 Mariana Aris
Mariana Aris José Mordoh
José Mordoh María Marcela Barrio
María Marcela Barrio- 1Centro de Investigaciones Oncológicas - Fundación Cáncer, Buenos Aires, Argentina
- 2Instituto Médico Especializado Alexander Fleming, Buenos Aires, Argentina
- 3Fundación Instituto Leloir, IIBBA-CONICET, Buenos Aires, Argentina
In the last few years, there has been a twist in cancer treatment toward immunotherapy thanks to the impressive results seen in advanced patients from several tumor pathologies. Cutaneous melanoma is a highly mutated and immunogenic tumor that has been a test field for the development of immunotherapy. However, there is still a way on the road to achieving complete and long-lasting responses in most patients. It is desirable that immunotherapeutic strategies induce diverse immune reactivity specific to tumor antigens, including the so-called neoantigens, as well as the blockade of immunosuppressive mechanisms. In this review, we will go through the role of promising monoclonal antibodies in cancer immunotherapy with immunomodulatory function, especially blocking of the inhibitory immune checkpoints CTLA-4 and PD-1, in combination with different immunotherapeutic strategies such as vaccines. We will discuss the rational basis for these combinatorial approaches as well as different schemes currently under study for cutaneous melanoma in the clinical trials arena. In this way, the combination of “push and release” immunomodulatory therapies can contribute to achieving a more robust and durable antitumor immune response in patients.
Introduction
An important role for the immune system in cancer biology has been proposed for decades. However, immunotherapy has been very recently recognized as an effective treatment to control tumor growth and dissemination. Cutaneous melanoma (CM) is a highly mutated and immunogenic tumor and has been a rich field for the development of tumor immunology and immunotherapy (1–3). There is strong evidence of common tumor antigens (Ags) as well as patient’s own tumor neoantigens (neoAgs) which are recognized by the immune system (4); of tumor infiltration by specific immune populations and their clinical correlation (5); and of tumor immunoediting including immune escape strategies (6). More recently, the discovery of multiple immune checkpoint mechanisms, either stimulatory or inhibitory pathways of the immune system, which can be targeted with monoclonal antibodies (mAbs), has burst into intense clinical research. In particular, immune checkpoint blockade (ICKB) with mAbs immunotherapy has proved for the first time to improve overall survival (OS) of CM metastatic patients (7). Nowadays, immunotherapeutic approaches including ICKB with mAbs is the fourth cancer treatment modality along with surgery, radiotherapy, and chemotherapy/targeted therapy. The use of ICKB has expanded beyond CM and these therapies are now approved for the treatment of several metastatic tumors, including renal cell carcinoma, non-small-cell lung carcinoma, squamous cell carcinoma of the head and neck, urothelial carcinoma, and Hodgkin lymphoma (www.fda.gov/drugs).
The inhibitory immune checkpoint molecules act as physiological brakes that prevent potentially harmful immune responses and autoimmunity. Cytotoxic T-lymphocyte antigen 4 (CTLA-4) is constitutively expressed in regulatory T cells (Tregs) but is only upregulated in T-cells after activation. Physiologically, CTLA-4 transmits an inhibitory signal to T cells to shut down immune responses by interaction with CD80 and CD86 molecules, which are expressed at the surface of Ag-presenting cells (APCs) such as dendritic cells (DCs) and macrophages; CTLA-4 also contributes to the inhibitory function of Tregs (8). Ipilimumab (Bristol–Myers Squibb) targets CTLA-4, blocking the inhibitory signal, unleashing cytotoxic T cells to eliminate the cancer cells (7). Another inhibitory immune-checkpoint that has been extensively targeted with mAbs is the axis programmed cell death-1/CD279 (PD-1) and their ligands PD-L1 (B7-H1/CD274) and PD-L2 (B7-DC/CD273) (9). PD-1 is expressed on the membrane of activated T lymphocytes; PD-L1 and PD-L2 are expressed in APCs and also in some tumor cells. PD-1 and PD-L1/PD-L2 binding induce a coinhibitory signal that limits the development of the T-cell response. Several blocking mAbs targeting PD-1 are currently indicated to treat different tumors, such as nivolumab (Bristol–Myers Squibb) (10) and pembrolizumab (Merck Sharp & Dohme) (11). Accordingly, there are mAbs targeting PD-L1, such as atezolizumab (Roche), avelumab (EMD, Serono), and durvalumab (Astra Zeneca). Blocking of CTLA-4 molecule would affect the initial priming phase while targeting the PD-1/PD-L1 axis would interfere more profoundly with the effector phase of the anti-immune response. Although ICKB has shown to potentiate long-lasting antitumor immune response in the metastatic setting, about only 30% of patients achieve durable responses to ICKB with mAbs, thus intense research is ongoing to unravel the mechanisms involved in both primary and acquired ICKB resistance.
Now that ICKB has proven to induce clinical responses for several tumor pathologies, it is currently being investigated in combination with other immunomodulatory strategies in clinical trials, in an attempt to improve antitumor immune responses. ICKB can be combined with antagonists of immunosuppressive molecules or different immunostimulatory treatments, such as active tumor vaccines, administration of cytokines, tumor-specific mAbs, and adoptive cell therapy (ACT). As shown in Figure 1, future immunotherapy can be seen from an integrative point of view, allowing the combination of different approaches that target both the tumor cells and the immune microenvironment, with impact in the systemic immune status. In this review, we have selected several examples of such combinatory strategies that are currently being investigated in clinical studies to discuss the rationale and potential results (www.clinicaltrials.gov) (Tables 1–3). Most of these trials are in the initial phases (I–II) and are directed toward advanced metastatic tumor patients including CM, mainly to optimize dose regimens and observe safety, side effects, and initial response in patients, including potential immunogenicity and antitumor immune response.
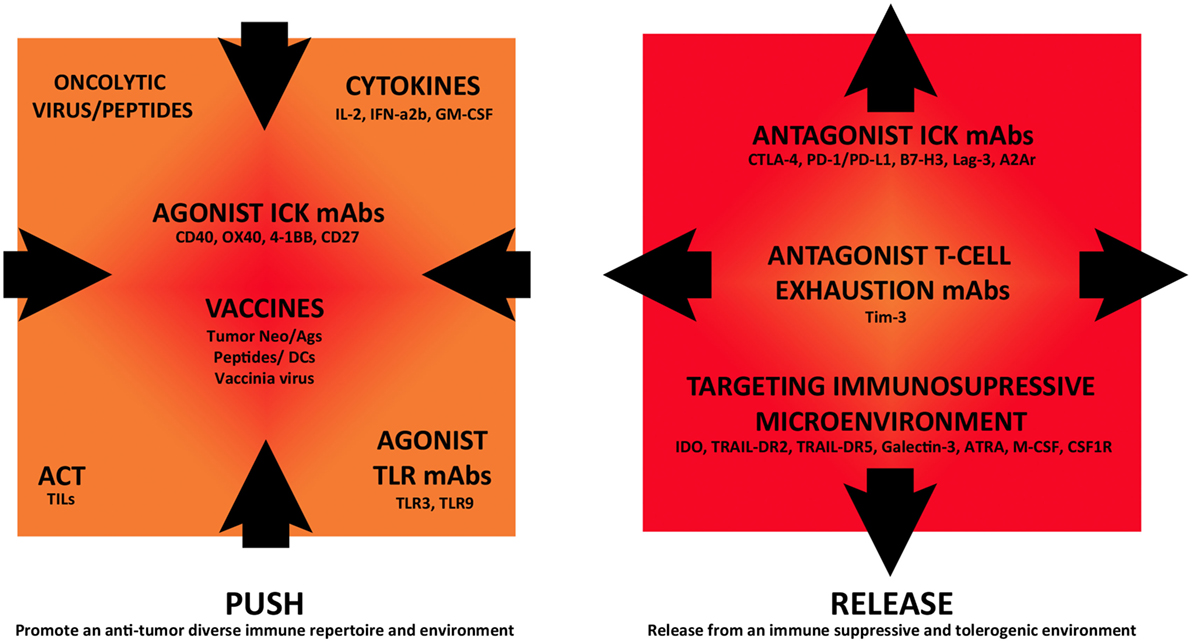
Figure 1. Immunomodulatory monoclonal antibodies and combination strategies in cutaneous melanoma immunotherapy. Different immunotherapeutic strategies are currently being assessed in combination in clinical trials that either “push” tumor immunoreactivity or “release” from inhibitory immune-regulatory mechanisms, fostering this way an antitumor immune response.
