 Shin-ichiro Fujii
Shin-ichiro Fujii Kanako Shimizu
Kanako Shimizu- Laboratory for Immunotherapy, RIKEN Center for Integrative Medical Sciences (IMS), Yokohama, Japan
Immune checkpoint blockade therapy has prevailed for several types of cancer; however, its effectiveness as a single therapy is still limited. In principle, dendritic cells (DCs) should be able to control the post-therapy immune response, in particular since they can link the two major arms of the immune system: innate and adaptive immunity. Therefore, DCs would be a logical and ideal target for the development of immunotherapies. Since DCs are not activated in the steady state, an adjuvant to convert their function from tolerogenic to immunogenic would be desirable. Upon ligand activation, invariant natural killer T (iNKT) cells simultaneously activate NK cells and also energize the DCs, resulting in their full maturation. To utilize such iNKT-licensed “fully” matured DCs as adjuvants, mechanisms of both intercellular communication between DC subsets and iNKT cells and intracellular molecular signaling in DCs have to be clarified and optimized. To generate both innate and adaptive immunity against cancer, a variety of strategies with the potential to target iNKT-licensed DCs in situ have been studied. The benchmark of success in these studies, each with distinct approaches, will be the development of functional NK cells and cytotoxic T cells (CTLs) as well as generation of long-term, memory CTL. In this review, we provide a framework for NKT-mediated immunotherapy through selective DC targeting in situ, describe progress in the design of licensed therapies for iNKT cell targeting of DCs, and highlight the challenge to provide maximal benefit to patients.
Introduction
Invariant natural killer T (iNKT) cells have several distinguishing characteristics, most notably they express an invariant TCRα chain, Vα14Jα18 in mice and Vα24Jα18 in human, paired with a TCRβ chain of limited diversity (1, 2). Unlike most αβTCRs, which recognize peptide MHC I/II complexes, iNKT TCRs recognize glycolipid antigens presented by the MHC class I-like molecule CD1d (3–5). The first NKT glycolipid ligand was identified by Kirin Pharmaceuticals (6). They extracted agelasphins as glycosphingolipid compounds from a marine sponge called Agelas mauritianus. They modified the structure of this compound and established a synthetic ligand with a branched galactosylceramide, commonly referred to as α-GalCer (7, 8). In addition to α-GalCer, iNKT cells recognize certain microbial ligands, for example cell wall sphingolipids from Sphingomonas, Borrelia, or Streptococcus (9–11). iNKT cells recognize such natural or synthetic glycolipids and promptly produce a broad range of cytokines. iNKT cells are not only stimulated by these glycolipid ligands directly via their invariant TCR but also indirectly. Since iNKT cells express IL-12 receptors, they can be stimulated by IL-12 released from dendritic cells (DCs) or macrophages. For example, Salmonella typhimurium does not express a glycolipid ligand, but can stimulate iNKT cells in vivo. In this case, the iNKT cells can be activated by both IL-12 and recognition of endogenous glycosphingolipid ligand on DCs (1, 12).
In the course of establishment or recurrence of tumor cells, genetic factors or immune related pressure may mediate the selective outgrowth of tumor cell clones lacking MHC or potentially immunogenic tumor associated antigens (TAA), thus leading to heterogeneous tumor cell evolution (13). In terms of cancer immunity, tumors are in general composed of two types of cells, some are MHC positive, but others are MHC negative. Based on our current understanding, the former can be eliminated by cytotoxic T cells (CTLs) and the latter can be eliminated by NK cells, thus both innate and adaptive immune responses are required for complete elimination of tumors. It is well-known that DCs can play a crucial role in activating both innate and adaptive immune responses (14–16). We and others demonstrated that iNKT cells, as well as many toll-like receptor (TLR) ligands, can be used for activation of DCs to bridge innate and adaptive immunity (17–20). In this review, we detail the rationale for modulation or optimization of iNKT cell-licensed antigen-expressing DCs and also describe various attempts that have so far been made for developing antitumor therapeutic strategies.
NKT Cell Subsets—Localization and Function
When TCRs on iNKT cells are stimulated by a ligand such as α-GalCer, they are capable of producing IFN-γ and IL-4. These Vα14+iNKT cells were recently shown to belong to three distinct functional subsets. The NKT1, NKT2, and NKT17 cells all express promyelocytic leukemia zinc finger, but can be distinguished because they are mainly regulated by transcription factors similar to those of helper T cells, i.e., T-bet, GATA-3, and RORγt, respectively (21, 22). Development of each type of iNKT cell is generally related to the cytokine milieu encountered upon activation (IFN-γ, IL-4, or IL-17). An intravenous injection of α-GalCer rapidly activates NKT1 cells in the red pulp of the spleen and liver, thus leading to systemic IFN-γ and IL-4 responses, but not iNKT cells in LN and thymus (23, 24). NKT2 cells are mainly located in the medullary area of the thymus and T cell zone of the spleen, as well as in mesenteric LNs. Oral administration of α-GalCer may induce the activation of NKT2 cells in mLN, resulting in local IL-4 production (23, 24). NKT17 cells which are capable of producing IL-17, but not IFN-γ, are particularly enriched in the lung and the subcapsular region of LNs (23, 24). Thus, the pattern and amount of cytokine production can be determined by the location of NKT cells, NKT cell type and routes of administration of NKT ligands.
