 James S. Foster1Angela D. Williams1Sallie Macy1Tina Richey1
James S. Foster1Angela D. Williams1Sallie Macy1Tina Richey1 Alan Stuckey2Daniel Craig Wooliver1
Alan Stuckey2Daniel Craig Wooliver1 Richa Koul-Tiwari1
Richa Koul-Tiwari1 Emily B. Martin1
Emily B. Martin1 Stephen J. Kennel1,2
Stephen J. Kennel1,2 Jonathan S. Wall1,2*
Jonathan S. Wall1,2*
- 1Department of Medicine, University of Tennessee Medical Center, Knoxville, TN, United States
- 2Department of Radiology, University of Tennessee Medical Center, Knoxville, TN, United States
There is a continuing need for therapeutic interventions for patients with the protein misfolding disorders that result in systemic amyloidosis. Recently, specific antibodies have been employed to treat AL amyloidosis by opsonizing tissue amyloid deposits thereby inducing cell-mediated dissolution and organ improvement. To develop a pan-amyloid therapeutic agent, we have produced an Fc-fusion product incorporating a peptide, p5, which binds many if not all forms of amyloid. This protein, designated Fcp5, expressed in mammalian cells, forms the desired bivalent dimer structure and retains pan-amyloid reactivity similar to the p5 peptide as measured by immunosorbent assays, immunohistochemistry, surface plasmon resonance, and pulldown assays using radioiodinated Fcp5. Additionally, Fcp5 was capable of opsonizing amyloid fibrils in vitro using a pH-sensitive fluorescence assay of phagocytosis. In mice,125 I-labeled Fcp5 exhibited an extended serum circulation time, relative to the p5 peptide. It specifically bound AA amyloid deposits in diseased mice, as evidenced by biodistribution and microautoradiographic methods, which coincided with an increase in active, Iba-1-positive macrophages in the liver at 48 h postinjection of Fcp5. In healthy mice, no specific tissue accumulation was observed. The data indicate that polybasic, pan-amyloid-targeting peptides, in the context of an Fc fusion, can yield amyloid reactive, opsonizing reagents that may serve as next-generation immunotherapeutics.
Introduction
The systemic amyloidoses are a family of rare diseases wherein normally soluble proteins form insoluble amyloid fibrils that deposit in various organs throughout the body resulting in dysfunction and severe morbidity (1). More than 30 distinct proteins, with dissimilar secondary structure and function, have been identified as components of visceral amyloid fibrils. The most common form, immunoglobulin light chain-associated (AL) amyloidosis, is a heterogeneous disease with respect to both the primary structure of the light chains involved and the varied anatomic distribution of the deposits (2). Other major forms of systemic amyloidosis are associated with the deposition of transthyretin, either wild-type (WT) (wtATTR) or mutant forms (ATTR) (3), leukocyte chemotactic factor 2 (Alect2) (4), and the acute phase protein serum amyloid protein A (AA) (5). Amyloid deposits are structurally complex and diverse, being comprised principally of proteinaceous fibrils as well as cell-derived, hypersulfated heparan sulfate proteoglycans, such as perlecan, and serum proteins, notably serum amyloid P component (SAP). Deposition of amyloid results in architectural damage to organs and tissues, particularly heart, kidney, and nerves, which leads to progressive disruption of organ function (6). Removal of tissue amyloid deposits would lead to improved organ performance, enhanced quality of life and likely a concomitant increase in survival rates for patients with these disorders. Such a therapeutic paradigm has emerged with the development of amyloid-reactive monoclonal antibodies (mAbs) capable of specifically binding the deposits, thereby opsonizing the otherwise non-immunogenic amyloid and facilitating dissolution of the amyloid by cells of the immune system (7). Early reports from the clinical evaluation of three such antibodies (NEOD001, 11-1F4 and the anti-SAP mAbs) have demonstrated improvement in organ performance as evidenced by positive changes in function-related biomarkers (8–11). Thus, opsonization of tissue amyloid by passive immunotherapy has become an important new strategy for treating patients with systemic amyloidosis. However, given the complexity and diversity of systemic amyloid diseases, the current anti-fibril antibody therapies may likely only be effective for patients with AL and AA amyloidosis. This may not be true of the humanized SAP-reactive immunoglobulin, which, in principle, could be used treat many types of systemic amyloid regardless of the type of precursor protein from which the fibrils are formed, due to the ubiquitous presence of SAP in all amyloid deposits. However, the universal efficacy of treatment using SAP-reactive Ab has yet to be demonstrated; in a preliminary report, only ~30% of enrolled patients demonstrated an improved SAP scan, indicating dissolution of tissue amyloid (8). Therefore, we posit that, given the heterogeneity of these diseases, no single immunotherapeutic mAb will be effective for treating all patients, and production of alternative reagents that are capable of pan-amyloid opsonization remains an important goal.
