 Rachel Byng-Maddick1,2†
Rachel Byng-Maddick1,2† Carolin T. Turner1†
Carolin T. Turner1† Gabriele Pollara1Matthew Ellis3
Gabriele Pollara1Matthew Ellis3 Naomi J. Guppy4Lucy C. K. Bell1
Naomi J. Guppy4Lucy C. K. Bell1 Michael R. Ehrenstein2,5†
Michael R. Ehrenstein2,5† Mahdad Noursadeghi1,5*†
Mahdad Noursadeghi1,5*†
- 1Division of Infection and Immunity, University College London, London, United Kingdom
- 2Division of Medicine, University College London, London, United Kingdom
- 3Division of Neuropathology, Institute of Neurology, University College London, London, United Kingdom
- 4UCL Advanced Diagnostics, London, United Kingdom
- 5National Institute for Health Research, University College London Hospitals, Biomedical Research Centre, London, United Kingdom
The impact of anti-tumor necrosis factor (TNF) therapies on inducible TNF-dependent activity in humans has never been evaluated in vivo. We aimed to test the hypothesis that patients responding to anti-TNF treatments exhibit attenuated TNF-dependent immune responses at the site of an immune challenge. We developed and validated four context-specific TNF-inducible transcriptional signatures to quantify TNF bioactivity in transcriptomic data. In anti-TNF treated rheumatoid arthritis (RA) patients, we measured the expression of these biosignatures in blood, and in skin biopsies from the site of tuberculin skin tests (TSTs) as a human experimental model of multivariate cell-mediated immune responses. In blood, anti-TNF therapies attenuated TNF bioactivity following ex vivo stimulation. However, at the site of the TST, TNF-inducible gene expression and genome-wide transcriptional changes associated with cell-mediated immune responses were comparable to that of RA patients receiving methotrexate only. These data demonstrate that anti-TNF agents in RA patients do not inhibit inducible TNF activity at the site of an acute inflammatory challenge in vivo, as modeled by the TST. We hypothesize instead that their therapeutic effects are limited to regulating TNF activity in chronic inflammation or by alternative non-canonical pathways.
Introduction
Tumor necrosis factor (TNF) is a pleiotropic cytokine that is transcriptionally activated in response to a variety of stimuli during inflammation, infection, and stress. Its non-redundant role in the immune system is apparent in TNF-deficient mice, which have no phenotypic abnormalities in the steady state, but are unable to mount organized immune responses and thus suffer from an increased susceptibility to infection (1–4). Upon cellular activation, TNF is mostly produced by mononuclear phagocytes but is also expressed by neutrophils, lymphocytes, endothelial cells, and fibroblasts (5, 6). The membrane-bound precursor and the cleaved, soluble form of TNF are both biologically active and exert their effects via two structurally distinct transmembrane glycoprotein receptors. TNF receptor (TNFR) 1 is expressed on almost all nucleated cells and TNFR2 (p75) expression is restricted to cells of the immune system (7). Engagement of these receptors initiates intracellular signaling pathways leading to transcription of TNF-responsive genes, which in turn regulate cell proliferation and apoptosis or induce pro-inflammatory mediators (5, 8, 9).
Excessive TNF activity contributes to the complex pathogenesis of rheumatoid arthritis (RA) (10), associated with a pro-inflammatory cascade that includes the production of IL-1 and IL-6, and drives tissue destruction (11). The use of anti-TNF therapies in RA has substantially improved the outcome and clinical course of the disease (12). The five licensed TNF inhibitors comprise the anti-TNF antibodies infliximab, adalimumab, and golimumab, the TNFR2 Fc fusion protein etanercept (ETN), and the pegylated Fab fragment certolizumab. All of these were developed to competitively inhibit the binding of TNF to its cognate cellular receptors and consequently block its biological activity. However, a comprehensive characterization of their in vivo inhibitory effect on TNF activity has yet to emerge. Variable effects on the level of TNF itself in serum or synovium of RA patients have been described, which do not necessarily correlate with the clinical response to anti-TNF therapy (13–16). In general, the level of pro-inflammatory mediators in serum and synovium, and pro-inflammatory cytokine production by peripheral blood mononuclear cells (PBMC) is reduced following anti-TNF therapy (17–19). While these data suggest that anti-TNF therapies ameliorate the immunopathogenesis of RA, they do not reveal the contexts in which anti-TNF therapies regulate TNF activity. We do not know if they block inducible TNF activity in both acute and chronically inflamed tissues or if they mediate their effects by blocking TNF in the circulation or hematopoetic compartments, where TNF may have important roles in shaping systemic immune responses. These gaps in our knowledge limit further refinement of biological therapies for inflammatory diseases. Moreover, we have described at least one indirect mechanism of action, in which anti-TNF antibodies unexpectedly promoted an interaction between membrane-bound TNF on monocytes and TNFR2 on regulatory T cells leading to enhanced Treg activity that may contribute to disease control (20, 21). ETN also binds and neutralizes lymphotoxin α (LTA) (22), suggesting another putative mechanism for non-canonical effects of anti-TNF agents.
A well-recognized complication of anti-TNF therapy is increased susceptibility to granulomatous infections, especially with Mycobacterium tuberculosis (Mtb) (23, 24), in which cell-mediated immune responses are thought to represent the principal mechanism of host defense (25). The role of TNF in immune protection against tuberculosis was primarily derived from observations in TNFR deficient mice, which do not assemble well-formed granuloma (26, 27). This observation was replicated by administration of anti-TNF agents in wild type mice (28, 29). Consequently, increased risk of tuberculosis associated with anti-TNF therapy is also widely interpreted to be due to deficient TNF activity in cell-mediated immune protection, but direct evidence for this is lacking. Interestingly, anti-TNF antibodies such as infliximab and adalimumab invoke significantly greater risk of active tuberculosis in man, than the soluble TNFR, ETN (30–32). Possible mechanisms for the differential risk is reported to be apoptosis of monocytes and activated T cells (33–35), or depletion of Mtb reactive CD8 T cells by antibody binding to membrane TNF (36).
We have previously described transcriptional profiling at the site of the tuberculin skin test (TST) to make molecular and systems level assessments of in vivo human immune responses at the site of a standardized experimental challenge (37, 38). Clinical inflammation in the TST has been widely used as a surrogate for T cell memory for mycobacterial antigens (39), but transcriptional profiling of biopsies from the injection site reflects all the components of integrated innate and adaptive immune responses, each of which can be quantified with independently derived transcriptional modules (38, 40). Importantly, this approach also revealed immune responses in the absence of clinically evident inflammatory induration, allowing unprecedented sensitivity to measure immune responses that were previously described as anergic (37, 38). In the present study, we aimed to test the hypothesis that anti-TNF treated RA patients will exhibit attenuated TNF-dependent transcriptional responses at the site of the TST, and consequently evaluate the role of TNF in genome-wide assessments of cell-mediated immune responses.
Materials and Methods
Study Approval
This study was approved by UK National Research Ethics Service (reference no: 11/LO/1863).
Study Population and Sampling
Healthy volunteers and adult patients with RA, treated with methotrexate (MTX), adalimumab, infliximab, or ETN, were invited to participate subject to selected criteria (Table 1). Written informed consent was obtained from all participants. Disease activity in RA patients was assessed using the Disease Activity Score of 28 commonly involved joints in RA (DAS28), comprising the number of swollen and tender joints, the erythrocyte sedimentation rate, and a visual analog scale of the patient’s subjective perception of pain marked out of 100 (41, 42).
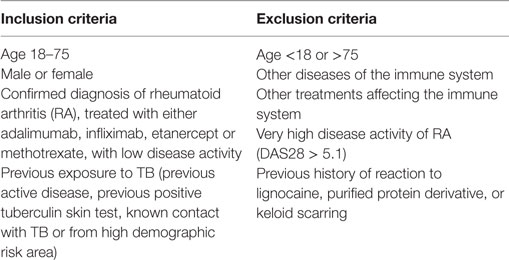
Table 1. Inclusion and exclusion criteria for study participants.