Induction of NK Cell Responses as an Adjunctive Effect of iNKT Cell Therapy
The critical role of iNKT cells in tumor immunosurveillance was shown in chemically induced spontaneous tumor models (25). The transfer of iNKT cells prevented the induction of methylcholanthrene sarcoma tumors in Jα18−/−, iNKT cell-deficient mice. A similar protocol involving transfer of iNKT cells had demonstrated a significant antitumor effect in p53-deficient mice (26) and in the transgenic adenocarcinoma of the mouse prostate tumor model (27).
Our first approach to understanding iNKT cell immunotherapy was to compare cell therapy (e.g., administration of CD1d+ cells loaded with α-GalCer) versus unbound glycolipid drug therapy (e.g., administration of soluble α-GalCer) (28). An injection of bone marrow-derived ex vivo DCs loaded with α-GalCer (BM-DC/Gal) induced iNKT cells capable of producing IFN-γ (28) (Figure 1), and this correlated with antitumor effects in B16 melanoma lung metastasis. In contrast, the iNKT cell response to unbound α-GalCer was more rapid, but transient and then the cells became anergic (28, 29). Thus, the glycolipid has different functional effects on iNKT cells when it is injected as a free glycolipid or in association with CD1d+ cells. When activated by the iNKT cell ligand, IFN-γ and IL-2 production by iNKT cells enhances the activation of NK cells as iNKT–NK axis (30) (Figure 2). The interaction between iNKT cells and DCs can also enhance NK cell activity. After activation by NKT cells, DCs express NKG2D ligands and CD70, thus leading to the activation of NK cells (31). In addition, since NK cells also express IL-12R, IL-12 released from DCs enhances NK cell-mediated IFN-γ production (Figure 2). Thus, iNKT cells efficiently stimulate NK cells. The near synchronous activation of these iNKT and NK cell can account for innate resistance to susceptible tumors.
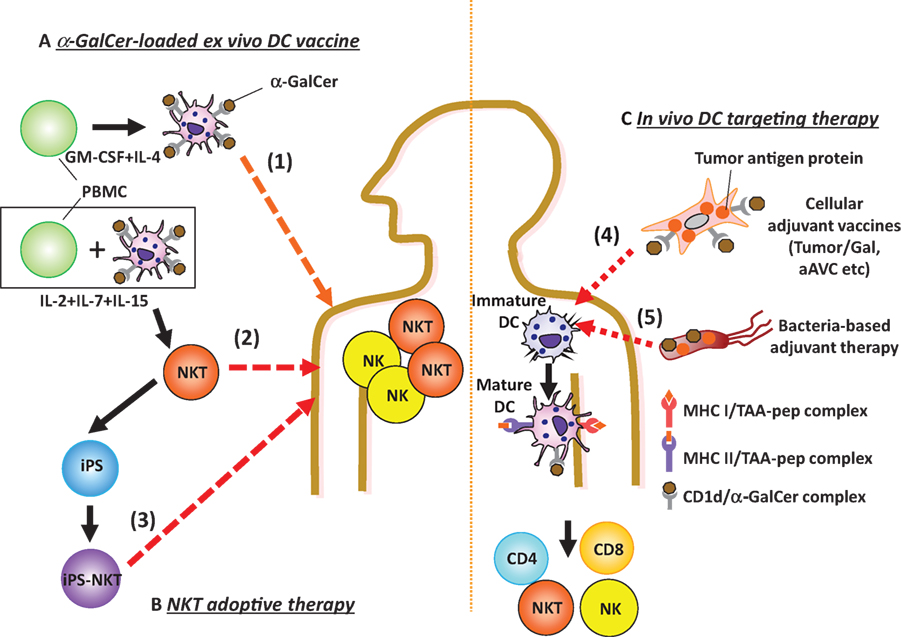
Figure 1. Ex vivo or in vivo glycolipid-based dendritic cell (DC) immunotherapy. (A,B) Ex vivo glycolipid-based DC therapy and NKT transfer therapy have been studied. (A) (1) Active immunization with ex vivo DCs: monocyte-derived DCs loaded with α-GalCer (DCs/Gal) or autologous PBMCs pulsed with α-GalCer are administered intravenously to cancer patients. The invariant natural killer T (iNKT) and NK cells are promptly activated in lung, liver, and spleen. (B) As passive immunization, effector cells are adoptively transferred. (2) For this approach, ex vivo iNKT cells are harvested after coculturing with autologous DC/Gal and then injected into cancer patients. (3) In the future, iPS-reprogrammed iNKT cells may be applicable for adoptive transfer therapy. (C) As new strategies of in vivo DC targeting therapies, (4) adjuvant vector cells, including tumor cells loaded with α-GalCer (Tumor/Gal) or tumor antigen mRNA-transfected, allogeneic CD1d+ cells loaded with α-GalCer (aAVC) or (5) non-somatic cell adjuvant (bacteria) will be candidates for the iNKT-triggered immunotherapy. When these agents are injected, both iNKT and NK cells will be activated. Host DCs can then prime antigen-specific CD4+ and/or CD8+ T cells.