We have developed a panel of synthetic, polybasic peptides that specifically bind to many forms of human amyloid, including AL, AA, ALect2, ATTR, Alzheimer’s disease-associated (Aβ), and islet amyloid polypeptide (AIAPP), found in patients with type 2 diabetes (12–14). One of these peptides, designated p5, is a non-natural 31-amino acid peptide with eight lysine residues spaced in a heptad repeat such that when an α-helical secondary structure is formed, the charged side chains align along one face of the helix (15). This conformation promotes electrostatic interactions with hypersulfated glycosaminoglycans, such as heparin, and with amyloid fibrils (16). We have shown that radioiodinated p5 rapidly binds amyloid deposits systemically in a murine model of AA amyloidosis with little evidence of binding to healthy organs and tissues (17). An elongated version of peptide p5, designated p5+14, is being translated for amyloid imaging (13); however, given the specific and pan-amyloid reactivity of these peptides, our goal now is to utilize them as therapeutic agents. To this end and to take advantage of the clinical efficacy of opsonizing mAb therapy in amyloid patients, we have developed a peptide p5-Fc fusion construct, designated Fcp5. Addition of the Fc to the peptide serves two notable functions: extension of the circulating half-life of p5 and provision of an immunologically active Fc moiety to mediate antibody-dependent cellular phagocytosis [ADCP; reviewed in Park et al. (18)]. Fusion of Fc domain proteins to biologically active proteins and receptors is an effective strategy to enhance the biological half-life by exploiting the pH-dependent interactions of the Fc with the neonatal Fc-receptor [FcRn (19, 20)]. An added advantage of the Fc-fusion proteins is that they generate multivalent reagents, thereby potentially enhancing avidity of the associated ligand for the target (21). The success of this approach is documented by the fact that numerous Fc-fusion proteins have been approved by the US Food and Drug Administration for indications such as rheumatoid arthritis (etanercept) and type 2 diabetes (dulaglutide) (22–24). Despite the increasing use of this strategy to enhance the half-life of small proteins and peptides, Fc fusion is not routinely employed as a means to generate immunologically active reagents capable of ADCP, which requires the use of specific Fc isotypes and glycoforms (25).
To generate a novel, immunotherapeutic reagent for targeting systemic amyloidosis, we synthesized a murine Fc-fusion construct that incorporated the synthetic pan-amyloid-reactive peptide p5 with a murine IgG2a Fc. The murine Fc was used in this preliminary fusion to allow proof of principle studies in mouse models of amyloidosis without the complication of immunogenicity of a human Fc component. In this study, we show that the Fcp5 protein can be readily produced and purified from cell culture medium, that the product retains the amyloid binding properties of the p5 molecule, and that the opsonizing function of the Fc is intact. When radiolabeled, Fcp5 exhibited specific binding to systemic amyloid in vivo with enhanced serum half-life relative to the p5 peptide.
Materials and Methods
Peptides and Antibodies
Peptide p5 was synthesized and purchased as a crude preparation (Anaspec, Fremont, CA, USA) and purified by reverse phase high performance liquid chromatography (RP-HPLC), lyophilized, and stored at −20°C. Before use, the peptide was hydrated in sterile PBS and the concentration determined by micro-BCA assay (Thermo-Pierce, Waltham, MA, USA). The Aβ(1–40), IAPP, and IAPP(Ile26Pro) peptides were purchased as 90% pure preparations (Anaspec), stored frozen, and prepared for use as previously described (26). The rVλ6Wil was produced as a recombinant protein in E. coli and purified from the periplasmic space extract by RP-HPLC, as described (27). MOPC 173 murine IgG2a was from Biolegend (San Diego, CA, USA) and murine IgG2a Fc fragment was purchased from Acros Biosystems (Newark, DE, USA). Murine 11-1F4 mAb was prepared and supplied in sterile PBS by SAIC (Frederick, MD, USA). Biotinylated goat anti-mouse IgG was purchased from Sigma-Aldrich (St. Louis, MO, USA) and the Iba-1-reactive rabbit pAb was from Wako (Richmond, VA, USA).
Fibrils and Amyloid Extracts
Amyloid-like fibrils were prepared in sterile PBS from purified rVλ6Wil, Aβ(1–40) and IAPP, as previously described (26). The fibrils were isolated by centrifugation at 15,000 × g for 5 min, and the presence of fibrils was confirmed by measuring the fluorescence emission (490 nm; excitation = 450 nm) of thioflavin T added to ~5 μg of fibril preparation. Human amyloid extracts were prepared from autopsy-derived organs using a modified water floatation method, as described elsewhere (28). Murine liver homogenates were prepared as previously described (26).
Cloning of Fcp5
The codon-optimized cDNA for peptide p5 (amino acid sequence: GGGYS KAQKA QAKQA KQAQK AQKAQ AKQAK Q), flanked by 5-amino acid (VTPTV) spacers, was purchased from Integrated DNA Technologies (Coralville, IA, USA). The cDNA was cloned into the Nco I and Bgl II sites of the pFUSE-mIgG2A-Fc vector (Invivogen, San Diego, CA, USA), via PCR-based In-Fusion cloning (Clontech, Mountain View, CA, USA), and in-frame with the IL-2 secretory leader and the CH2 and CH3 domains of the murine IgG2a heavy chain. The plasmid sequence was verified by sequencing, using standard techniques, at the University of Tennessee Molecular Biology Core Facility.
Production and Purification of Fcp5 Protein
Fcp5 protein was produced by transient transfection of HEK293T/17 cells (ATCC, Manassas, VA, USA) in 100-mm tissue culture dishes using 6 µg of plasmid DNA with 20 µg of linear polyethylenimine (Polysciences, Warrington, PA, USA) per dish and culturing 9 days in DMEM/F12 (Lonza, Walkersville, MD, USA) with 2% (v/v) immunoglobulin-depleted fetal bovine serum (Thermo-Hyclone, Logan, UT, USA) and penicillin–streptomycin (Lonza), with media changes every 3 days. The collected cell supernatants were clarified by centrifugation at 1,500 × g for 10 min and the secreted Fcp5 purified by affinity chromatography using protein A-conjugated Sepharose (GE Healthcare, Pittsburg, PA, USA) with elution by 0.1 M glycine, pH 3.0, followed by dialysis in PBS and quantitation by Coomassie blue protein assay (Thermo-Pierce, Dallas, TX, USA). The integrity of the purified Fcp5 was documented by SDS-PAGE on native and reduced samples with staining by Coomassie brilliant blue, or by periodic acid Schiff staining for carbohydrate (Thermo-Pierce).