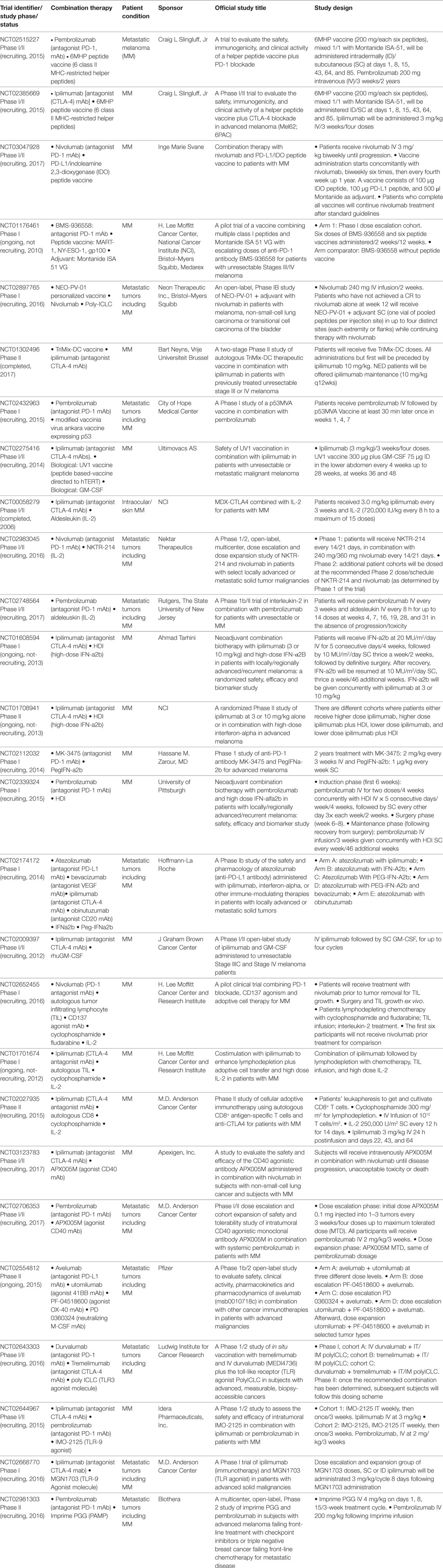
Table 1. Clinical trials combining immune checkpoint blockade with immunostimulatory strategies.
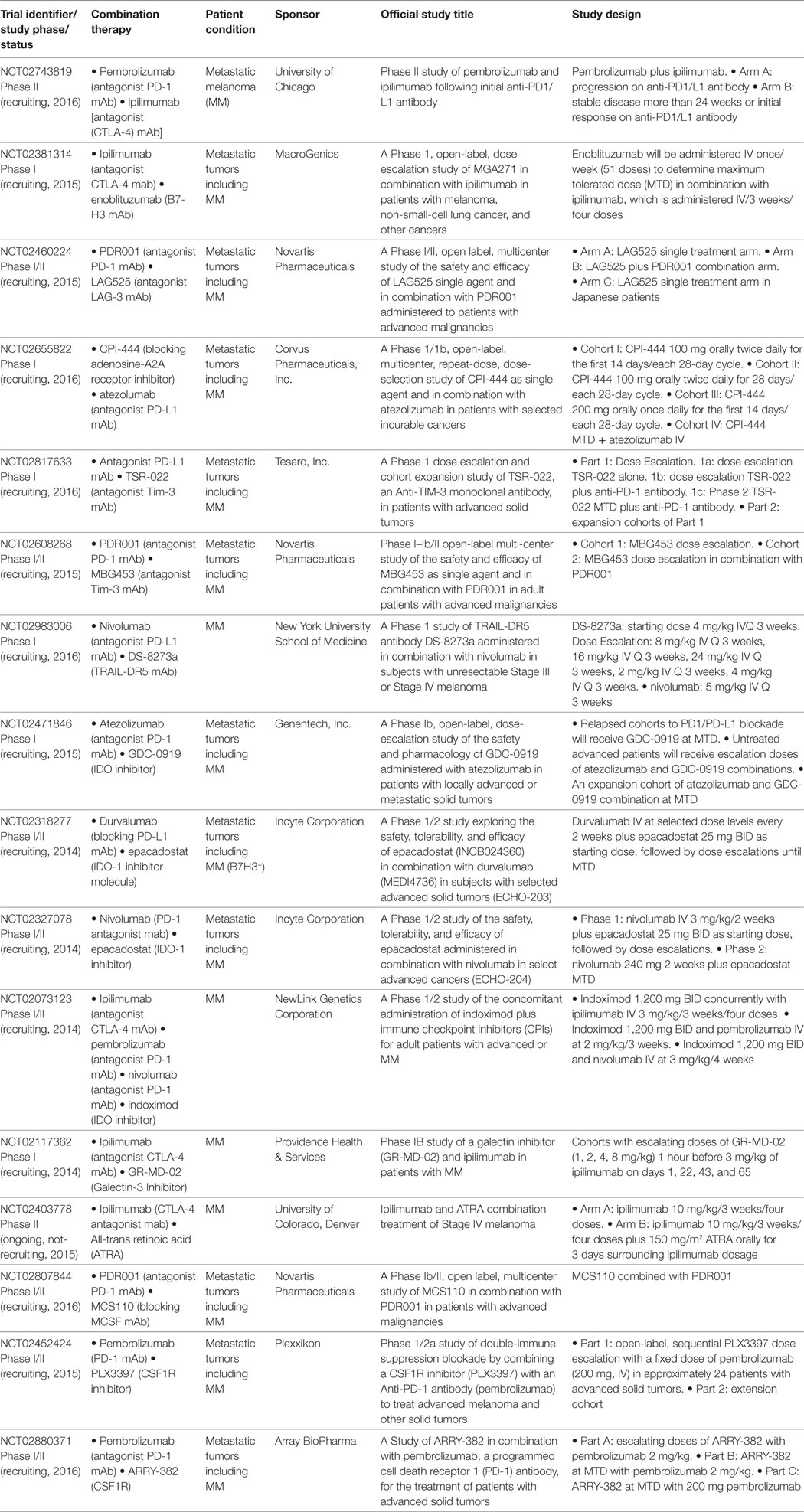
Table 2. Clinical trials combining immune checkpoint blockade with targeting of immunosuppressive molecules.