Participants with positive peripheral blood IFNγ release assays as evidence of antimycobacterial T cell memory (see below) received either intradermal TST or saline injections in the volar aspect of the forearm as previously described (37, 38). At 72 h, the maximum diameter of inflammatory induration was measured, and two 3 mm adjacent punch biopsies were obtained over the center of the injection sites. In addition, whole blood was taken from further participants in each study group for ex vivo stimulation with 10 ng/mL recombinant human TNF (Life Technologies). Demographic, clinical, and laboratory data for each study group are summarized in Table 2 and for each participant in Data Sheet S3 in Supplementary Material, including all available data on previous blood IFNγ release assays or TB treatment. No participants had any evidence of active TB or received any treatment for TB during their participation in the study.
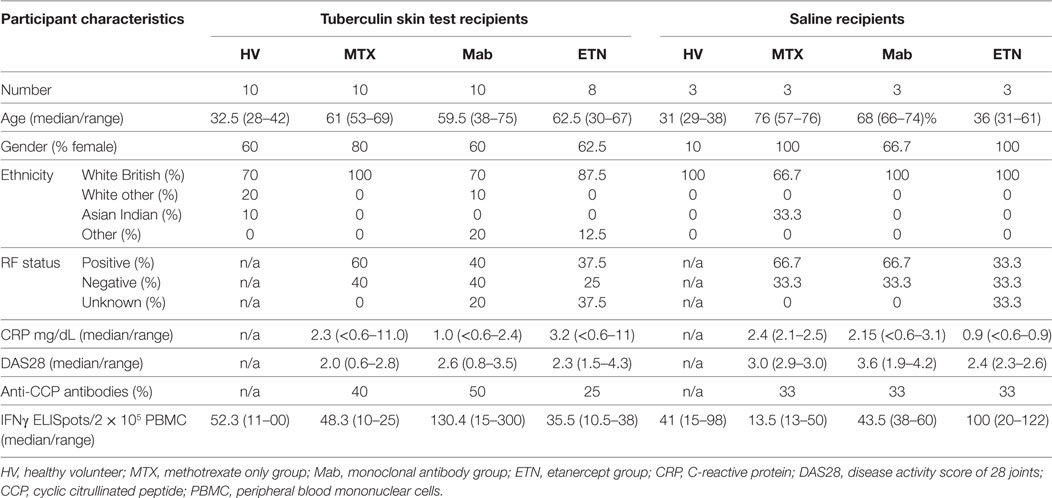
Table 2. Summary data for study participants undergoing skin tests.
Peripheral Blood IFNγ Release Assays
Participants were screened for antimycobacterial T cell memory by peripheral blood IFNγ release assays using QuantiFERON-TB Gold tests (Qiagen) according to the manufacturer’s instructions or IFNγ ELISpot responses to purified protein derivative (PPD, Statens Serum Institute). Briefly, PBMC were obtained by density gradient centrifugation of heparinized whole blood with Ficoll-Paque PLUS (GE Healthcare Biosciences). Sterile, clear 96-well filter plate with a PVDF base membrane (Merck Millipore) were coated with 1 µg/mL antihuman IFNγ antibody (eBioscience) for 24 h at 4°C, prior to the addition of 2 × 105 PBMC per well and PPD 10 µg/mL (SSI, Denmark), soluble anti-CD3 (HIT 3a) (eBioscience) as a positive control, or vehicle control. Plates were incubated for 48 h at 37°C. IFNγ production was detected using 1 µg/mL of biotinylated detection antibody (eBioscience), 1 µg/mL of streptavidin alkaline phosphatase (ALP) conjugate (Calbiochem) and BCIP/NBT substrate for ALP (Merck). The reaction was allowed to develop in the dark and stopped by addition of distilled water. Reactive foci were counted using AID ELISpot Software version 5.0 on the AID ELISpot Reader.
Whole Genome Transcriptional Profiling and Analysis
Total RNA from peripheral blood, monocyte-derived macrophages (MDM), and skin biopsy samples was purified for genome-wide transcriptional profiling as previously described (37, 38). All novel gene expression data are available on ArrayExpress1 under accession numbers E-MTAB-5095 [lipopolysaccharide (LPS)/ETN-MDM module], E-MTAB-5094 (whole blood stimulation), and E-MTAB-5093 (skin biopsies). Differential gene expression was assessed in MultiExperiment Viewer v4.9.0.2 Two-tailed t-tests were used to derive TNF modules from ex vivo and cell culture experiments. Mann–Whitney tests were used to compare TST and saline injection sites. A p value of <0.05 was considered significant. This statistical threshold to identify significant differentially expressed genes was complemented by using a twofold change filter and statistical enrichment in upstream regulator analysis, performed using Ingenuity Pathway Analysis (Qiagen); and pathway analysis using InnateDB,3 visualized as network diagram in Gephi v0.8.2 beta.4 Principal component analysis was performed using the prcomp function in R.5
Derivation of Transcriptional Modules
Derivation and validation of the TNF-MDM module was previously described (38). To generate the LPS/ETN-MDM module, human blood MDM cultures (43) were primed for 1 h ± 10 μg/mL ETN (Pfizer) before ultra-pure LPS (100 ng/mL; Invivogen) stimulation for 24 h (n = 3 per group). Transcriptional profiling of stimulated MDM was then performed as previously reported (38). LPS-inducible genes (>2-fold) were defined by comparison with transcriptional data from unstimulated MDM (n = 17). Among these LPS response genes, those that were attenuated significantly (>2-fold) in the presence of ETN were used as the LPS/ETN-MDM module. To derive a TNF-specific module in keratinocytes (TNF-KC module), we made use of previously published transcriptomic data from primary human keratinocytes (KCs) stimulated with a selection of cytokines, including TNF (44). Significant transcriptional responses (>4-fold) to 24 h TNF stimulation (10 ng/mL) were identified by comparison to unstimulated KCs, excluding genes that were also upregulated (>2-fold) by IFNγ, IL17 or IL22. The TNF-blood module was obtained by comparison of transcriptomes from whole blood (n = 4 healthy volunteers) incubated at 37°C for 3 h ±10 ng/mL recombinant human TNF, to identify transcripts that were significantly upregulated by >2-fold. The gene lists that make up each TNF module are listed in Data Sheet S1 in Supplementary Material. A human T cell-specific transcriptional signature (module M19) (45) and gene lists representing IFNγ activity and responses to Mtb (38, 40) have been published previously. Module scores represent the geometric mean log2 expression of all contributing genes and module enrichment represents the difference in module scores between data from TST and saline injection sites.
Histology and Immunohistochemistry of Skin Biopsy Specimens
Punch skin biopsies for histological analysis were snap frozen in OCT Compound (Tissue-Tek). Frozen sections were carefully thawed and fixed in 4% neutral buffered formalin, then embedded in paraffin wax (Sakura). 3 µM sections were cut and stained using the Leica Bond III automated immunostaining platform, with the Leica Bond Polymer Refine detection kit (Leica DS9800) and a DAB chromogen. More specifically, antihuman antibodies against CD3 (clone LN10) (Leica NCL-L-CD3-565) and matrix metalloproteinase (MMP) 9 (clone 56-2A4) (Millipore MAB3309) were used. Whole slide images of the histology sections were acquired with an Axio-Scan microscope using Zen 2 core software at ×20 magnification and are presented without any subsequent processing. Digital image analysis was performed using Definiens AG (Munich) Tissue Studio 4.3 software. Tissue detection automatically identified all the tissue within each image, then a machine learning method was used to separate the sample from background and non-tissue regions, and segment the sample into dermis and epidermis; manual correction was used to ensure valid separation of these regions of interest (ROI). A fixed threshold was then applied to each ROI to identify the chromogen positive areas (μm2), which is represented as a percentage of the total tissue/ROI area.