In Vitro Binding Studies—Europium-Linked Immunosorbent Assay (EuLISA)
Bioactivity of the Fcp5, peptide p5, and Fc2a control was assessed using a EuLISA. The wells of a 96-well polystyrene microplate (Corning, Corning, NY, USA) were coated either with poly-l-lysine (Sigma-Aldrich) followed by low molecular weight heparin (Sigma-Aldrich), amyloid-like fibrils (0.83 µM), or monomeric forms of rVλ6Wil, Aβ(1–40) or human IAPP(Ile26Pro). Wells coated with fibrils or monomeric proteins were incubated overnight at 37 or 4°C, respectively. The wells were then treated with 200 µL of blocking buffer (PBS containing 1% bovine serum albumin; BSA) for 1 h at room temperature before washing with PBS and addition of the appropriate concentration of Fcp5, biotinylated-p5, or Fc2a in PBS with 1% (w/v) BSA and 0.05% (v/v) tween 20. Following a wash step, the bound Fcp5 and Fc2a were detected by addition of biotinylated goat anti-mouse IgG (Sigma-Aldrich). After washing with PBS/tween, wells were incubated with 100 µL of europium–streptavidin (Perkin Elmer, Waltham, MA, USA) and washed, followed by addition of 100 µL enhancement solution (Perkin Elmer) before measurement of time-resolved fluorescence emission using a Wallac Victor 3 plate reader (Perkin Elmer).
Radioiodination
Briefly, Fcp5, p5, Fc2a, and mAb 11-1F4 were radioiodinated with 2 mCi iodine-125 (125I; Perkin Elmer) in the presence of 10 µg of chloramine T. The radiolabeled products were purified by size-exclusion gel filtration using either; Sephadex G-25 (PD10; GE Healthcare, Pittsburgh, PA, USA), Aca44 (Sigma-Aldrich), or Aca34 (Sigma-Aldrich) with a mobile phase of PBS containing 0.1% (w/v) gelatin, as previously described (29). The radiochemical yield was estimated by measuring the amount of 125I recovered in the purified pooled product relative to the amount of added 125I. The radiochemical purity and integrity of the purified product was assessed by SDS gel electrophoresis using 10% polyacrylamide gels followed by phosphor imaging (Cyclone Storage Phosphor System, Perkin Elmer, Shelton, CT, USA).
In Vitro Binding Studies—Pulldown Assay
Binding of radioiodinated Fcp5, Fc2a, p5, and mAb 11-1F4 with amyloid-like fibrils, amyloid extracts, and murine liver homogenates was measured using a pulldown assay, as previously described (29).
Surface Plasmon Resonance Measurements
Kinetic binding of Fcp5 and p5 to rVλ6Wil amyloid-like fibrils was assessed using surface plasmon resonance (BIAcore X; GE Healthcare, Pittsburgh, PA, USA). Experiments were performed as described previously (26) using a CM5 chip coated with rVλ6Wil fibrils (channel 1) and rVλ6Wil monomer (channel 2), as a control. Sensorgrams were recorded as the difference in resonance units of channel 1 minus channel 2. Seventy microliters samples of Fcp5 (2 µg/mL) or peptide p5 (0.2 µg/mL) were loaded and the data collected for 400 s (200 s binding-phase and 200 s dissociation-phase) with a flow rate of 20 µL/min. Binding and washout data were extracted from the sensorgram, aligned, and analyzed with the BIAevaluation software (Ver. 3) by fitting to the provided two-state binding algorithm with conformational change [A + B = AB = AB*].
Immunohistochemistry and Histology
Formalin-fixed, paraffin-embedded tissues sections were cut at 6 µM and placed on Plus slides (Fisher Scientific, Norcross, GA, USA). Antigen retrieval was performed using Target Retrieval Solution™, pH 9 (Dako Corporation, Carpenteria, CA, USA), according to the manufacturer’s instructions. The tissue sections were then incubated with Fcp5 or control Fc2a protein at 0.15 µg/mL in PBS overnight at 4°C. The slides were then washed in water and further processed by antibody avidin–biotin detection of the bound Fc fragments (Elite Mouse IgG kit; Vector Laboratories, Burlingame, CA, USA) and development with diaminobenzidene reagent (ImmPACT™ Peroxidase Substrate kit; Vector laboratories). Tissue staining with Congo red (CR) and Iba-1-reactive pAb was performed as previously described (29, 30). Images were acquired using a Leica DMR microscope fitted with a SPOT-RT cooled CCD camera (Diagnostic Instruments, Sterling Heights, MI, USA). Image segmentation of the Iba-1-stained tissues was performed using the “count” feature in Image Pro Plus (v. 9.0.4) (Media Cybernetics, Rockville, MD, USA).
Ex Vivo Phagocytosis Assay
The rVλ6Wil monomer and human ATTR extracts were labeled with the pH-sensitive dye, pHrodo green-STP ester (Life Technologies), according to manufacturer’s instructions. Amyloid-like fibrils were prepared by angled shaking (225 rpm) of a solution of 100 µg pHrodo green-labeled rVλ6Wil monomer and 900 µg of unlabeled monomer for ~72 h at 37°C. Free pHrodo green dye was removed from the fibril and extract preparations by centrifugation at 10,000 × g for 15 min and washing in PBS. For the phagocytosis assay, ~1 × 106 RAW 264.7 cells (ATCC) were added to each well of a 12-well culture dish, containing a glass coverslip, and incubated overnight. The following day, 4 µg of pHrodo green-labeled human ATTR extract or rVλ6Wil fibril preparation was mixed with 20 µg of Fcp5, MOPC2a, Fc2a, or, as a negative control, an equal volume of PBS. The sample was incubated for 30 min, unbound protein removed by washing each preparation in PBS with 2× centrifugation at 10,000 × g and the treated fibrils or extract added to the wells containing RAW 264.7 cells.