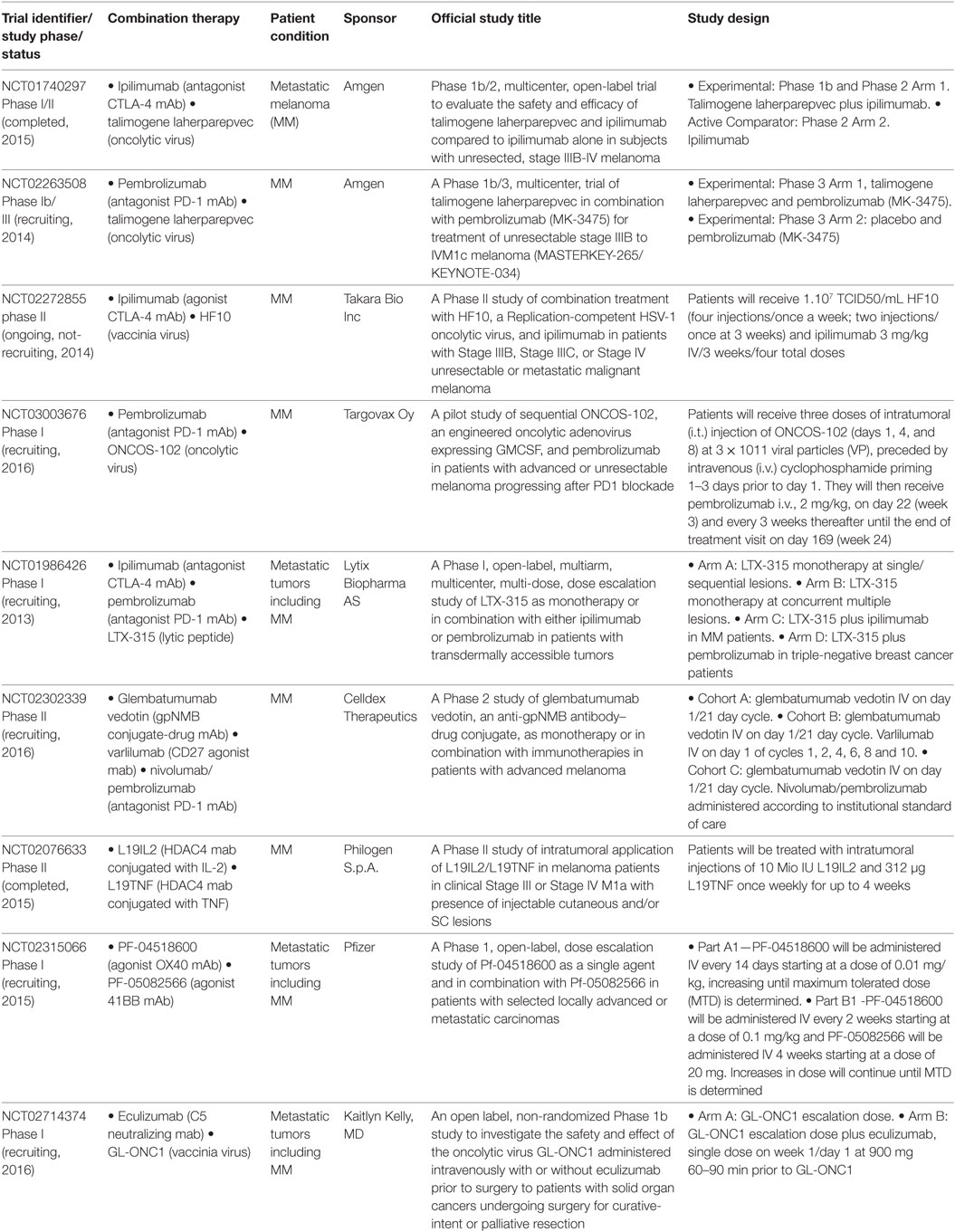
Table 3. Other combinations in clinical trials with immunomodulatory monoclonal antibodies.
Clinical Trials
ICKB Combined with Immunostimulatory Strategies
Vaccines
Therapeutic vaccines are under investigation but still have shown only limited clinical benefit for patients with CM and other tumors. The rationale of therapeutic vaccination is to boost the patient’s immune system to induce a pro-inflammatory TH1 immune response targeting both shared common tumor Ags and patient-specific neoAgs generated by somatic mutations in a personalized fashion. Vaccines can be prepared as peptides, tumor lysates, or irradiated whole tumor cells, administered with adjuvants to potentiate the immune response or as autologous DCs loaded with the tumor-Ag source.
The Phase III pivotal study that allowed FDA approval of ipilimumab to treat unresectable Stage III–IV melanoma patients was in fact designed to determine the safety and efficacy of ipilimumab in combination with BMS-734019 vaccine, a tumor-associated Ag (TAA) gp100-peptide vaccine, versus vaccine or ipilimumab alone (NCT00094653) (7). The main results of that study were that ipilimumab, with or without a vaccine, in comparison to vaccine alone, improved OS of metastatic CM patients. Severe adverse events were observed but most of them were reversible and manageable. In 2015, a pooled analysis was performed of long-term survival data of 1861 patients from Phase II and III studies (NCT00032045, NCT00058279, NCT00077532, NCT00094653, NCT00135408, NCT00261365, NCT00289627, NCT00289640, NCT00324155, and NCT00623766), some of them including combined vaccination with peptides, the majority of patients receiving the 3 mg/kg regimen. Also, data from additional 2,985 patients from an expanded access program were analyzed. A plateau in the survival curve was observed, beginning at approximately 3 years, which was independent of prior therapy or ipilimumab dose, supporting the impact of ipilimumab in long-term survival for advanced CM patients (12).
Recent combination trials of ICKB with peptide vaccines are shown in Table 1. 6MHP, a melanoma vaccine comprised of 6 MHC-II-restricted helper peptides administered subcutaneous (SC) and intradermally (ID) as water-in-oil emulsions with Montanide ISA-51, in combination with pembrolizumab (NCT02515227) or ipilimumab (NCT02385669), is under investigation. NCT03047928 trial will combine nivolumab with a peptide vaccine consisting of PD-L1 and indoleamine 2,3-dioxygenase (IDO) peptides. T-cell reactivity against PD-L1 and IDO in the tumor microenvironment and in the peripheral blood of CM patients with cytotoxic activity has been reported (13). Thus boosting specific T cells that recognize immune regulatory proteins such as IDO and PD-L1 may directly modulate immune regulation. In the protocol, patients will be treated with nivolumab every second week as long as there is a clinical benefit. The PD-L1/IDO peptide vaccine is given from the start of nivolumab and every second week for the first six vaccines and thereafter every fourth week up to 1 year. NCT01176461 tested the side effects of an investigational vaccine consisting of gp100280-288 and NY-ESO-1157-165 MHC-I peptides and adjuvant Montanide ISA-51-VG, combined with escalating doses of antagonist PD-1 mAb BMS-936558. A cohort of patients will not receive the vaccine. Phase I trial results indicated that combination was well tolerated and induced responses lasting up to 140 weeks (14).
Another interesting strategy is being tested in NCT02897765, combining nivolumab with NEO-PV-01, a personalized vaccine therapy designed to target mutated proteins which are present uniquely on an individual’s tumor neoAgs, with poly-ICLC as the adjuvant. Patients will receive 240 mg IV nivolumab every 2 weeks and those patients who have not achieved a complete response to nivolumab alone at week 12 will receive NEO-PV-01+ adjuvant SC, in up to four distinct sites while continuing therapy with nivolumab. The study will monitor side effects and Ag-specificity in peripheral CD8+ and CD4+ T cell responses and tumor biopsies, which will be assessed after treatment.