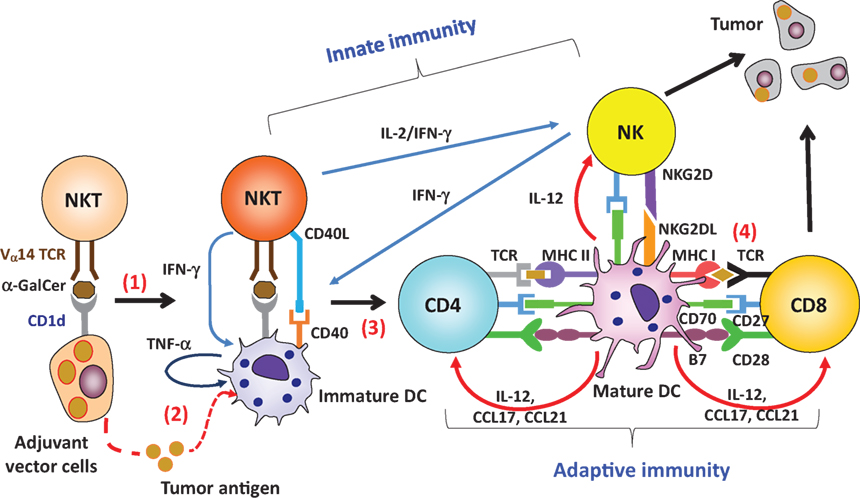
Figure 2. Adjuvant effect by invariant natural killer T (iNKT) cell-triggered dendritic cells (DCs) on protective antitumor responses. (1) Administration of adjuvant vector cells, including Tumor/Gal or aAVC initially stimulate iNKT cells. (2) The adjuvant vector cells are killed by iNKT cells and NK cells, and then tumor antigen released from them can be captured by endogenous CD11c+DCs. (3) The CD11c+ DCs then undergo iNKT cell-induced maturation. (4) The activated DCs can then induce an antigen-specific T cell response in the lymphoid tissues. Thus, the CD11c+DCs in situ are able to cross present tumor antigen, derived from phagocytosed adjuvant vector cells, to CD4+ or CD8+ T cells in an MHC-dependent manner.
Efficient Induction of Antitumor CTLs by iNKT Cell-Licensed DCs
In situ DCs activated by iNKT cells act as a cellular adjuvant for T-cell priming. The licensing of DCs by iNKT cells occurs by several molecular mechanisms. When activated iNKT cells encounter DCs in situ, co-stimulatory molecules on DCs are upregulated, indicative of DC maturation (32, 33). Activated iNKT cells can promote conversion of DCs from a tolerogenic to an immunogenic state. The DC surface remodeling of these mature DCs is driven by two inflammatory cytokines, TNF-α and IFN-γ. Cytokine production by the innate lymphocytes is tightly regulated by the interactions among unique cell types. TNF-α is produced by endogenous DCs, whereas IFN-γ is produced by iNKT or NK cells (Figure 2). It was previously reported that IFN-γ-producing DCs are important for priming of the gut intraepithelial lymphocyte response against intracellular parasitic infection (34). We also tested the possibility of IFN-γ production by CD11c+DCs after administration of α-GalCer and found that isolated CD11c+cells produce IFN-γ, but that these were CD11c+NK cells and not any DC subset (35).
Co-stimulatory molecules, CD40, CD80, and CD86 on DCs, which are important for priming T cells, are upregulated during the early phase (from 4 h). In addition, we and others recently demonstrated upregulation of CD70, 4-1-BBL, and IL-15Ra on DCs, which are important for generation of memory T cells, at the late phase (from 40 h) (36, 37).
Other innate lymphocytes, such as γδT cells (38, 39) and NK cells (40, 41) may also have adjuvant effects. They produce IFN-γ and TNF-α, thus promoting the maturation of DCs and help for the generation of CTLs. We also compared the magnitude of T cell responses after priming with the iNKT cell ligand α-GalCer versus NK cell ligands, such as retinoic acid early inducible-1ε (Rae1ε), Rae1γ, CD70 and murine UL16-binding protein-like transcript 1 (Mult-1). CTL induction triggered by iNKT cells is apparently more powerful than that triggering by NK cells (42).