Following a 110-min incubation at 37°C, Hoescht 33342 dye (Life Technologies) was added to each well to stain the cell nuclei, and the mixture was incubated for a further 10 min. The coverslips were then washed with cell culture medium and live-mounted on a slide for image acquisition using a Leica DMR 500 epifluorescent microscope fitted with a SPOT-RT digital camera and blue or green fluorescence filter sets. Image segmentation was performed using Image Pro Plus software and the area of green fluorescence measured and expressed per cell (phagocytosis index) using 4 or 6 low magnification (5× objective) fields of view (FOV) from each coverslip. Approximately 2,000 cells were analyzed for each sample.
Biodistribution Measurements
In vivo biodistribution and microautoradiography were performed, as previously described (13), in H2-Ld-huIL-6 Tg Balb/c mice with systemic AA amyloid deposition at 5 wk post IV injection of 100 µg amyloid-enhancing factor [AEF (31)]. Amyloid-free WT mice served as the control. Mice were administered, IV in the lateral tail vein, ~100 μCi 125I-Fcp5 [either 3 µg bolus (pharmacokinetics) or 100 µg bolus (biodistribution) of Fcp5 with 10% (w/w) 125I-Fcp5] in a 200 µL-volume of sterile PBS containing 0.1% gelatin. The Fcp5-injected mice were left from 1 h and up to 144 h postinjection before being euthanized by isoflurane inhalation overdose. Thereafter, the organs were harvested at necropsy for biodistribution and microautoradiographic analyses, as described (13).
Measurement of serum radioactivity was performed using cohorts of three mice bled at 1, 4, and 24 h post IV injection of ~30 μCi 125I-Fcp5. Blood was obtained from the retro-orbital sinus immediately before euthanasia. The blood was allowed to clot at 37°C for 15 min before being centrifuged at 15,000 × g for 2 min and serum collected.
Ethics Statement
All patient-derived tissue samples were used in accordance with an Institutional Review Board-approved application. Animal studies were carried out in strict accordance with a protocols approved by the University of Tennessee Institutional Animal Care and Use Committee. All procedures were approved by the IACUC and were performed in accordance with the guidelines provided by OLAW and the Guide for the Care and Use of Laboratory Animals. The University of Tennessee Medical Center animal program is AAALAC-i-accredited.
Statistical Methods
The binding of Fcp5 to substrates are shown as the mean ± SD of six replicates. Data points for the phagocytosis assay were compared between each respective using ANOVA with Dunnett correction for multiple comparisons; mean ± SD, n = 3. Evaluation of the macrophage distributions were performed using a two-tailed t-test with a significance value of p < 0.05. Analyses were performed using Prism ver. 6.07 (Graphpad Inc., La Jolla, CA, USA).
Results
Fcp5 Production and Characterization
The Fcp5 expression construct was prepared by cloning the cDNA encoding peptide p5 with the addition of a five amino acid linker into, and in frame with, a vector encoding the murine IgG2aFc. Transient transfection of the Fcp5 encoding vector into HEK293T cells resulted in demonstrable Fcp5 protein production and secretion into the culture media at 48 h post transfection. The Fcp5 protein was subsequently purified from immunoglobulin-depleted cell culture medium supernatants of transiently transfected HEK293T cells by Protein A affinity chromatography with a yield of ~50 μg per 1 × 106 cells. Analysis of the isolated Fcp5 by SDS-PAGE demonstrated a single band in the non-reduced form (Figure 1A) at ~85 kDa and, when reduced, at ~42 kDa consistent with the presence of a disulfide-linked glycosylated Fcp5 dimer (theoretical protein dimer Mw = 63,106) and monomer (Mw = 31,553), respectively. Staining the gel using periodic acid Schiff’s reagent indicated that Fcp5 was glycosylated, which likely accounted for the increase in the apparent molecular weight seen on the gel (Figure 1B).
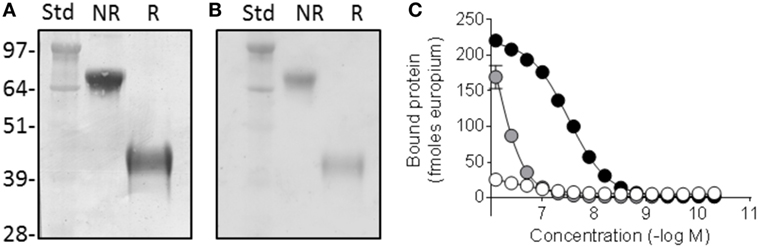
Figure 1. Fcp5 is secreted as a glycosylated dimer with heparin-binding activity. (A) Coomassie-stained SDS-PAGE gel showing dimeric construct in the non-reduced (NR) form which is reduced to monomer when reduced (R). (B) Glycosylation of Fcp5 shown by periodic acid Schiff-stained gel. (C) Heparin binding was observed with Fcp5 (black) and peptide p5 (gray) but not Fc2a control (white).
Since p5 was developed as a heparin-reactive peptide, the bioactivity of purified Fcp5 was initially examined by studying the interaction with low molecular weight heparin (Figure 1C). The Fcp5 (black) bound heparin with an EC50 of ~30 nM, which was ~30-fold higher than the binding of the p5 peptide alone (~850 nM; gray). No reactivity of an IgG2a Fc fragment (Fc2a) control protein with heparin was observed (white).
Fcp5 Reactivity with Amyloid-Like Fibrils and Monomeric Protein
The binding of Fcp5 with amyloid-like fibrils was compared to that of p5 using fibrils made from: an immunoglobulin light chain variable domain (rVλ6Wil), the Aβ(1–40) peptide, and IAPP (Figure 2). The Fcp5 (black), peptide p5 (gray), or Fc2a control (white) was added to fibrils immobilized in the wells of a microplate (Figure 2A). The Fcp5 bound rVλ6Wil, Aβ(1–40) and IAPP amyloid-like fibrils with estimated EC50 values of 1, 4, and 15 nM, respectively, which were higher affinities than those calculated for peptide p5 (14, 230, and 200 nM, respectively; Figure 2A). The control Fc2a, again, exhibited no detectable binding. Since only Fcp5 and p5 exhibited binding to the fibrils, the reactivity of these reagents was tested using monomeric forms of rVλ6Wil, Aβ(1–40) and a non-fibrillogenic form of human IAPP with a Pro substitution at position 26 (Figure 2B). In all cases, there was no reactivity of Fcp5 (black) or p5 (gray) with the non-fibrillar substrates.