NCT01302496 studies ipilimumab combined with TriMix vaccine. The TriMix-DC vaccine is a DC-based vaccine that can induce a T-cell repertoire that recognizes the TAA MAGE-A3, MAGE-C2, tyrosinase, and gp100 in an HLA-restricted way, in unresectable stage III-IV melanoma patients (15, 16). To prepare TriMix-Dc vaccine, autologous DCs are coelectroporated with TriMix mRNA (a combination of CD40L, caTLR4, and CD70 encoding mRNA) in combination with one of four TAA mRNAs linked to an HLA-II targeting signal. After electroporation, the four different TriMixDC-MEL cellular constituents (i.e., DCs expressing one of the four Ags) are mixed at equal ratios and cryopreserved until vaccination. The study is complete but results are still unpublished.
NCT02432963 proposes another combination strategy with ICKB and vaccines, testing a modified Vaccinia Virus Ankara Vaccine expressing tumor protein p53 (p53MVA Vaccine) in combination with pembrolizumab, in patients with tumors with overexpression or mutation of p53. Immune monitoring in peripheral blood samples obtained through the protocol will analyze T-cell reactivity to p53, myeloid-derived suppressor cell (MDSC) and Tregs immunosuppressive populations, and other selected subsets including PD-1+, PDL-1+, and PDL-2+ cells.
Finally, in a Phase I/II study (NCT02275416), ipilimumab (3 mg/kg, every 3 weeks for a total of four doses) in combination with GM-CSF (75 µg) and UV1 peptide based-vaccine directed to hTERT is being tested in unresectable Stage III or Stage IV melanoma patients. UV1 vaccine (300 µg) will be administered by injecting ID in the lower abdomen before and between treatments with ipilimumab, thereafter every 4th week up to 28 weeks, and then at weeks 36 and 48. This study will analyze safety and tolerability of the combination and also will measure specific T-cell responses, quality of life, and treatment response by CT scans. A search for potential biomarkers of efficacy and safety studies will be also performed.
Cytokines
There are several trials combining ICKB with typical cytokines first assessed in CM patients as non-specific immunotherapy treatments, such as IL-2, IFN-α2b, and GM-CSF (Table 1). Aldesleukin is a recombinant analog of the endogenous cytokine IL-2 that has immunoregulatory and antineoplastic activities. It promotes activation of T, B, and NK cells; however, serious related adverse events were seen upon IL-2 administration (17). IL-2 was approved for treatment of metastatic renal cell carcinoma in 1992 and for metastatic melanoma (MM) in 1998 by FDA. Nowadays, IL-2 monotherapy is not the optimal and standard treatment for both metastatic renal cell carcinoma and MM but efforts to further improve the efficacy of IL-2 therapy are focused on combined therapies. Results from NCT00058279 combining ipilimumab with IL-2 revealed immune-related adverse events. A non-synergistic effect was observed, since the 22% objective response rate observed, results from the additive effect of the expected response rate for each therapy; however, long-term responses were still observed (12). NCT02983045 ongoing study will analyze ICKB in combination with NKTR-214, a prodrug for IL-2, conjugated to six releasable PEG chains. In a preclinical CM mouse model, this molecule showed 20 times preferential activation of CD8+ T cells (IL2Rβ) over Treg cells (IL2Rα) in comparison to aldesleukin. In this model, NKTR-214 proved efficacy as a single agent, and long-term immunity when combined with antagonist CTLA-4 mAb, in addition to resistance to tumor rechallenge (18). NCT02748564 Phase Ib/II study will evaluate the safety and tolerability of IL-2 when given in combination with pembrolizumab to patients with advanced CM.
Adjuvant IFN-α2b increases disease-free survival (DFS) although not OS in CM patients but it is accompanied with considerable toxicity (19, 20); it is not universally considered as a gold standard treatment (21). Besides, the optimal dose and duration of IFN-α2b treatment are still unclear (22, 23). IFN-α2b would have several mechanisms of action, from induction of apoptosis in tumor cells to activation of monocytes and macrophages favoring Ag processing. There are several ongoing trials combining ICKB with IFN-α2b or PEG-IFN-α2b (NCT01608594, NCT01708941, NCT02112032, NCT02339324, and NCT02174172). Low doses of cytokine GM-CSF proved to be a strong monocyte attractant and necessary to differentiate monocytes into DCs promoting a TH1 response (24); a combination of ipilimumab with this cytokine is also on the way (NCT02009397).
Adoptive Cell Therapy
One approach to restore the functionality of effector immune cells is to cultivate autologous tumor infiltrating lymphocyte (TIL) ex vivo after tumor resection and infused them back into the patient (25); this is defined as ACT. Combination of ACT with ICKB may counteract any inhibitory immune checkpoint signal from the tumor microenvironment, provided that T cell effectors have been expanded and activated in vitro in the presence of tumor Ags previous to treatment (Table 1). NCT02652455 will compare the effect of nivolumab administration prior to tumor resection and in vitro culture of TILs. These will be cultivated ex vivo with agonist CD137 mAb to augment T cell proliferation and infused them after chemotherapy-induced lymphodepletion of patients. They will be treated in vivo with IL-2 to support T cell proliferation. NCT01701674 will study the effect of ipilimumab before leukapheresis, while NCT02027935 will do it afterward.
Stimulatory Immune Checkpoints
CD40 is a costimulatory receptor that is essential for activating both innate and adaptive immune systems (26). CD40 binds its ligand CD40L, which is transiently expressed on T cells and other non-immune cells under inflammatory conditions. A wide spectrum of molecular and cellular processes is regulated by CD40 engagement including the initiation and progression of cellular and humoral adaptive immunity. Use of agonist CD40 mAbs with high-affinity fosters activation of APCs (DCs, monocytes, and B cells), leading to stimulation of tumor-specific immune responses. Recently, it was reported in a mouse tumor model that use of agonist CD40 mAb reversed resistance to PD-1, downregulating PD-1 levels in T cells via IL-12 production (27). Agonist CD40 mAb APX005M is currently being evaluated in Phase I/II trials in combination with ipilimumab (NCT03123783) or pembrolizumab (NCT02706353) (Table 1). NCT02554812 trial combines avelumab in different cohorts with agonist mAbs toward T cells costimulatory molecules 4-1BB and OX-40 (28) or neutralizing mAb toward M-CSF/CSF1 (macrophage colony-stimulating factor) (29).
Toll-Like Receptors (TLRs)/PAMP
Toll-like receptors can detect a broad range of human pathogens, as well as a variety of molecules such as PAMP (pathogen-associated molecular patterns) that indicate tissue damage. This recognition triggers a cascade of innate and adaptive immune responses that fully activate the immune system. Agonist TLR mAbs support this response. It was reported that triggering of TLR3 induces T-cell activation and a strong upregulation of HLA-I and PD-L1 in neuroblastoma and melanoma cells (30, 31). Therefore, ICKB will counteract limitation of the T cell response induced by TLR signaling. Ongoing trials include combinations of PD-1 and CTLA-4 ICKB with agonist TLR3 and TLR9 mAbs (NCT02643303, NCT02644967, and NCT02668770). Trial NCT02981303 will assess Imprime PGG, a β-1,3/1,6 glucan PAMP molecule isolated from the cell wall of a proprietary Saccharomyces, in combination with pembrolizumab (Table 1).