An important difference between these other innate lymphocytes and iNKT cells lies in the CD40L signal to DCs. Bennett et al. reported that CD40L on helper T cells plays an crucial role in licensing DCs (43). Activated iNKT cells express CD40L transiently (44), but other lymphocytes do not. In fact, the adjuvant effect of DCs triggered by iNKT cells is eliminated when CD40−/− mice are used as recipients (44), even though the iNKT cell response is still present. In a reciprocal study, co-administration of antigen plus soluble TNF-α and soluble IFN-γ induces the phenotypic maturation of DCs in situ, but does not generate antigen-specific T cells. Thus, phenotypic maturation of DCs does not always correspond to an antigen-specific T cell response, whereas functional maturation of DCs does. CD40–CD40L interaction during DC-iNKT cell cross talk is critical for DC maturation, resulting in IL-12 production (Figure 2), whereas inflammatory cytokines (TNF-α and IFN-γ) serve as co-factors for full maturation of DCs. In addition, iNKT cell-licensed DCs apparently use a different mechanism rather than that used during TLR signaling. The adjuvant effect of TLR ligands depends on either MyD88 or TRIF (45), or both, but that by iNKT-licensed DCs does not (32). As discussed above, NKT licensed-DCs depend on CD40/CD40L signaling and inflammatory signals (TNF-α and IFN-γ). CD40/CD40L signaling may involve the TRAFs (TRAF1, 2, 3, 5, 6), whereas TNF-α and IFN-γ signaling may involve the TRAF2 and JAK1/2-STAT1 pathway respectively (46, 47).
The location of DCs and iNKT cells in spleen is another important factor in their mutual activation. After their activation, iNKT cells accumulate in the marginal zone, where they co-interact with DCs. After activation by iNKT cells, XCR1+ DCs can traffic to the PALS area and then prime T cells (48). These responses are orchestrated by chemokines, cytokines, and cell surface molecules. iNKT cell-licensed DCs produce CCL17 (37, 49), which attracts CCR4+ CD8+ T cells for subsequent activation.
Several factors during the initiation of innate immunity determine the subsequent flavor of the adaptive immune response: (1) the number and function of iNKT cells and APCs, (2) the nature of the ligands (i.e., OCH, α-GalCer, or α-C-GalCer) (35), (3) the properties of host APCs (DC location or subset) (44), and (4) the level of CD1d expression (50). Thus, the magnitude of the innate immune response generated by all these factors can be directly correlated with the subsequent adaptive immune response.
Development of iNKT-Triggered Antitumor Strategies Linking Innate and Adaptive Therapy
Cellular Vaccines Acting As Immunological Adjuvant and Tumor Antigen Carrying Vector
We and others demonstrated that co-administration of antigen and iNKT ligand generates antigen-specific T cells, in addition to activating iNKT and NK cells (32, 33, 51). However, several optimal conditions are limited. It has been reported that DCs cannot phagocytize antigen after their maturation. Indeed, administration of tumor antigen 4 h after an administration of NKT cell ligand did not lead to a T cell response (32, 33). Therefore, tumor antigen and NKT cell ligands have to be delivered simultaneously to in vivo DCs (32, 33). In addition, we also found that this co-administration protocol generates CTL, but not memory T cells easily.
CD1d+ cells loaded with α-GalCer can activate iNKT cells directly in vitro, but it was not known whether adaptive immunity was generated after initiating this innate immune response. We first showed conclusively that CD1d+tumor/Gal can induce antigen-specific CD4+ and CD8+ T cell immunity (50) (Figure 1). From this finding, we proposed a strategy using all-in one cell type that expressed tumor antigen as well as CD1d that was loaded with the NKT cell ligand α-GalCer simultaneously, a cell that we reported an adjuvant vector cell (50, 52) (Figure 1). CD1d is generally expressed on most hematopoietic cells, e.g., DCs, B cells, T cells, and macrophages, and on some non-hematopoietic cells, e.g., intestinal epithelium and hepatocytes, including multiple tumor types (53). Therefore, approaches using adjuvant vector cells may be applicable not only for most hematological disorders, where they can be applied relatively easily, but also for many solid tumor cells, a more difficult therapeutic target. In fact, the therapeutic strategy using tumor/Gal has been extended to many types of CD1d+ tumor cells, including in our own studies, e.g., B16 melanoma, EL4 thymoma, WEHI3B leukemia, and J558 plasmacytoma. In addition, Hunn et al. reported that irradiated Glioma/Gal was effective in a prophylactic setting and also in a therapeutic setting together with Treg depletion of intracranial glioma model (54). Kobayashi et al. have reported on an α-GalCer-loaded B cell lymphoma (Eμ-myc tumor) combined with an agonistic antibody targeting 4-1BB (CD137) (55). The studies as above demonstrated the antigen-specific effector T cell-mediated survival. These tumor/Gal vaccines would be useful in an autologous setting.