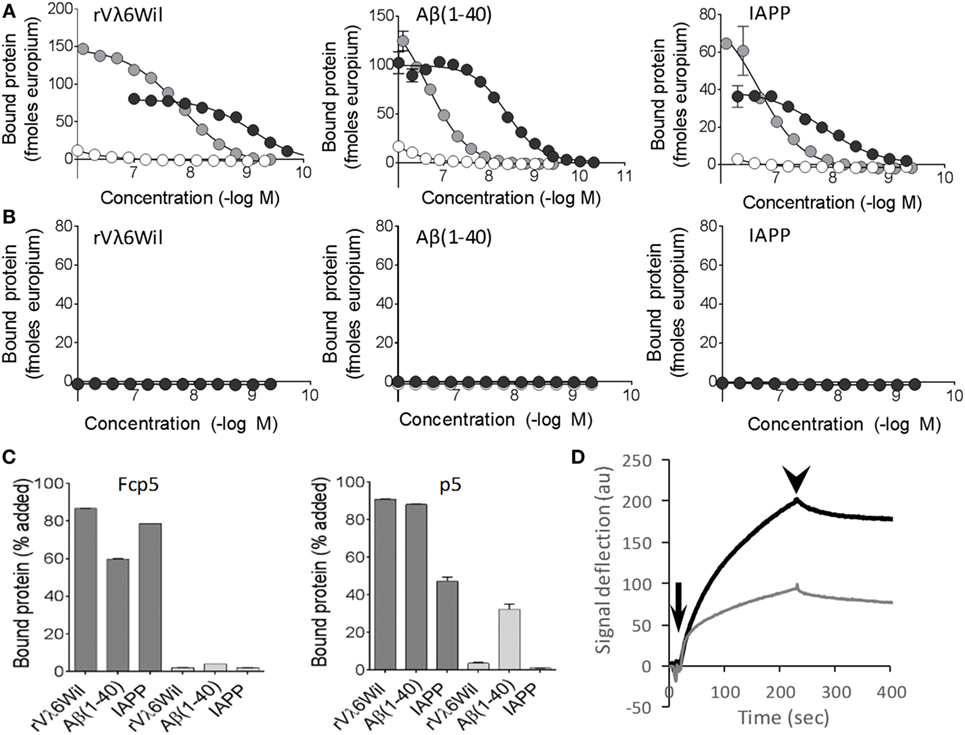
Figure 2. Fcp5 binds the three major forms of amyloid-like fibrils. (A) The Fcp5 construct (black) and peptide p5 (gray), but not Fc2a (white), bound surface-adsorbed amyloid-like fibrils composed of rVλ6Wil, Aβ(1–40), and IAPP. In each case, Fcp5 bound with greater affinity. (B) No reactivity of Fcp5 (black) or p5 (gray) was observed when non-fibrillar rVλ6Wil, Aβ(1–40), or human IAPP(Ile26Pro) were adsorbed to the surface of microplate wells. (C) Pulldown assays were performed using 125I-Fcp5 and peptide 125I-p5 in PBS (dark gray) and 1 M NaCl (light gray) to assess the importance of electrostatic interactions. (D) Kinetic binding of Fcp5 (black) and p5 (gray) to rVλ6Wil fibrils by surface plasmon resonance. Injection (arrow) and washout (arrowhead) phases occurred over 200 s.
The reactivity of Fcp5 and p5 with amyloid-like fibrils was further interrogated following radioiodination with iodine-125 (125I) in a “solution phase” pulldown assay, in PBS or in 1 M NaCl (Figure 2C). High salt milieu was used to examine the relative importance of electrostatic interactions in the binding. The interaction of 125I-labeled Fcp5 and p5 with the fibrils in PBS was comparable (Figure 2C, dark gray) and, with the exception of 125I-p5 binding to Aβ(1–40), the binding was completely abrogated in 1 M NaCl (Figure 2C, light gray).
Kinetic analysis of the interaction of Fcp5 (black) and p5 (gray) with rVλ6Wil fibrils was performed by using surface plasmon resonance (Figure 2D). Evaluation of the Fcp5 kinetic profile, using a 2-state binding algorithm, yielded a KD of ~3 nM and a signal deflection of ~200 au. Similar analysis of peptide p5 gave an approximately equivalent KD (~5 nM) but a deflection of only 100 au, consistent with the lower molecular weight of the p5 vs Fcp5.
Human Tissue Amyloid Reactivity
Peptide p5 can be used to immunostain many forms of human amyloid in tissue samples. To investigate whether this property was retained by the peptide in the context of the Fc fusion, formalin-fixed paraffin-embedded tissues obtained at autopsy from patients with AL (κ or λ), ATTR, ALect2, AA, or AIAPP-associated amyloidoses were immunostained using Fcp5 or with Fc2a as a negative control (Figure 3). In all cases, Fcp5 co-localized with amyloid deposits in the tissue, as evidenced by the brown diaminobenzidene staining that correlated with the tissue distribution of amyloid, shown as green birefringent material in CR-stained consecutive tissue sections.