ICKB Combined with Targeting of Immunosuppressive Molecules/Pathways
Other ICKB
Immune checkpoint blockade is also being assessed in combinations with the targeting of other molecules/pathways that promote an immunosuppressive environment (Table 2). For instance, there are trials targeting several ICKB. NCT02743819 trial combines pembrolizumab with ipilimumab in advanced patients which, following treatment with PD-1/PD-L1 mAb, either progress or present stable disease/initial response for more than 24 weeks. NCT02381314 studies in B7-H3 expressing tumors such as CM, the combination of ipilimumab with enoblituzumab, a B7-H3 mAb was designed to improve ADCC by increasing FcR affinity. NCT02460224 analyzes the combination of LAG525 and PDR001, antagonist mAbs for LAG-3 and PD-1, respectively. LAG-3 is an immune checkpoint that binds a non-holomorphic region of the MHC-II molecule and has an important role in the tumor microenvironment. It was reported that soluble-LAG-3 binds to immature DCs, promoting their maturation (32). However, LAG-3 is involved in alternative activation of plasmacytoid DCs in melanoma lesions (33). Interaction of MHC-II on APCs with LAG3 downregulates T-cell proliferation and activation (34). In agreement, LAG-3 mediates resistance to apoptosis on MHC-II expressing melanoma cells (35). LAG-3 is substantially expressed on melanoma TILs, including those with potent immunosuppressive activity. LAG3 was shown to have a synergistic action with the PD-1/PD-L1 axis, critical for releasing an antitumor immune response. In a mouse melanoma model, tumor-specific CD4+ effector T-cells showed traits of chronic exhaustion, with high expression levels of PD-1, TIM-3, 2B4, TIGIT, and LAG-3 inhibitory molecules. Blockade with a combination of anti-PD-L1 and anti-LAG-3 mAbs overcame the requirement to deplete tumor-specific Tregs in this model (36). The PD-1 expression on CD8+ TILs identified a repertoire of clonally expanded tumor-reactive cells, including mutated neoAg-specific CD8+ T-cells; these cells expressed LAG-3 and Tim-3 (37). It has recently been described that ipilimumab expanded T cells in patients with higher expression levels of CD27, intracellular CTLA-4, TIM-3, and LAG-3, which can be taken into account for future combination trials (38).
Adenosine-A2A receptor (A2Ar), an ectonucleotidase that catabolizes the hydrolysis of extracellular adenosine monophosphate (AMP) to adenosine, is a novel metabolic target for ICKB. In preclinical models, it was shown that expression of A2Ar on myeloid cells suppressed T and NK cell responses in the solid tumor microenvironment (39). Also, A2Ar blockade enhanced antitumor activity of PD-1 and CTLA-4 ICKB (40). NCT02655822 is on the way combining PD-1 ICKB with an A2Ar inhibitor molecule.
T-Cell Exhaustion
TIM-3 was first identified as a specific TH1 receptor. When it binds to galectin-9, it generates an inhibitory signal that results in apoptosis of TH1 cells (41). TIM-3 was also described as a marker of T-cell exhaustion (38). TIM-3 is also expressed by NK cells and naive DCs, acting in synergy with TLR signaling to induce inflammation. Expression of TIM-3 in monocytes and macrophages promotes phagocytosis of apoptotic cells through interaction with phosphatidylserine, which enhances Ag cross-presentation (42). Also, TIM-3 binds HMGB1, impairing its recruitment of nucleic acids into endosomes, a key step in the sensing of DNA by the innate immune system, promoting tumor escape (43). Notably, it was shown that PD-1 and Tim-3 limited the expansion of tumor Ag-specific CD8+ T cells induced by a melanoma peptide vaccine, as dual blockade enhanced the expansion and cytokine production of vaccine-induced CD8+ T cells in vitro (44). NCT02817633 and NCT02608268 are ongoing combining ICKB with an antagonist Tim-3 mAb (Table 2).
Tumor Immune Microenvironment
NCT02983006 trial is testing the combination of nivolumab with DS-8273a (Table 2). This agonist mAb showed selective targeting of MDSC through TNF-receptor TRAIL-DR2, without affecting other mature myeloid or lymphoid cells (45). As an agonist of TRAIL-DR5 to induce apoptosis in tumor cells, in a Phase I trial DS-8273 as monotherapy was well tolerated but no objective responses were observed, although decreases in MDSC temporally associated with DS-8273a exposure were observed (46).
Indoleamine 2,3-dioxygenase is the first and rate-limiting enzyme involved in tryptophan catabolism, which can halt T-cells growth. In cancer, IDO is expressed within the tumor itself as well as in the tumor microenvironment, where it promotes the establishment of peripheral immune tolerance to tumor Ags. On the tumor side, lymph node CM cells express IDO, recruiting Treg, which is associated with a poor outcome (47). At the tumor microenvironment, IDO promotes MDSC recruitment by a mechanism dependent on Tregs (48); it also inhibits NK cell function along with PGE-2 (49). Activated T cells in vitro induce MDSC function through IL-10; these MDSC secrete ARG-1 and IDO and express PD-L1 and MHC-II, leading to upregulation of PD-1 and LAG-3 on T-cells, promoting an immunosuppressive tumor microenvironment (50). In CM patients, high levels of circulating PD-L1+ cytotoxic T-cells were associated with increased expression levels of CTLA-4 in Tregs and IDO in MDSC and plasmacytoid DCs. All these parameters were related to a negative outcome, independent of disease stage (51).
It is interesting to note that IDO is an immunogenic protein, therefore, activation of pro-inflammatory IDO-specific CD4+ responses may delay or overcome the immunosuppressive actions of IDO, consequence of early expression in maturing APCs; however, IDO-specific Tregs may enhance IDO-mediated immune suppression (47). In mouse melanoma models, IDO is an essential mechanism of resistance to ICKB, including CTLA-4 and PD-1. CTLA-4 blockade combined with IDO inhibitors strongly synergizes to mediate tumor rejection (49). It is postulated that following melanoma infiltration by lymphocytes, upregulation of PD-L1, IDO, and Tregs is regulated by an intrinsic immune mechanism (52). And that combination of CTLA-4, PD-1/PD-L1, and IDO blockade restores IL-2 production and proliferation of CD8+ T-cells (53). It was recently reported that melanoma expresses high levels of IDO and galectin-3, which upregulate Tregs, suppressing the expansion of tumor-specific T cells cultivated for ACT, which could be reversed by blockade of IDO and galectin-3 (54). Trials combining ICKB with inhibitors of IDO (NCT02471846, NCT02318277, NCT02327078, and NCT02073123) or galectin-3 (NCT02117362) are currently on the way (Table 2).
NCT02403778 trial proposes the combination of ipilimumab with all-trans retinoic acid (ATRA), a derivative of vitamin A (Table 2). ATRA induces maturation of immunosuppressive MDSCs into myeloid cells (55, 56); this was shown to be of benefit in a lung cancer vaccine and ACT for sarcomas (57, 58). Thus, this combination is designed to decrease MDSCs and differentiate immature monocytes into mature DCs and increase tumor Ag-specific T-cell responses. In this trial, ATRA single-arm versus ATRA with ipilimumab combined-arm will be compared.
Finally, other combinations of ICKB and targeting the tumor microenvironment include antagonist mAbs for M-CSF/CSF1 (NCT02807844) or its receptor MCSFR/CSF1R (NCT02452424 and NCT02880371) (Table 2). Interaction of α4β1 integrin from extracellular matrix with MCSF receptor leads to the activation of Rac2 and regulation of macrophage toward a M2 immunosuppressive phenotype (59). In a melanoma mouse model, targeting of CSF1R on MDSCs overcomes resistance to IDO-expressing melanoma cells (60).