Instead of such syngeneic tumor/Gal therapy, we newly established the concept of an artificial adjuvant vector cell (aAVC) as a new type of cancer vaccine platform that incorporates in vivo iNKT-licensed DC therapy (Figure 1). These cells (aAVC), NIH3T3 cells for mouse and HEK293 for humans, have been transfected with a CD1d and tumor antigen mRNA and then loaded with α-GalCer (37, 42, 56). The aAVC express the α-GalCer-CD1d complex on their surface and tumor antigen protein inside of the allogeneic cells. The aAVC treatment reduces the number of metastases, and eliminated grossly large tumors (37, 42, 56).
As the mechanism of adjuvant vector cells (tumor/Gal or aAVC), four immunological steps take place (Figure 2). Initially, these cells directly activate iNKT cells. iNKT cells producing IFN-γ can then simultaneously activate NK cells. These innate killer iNKT/NK cells capable of producing IFN- γ reject the adjuvant vector cells, but some of the killed adjuvant vector cells are taken up by DCs in situ, thereby several immunogenic features of DCs are engaged. The adjuvant vector cells-capturing DCs in lung, liver, and spleen become matured by their interaction with iNKT cells, resulting from CD40L–CD40 interactions and production of inflammatory cytokines. Next, the mature DCs present the TAAs to T cells on both MHC class I and II in situ. Particularly, the XCR1+DCs are specialized to cross-present antigens on MHC class I. Notably, when mice were vaccinated with adjuvant vector cells, they became resistant to the parental tumor cells. In fact, administration of adjuvant vector cells induces CTL and long-term memory T cells efficiently in vivo (48, 50, 52).
Bacteria-Based Adjuvant Therapy
Listeria monocytogenes (LM) is a Gram-positive intracellular bacterium. Several groups have investigated whether recombinant LM lacking virulence genes, but expressing several TLR ligands such as lipoteichoic acid, would be useful for delivering TAA in vivo (57, 58). After infecting the target cells with LM, there was active phagocytosis and lysis of the bacteria in the phagosome. The recombinant LM allowed for the delivery of the TAA directly into macrophages and DCs, which can present TAA peptides to CD4+ and CD8+ T cells. In practice, a live attenuated, LM-based tumor vaccine expressing TAA-Mage-b (Mb) and α-GalCer has been studied (59) (Figure 1). The T cell-mediated antitumor efficacy resulting from direct incorporation of α-GalCer into live LM-Mb was found to be more powerful and safer than co-administration of the LM-Mb vaccine and α-GalCer, but the iNKT cell response was weaker.
Bacille Calmette–Guerin (BCG) was derived by attenuating Mycobacterium bovis and is widely used in many countries as a tuberculosis vaccine, although its efficacy has been contested. Recombinant BCG (rBCG) strains expressing either Listeriolysin-O from LM or perfringolysin from Clostridium perfringens have been investigated as candidate tumor vaccines (60). In simple rBCG-based vaccination models, skin CD11bhigh DC subsets present antigen to CD4+ T cells (61). Using an approach of incorporating glycolipids into rBCG strains, rBCG strains expressing an SIV Gag antigen (rBCG-SIV gag) together with α-GalCer enhanced CTL more efficiently compared to responses primed by simple rBCG-SIV gag (62) (Figure 1). Similar to the concept of adjuvant vector cells or aAVC, these two types of bacteria vaccine expressing antigen and NKT ligand showed CTL induction more efficiently than that of co-administration approach.
iNKT Cell Transfer Immunotherapy
As the other option, complementation of iNKT cell therapy may be an approach for cancer patients with decreased iNKT cell frequencies in target organs. This type of iNKT cell-based immunotherapy may be able to take advantage of the recent iPS reprograming technology. We recently established a protocol to reprogram human Vα24+iNKT cells and then to re-differentiate them into functional iNKT cells, so called iPS-iNKT cells (63). Similar to conventional human Vα24+iNKT cells, iPS-iNKT cells can produce IFN-γ upon NKT ligand activation and kill several types of human tumor cell lines (leukemia, lung cancer, and head and neck cancers) in vitro and in vivo. As already discussed in this review, we demonstrated that NK cell activity but not T cell induction is induced after the iNKT cell activation, mainly through IFN-γ production as iNKT–NK axis. In fact, once activated in vivo, human iPS-iNKT cells have been shown, in “humanized” NOG mice with human peripheral blood cells, to mediate adjunctive activity by activating autologous NK cells (Figure 1). We, therefore, suggest that human iPS-Vα24+iNKT cells could exert antitumor activity in vivo.
