance. Proc Natl Acad Sci U S A (2007) 104:20920–5. doi:10.1073/pnas.0710265105
50. Zafirova B, Mandaric S, Antulov R, Krmpotic A, Jonsson H, Yokoyama WM, et al. Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-deficient mice. Immunity (2009) 31:270–82. doi:10.1016/j.immuni.2009.06.017
51. Cheng J, Dutra A, Takesono A, Garrett-Beal L, Schwartzberg PL. Improved generation of C57BL/6J mouse embryonic stem cells in a defined serum-free media. Genesis (2004) 39:100–4. doi:10.1002/gene.20031
52. Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A (1993) 90:8424–8. doi:10.1073/pnas.90.18.8424
53. Lührig S, Siamishi I, Tesmer-Wolf M, Zechner U, Engel W, Nolte J. Lrrc34, a novel nucleolar protein, interacts with Npm1 and Ncl and has an impact on pluripotent stem cells. Stem Cells Dev (2014) 23:2862–74. doi:10.1089/scd.2013.0470
54. Streckfuss-Bömeke K, Wolf F, Azizian A, Stauske M, Tiburcy M, Wagner S, et al. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur Heart J (2013) 34:2618–29. doi:10.1093/eurheartj/ehs203
55. Boukouaci W, Lauden L, Siewiera J, Dam N, Hocine HR, Khaznadar Z, et al. Natural killer cell crosstalk with allogeneic human cardiac-derived stem/progenitor cells controls persistence. Cardiovasc Res (2014) 104:290–302. doi:10.1093/cvr/cvu208
56. Kärre K. Natural killer cell recognition of missing self. Nat Immunol (2008) 9:477–80. doi:10.1038/ni0508-477
57. Kasahara M, Sutoh Y. Comparative genomics of the NKG2D ligand gene family. Immunol Rev (2015) 267:72–87. doi:10.1111/imr.12320
58. Ullrich E, Koch J, Cerwenka A, Steinle A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology (2013) 2:e26097. doi:10.4161/onci.26097
59. Isernhagen A, Malzahn D, Viktorova E, Elsner L, Monecke S, von Bonin F, et al. The MICA-129 dimorphism affects NKG2D signaling and outcome of hematopoietic stem cell transplantation. EMBO Mol Med (2015) 7:1480–502. doi:10.15252/emmm.201505246
60. Isernhagen A, Malzahn D, Bickeböller H, Dressel R. Impact of the MICA-129Met/Val dimorphism on NKG2D-mediated biological functions and disease risks. Front Immunol (2016) 7:588. doi:10.3389/fimmu.2016.00588
61. Carapito R, Aouadi I, Ilias W, Bahram S. Natural killer group 2, member D/NKG2D ligands in hematopoietic cell transplantation. Front Immunol (2017) 8:368. doi:10.3389/fimmu.2017.00368
62. Gam R, Shah P, Crossland RE, Norden J, Dickinson AM, Dressel R. Genetic association of hematopoietic stem cell transplantation outcome beyond histocompatibility genes. Front Immunol (2017) 8:380. doi:10.3389/fimmu.2017.00380
63. Baranwal AK, Mehra NK. Major histocompatibility complex class I chain-related A (MICA) molecules: relevance in solid organ transplantation. Front Immunol (2017) 8:182. doi:10.3389/fimmu.2017.00182
64. Risti M, Bicalho MD. MICA and NKG2D: is there an impact on kidney transplant outcome? Front Immunol (2017) 8:179. doi:10.3389/fimmu.2017.00179
65. Phillips LK, Gould EA, Babu H, Krams SM, Palmer TD, Martinez OM. Natural killer cell-activating receptor NKG2D mediates innate immune targeting of allogeneic neural progenitor cell grafts. Stem Cells (2013) 31:1829–39. doi:10.1002/stem.1422
66. Wang D, Quan Y, Yan Q, Morales JE, Wetsel RA. Targeted disruption of the beta2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl Med (2015) 4:1234–45. doi:10.5966/sctm.2015-0049
67. Itakura G, Kobayashi Y, Nishimura S, Iwai H, Takano M, Iwanami A, et al. Controlling immune rejection is a fail-safe system against potential tumorigenicity after human iPSC-derived neural stem cell transplantation. PLoS One (2015) 10:e0116413. doi:10.1371/journal.pone.0116413
Keywords: teratoma, autologous transplantation, embryonic stem cells, induced pluripotent stem cells, multipotent adult germline stem cells, natural killer cells, cytokine-activated natural killer cells, natural killer receptor ligands
Citation: Gröschel C, Hübscher D, Nolte J, Monecke S, Sasse A, Elsner L, Paulus W, Trenkwalder C, Polić B, Mansouri A, Guan K and Dressel R (2017) Efficient Killing of Murine Pluripotent Stem Cells by Natural Killer (NK) Cells Requires Activation by Cytokines and Partly Depends on the Activating NK Receptor NKG2D. Front. Immunol. 8:870. doi: 10.3389/fimmu.2017.00870
Received: 14 May 2017; Accepted: 10 July 2017;
Published: 26 July 2017
Edited by:
Martin Johannes Hoogduijn, Erasmus University Rotterdam, NetherlandsReviewed by:
Reem Al-Daccak, Institut National de la Santé et de la Recherche Médicale, FranceGabriele Multhoff, Technische Universität München, Germany
Copyright: © 2017 Gröschel, Hübscher, Nolte, Monecke, Sasse, Elsner, Paulus, Trenkwalder, Polić, Mansouri, Guan and Dressel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf Dressel, cmRyZXNzZUBnd2RnLmRl
†These authors have shared first authorship.
‡These authors have shared senior authorship.