Other Combinations with Immunomodulatory mAbs
There are several oncolytic viruses that have shown to promote an immunogenic cell death leading to a TH1 response; combination with ICKB is aimed to sustain in time this tumor microenvironment (61). Talimogene laherparepvec (T-VEC, Imlygic) is a genetically modified, attenuated, herpes simplex virus type 1 designed to promote an antitumor response through selective viral replication in tumor cells and stimulation of systemic antitumor immunity through GM-CSF (62). This was the first oncolytic viral therapy to be approved by the FDA in 2015 for intralesional treatment of unresectable lesions in patients with melanoma recurrent after the initial surgery. The combination of T-VEC with ipilimumab in a Phase I trial proved to be safe and appeared to have greater efficacy than single agents (NCT01740297) (63). NCT02263508 is a Phase 1b/3 study that will assess the combination of talimogene laherparepvec with pembrolizumab in unresectable CM patients. Combination studies with other oncolytic virus include NCT02272855 trial, which will analyze CTLA-4 ICKB with HF10, an oncolytic virus that has shown to induce angiogenesis and affluence of CD8+ T-cells at the tumor microenvironment (64). Finally, in the NCT03003676 study the combination of pembrolizumab with ONCOS-102, an engineered oncolytic Adenovirus expressing GM-CSF, will be analyzed in CM patients that have progressed to the PD-1 blockade.
The NCT01986426 study is designed to assess the safety, tolerability, and efficacy of different intratumoral dosing regimens of LTX-315; a lytic-peptide that induces immunogenic cell death; it will be assessed as monotherapy or in combination with ipilimumab or pembrolizumab. It was shown in mouse models that this peptide overcomes tumor resistance to CTLA-4 ICKB (65) (Table 1).
The NCT02302339 study will examine the effectiveness and safety of glembatumumab vedotin as monotherapy and in combination with either nivolumab or pembrolizumab (Table 3). Glembatumumab vedotin mAb is conjugated to the cytotoxic drug monomethyl auristatin E. This mAb targets glycoprotein NMB, expressed on the surface of tumor cells, releasing the drug and inducing tumor cell death. Combinations with immunotherapy include pembrolizumab/nivolumab and varlilumab, an agonist mAb of the T-cell costimulatory receptor CD27. NCT02076633 trial also examines conjugated mAbs for treatment of metastatic CM patients; L19IL2 targets melanoma cells through HDAC4 and it is conjugated to IL-2; instead L19TNF is conjugated to TNFα, exerting its major effects via a preferential toxicity for the endothelial cells of the tumor-associated vasculature, therefore, increasing an antitumor immune response. Preclinical data suggest that intratumoral administration of these conjugates can be more effective.
Another approach explores the synergy of two agonist mAbs targeting the T-cells costimulatory molecules OX40 and 41BB (25, 66) (NCT02315066) (Table 3). Finally, NCT02714374 ongoing trial is combining GL-ONC1, a genetically modified oncolytic vaccinia virus, with eculizumab, a neutralizing C5 mAb, with the goal that GL-ONC1 remains in the body long before being cleared by the immune system.
Discussion
Combined immunotherapy involving mAbs and other immunomodulatory strategies is an emerging field. There is no still any such combination therapy approved. The immune system should be considered as one interrelated signaling network where targeting different points may act synergistically to promote anticancer effects. Proper immune stimulation and blockade of immunosuppression can be seen as a “push and release” strategy, where both are critical for the efficacy of an anti-cancer immunotherapy (Figure 1). ICKB is currently being assessed in combination with immune stimulatory strategies, such as vaccination, cytokines, ACT, stimulatory immune checkpoint agonists, and targeting of TLRs. Other approaches combine ICKB with targeting of several immune suppressive mechanisms, such as blocking other immune checkpoints, T-cell exhaustion inhibition, and promotion of an antitumor microenvironment.
The idea of combining ICKB with immunomodulatory strategies such as vaccines is very attractive, given that in some cases amplification of Ag-specific T cells, as well as the induction of antibodies recognizing tumor Ags are observed after vaccination. Blocking of immune checkpoints may result in effector T cells that could attain potent tumor destruction more potently; a useful T helper function and modulation of Tregs will result in the expansion of cytotoxic T cell effectors. However, vaccines, in general, have shown a low 10–15% rate of clinical responses, with still a lack of efficacy to eradicate tumor masses and avoid further dissemination in metastatic patients (67). Most of the clinical trials revised in this work are being assessed in advanced CM patients, thus immune suppression, both systemic and local, can be hard to overcome even with ICKB, since there are patients that do not respond at all. Vaccination strategies may be more useful when administered in the adjuvant setting to control tumor relapse in high-risk stage II-III CM patients (68), and thus, their combination with ICKB may hold the promise of durable clinical benefit avoiding metastases to distant organs and achieving prolonged OS.
Assessment of clinical responses in ICKB cancer treatments can be challenging since traditional Response Evaluation Criteria In Solid Tumors, RECIST, may underestimate the actual response that can be delayed and atypical, as evidenced in patients treated with ICKB (69). The immune-related response criteria have been established to allow patients to continue treatment after the first progression until a new progression is presented, given the chance of eventual clinical benefit to more patients (70). It should be taken into account that there are both constitutive and acquired IKCB resistance mechanisms compromising treatment outcome (71–73).
Identification of early predictors of response is desirable to identify patients that would benefit the most and avoid unnecessarily prolonged treatments. Regarding ICKB biomarkers at the local level, an association of PD-L1 expression in pretreatment tumor biopsies with objective response to anti-PD-1/PD-L1 therapy has been observed given the constant finding that PD-L1 expression is enriched in anti-PD-1/PD-L1 therapy responders in several tumors (74). Weighted-average ORR across several studies for patients whose tumors were tested for PD-L1 is 29%; if the tumor expresses PD-L1, these increases to 48%. However, a significant proportion of responding patients with PD-L1 negative-tumors were observed. Also, high-density infiltration of CD8 T-cells in tumor biopsies has been associated with PD-L1 expression in tumor cells; it was associated independently with an improved prognosis, with increased time to development of brain metastases in CM patients (75). Also, it was recently reported that PD-L2 expression in metastatic CM was associated with immune infiltration and a better prognosis independently of therapy of choice (76). Other proposed biomarkers for selecting which patients are likely to benefit from cancer immunotherapies are the tumor mutational load and microsatellite in the stability. CM is a tumor with a high rate of mutation (1), and thus with a higher probability of neoAg generation, increasing the number of immunogenic structures that could stimulate a more potent and broad repertoire of antitumor immune effectors. In the same way, microsatellite instability is a frequent event in CM which accounts for tumor immunogenicity, contributing to making of CM a pathology suitable for immunotherapy (77).
At the peripheral level, serum IL-8 concentrations actually reflect tumor burden (78). It was recently reported that measurement of serum IL-8 levels 2–3 weeks following starting therapy can predict response and OS in metastatic CM patients treated with PD-1 ICKB, even before imaging evaluation (79). Also, there are recent publications by two independent groups which reported that assessment of circulating cell-free DNA from tumors in CM patients receiving ICKB treatment is an accurate predictor of tumor response, PFS and OS, as patients with elevated ctDNA on therapy had a poor prognosis (80, 81).
In the combination of several immunotherapy strategies, identifying which patients are likely to respond to therapy will be even more challenging. This is given the complexity of the immune system, and the limited understanding of its regulation and multiple interactions between immune cells, immune-modulating molecules, tumor cells, and other compartments of the tumor microenvironment, such as the lymphatic and blood systems.