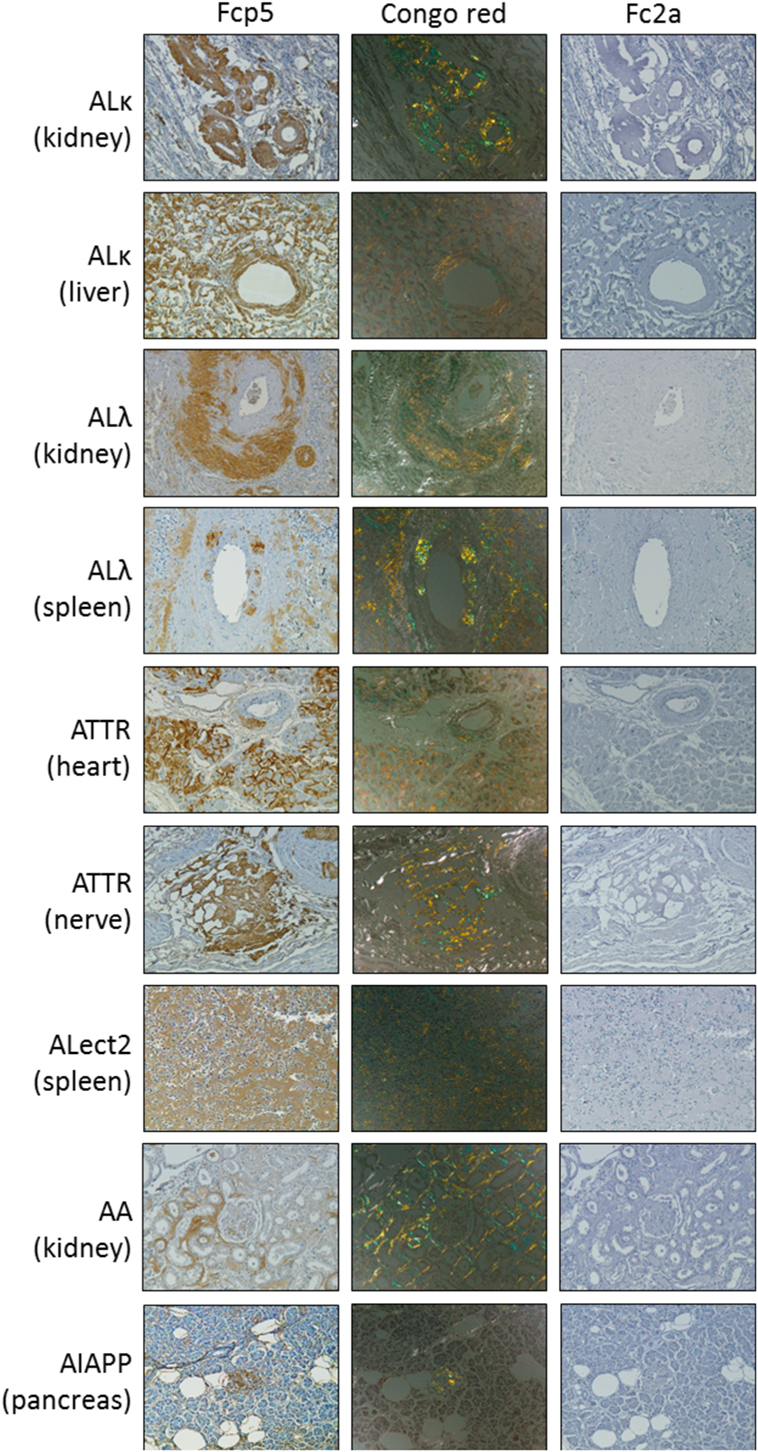
Figure 3. The Fcp5 specifically binds human amyloid deposits in formalin-fixed tissues. Fcp5 immunostained human ALκ, ALλ, ATTR, ALect2, AA, and AIAPP amyloid in formalin-fixed paraffin-embedded tissues as evidenced by the brown DAB staining which co-localized with the amyloid seen as green-gold birefringent material in Congo red-stained consecutive tissue sections. There was no binding of Fc2a. Original objective magnification 20×.
Amyloid Extract Reactivity
To further study the reactivity of Fcp5 with a more complex form of amyloid, pulldown experiments were conducted using radioiodinated Fcp5, p5, Fc2a, and the clinically relevant mAb, 11-1F4 (Figure 4), using as a substrate human ALκ, ALλ, and ATTR amyloid extracts as well as murine AA amyloid-laden liver homogenate, and a control healthy liver homogenate. As compared to the 125I-p5 peptide, the reactivity of 125I-Fcp5 with all amyloid extracts, with the exception of one ALκ (Cab) and one ALλ (Bal), was reduced by ~10-fold to 20-fold, but it remained positive (Figures 4A,B). The pattern of reactivity with each amyloid extract was similar for both 125I-Fcp5 and 125I-p5. The 125I-11-1F4 murine mAb did not avidly bind any of the amyloid extracts or tissue homogenates in this assay, with <2% of the 125I mAb bound in all cases, except ALκ (Cab; Figure 4C). The 125I-Fc2a, which served as a negative control, did not bind any of the substrates (<0.1% bound; Figure 4D).