Results
Derivation of TNF-Inducible Transcriptional Modules
Current anti-TNF therapies aim to block the bioactivity of TNF rather than expression or secretion of TNF. Therefore, measurement of TNF function rather than TNF levels is required to assess the effects of anti-TNF therapies. TNF exerts its biological functions via stimulation of the TNFRs and intracellular signaling cascades that converge on regulation of transcription factors and consequently gene expression (46). Our first aim was to derive and validate gene expression modules, which could be used to detect and quantify functional TNF activity. In MDM, we had previously reported one such module that discriminated cellular responses to stimulation with recombinant TNF (TNF-MDM module) from a selection of other cytokines including IFNγ, IL4, and IL13 (38). In the present study we reasoned that the transcriptional response to TNF stimulation may be influenced by its cellular context or the presence of other inflammatory mediators. Any individual gene expression module may be inadequate to detect all TNF activity. Therefore, we derived three novel TNF-inducible gene expression modules by alternative approaches.
We first sought to identify gene expression attributable to TNF in the presence of other bioactive cytokines. We used innate immune stimulation of MDM with bacterial LPS as a prototypic stimulus to invoke secretion of a wide range of immunologically active factors, including TNF, contributing to LPS-inducible gene expression by their downstream autocrine and paracrine activity. To identify gene expression attributable to LPS-induced TNF, we compared the transcriptome of MDM 24 h after LPS stimulation in the presence and absence of ETN. Since ETN blocks the activity of LTA as well as that of TNF, we analyzed previously published data obtained from LPS stimulation in the same MDM model for induction of TNF and LTA expression (47, 48). As expected LPS induced robust upregulation of TNF expression in the primary transcriptional response (Figure 1A). This response would be expected to drive TNF-dependent gene expression changes at subsequent time points. In contrast, there was no induction of LTA. Therefore, gene expression attenuated by ETN in LPS-stimulated MDM unequivocally reflected endogenous TNF activity. We found significantly lower expression of 77 genes in the presence of ETN (Figure 1B), which we infer to be TNF responsive. We refer to these genes as the LPS/ETN-MDM module. In addition, we tested the effect of anti-TNF therapies on functional TNF activity in blood and derived a TNF gene expression module by identifying significantly upregulated genes following ex vivo whole blood stimulation with TNF (Figure 1C). We refer to this as the TNF-blood module. Our ultimate aim was to evaluate TNF activity in response to an in vivo immunological challenge using the TST (37, 38). Differential transcriptional responses to the same stimulus in different cell types are widely recognized (49). Therefore, we took advantage of previously published transcriptomic data from KCs stimulated with a selection of cytokines including TNF (44) to derive a TNF-specific module in keratinocytes (TNF-KC module) (Figure 1D).
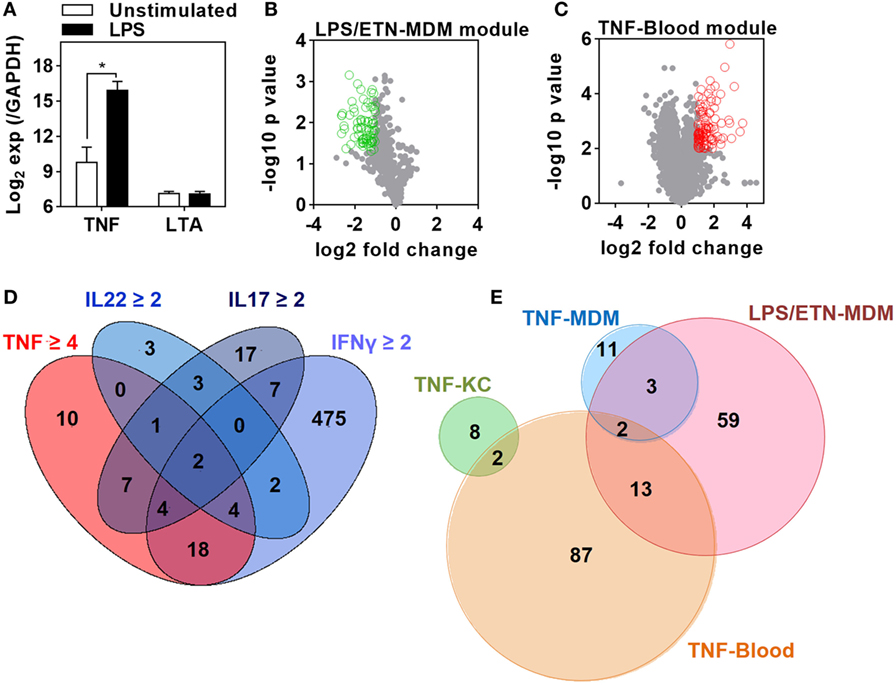
Figure 1. Derivation of transcriptional modules to measure tumor necrosis factor (TNF) activity. (A) Relative expression of TNF and lymphotoxin α (LTA) in monocyte-derived macrophage (MDM) cultures before and after 3 h stimulation with lipopolysaccharide (LPS) 100 ng/mL derived from previously published microarray data (ArrayExpress accession No: E-MEXP-1904 and E-MEXP-2032). Bars represent mean + SD (n = 3–5) and *p < 0.05, t-test. (B) Volcano plot of the effect of etanercept (ETN) on LPS-induced genes in MDM after 24 h. Open circles (green) represent 77 genes that were significantly (p < 0.05, t-test) attenuated (>2-fold) in the presence of ETN to give the “LPS/ETN-MDM” module. (C) Transcriptional response to TNF (10 ng/mL for 24 h) in ex vivo whole blood stimulation experiments. Open circles (red) represent 104 genes that were significantly (p < 0.01, t-test) upregulated (>2-fold) in response to TNF to give the “TNF-blood” module. (D) 4-way Venn diagram showing the overlap of keratinocyte (KC) genes significantly (p < 0.05, t-test) upregulated (>2-fold by IFNγ, IL22, IL17, or 4-fold by TNF) after 24 h stimulation. The 10 TNF-specific genes were used to give the “TNF-KC” module. (E) Venn diagram of the genes making up each TNF transcriptional modules, derived as described in panels (B–D) and by Bell et al. (“TNF-MDM”).
Direct comparison of the genes included in each of the four modules (Data Sheet S1 in Supplementary Material) associated with TNF activity showed only modest overlap (Figure 1E). Cross-validation of each gene list in the different experiments which were used to derive them also highlighted their context specificity. Modules derived in MDM were not enriched in TNF-stimulated KCs and modules derived in KCs were not enriched in TNF- or LPS-stimulated MDM (Figure 2). Importantly, however, independent bioinformatic analysis of predicted upstream regulators of each of these gene lists showed statistically robust associations with TNF (Data Sheet S2 in Supplementary Material).