Conclusion
Monoclonal antibodies have gained evidence of their effectiveness for cancer treatment. This seems to be the tip of an iceberg, as we are learning that not only targeting the tumor but also modulating the immune response, may be a powerful way to achieve long-term clinical responses. In this way, mAbs can be again considered “magic bullets” targeting molecules with different immunomodulatory effects. We have revisited most of the current clinical trials that explore combined use of immunomodulatory mAbs with different immunotherapeutic approaches, with the aim to improve and/or potentiate clinical responses in CM patients. This is an exciting and expanding research field that is rapidly spreading to other tumor types.
Author Contributions
MA and MB: conception and design of the review; manuscript writing. JM: conception and design of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MA, JM, and MB are members of CONICET, Argentina. This work was supported by grants from CONICET, Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), Instituto Nacional del Cáncer—Ministerio de Salud de la Nación Argentina (INC-MSal), Fundación Sales, Fundación Cáncer, and Fundación Pedro F. Mosoteguy, Argentina.
Abbreviations
Ags, antigens; APCs, Ag-presenting cells; CM, cutaneous melanoma; CTLA-4, cytotoxic T-lymphocyte antigen 4; DCs, dendritic cells; DFS, disease-free survival; ICKB, immune checkpoint blockade; ID, intradermally; IV, intravenous; IDO, indoleamine 2,3-dioxygenase; MDSC, myeloid-derived suppressor cell; MM, metastatic melanoma; mAb, monoclonal antibodies; neoAgs, neoantigens; OS, overall survival; PAMP, pathogen-associated molecular patterns; PD-1, programmed cell death-1/CD279; SC, subcutaneous; Tregs, regulatory T cells; TLR, toll-like receptors; TAA, tumor-associated Ag; TIL, tumor infiltrating lymphocytes.
References
1. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi:10.1038/nature12477
2. Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med (2015) 373:1926–36. doi:10.1056/NEJMoa1502583
3. Madorsky Rowdo FP, Baron A, Urrutia M, Mordoh J. Immunotherapy in cancer: a combat between tumors and the immune system; You Win Some, You Lose Some. Front Immunol (2015) 6:127. doi:10.3389/fimmu.2015.00127
4. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi:10.1126/science.aaa4971
5. Galon J, Fox BA, Bifulco CB, Masucci G, Rau T, Botti G, et al. Immunoscore and immunoprofiling in cancer: an update from the melanoma and immunotherapy bridge 2015. J Transl Med (2016) 14:273. doi:10.1186/s12967-016-1029-z
6. Aris M, Barrio MM, Mordoh J. Lessons from cancer immunoediting in cutaneous melanoma. Clin Dev Immunol (2012) 2012:192719. doi:10.1155/2012/192719
7. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
8. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1988) 7:1734–6.
9. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
10. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372:320–30. doi:10.1056/NEJMoa1412082
11. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 6736:1–9. doi:10.1016/S0140-6736(14)60958-2
12. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol (2015) 33:1889–94. doi:10.1200/JCO.2014.56.2736
13. Andersen MH. Anti-regulatory T cells. Semin Immunopathol (2017) 39:317–26. doi:10.1007/s00281-016-0593-x
14. Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol (2013) 31:4311–8. doi:10.1200/JCO.2013.51.4802
15. Wilgenhof S, Van Nuffel AMT, Corthals J, Heirman C, Tuyaerts S, Benteyn D, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother (2011) 34:448–56. doi:10.1097/CJI.0b013e31821dcb31
16. Benteyn D, Van Nuffel AMT, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL). Biomed Res Int (2013) 2013:976383. doi:10.1155/2013/976383
17. Shaker MA, Younes HM. Interleukin-2: evaluation of routes of administration and current delivery systems in cancer therapy. J Pharm Sci (2009) 98:2268–98. doi:10.1002/jps.21596
18. Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res (2016) 22. doi:10.1158/1078-0432.CCR-15-1631 Available from: http://clincancerres.aacrjournals.org/content/22/3/680,
19. Kirkwood JM, Bender C, Agarwala S, Tarhini A, Shipe-Spotloe J, Smelko B, et al. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol (2002) 20:3703–18. doi:10.1200/JCO.2002.03.052
20. Eggermont AMM, Suciu S, Testori A, Santinami M, Kruit WHJ, Marsden J, et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J Clin Oncol (2012) 30:3810–8. doi:10.1200/JCO.2011.41.3799
21. Mocellin S, Lens MB, Pasquali S, Pilati P, Chiarion Sileni V. Interferon alpha for the adjuvant treatment of cutaneous melanoma. Cochrane Database Syst Rev (2013) 6:CD008955. doi:10.1002/14651858.CD008955.pub2
22. Eggermont AMM, Suciu S, MacKie R, Ruka W, Testori A, Kruit W, et al. Post-surgery adjuvant therapy with intermediate doses of interferon alfa 2b versus observation in patients with stage IIb/III melanoma (EORTC 18952): randomised controlled trial. Lancet (2005) 366:1189–96. doi:10.1016/S0140-6736(05)67482-X
23. Tarhini AA, Kirkwood JM, Gooding WE, Cai C, Agarwala SS. Durable complete responses with high-dose bolus interleukin-2 in patients with metastatic melanoma who have experienced progression after biochemotherapy. J Clin Oncol (2007) 25:3802–7. doi:10.1200/JCO.2006.10.2822
24. Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol (2007) 18:226–32. doi:10.1093/annonc/mdl158
25. Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology (2012) 1:409–18. doi:10.4161/onci.18851
26. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev (2009) 229:152–72. doi:10.1111/j.1600-065X.2009.00782.x
27. Ngiow SF, Young A, Blake SJ, Hill GR, Yagita H, Teng MWL, et al. Agonistic CD40 mAb-driven IL12 reverses resistance to anti-PD1 in a T-cell–rich tumor. Cancer Res (2016) 76:6266–77. doi:10.1158/0008-5472.CAN-16-2141
28. Kumari A, Garnett-Benson C. Effector function of CTLs is increased by irradiated colorectal tumor cells that modulate OX-40L and 4-1BBL and is reversed following dual blockade. BMC Res Notes (2016) 9:92. doi:10.1186/s13104-016-1914-9
29. Masek-Hammerman K, Peeva E, Ahmad A, Menon S, Afsharvand M, Peng Qu R, et al. Monoclonal antibody against macrophage colony-stimulating factor suppresses circulating monocytes and tissue macrophage function but does not alter cell infiltration/activation in cutaneous lesions or clinical outcomes in patients with cutaneous lupus ery. Clin Exp Immunol (2016) 183:258–70. doi:10.1111/cei.12705
30. Boes M, Meyer-Wentrup F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett (2015) 361:49–56. doi:10.1016/j.canlet.2015.02.027
31. Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: implications for PD-1 pathway blockade. Clin Cancer Res (2015) 21:3969–76. doi:10.1158/1078-0432.CCR-15-0244
32. Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood (2003) 102:2130–7. doi:10.1182/blood-2003-01-0273
33. Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol (2014) 134:1893–902. doi:10.1038/jid.2014.29
34. Huard B, Prigent P, Pagès F, Bruniquel D, Triebel F. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur J Immunol (1996) 26:1180–6. doi:10.1002/eji.1830260533
35. Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol (2011) 186:5173–83. doi:10.4049/jimmunol.1002050
36. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol (2013) 190:4899–909. doi:10.4049/jimmunol.1300271
37. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124:2246–59. doi:10.1172/JCI73639