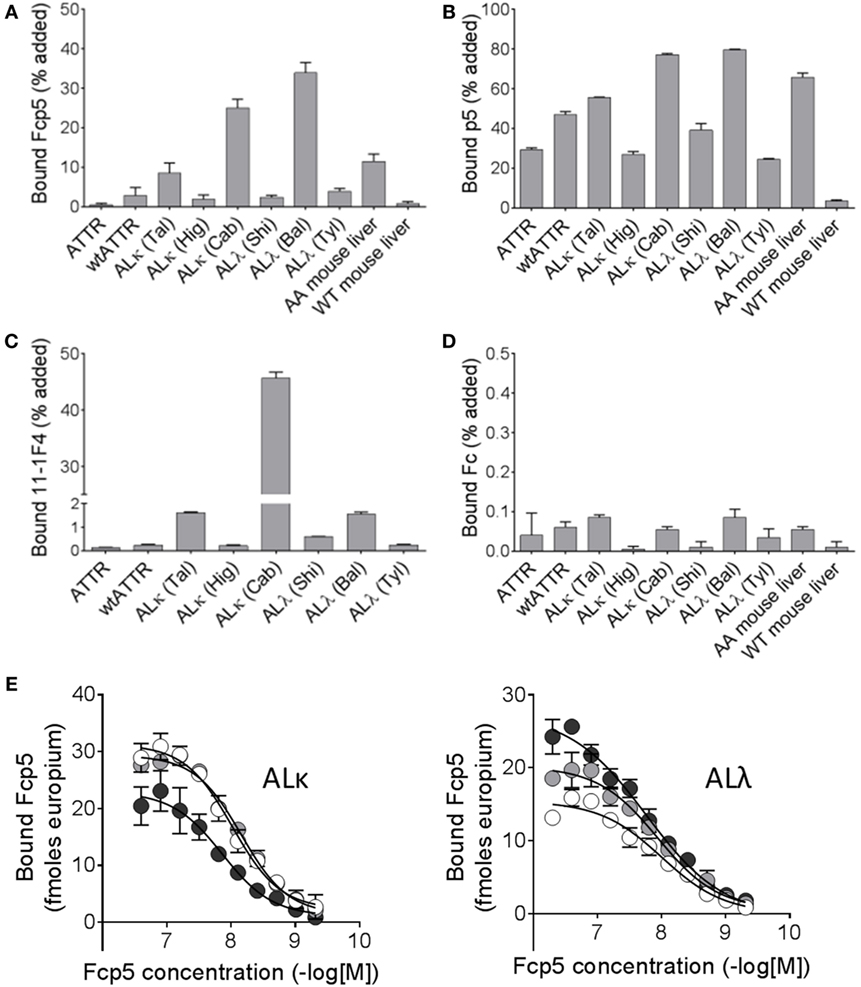
Figure 4. Radioiodinated Fcp5 binds human and murine amyloid extracts. Proteins Fcp5 (A), p5 (B), 11-1F4 (C), and Fc2a (D) were radiolabeled and used in a pulldown assay to assess binding to human ATTR extracts, AL extracts, and mouse AA homogenates. (E) Binding of Fcp5 to surface-adsorbed human ALκ and ALλ amyloid extracts (three different patients per isotype) was assessed by europium-linked immunosorbent assay.
A EuLISA was also used to evaluate the relative affinity of the binding of Fcp5 with human AL amyloid extracts (Figure 4E). Fcp5 was titrated into wells containing surfaced-adsorbed AL amyloid extracts (n = 3 ALκ and 3 ALλ). Binding to both ALκ and ALλ was similar in intensity with EC50 values, estimated from fitting a sigmoidal dose-response algorithm, ranged from 7 to 17 nM (mean EC50 = 11.5 nM, n = 6).
Ex Vivo Opsonization Assay
To examine the bioactivity of the Fc component in the Fcp5 construct, ADCP was studied using an ex vivo phagocytosis assay. Phagocytosis of rVλ6Wil amyloid-like fibrils (Figure 5A) and human ATTR amyloid extracts (Figure 5B) labeled with the pH-sensitive dye, pHrodo green, by RAW.264.7 macrophages was measured. Labeled fibrils or ATTR amyloid extract were incubated in the presence of Fcp5 or, as negative control, MOPC IgG2a, Fc2a, or PBS alone in the presence of coverslip-bound macrophages. Cellular uptake of the labeled fibrils or extract into acidified phagolysosomes resulted in an increase in the fluorescence intensity of the pHrodo green fluorophore. The mean area of green fluorescence (n = 4 microscope FOV) was quantified and expressed per cell, based on a count of Hoescht-stained nuclei in each FOV (Figure 5). Using this technique, we observed a significant increase in phagocytosis of the fibrils and extracts in the presence of Fcp5, relative to all control samples. Digital images of the RAW.264.7 macrophages incubated with Fc2a- and Fcp5-treated substrate demonstrated the increased green fluorescence emission from the Fcp5 opsonized fibrils (Figure 5A) and human ATTR amyloid, relative to the control (Figure 5B).
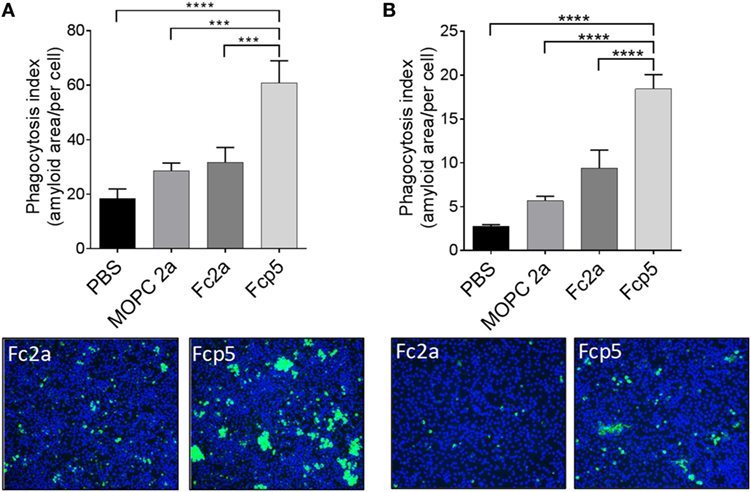
Figure 5. Phagocytosis of AL amyloid-like fibrils and human ATTR extracts is enhanced by opsonization by Fcp5. Quantitative estimation of phagocytosis of pHrodo green-labeled rVλ6Wil amyloid-like fibrils (A) and human ATTR amyloid extract (B). Addition of Fcp5 significantly enhanced phagocytosis of the substrate by adherent RAW264.7 cells. Images of the engulfed (fluorescent) substrate demonstrate the qualitative difference between Fc2a and Fcp5 incubation. Original objective magnification 10×. ***p < 0.0005 and ****p < 0.0001 using ANOVA with Dunnett correction for multiple comparisons; Mean ± SD, n = 3.