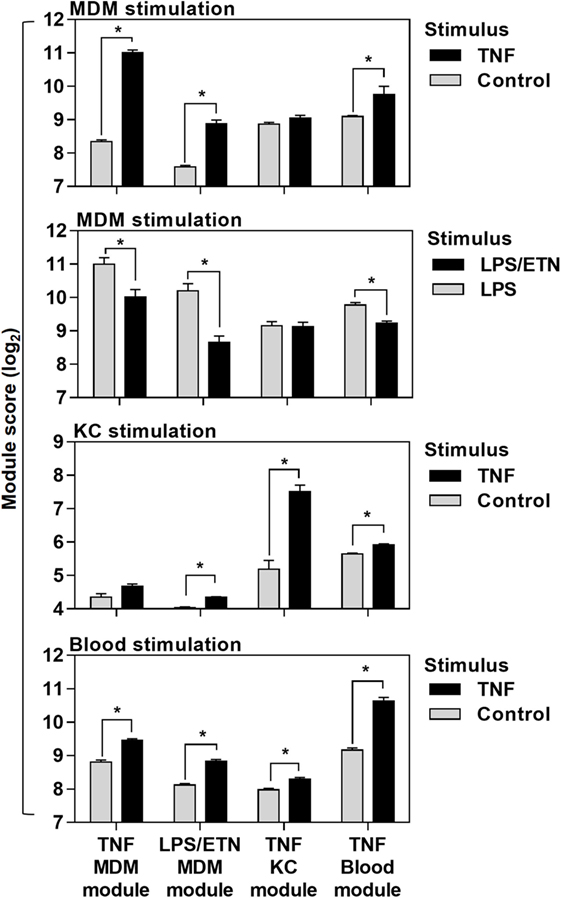
Figure 2. Cross-validation of transcriptional tumor necrosis factor (TNF) modules in the different experiments they were derived from. Module scores, calculated as geometric mean expression of the genes contributing to each module, in genome-wide transcriptomic data from monocyte-derived macrophages (MDM), keratinocytes (KC), and blood stimulated with ±TNF or lipopolysaccharide (LPS)-stimulated MDM ± etanercept (ETN). Bars represent mean + SD (n = 3–5). *p < 0.05, t-test.
Anti-TNF Therapy Attenuates TNF-Inducible Gene Expression in Blood
Previous reports have interpreted the reduction in circulating pro-inflammatory mediators following initiation of anti-TNF therapies in RA as evidence for inhibition of TNF activity (17–19). Therefore, we reasoned that anti-TNF therapy would be associated with steady state reduction of TNF-dependent gene expression in blood and would attenuate upregulation of these gene expression modules following ex vivo stimulation with TNF. We quantified expression of the blood-TNF module before and after ex vivo TNF stimulation of blood from RA patients treated with anti-TNF therapies and compared these to blood samples from RA patients on MTX only. To avoid the confounding of differential disease activity, we restricted recruitment to patients on a stable treatment regimen for at least three months and who showed low levels of background inflammation, using C-reactive protein and the RA disease activity score of 28 joints (DAS28) (Table 2; Data Sheet S3 in Supplementary Material). There were no significant differences in DAS28 scores between RA patients in the different treatment groups (Figure 3A), as a biomarker of disease activity (42). All had a reduction of DAS28 score of >1.2 from pretreatment levels, thereby allowing them to continue on their current treatment regimen. None had a residual DAS28 score >5.1, which would represent ongoing highly active disease. In addition, the proportions of anti-cyclic citrullinated peptide antibody as a prognostic biomarker of disease (50) were not significantly different between the groups (Table 2).
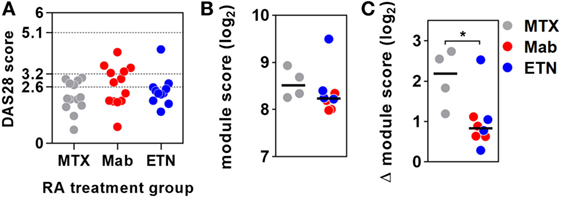
Figure 3. Anti-tumor necrosis factor (TNF) therapy attenuates TNF-inducible gene expression in blood. (A) Distribution of DAS28 scores in rheumatoid arthritis (RA) patients treated with methotrexate (MTX) only, anti-TNF antibodies (Mab), or etanercept (ETN). Dotted lines represent threshold values for disease in remission (<2.6), mild disease activity (<3.2), and moderate disease activity (<5.1). Transcriptional TNF activity, represented by the blood-derived TNF gene expression module score, (B) in peripheral blood of RA patients treated with MTX (n = 4), monoclonal anti-TNF antibodies (n = 4), or ETN (n = 4) at baseline, and (C) the change (Δ) in module score associated with TNF stimulation (10 ng/mL for 3 h). Data points were derived from individual experiments. Line represents the median. *p < 0.05, Mann–Whitney test.
Baseline levels of TNF-dependent gene expression were not significantly different in blood from RA patients on anti-TNF therapies compared to that of patients on MTX (Figure 3B). Ex vivo whole blood stimulation with TNF upregulated the expression of each of the TNF-dependent modules in all groups of patients, but this increase was significantly attenuated in patients on anti-TNF therapies (Figure 3C). In addition, we found that TNF was among the top predicted upstream regulators of all genes significantly attenuated by anti-TNF therapy in TNF-stimulated blood (Data Sheet S2 in Supplementary Material). Therefore, we concluded that in keeping with their expected effect, anti-TNF therapies attenuated the functional response to circulating TNF, which we modeled by ex vivo stimulation of peripheral blood.
Quantitation of TNF Activity in the TST
Next, we tested the effect of anti-TNF therapies on TNF activity at the site of an immune challenge in vivo. To do so, we undertook transcriptional profiling of skin biopsies from the site of TSTs in individuals with antimycobacterial T cell memory (Table 2; Data Sheet S3 in Supplementary Material). In healthy volunteers, the TST invokes a multivariate immune response that comprises innate immune responses, immune cell recruitment, and cytokine responses (Figure 4; Data Sheet S4 in Supplementary Material) (37, 38). This includes robust enrichment of TNF gene expression itself and of all four TNF-dependent gene expression modules derived from MDM, KCs, and blood cells, representing a broad range of TNF-responsive cells in the context of the TST (Figures 5A,B).
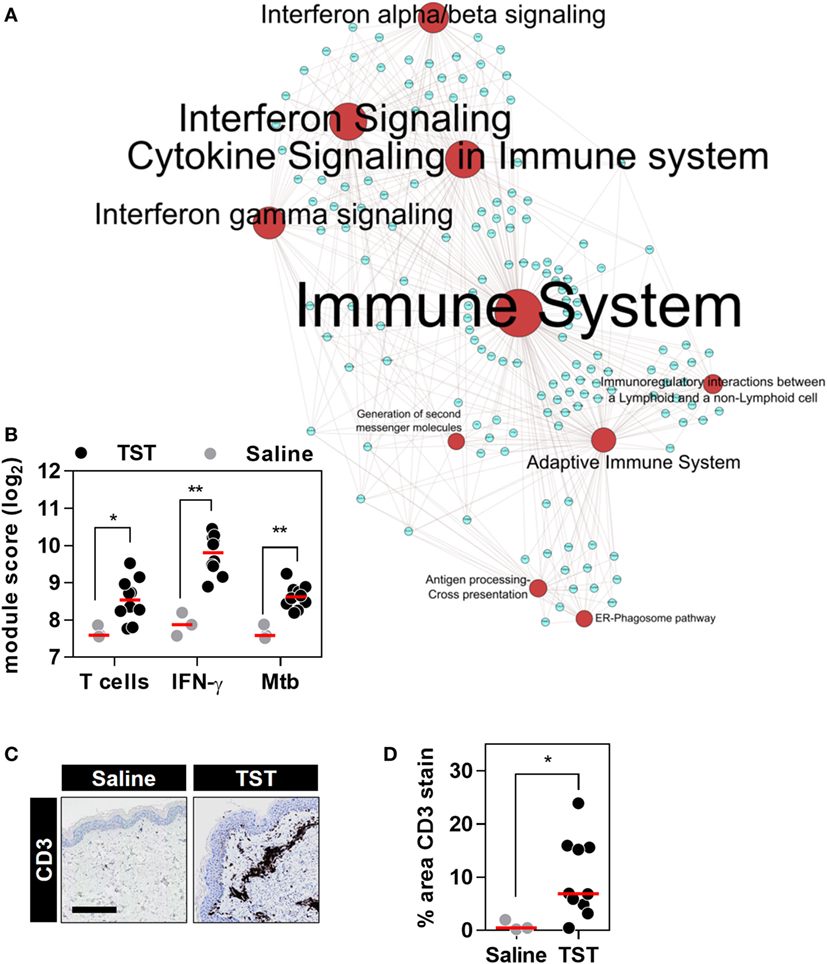
Figure 4. The transcriptional tuberculin skin test (TST) response of healthy volunteers. (A) Network graph of the top 10 Reactome pathways enriched among the genes upregulated in TST (n = 10) compared to saline (n = 3) samples from healthy volunteers. Red nodes represent pathways, and blue nodes represent genes. Node and label font size denote the significance of the respective pathway and are proportional to its −log10 p value. (B) Transcriptional module scores representing T cell recruitment, IFNγ activity, and responses to Mycobacterium tuberculosis (Mtb) in each biopsy. (C) Representative immunostaining of CD3 in skin biopsy samples from the site of TST and saline injections (scale bar = 500 µM). (D) Quantitation of CD3 immunostaining in each biopsy. Data points represent individual participants, and group medians are indicated by red lines. *p < 0.05, **p < 0.01 in Mann–Whitney tests.