Conclusion
When iNKT cells are activated by the ligand, they subsequently have the power to activate NK cells by producing IFN-γ as iNKT–NK axis. In fact, several immunotherapies using autologous DC/Gal in clinical trials of solid tumor and hematological malignancies have indicated the importance of IFN-γ-producing iNKT cells and NK cell activation (Figure 1). Although iNKT cells can mature DCs in vivo, IFN-γ alone is not sufficient to DC maturation and T cell induction. Therefore, to further develop iNKT cell-mediated therapy, many groups have focused on the interaction between DC and iNKT cells. We summarized details of the mechanism of the interaction between these two cell types and also introduced several iNKT cell-triggered DC approaches that are candidates for potential new therapies for cancer treatment. Adjuvant vector cells, including Tumor/Gal or aAVC as well as non-somatic cell adjuvants (bacteria) are all candidates for iNKT-triggered DC mediated immunotherapy.
Author Contributions
SF and KS conceptualized, wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dr. Peter Burrows for peer-reviewing and helpful comments in the preparation of the manuscript. This work is supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to KS and SF, grant numbers 15K09590 and 26460583 respectively) and the Japan Agency for Medical Research and Development (translational research network program) (to SF, grant number 15lm0103002j0004).
References
1. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13:101–17. doi:10.1038/nri3369
2. McEwen-Smith RM, Salio M, Cerundolo V. The regulatory role of invariant NKT cells in tumor immunity. Cancer Immunol Res (2015) 3:425–35. doi:10.1158/2326-6066.CIR-15-0062
3. Bendelac A. CD1: presenting unusual antigens to unusual T lymphocytes. Science (1995) 269:185–6. doi:10.1126/science.7542402
4. Bendelac A, Lantz O, Quimby ME, Yewdell JW, Bennink JR, Brutkiewicz RR. CD1 recognition by mouse NK1+ T lymphocytes. Science (1995) 268:863–5. doi:10.1126/science.7538697
5. Exley M, Garcia J, Balk SP, Porcelli S. Requirements for CD1d recognition by human invariant Valpha24+ CD4-CD8- T cells. J Exp Med (1997) 186:109–20. doi:10.1084/jem.186.1.109
6. Natori T, Morita M, Akimoto K, Koezuka Y. Agelasphins, novel antitumor and immunostimulatory cerebrosides from the sponge Agelas mauritanus. Tetrahedron (1994) 50:2771–84. doi:10.1016/S0040-4020(01)86991-X
7. Morita M, Motoki K, Akimoto K, Natori T, Sakai T, Sawa E, et al. Structure-activity relationship of a-galactosylceramides against B16-bearing mice. J Med Chem (1995) 38:2176. doi:10.1021/jm00012a018
8. Kobayashi E, Motoki K, Uchida T, Fukushima H, Koezuka Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol Res (1995) 7:529–34.
9. Sriram V, Du W, Gervay-Hague J, Brutkiewicz RR. Cell wall glycosphingolipids of Sphingomonas paucimobilis are CD1d-specific ligands for NKT cells. Eur J Immunol (2005) 35:1692–701. doi:10.1002/eji.200526157
10. Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol (2006) 7:978–86. doi:10.1038/ni1380
11. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol (2011) 12:966–74. doi:10.1038/ni.2096
12. Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol (2003) 4:1230–7. doi:10.1038/ni1002
13. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell (2017) 168:613–28. doi:10.1016/j.cell.2017.01.018
14. Shimizu K, Fujii S. An adjuvant role of in situ dendritic cells (DCs) in linking innate and adaptive immunity. Front Biosci (2008) 13:6193–201. doi:10.2741/3147
15. Durai V, Murphy KM. Functions of murine dendritic cells. Immunity (2016) 45:719–36. doi:10.1016/j.immuni.2016.10.010
16. Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res (2017) 27:96–108. doi:10.1038/cr.2016.149
17. Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med (2005) 202:203–7. doi:10.1084/jem.20050810
18. Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Va14+ natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev (2007) 220:183–98. doi:10.1111/j.1600-065X.2007.00561.x
19. Fujii S, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, et al. NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol (2013) 4:409. doi:10.3389/fimmu.2013.00409
20. Faveeuw C, Trottein F. Optimization of natural killer T cell-mediated immunotherapy in cancer using cell-based and nanovector vaccines. Cancer Res (2014) 74:1632–8. doi:10.1158/0008-5472.CAN-13-3504