38. Bjoern J, Lyngaa R, Andersen R, Rosenkrantz LH, Hadrup SR, Donia M, et al. Influence of ipilimumab on expanded tumour derived T cells from patients with metastatic melanoma. Oncotarget (2017) 8:27062–74. doi:10.18632/oncotarget.16003
39. Cekic C, Day Y-J, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res (2014) 74:7250–9. doi:10.1158/0008-5472.CAN-13-3583
40. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Stagg J, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia (2017) 19:530–6. doi:10.1016/j.neo.2017.02.004
41. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi:10.1038/ni1271
42. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood (2009) 113:3821–30. doi:10.1182/blood-2008-10-185884
43. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13:832–42. doi:10.1038/ni.2376
44. Fourcade J, Sun Z, Pagliano O, Chauvin J-M, Sander C, Janjic B, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res (2014) 74:1045–55. doi:10.1158/0008-5472.CAN-13-2908
45. Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res (2016) 23(12):2942–50. doi:10.1158/1078-0432.CCR-16-1784
46. Forero A, Bendell JC, Kumar P, Janisch L, Rosen M, Wang Q, et al. First-in-human study of the antibody DR5 agonist DS-8273a in patients with advanced solid tumors. Invest New Drugs (2017) 35:298–306. doi:10.1007/s10637-016-0420-1
47. Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, et al. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle (2009) 8:1930–4. doi:10.4161/cc.8.12.8745
48. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep (2015) 13:412–24. doi:10.1016/j.celrep.2015.08.077
49. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res (2012) 72:1407–15. doi:10.1158/0008-5472.CAN-11-2544
50. Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget (2016) 7:1168–84. doi:10.18632/oncotarget.6662
51. Chevolet I, Speeckaert R, Schreuer M, Neyns B, Krysko O, Bachert C, et al. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. Oncoimmunology (2015) 4:e982382. doi:10.4161/2162402X.2014.982382
52. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med (2013) 5:200ra116. doi:10.1126/scitranslmed.3006504
53. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J Immunother Cancer (2014) 2:3. doi:10.1186/2051-1426-2-3
54. Melief SM, Visser M, van der Burg SH, Verdegaal EME. IDO and galectin-3 hamper the ex vivo generation of clinical grade tumor-specific T cells for adoptive cell therapy in metastatic melanoma. Cancer Immunol Immunother (2017) 66(7):913–26. doi:10.1007/s00262-017-1995-x
55. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67:11021–8. doi:10.1158/0008-5472.CAN-07-2593
56. Lee J-M, Seo J-H, Kim Y-J, Kim Y-S, Ko H-J, Kang C-Y. The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int J Cancer (2012) 131:741–51. doi:10.1002/ijc.26411
57. Iclozan C, Antonia S, Chiappori A, Chen D-T, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother (2013) 62:909–18. doi:10.1007/s00262-013-1396-8
58. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res (2016) 4:869–80. doi:10.1158/2326-6066.CIR-15-0230
59. Joshi S, Singh AR, Zulcic M, Bao L, Messer K, Ideker T, et al. Rac2 controls tumor growth, metastasis and M1-M2 macrophage differentiation in vivo. PLoS One (2014) 9:e95893. doi:10.1371/journal.pone.0095893
60. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine (2016) 6:50–8. doi:10.1016/j.ebiom.2016.02.024
61. Guo ZS, Liu Z, Kowalsky S, Feist M, Kalinski P, Lu B, et al. Oncolytic immunotherapy: conceptual evolution, current strategies, and future perspectives. Front Immunol (2017) 8:555. doi:10.3389/fimmu.2017.00555
62. Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33:2780–8. doi:10.1200/JCO.2014.58.3377
63. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol (2016) 34:2619–26. doi:10.1200/JCO.2016.67.1529
64. Sahin TT, Kasuya H, Nomura N, Shikano T, Yamamura K, Gewen T, et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther (2012) 19:229–37. doi:10.1038/cgt.2011.80
65. Yamazaki T, Pitt JM, Vétizou M, Marabelle A, Flores C, Rekdal Ø, et al. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ (2016) 23:1004–15. doi:10.1038/cdd.2016.35
66. Bansal-Pakala P, Gebre-Hiwot Jember A, Croft M. Signaling through OX40 (CD134) breaks peripheral T-cell tolerance. Nat Med (2001) 7:907–12. doi:10.1038/90942
67. Aris M, Barrio MM. Combining immunotherapy with oncogene-targeted therapy: a new road for melanoma treatment. Front Immunol (2015) 6:46. doi:10.3389/fimmu.2015.00046
68. Mordoh J, Pampena MB, Aris M, Blanco PA, Lombardo M, von Euw EM, et al. Phase II study of adjuvant immunotherapy with the CSF-470 vaccine plus bacillus Calmette–Guerin plus recombinant human granulocyte macrophage-colony stimulating factor vs medium-dose interferon alpha 2B in stages IIB, IIC, and III cutaneous melanoma patie. Front Immunol (2017) 8:625. doi:10.3389/fimmu.2017.00625
69. Hoos A, Eggermont AMM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst (2010) 102:1388–97. doi:10.1093/jnci/djq310
70. Hodi FS, Hwu W-J, Kefford R, Weber JS, Daud A, Hamid O, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol (2016) 34:1510–7. doi:10.1200/JCO.2015.64.0391
71. Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-KB. PLoS One (2015) 10:e0123410. doi:10.1371/journal.pone.0123410
72. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375:819–29. doi:10.1056/NEJMoa1604958
73. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov (2017) 7:188–201. doi:10.1158/2159-8290.CD-16-1223
74. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol (2015) 23:32–8. doi:10.1016/j.coph.2015.05.011
75. Kluger HM, Zito CR, Barr ML, Baine MK, Chiang VLS, Sznol M, et al. Characterization of PD-L1 expression and associated T-cell infiltrates in metastatic melanoma samples from variable anatomic sites. Clin Cancer Res (2015) 21:3052–60. doi:10.1158/1078-0432.CCR-14-3073
76. Obeid JM, Erdag G, Smolkin ME, Deacon DH, Patterson JW, Chen L, et al. PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: correlation with tumor-infiltrating immune cells and clinical outcome. Oncoimmunology (2016) 5:e1235107. doi:10.1080/2162402X.2016.1235107
77. Kubeček O, Kopecký J. Microsatellite instability in melanoma. Melanoma Res (2016) 26:545–50. doi:10.1097/CMR.0000000000000298
78. Sanmamed MF, Carranza-Rua O, Alfaro C, Oñate C, Martín-Algarra S, Perez G, et al. Serum interleukin-8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin Cancer Res (2014) 20:5697–707. doi:10.1158/1078-0432.CCR-13-3203
79. Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small cell lung cancer patients. Ann Oncol (2017) 28(8):1988–95. doi:10.1093/annonc/mdx190
80. Cabel L, Riva F, Servois V, Livartowski A, Daniel C, Rampanou A, et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: a proof-of-concept study. Ann Oncol (2017) 28(8):1996–2001. doi:10.1093/annonc/mdx212
Keywords: monoclonal antibodies, immune checkpoint blockade, combined tumor immunotherapy, clinical trials, cutaneous melanoma
Citation: Aris M, Mordoh J and Barrio MM (2017) Immunomodulatory Monoclonal Antibodies in Combined Immunotherapy Trials for Cutaneous Melanoma. Front. Immunol. 8:1024. doi: 10.3389/fimmu.2017.01024
Received: 30 June 2017; Accepted: 08 August 2017;
Published: 25 August 2017
Edited by:
Leonor Kremer, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Kerry S. Campbell, Fox Chase Cancer Center, United StatesZong Sheng Guo, Harvard University, United States
Copyright: © 2017 Aris, Mordoh and Barrio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariana Aris, bWFyaWFuYS5hcmlzQGNvbmljZXQuZ292LmFy