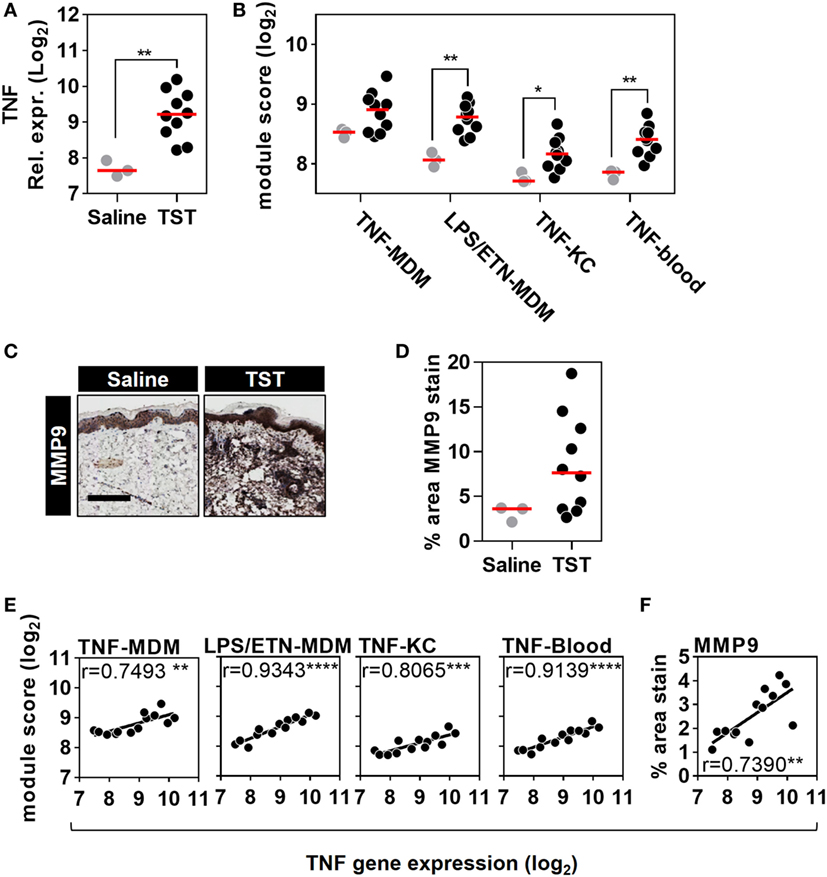
Figure 5. Quantitation of tumor necrosis factor (TNF) activity in the tuberculin skin test (TST) among healthy volunteers. (A) Relative TNF gene expression, (B) module scores of four separate TNF modules derived from genome-wide microarrays, and (C) representative immunostaining of matrix metalloproteinase (MMP) 9 in skin biopsy samples from the site of TST and saline injections (scale bar = 500 µM). (D) Positive immunostaining for matrix metalloproteinase (MMP) 9 in skin biopsies from the site of TST (n = 10) or control saline injections (n = 3) in healthy volunteers. Data points represent individual experiments. *p < 0.05, **p < 0.01, Mann–Whitney test. (E,F) Correlation between TNF expression and each of the TNF module scores or positive log2 MMP9 immunostaining. Correlation coefficients (r) and p values were derived from Pearson correlation analyses. **p < 0.01, ***p < 0.001, ****p < 0.0001.
To extend the assessment of TNF activity beyond transcriptional responses to the protein level, we also immunostained TST biopsy sites to quantify expression of the MMP9 (Figure 5C). MMP9 has been associated with TNF activity in previous studies (51). It is also a component of the TNF-KC module and therefore a specific protein target in the skin to discriminate between TNF and IFNγ activity (Figure 1D; Data Sheet S1 in Supplementary Material). Six of the ten healthy volunteers undergoing TST injection showed increased MMP9 protein expression compared to participants receiving control saline injections (Figure 5D). The variability in TNF modules and MMP9 immunostaining each demonstrated a statistically significant correlation with TNF transcript levels among different individuals (Figures 5E,F). These correlations consolidated the evidence for a functional relationship between TNF expression and the independently derived TNF-inducible transcriptional modules or MMP9 immunostaining. Hence, we conclude that the TST invokes a functional TNF response, within which we can quantify variation in TNF bioactivity.
Functional TNF Activity Is Preserved at the Site of an Immunological Challenge In Vivo Despite Anti-TNF Therapy
Rheumatoid arthritis patients treated with anti-TNF therapies or MTX only, had comparable peripheral blood IFNγ ELISpot responses to tuberculin/PPD stimulation (Figure 6A), indicating that these study groups had equivalent T cell memory responses for tuberculin irrespective of any differences in their past history of TB exposure or treatment. In these participants, we defined an integrated TST transcriptional signature. This signature comprised 595 genes that were significantly enriched in the TST compared to saline injection sites in at least one study group (Figure 6B). Bioinformatic analysis of these genes reflected the same immune pathways evident in the TST transcriptome of healthy individuals (Figure 6C).
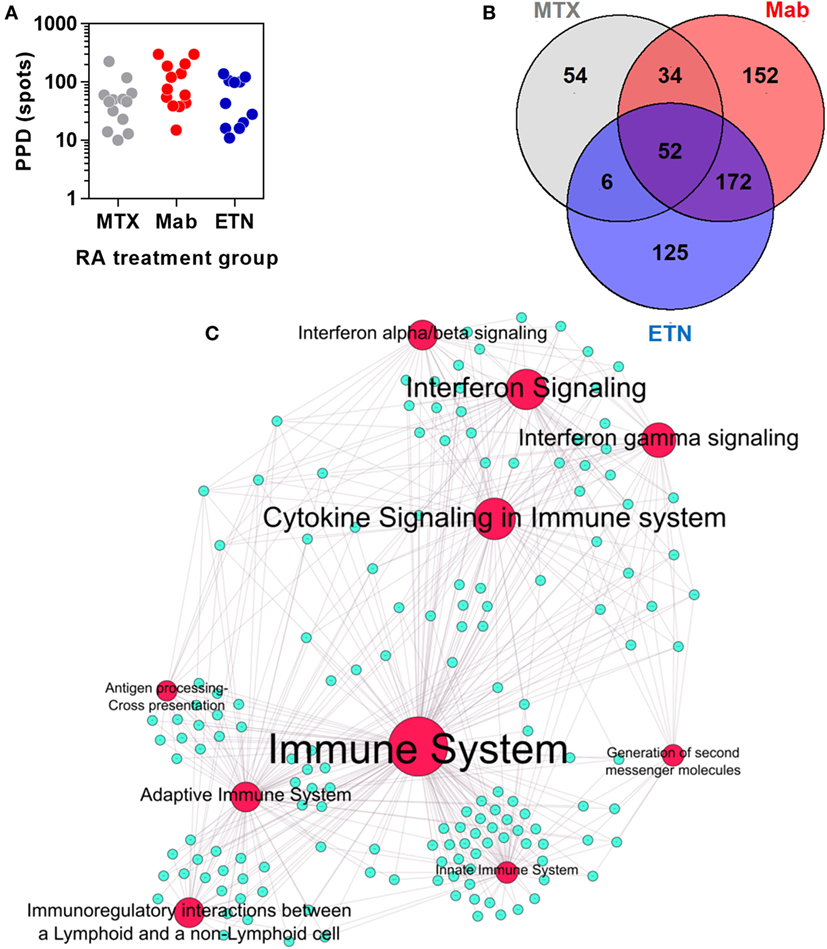
Figure 6. The transcriptional tuberculin skin test (TST) response of rheumatoid arthritis (RA) patients. (A) Distribution of quantitative IFNγ ELISpot results in RA patients treated with methotrexate (MTX) only, anti-TNF antibodies (Mab), or etanercept (ETN). Each data point represents an individual patient. (B) Number and overlap of genes upregulated in TST compared to saline samples in patients treated with MTX, monoclonal anti-TNF antibodies (Mab), or the soluble tumor necrosis factor (TNF) receptor ETN. The total number of genes displayed in this Venn diagram was combined in the integrated TST module. (C) Network graph of the top 10 Reactome pathways enriched in the integrated TST signature genes. Red nodes represent pathways, and blue nodes represent genes. Node and label font size denote the significance of the respective pathway and are proportional to its −log10 p value.