21. Constantinides MG, Bendelac A. Transcriptional regulation of the NKT cell lineage. Curr Opin Immunol (2013) 25:161–7. doi:10.1016/j.coi.2013.01.003
22. Engel I, Seumois G, Chavez L, Samaniego-Castruita D, White B, Chawla A, et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol (2016) 17:728–39. doi:10.1038/ni.3437
23. Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity (2015) 43:566–78. doi:10.1016/j.immuni.2015.06.025
24. Slauenwhite D, Johnston B. Regulation of NKT cell localization in homeostasis and infection. Front Immunol (2015) 6:255. doi:10.3389/fimmu.2015.00255
25. Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med (2002) 196:119–27. doi:10.1084/jem.20020092
26. Swann JB, Uldrich AP, van Dommelen S, Sharkey J, Murray WK, Godfrey DI, et al. Type I natural killer T cells suppress tumors caused by p53 loss in mice. Blood (2009) 113:6382–5. doi:10.1182/blood-2009-01-198564
27. Bellone M, Ceccon M, Grioni M, Jachetti E, Calcinotto A, Napolitano A, et al. iNKT cells control mouse spontaneous carcinoma independently of tumor-specific cytotoxic T cells. PLoS One (2010) 5:e8646. doi:10.1371/journal.pone.0008646
28. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged interferon-g producing NKT response induced with a-galactosylceramide-loaded dendritic cells. Nat Immunol (2002) 3:867–74. doi:10.1038/ni827
29. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest (2005) 115:2572–83. doi:10.1172/JCI24762
30. Hayakawa Y, Takeda K, Yagita H, Kakuta S, Iwakura Y, Van Kaer L, et al. Critical contribution of IFN-gamma and NK cells, but not perforin-mediated cytotoxicity, to anti-metastatic effect of alpha-galactosylceramide. Eur J Immunol (2001) 31:1720–7. doi:10.1002/1521-4141(200106)
31. Arora P, Baena A, Yu KO, Saini NK, Kharkwal SS, Goldberg MF, et al. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity (2014) 40:105–16. doi:10.1016/j.immuni.2013.12.004
32. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a co-administered protein. J Exp Med (2003) 198:267–79. doi:10.1084/jem.20030324
33. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol (2003) 171:5140–7. doi:10.4049/jimmunol.171.10.5140
34. Moretto MM, Weiss LM, Combe CL, Khan IA. IFN-gamma-producing dendritic cells are important for priming of gut intraepithelial lymphocyte response against intracellular parasitic infection. J Immunol (2007) 179:2485–92. doi:10.4049/jimmunol.179.4.2485
35. Fujii S, Shimizu K, Hemmi H, Fukui M, Bonito AJ, Chen G, et al. Glycolipid a-C-galactosylceramide is a distinct inducer of dendritic cell function during innate and adaptive immune responses of mice. Proc Natl Acad Sci U S A (2006) 103:11252–7. doi:10.1073/pnas.0604812103
36. Taraban VY, Martin S, Attfield KE, Glennie MJ, Elliott T, Elewaut D, et al. Invariant NKT cells promote CD8+ cytotoxic T cell responses by inducing CD70 expression on dendritic cells. J Immunol (2008) 180:4615–20. doi:10.4049/jimmunol.180.7.4615
37. Shimizu K, Yamasaki S, Shinga J, Sato Y, Watanabe T, Ohara O, et al. Systemic DC activation modulates the tumor microenvironment and shapes the long-lived tumor-specific memory mediated by CD8+ T cells. Cancer Res (2016) 76:3756–66. doi:10.1158/0008-5472.CAN-15-3219
38. Leslie DS, Vincent MS, Spada FM, Das H, Sugita M, Morita CT, et al. CD1-mediated γ/δ T cell maturation of dendritic cells. J Exp Med (2002) 196:1575–84. doi:10.1084/jem.20021515
39. Conti L, Casetti R, Cardone M, Varano B, Martino A, Belardelli F, et al. Reciprocal activating interaction between dendritic cells and pamidronate-stimulated gammadelta T cells: role of CD86 and inflammatory cytokines. J Immunol (2005) 174:252–60. doi:10.4049/jimmunol.174.1.252
40. Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med (2002) 195:327–33. doi:10.1002/ijc.23962
41. Mocikat R, Braumuller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity (2003) 19:561–9. doi:10.1016/S1074-7613(03)00264-4
42. Fujii S, Goto A, Shimizu K. Antigen mRNA-transfected, allogeneic fibroblasts loaded with NKT-cell ligand confer antitumor immunity. Blood (2009) 113:4262–72. doi:10.1182/blood-2008-08-176446
43. Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature (1998) 393:478–80. doi:10.1038/30996
44. Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med (2004) 199:1607–18. doi:10.1084/jem.20040317
45. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
46. Senhaji N, Kojok K, Darif Y, Fadainia C, Zaid Y. The contribution of CD40/CD40L axis in inflammatory bowel disease: an update. Front Immunol (2015) 6:529. doi:10.3389/fimmu.2015.00529