Principal component analysis of the integrated TST signature genes, as well as enrichment of T cell-associated and IFNγ-dependent gene expression modules, indicated that the major features of transcriptional responses in the TST of RA patients were not affected by anti-TNF therapy (Figure 7).
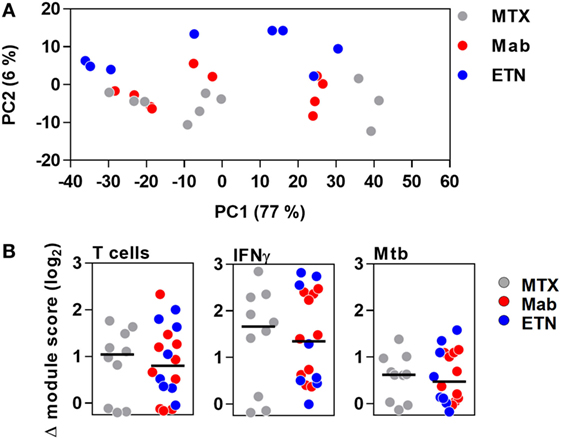
Figure 7. The transcriptional tuberculin skin test (TST) response in patients with rheumatoid arthritis. (A) Principal component (PC) analysis of integrated TST signature genes showed no treatment-specific clustering in PC1 and PC2, which together explained 83% of variation in this data set. Data points represent individual experiments. (B) Transcriptional modules representing enrichment of T cells, IFNγ activity, and innate immune responses to Mycobacterium tuberculosis (Mtb) in TST compared to saline baseline as shown by log2 module enrichment scores. Individual patients are represented with separate data points, and group medians are indicated by black lines. MTX, methotrexate; Mab, monoclonal anti-TNF antibodies; ETN, etanercept.
The TST response in RA patients included enrichment of TNF gene expression, which was similar in all patient groups and therefore not affected by anti-TNF therapy (Figure 8A). Surprisingly, however, comparable levels of the TNF-dependent gene expression modules were also observed in all the study groups (Figure 8B). Likewise, MMP9 immunostaining representing a protein biomarker of TNF activity was comparable in anti-TNF treated and control patients (Figures 8C,D). Therefore, we conclude that anti-TNF therapy in RA patients does not inhibit inducible TNF function during a prototypic cell-mediated immune response at the site of immune challenge.
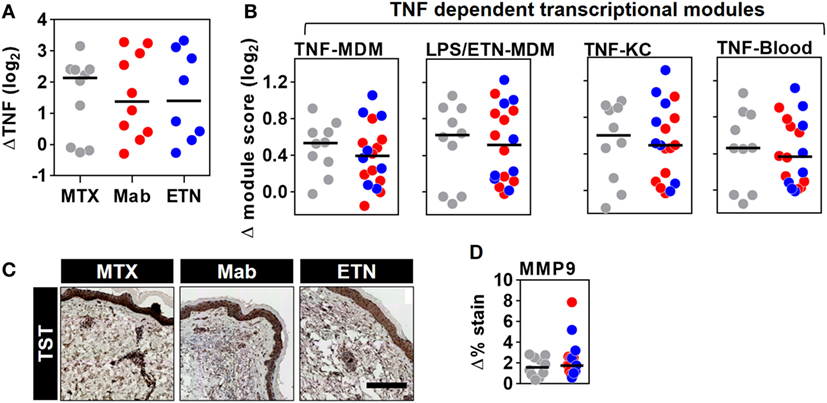
Figure 8. Inducible tumor necrosis factor (TNF) activity is preserved in the TST of rheumatoid arthritis (RA) patients despite anti-TNF therapy. The change in (A) TNF gene expression and (B) each of the four TNF-dependent gene expression modules in TST skin biopsies from RA patients treated with methotrexate (MTX, n = 10), monoclonal anti-TNF antibodies (Mab, n = 10), or etanercept (ETN, n = 8), compared to control saline injections (n = 3 per group). (C) Representative images of MMP9 immunohistochemical staining (scale bar = 500 μm) and (D) positive MMP9 staining in skin biopsies from the same patients. Data points represent individual experiments. Line represents the median. TST, tuberculin skin test; MDM, monocyte-derived macrophage; LPS, lipopolysaccharide; KC, keratinocyte; MMP9, matrix metalloproteinase 9.
Discussion
The rationale for the development of anti-TNF therapies in RA was based on local expression of TNF at the site of disease, our understanding of its functional role in pro-inflammatory immune cell recruitment to tissues and data from animal models in which over expression of human TNF caused a chronic inflammatory arthritis that could be ameliorated by anti-TNF therapy (52). In this context, the unequivocal therapeutic effects of anti-TNF antibodies and ETN in RA led to two conclusions. First, anti-TNF therapies mediate their beneficial effect by inhibiting TNF activity at the site of disease and consequently, TNF function makes an essential contribution to the immunopathogenesis of disease. Second, the same rationale applied to the detrimental effect of TNF inhibition, as increased risk of certain infections among patients on anti-TNF therapies (31, 53) suggested that TNF activity is an essential component of protective immune responses.
Several lines of evidence suggest that the effects of anti-TNF agents are not wholly understood. For example, it has become evident that anti-TNF antibodies and ETN have differential biological activity. Although they mediate similar therapeutic effects in RA, anti-TNF antibodies but not ETN are effective in other chronic inflammatory disease such as Crohn’s colitis (54). Likewise, the use of anti-TNF antibodies incurs significantly greater risk of active tuberculosis than that of ETN (30–32). In addition, we have previously shown that anti-TNF antibodies, but not ETN, induce increased emergence of functionally active regulatory T cells (20). Counterintuitively, this last example may be mediated by augmentation of TNF function through enhancing the interaction of membrane-bound TNF and TNFR2 (21). Therefore, the biological effects of anti-TNF therapies may extend beyond the canonical view that they block TNF activity at the site of immune responses in tissue. In fact, there are no experimental data that conclusively show this effect in man.
To test this question directly, we adopted a human experimental challenge approach using the TST as a standardized stimulus with which to invoke a focus of cell-mediated immune responses (37). Transcriptional profiling from skin punch biopsies from the site of the TST afforded us the opportunity to make comprehensive genome-wide assessments of immune responses at the molecular level with unprecedented sensitivity. We complemented this approach with the generation of independent experimentally derived TNF-dependent gene expression modules representing the functional bioactivity of TNF. Our evaluation of TNF activity at the transcriptional level revealed striking differences between TNF-dependent transcriptional responses in macrophages, blood cells, and KCs. The mechanisms underlying the context specificity in the functional activity of TNF likely reflect distinct epigenetic landscapes in different cell types (55). Despite the limited overlap between each of the four TNF-dependent gene expression modules we derived, in silico bioinformatics analysis yielded TNF as one of the top predicted upstream transactivator of each list of genes. These independently derived transcriptional modules associated with different cell types present at the site of the TST, namely, KCs, macrophages, and recruited blood leukocytes enhanced the sensitivity of our analysis and provided cross-validation.