47. Summers deLuca L, Gommerman JL. Fine-tuning of dendritic cell biology by the TNF superfamily. Nat Rev Immunol (2012) 12:339–51. doi:10.1038/nri3193
48. Shimizu K, Asakura M, Shinga J, Sato Y, Kitahara S, Hoshino K, et al. Invariant NKT cells induce plasmacytoid dendritic cell (DC) cross-talk with conventional DCs for efficient memory CD8+ T cell induction. J Immunol (2013) 190:5609–19. doi:10.4049/jimmunol.1300033
49. Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol (2010) 11:313–20. doi:10.1038/ni.1848
50. Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S. Cross-presentation of glycolipid from tumor cells loaded with a-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med (2007) 204:2641–53. doi:10.1084/jem.20070458
51. Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, Schmieg J, Kronenberg M. Natural killer T cell ligand alpha-galactosylceramide enhances protective immunity induced by malaria vaccines. J Exp Med (2002) 195:617–24. doi:10.1084/jem.20011889
52. Shimizu K, Goto A, Fukui M, Taniguchi M, Fujii S. Tumor cells loaded with α-galactosylceramide Induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J Immunol (2007) 178:2853–61. doi:10.4049/jimmunol.178.5.2853
53. Porcelli SA, Modlin RL. The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol (1999) 17:297–329. doi:10.1146/annurev.immunol.17.1.297
54. Hunn MK, Farrand KJ, Broadley KW, Weinkove R, Ferguson P, Miller RJ, et al. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin Cancer Res (2012) 18:6446–59. doi:10.1158/1078-0432.CCR-12-0704
55. Kobayashi T, Doff BL, Rearden RC, Leggatt GR, Mattarollo SR. NKT cell-targeted vaccination plus anti-4-1BB antibody generates persistent CD8 T cell immunity against B cell lymphoma. Oncoimmunology (2015) 4:e990793. doi:10.4161/2162402X.2014.990793
56. Shimizu K, Mizuno T, Shinga J, Asakura M, Kakimi K, Ishii Y, et al. Vaccination with antigen-transfected, NKT cell ligand-loaded, human cells elicits robust in situ immune responses by dendritic cells. Cancer Res (2013) 73:62–73. doi:10.1158/0008-5472.CAN-12-0759
57. Sun R, Liu Y. Listeriolysin O as a strong immunogenic molecule for the development of new anti-tumor vaccines. Hum Vaccin Immunother (2013) 9:1058–68. doi:10.4161/hv.23871
58. Wood LM, Paterson Y. Attenuated Listeria monocytogenes: a powerful and versatile vector for the future of tumor immunotherapy. Front Cell Infect Microbiol (2014) 4:51. doi:10.3389/fcimb.2014.00051
59. Singh M, Quispe-Tintaya W, Chandra D, Jahangir A, Venkataswamy MM, Ng TW, et al. Direct incorporation of the NKT-cell activator alpha-galactosylceramide into a recombinant Listeria monocytogenes improves breast cancer vaccine efficacy. Br J Cancer (2014) 111:1945–54. doi:10.1038/bjc.2014.486
60. Ng TW, Saavedra-Avila NA, Kennedy SC, Carreno LJ, Porcelli SA. Current efforts and future prospects in the development of live mycobacteria as vaccines. Expert Rev Vaccines (2015) 14:1493–507. doi:10.1586/14760584.2015.1089175
61. Bollampalli VP, Harumi Yamashiro L, Feng X, Bierschenk D, Gao Y, Blom H, et al. BCG skin infection triggers IL-1R-MyD88-dependent migration of EpCAMlow CD11bhigh skin dendritic cells to draining lymph node during CD4+ T-Cell priming. PLoS Pathog (2015) 11:e1005206. doi:10.1371/journal.ppat.1005206
62. Venkataswamy MM, Ng TW, Kharkwal SS, Carreno LJ, Johnson AJ, Kunnath-Velayudhan S, et al. Improving Mycobacterium bovis bacillus Calmette-Guerin as a vaccine delivery vector for viral antigens by incorporation of glycolipid activators of NKT cells. PLoS One (2014) 9:e108383. doi:10.1371/journal.pone.0108383
Keywords: dendritic cells, invariant NKT cells, innate immunity, adaptive immunity, cross presentation, immunotherapy
Citation: Fujii S and Shimizu K (2017) Exploiting Antitumor Immunotherapeutic Novel Strategies by Deciphering the Cross Talk between Invariant NKT Cells and Dendritic Cells. Front. Immunol. 8:886. doi: 10.3389/fimmu.2017.00886
Received: 29 May 2017; Accepted: 11 July 2017;
Published: 31 July 2017
Edited by:
Tonya J. Webb, University of Maryland, Baltimore, United StatesReviewed by:
Pooja Arora, Albert Einstein College of Medicine, United StatesUrsula Grohmann, University of Perugia, Italy
Copyright: © 2017 Fujii and Shimizu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shin-ichiro Fujii, c2hpbi1pY2hpcm8uZnVqaWlAcmlrZW4uanA=