In healthy subjects, we showed that TNF expression was enriched in the TST and that the variability in its enrichment correlated with concordant enrichment of each of the four TNF-dependent transcriptional modules. In addition, we extended this analysis to the protein level, by showing correlation with MMP9 as a TNF-inducible protein (56). Taken together, these data showed unequivocally that the TST invokes a functional TNF response and that we can quantify variability in TNF bioactivity in situ within this response.
To test the effects of anti-TNF agents, we sought to compare RA patients with quiescent disease on MTX to those receiving anti-TNF agents. In blood, baseline TNF bioactivity represented by the expression of TNF modules was not significantly different between RA patients on MTX and those on anti-TNF therapies. Therefore, our data suggest that the widely reported diminution of pro-inflammatory mediators in RA patients with quiescent disease following anti-TNF therapy may not be related to a targeted reduction in steady state expression of TNF-dependent gene expression but could simply represent successful control of active disease in patients on treatment with MTX or anti-TNF therapies, albeit through possibly divergent mechanisms. Nonetheless, we confirmed that enrichment of TNF-dependent gene expression following ex vivo TNF stimulation of whole blood was significantly attenuated in blood from patients on anti-TNF agents. These findings are in keeping with previous reports of neutralization of TNF bioactivity by plasma from patients receiving infliximab (57, 58). However, in the same groups of patients, receipt of anti-TNF agents did not attenuate the enrichment of the TNF-dependent transcriptional modules or MMP9 immunostaining within the TST. Hence, we concluded that although anti-TNF agents can inhibit TNF bioactivity in the circulation, they have no discernible effect on in vivo TNF activity at the site of an acute immune challenge.
Our data fundamentally challenge the long held view that anti-TNF agents mediate their therapeutic effect in RA by targeting TNF activity at the site of disease. We speculate that anti-TNF agents do not reach sufficient concentration within the tissue microenvironment to neutralize the level of TNF produced in an acute cell-mediated immune response. It remains possible that they can inhibit lower levels of TNF at foci of chronic inflammation. Alternatively, their therapeutic effects may be mediated by neutralizing the actions of circulating TNF, suggesting a previously unrecognized model by which TNF may contribute to the pathogenesis of RA. Further evaluation of this hypothesis may yield new mechanistic insights into the systemic immunopathology of RA and other inflammatory diseases amenable to anti-TNF therapies.
Ethics Statement
This study was approved by UK National Research Ethics Service (reference no: 11/LO/1863), and written informed consent was obtained from all participants.
Author Contributions
RB-M, MRE, and MN designed the experiments. RB-M, CT, NG, and LB conducted the experiments. RB-M, CT, GP, ME, and MN undertook the analysis. RB-M, CT, MRE, and MN wrote the paper with input from all other authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Joanne Williams and Victoria Howard (UCL) and the Clinical Research Facility at University College London Hospital for their help in patient recruitment and sampling. The authors also thank Dr. Henry Penn and Dr. Shahir Hamdulay for their help in patient recruitment from Northwick Park Hospital.
Funding
This work was supported by an Arthritis Research UK Clinical Research Fellowship awarded to RB-M (20131), a Wellcome Trust Fellowship to GP (WT101766/Z/13/Z), the Medical Research Council (MR/M003833/1), and the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.00932/full#supplementary-material.
Footnotes
References
1. Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med (1996) 184(4):1397–411. doi:10.1084/jem.184.4.1397
2. Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci U S A (1997) 94(15):8093–8. doi:10.1073/pnas.94.15.8093
3. Wellmer A, Gerber J, Ragheb J, Zysk G, Kunst T, Smirnov A, et al. Effect of deficiency of tumor necrosis factor alpha or both of its receptors on Streptococcus pneumoniae central nervous system infection and peritonitis. Infect Immun (2001) 69(11):6881–6. doi:10.1128/IAI.69.11.6881-6886.2001
4. Botha T, Ryffel B. Reactivation of latent tuberculosis infection in TNF-deficient mice. J Immunol (2003) 171(6):3110–8. doi:10.4049/jimmunol.171.6.3110
5. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther (2008) 117(2):244–79. doi:10.1016/j.pharmthera.2007.10.001
6. Brzustewicz E, Bryl E. The role of cytokines in the pathogenesis of rheumatoid arthritis – practical and potential application of cytokines as biomarkers and targets of personalized therapy. Cytokine (2015) 76(2):527–36. doi:10.1016/j.cyto.2015.08.260
7. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov (2010) 9(6):482–93. doi:10.1038/nrd3030
8. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol (2003) 3(9):745–56. doi:10.1038/nri1184
9. Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol (2005) 23:787–819. doi:10.1146/annurev.immunol.23.021704.115719
10. Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell (1996) 85(3):307–10. doi:10.1016/S0092-8674(00)81109-5
11. Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) (2012) 51(Suppl 5):v3–11. doi:10.1093/rheumatology/kes113
12. Ackermann C, Kavanaugh A. Tumor necrosis factor as a therapeutic target of rheumatologic disease. Expert Opin Ther Targets (2007) 11(11):1369–84. doi:10.1517/14728222.11.11.1369
13. Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Ishida H, et al. Long-term follow-up of the changes in circulating cytokines, soluble cytokine receptors, and white blood cell subset counts in patients with rheumatoid arthritis (RA) after monoclonal anti-TNF alpha antibody therapy. J Clin Immunol (1999) 19(5):305–13. doi:10.1023/A:1020543625282
14. Ulfgren AK, Andersson U, Engstrom M, Klareskog L, Maini RN, Taylor PC. Systemic anti-tumor necrosis factor alpha therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor alpha synthesis. Arthritis Rheum (2000) 43(11):2391–6. doi:10.1002/1529-0131(200011)43:11<2391:AID-ANR3>3.0.CO;2-F
15. Barrera P, Joosten LA, den Broeder AA, van de Putte LB, van Riel PL, van den Berg WB. Effects of treatment with a fully human anti-tumour necrosis factor alpha monoclonal antibody on the local and systemic homeostasis of interleukin 1 and TNFalpha in patients with rheumatoid arthritis. Ann Rheum Dis (2001) 60(7):660–9. doi:10.1136/ard.60.7.660
16. Edrees AF, Misra SN, Abdou NI. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: correlation of TNF-alpha serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin Exp Rheumatol (2005) 23(4):469–74.
17. Charles P, Elliott MJ, Davis D, Potter A, Kalden JR, Antoni C, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol (1999) 163(3):1521–8.
18. Schuerwegh AJ, Van Offel JF, Stevens WJ, Bridts CH, De Clerck LS. Influence of therapy with chimeric monoclonal tumour necrosis factor-alpha antibodies on intracellular cytokine profiles of T lymphocytes and monocytes in rheumatoid arthritis patients. Rheumatology (Oxford) (2003) 42(4):541–8. doi:10.1093/rheumatology/keg171
19. Schotte H, Schluter B, Willeke P, Mickholz E, Schorat MA, Domschke W, et al. Long-term treatment with etanercept significantly reduces the number of proinflammatory cytokine-secreting peripheral blood mononuclear cells in patients with rheumatoid arthritis. Rheumatology (Oxford) (2004) 43(8):960–4. doi:10.1093/rheumatology/keh178
20. Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNFa therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-B. J Exp Med (2007) 204(1):33–9. doi:10.1084/jem.20061531
21. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med (2016) 213(7):1241–53. doi:10.1084/jem.20151255
22. Mitoma H, Horiuchi T, Tsukamoto H, Ueda N. Molecular mechanisms of action of anti-TNF-alpha agents – comparison among therapeutic TNF-alpha antagonists. Cytokine (2016). doi:10.1016/j.cyto.2016.08.014
23. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha neutralising agent. N Engl J Med (2001) 345(15):1098–104. doi:10.1056/NEJMoa011110
24. Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis (2004) 38(9):1261–5. doi:10.1086/383317
25. van Crevel R, Ottenhoff TH, van der Meer JW. Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev (2002) 15(2):294–309. doi:10.1128/CMR.15.2.294-309.2002
26. Bean AGD, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol (1999) 162:3504–11.
27. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol (2002) 168(9):4620–7. doi:10.4049/jimmunol.168.9.4620
28. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity (1995) 2(6):561–72. doi:10.1016/1074-7613(95)90001-2
29. Plessner HL, Lin PL, Kohno T, Louie JS, Kirschner D, Chan J, et al. Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J Infect Dis (2007) 195(11):1643–50. doi:10.1086/517519
30. Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Breban M, et al. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: the three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum (2009) 60(7):1884–94. doi:10.1002/art.24632
31. Dixon WG, Hyrich KL, Watson KD, Lunt M, Galloway J, Ustianowski A, et al. Drug-specific risk of tuberculosis in patients with rheumatoid arthritis treated with anti-TNF therapy: results from the British Society for Rheumatology Biologics Register (BSRBR). Ann Rheum Dis (2010) 69:522–8. doi:10.1136/ard.2009.118935
32. Salmon-Ceron D, Tubach F, Lortholary O, Chosidow O, Bretagne S, Nicolas N, et al. Drug-specific risk of non-tuberculosis opportunistic infections in patients receiving anti-TNF therapy reported to the 3-year prospective French RATIO registry. Ann Rheum Dis (2011) 70(4):616–23. doi:10.1136/ard.2010.137422
33. Lugering A, Schmidt M, Lugering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology (2001) 121(5):1145–57. doi:10.1053/gast.2001.28702
34. ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut (2002) 50(2):206–11. doi:10.1136/gut.50.2.206
35. Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y, et al. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum (2005) 52(1):61–72. doi:10.1002/art.20764
36. Bruns H, Meinken C, Schauenberg P, Harter G, Kern P, Modlin RL, et al. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J Clin Invest (2009) 119:1167–77. doi:10.1172/JCI38482
37. Tomlinson GS, Cashmore TJ, Elkington PTG, Yates J, Lehloenya RJ, Tsang J, et al. Transcriptional profiling of innate and adaptive human immune responses to mycobacteria in the tuberculin skin test. Eur J Immunol (2011) 41:3253–60. doi:10.1002/eji.201141841
38. Bell LC, Pollara G, Pascoe M, Tomlinson GS, Lehloenya RJ, Roe J, et al. In vivo molecular dissection of the effects of HIV-1 in active tuberculosis. PLoS Pathog (2016) 12(3):e1005469. doi:10.1371/journal.ppat.1005469
39. Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA (1999) 282(7):677–86. doi:10.1001/jama.282.7.677
40. Pollara G, Murray MJ, Heather JM, Byng-Maddick R, Guppy N, Ellis M, et al. Validation of immune cell modules in multicellular transcriptomic data. PLoS One (2017) 12(1):e0169271. doi:10.1371/journal.pone.0169271
41. Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum (1995) 38(1):44–8. doi:10.1002/art.1780380107
42. van Riel PL, Renskers L. The disease activity score (DAS) and the disease activity score using 28 joint counts (DAS28) in the management of rheumatoid arthritis. Clin Exp Rheumatol (2016) 34(5 Suppl 101):S40–4.
43. Tomlinson GS, Bell LC, Walker NF, Tsang J, Brown JS, Breen R, et al. HIV-1 infection of macrophages dysregulates innate immune responses to Mycobacterium tuberculosis by inhibition of interleukin-10. J Infect Dis (2014) 209(7):1055–65. doi:10.1093/infdis/jit621
44. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol (2008) 159(5):1092–102. doi:10.1111/j.1365-2133.2008.08769.x
45. Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol (2014) 15(2):195–204. doi:10.1038/ni.2789
46. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol (2016) 12(1):49–62. doi:10.1038/nrrheum.2015.169
47. Noursadeghi M, Tsang J, Miller RF, Straschewski S, Kellam P, Chain BM, et al. Genome-wide innate immune responses in HIV-1-infected macrophages are preserved despite attenuation of the NF-kappa B activation pathway. J Immunol (2009) 182(1):319–28. doi:10.4049/jimmunol.182.1.319
48. Tsang J, Chain BM, Miller RF, Webb BL, Barclay W, Towers GJ, et al. HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS (2009) 23(17):2255–63. doi:10.1097/QAD.0b013e328331a4ce
49. Ishihara K, Hirano T. Molecular basis of the cell specificity of cytokine action. Biochim Biophys Acta (2002) 1592(3):281–96. doi:10.1016/S0167-4889(02)00321-X
50. Kastbom A, Strandberg G, Lindroos A, Skogh T. Anti-CCP antibody test predicts the disease course during 3 years in early rheumatoid arthritis (the Swedish TIRA project). Ann Rheum Dis (2004) 63(9):1085–9. doi:10.1136/ard.2003.016808
51. Tsai CL, Chen WC, Hsieh HL, Chi PL, Hsiao LD, Yang CM. TNF-alpha induces matrix metalloproteinase-9-dependent soluble intercellular adhesion molecule-1 release via TRAF2-mediated MAPKs and NF-kappaB activation in osteoblast-like MC3T3-E1 cells. J Biomed Sci (2014) 21:12. doi:10.1186/1423-0127-21-12
52. Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J (1991) 10(13):4025–31.
53. Galloway JB, Hyrich KL, Mercer LK, Dixon WG, Fu B, Ustianowski AP, et al. Anti-TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: updated results from the British Society for Rheumatology Biologics Register with special emphasis on risks in the elderly. Rheumatology (Oxford) (2011) 50(1):124–31. doi:10.1093/rheumatology/keq242
54. Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, et al. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology (2001) 121(5):1088–94. doi:10.1053/gast.2001.28674
55. Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res (2013) 23(3):555–67. doi:10.1101/gr.147942.112
56. Aota K, Azuma M. Targeting TNF-alpha suppresses the production of MMP-9 in human salivary gland cells. Arch Oral Biol (2013) 58(12):1761–8. doi:10.1016/j.archoralbio.2013.09.004
57. Marotte H, Maslinski W, Miossec P. Circulating tumour necrosis factor-alpha bioactivity in rheumatoid arthritis patients treated with infliximab: link to clinical response. Arthritis Res Ther (2005) 7(1):R149–55. doi:10.1186/ar1465
Keywords: tumor necrosis factor, anti-tumor necrosis factor, rheumatoid arthritis, tuberculin skin test, transcriptional profiling
Citation: Byng-Maddick R, Turner CT, Pollara G, Ellis M, Guppy NJ, Bell LCK, Ehrenstein MR and Noursadeghi M (2017) Tumor Necrosis Factor (TNF) Bioactivity at the Site of an Acute Cell-Mediated Immune Response Is Preserved in Rheumatoid Arthritis Patients Responding to Anti-TNF Therapy. Front. Immunol. 8:932. doi: 10.3389/fimmu.2017.00932
Received: 01 April 2017; Accepted: 21 July 2017;
Published: 04 August 2017
Edited by:
Pierre Miossec, Claude Bernard University Lyon 1, FranceCopyright: © 2017 Byng-Maddick, Turner, Pollara, Ellis, Guppy, Bell, Ehrenstein and Noursadeghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahdad Noursadeghi, bS5ub3Vyc2FkZWdoaUB1Y2wuYWMudWs=
†These authors have contributed equally to this work.