In Vivo Pharmacokinetics and Amyloid Reactivity
The in vivo characteristics of Fcp5 were studied following radiodionation of the protein (Figure 6). The reagent was readily radiolabeled with 125I with a radiochemical yield of ~50% (n = 3 reactions) and a radiochemical purity of >90% based on estimation of free radioiodide in the purified product by analysis of SDS-PAGE phosphor images (data not shown). The 125I-Fcp5, stored at 4°C in PBS with 0.1% gelatin, retained its affinity for rVλ6Wil amyloid-like fibrils, as evidenced using a pulldown assay, for more than 30 days with an estimated loss of total binding activity of only 1.8% per day over this period (Figure 6A). Estimation of the circulating half-life of 125I-Fcp5 was performed in healthy, WT mice, and animals with systemic AA amyloidosis by measuring the percent injected dose (%ID/g) in serum samples collected from cohorts of mice over 24 h postinjection (Figure 6B). The data were analyzed using a one-phase exponential decay equation, which yielded a serum t1/2 for 125I-Fcp5 in WT mice of 3.4 h (white) and in AA mice of 2.4 h (gray). The pharmacokinetics of 124I-p5 have been previously studied in WT mice, by using dynamic PET/CT (32), and is shown here by way of a comparison (black). Analysis of serum samples collected from WT and AA mice at 1, 4, and 24 h postinjection, by SDS-PAGE followed by phosphor imaging, indicated that the circulating 125I-Fcp5 was structurally intact with no evidence of radiolabeled proteolytic fragments and no indication of a truncated 125I-Fcp5 (Figure 6C). The gel profile for each serum sample was indistinguishable from the injected material (time 0; Figure 6C).
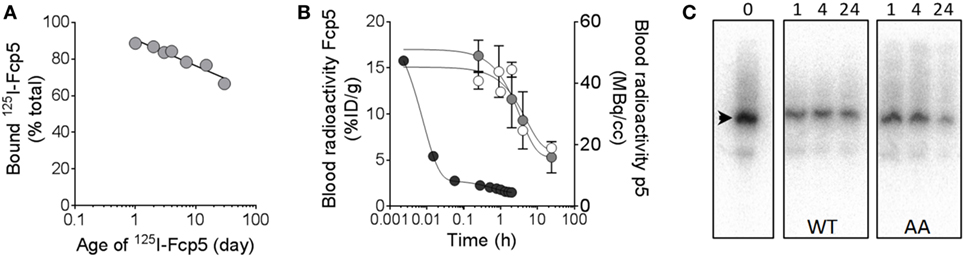
Figure 6. Radioiodinated Fcp5 is stable in vitro and in vivo with an extended serum half-life as compared to p5. (A) Binding of a preparation of 125I-Fcp5 to rVλ6Wil fibrils was assessed using a pulldown assay for 30 days post labeling. (B)125I-Fcp5 in the blood of wild-type (WT) (white) and AA (gray) mice was measured (%ID/g) over 48 h postinjection by gamma counting and compared to historical data for 124I-p5 in WT mice (black; MBq/cc). (C) Serum stability of 125I-Fcp5 in AA and WT mice, up to 24 h postinjection, was assessed by SDS-PAGE and phosphorimaging. There was no evidence of low MW radiolabeled metabolites nor a reduction in the MW of the 125I-Fcp5 (arrowhead).
The biodistribution of 125I-Fcp5 (10 µg doped in a therapeutic IV bolus of ~4 mg/Kg of non-radiolabeled Fcp5) was studied in AA and WT mice (n = 3 per time point; Table 1). Consistent with the serum pharmacokinetics data, there was a high concentration of 125I-Fcp5 in the blood at each time point in both AA and WT animals. In the AA mice, there was evidence of retention of 125I-Fcp5 in the liver (~10% ID/g at 1 h postinjection), relative to the WT mice (~8% ID/g); however, this was not significantly different (Table 1).
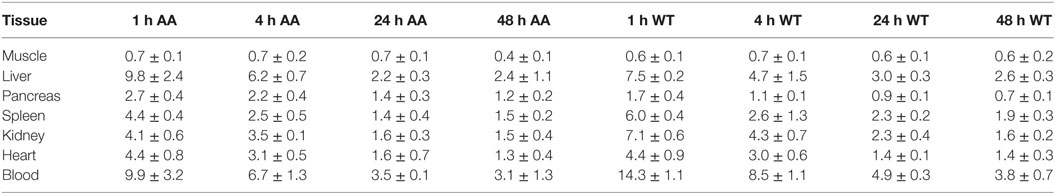
Table 1. Biodistribution of 125I-Fcp5 in AA and wild-type (WT) mice expressed as percent injected dose per gram (%ID/g).
To assess the specific reactivity of 125I-Fcp5 with AA amyloid deposits in the liver and spleen, the sites of major amyloid deposition in the transgenic mouse model of AA amyloidosis, tissue sections were prepared and microautoradiography performed (Figure 7). At each time point (1–48 h postinjection), there was evidence of specific accumulation of 125I-Fcp5 in amyloid deposits, most notably within the liver, as evidenced by the presence of black silver grains associated with the presence of radiolabeled Fcp5 (arrows, Figure 7). The deposition of 125I-Fcp5 correlated anatomically with sites of amyloid deposition in the organs seen as green-gold birefringent material in CR-stained consecutive tissue sections. The binding of 125I-Fcp5, particularly with hepatic amyloid lesions, persisted, albeit weakened, even at 48 h postinjection. The density of silver grains in the splenic amyloid deposits was less than that seen in the liver; however, specific amyloid binding of 125I-Fcp5 at 48 h was still detectable (Figure 7). There was no evidence, in the microautoradiographs, of specific cell-associated focal binding of 125I-Fcp5 in WT liver or spleen tissues (Figure 7).
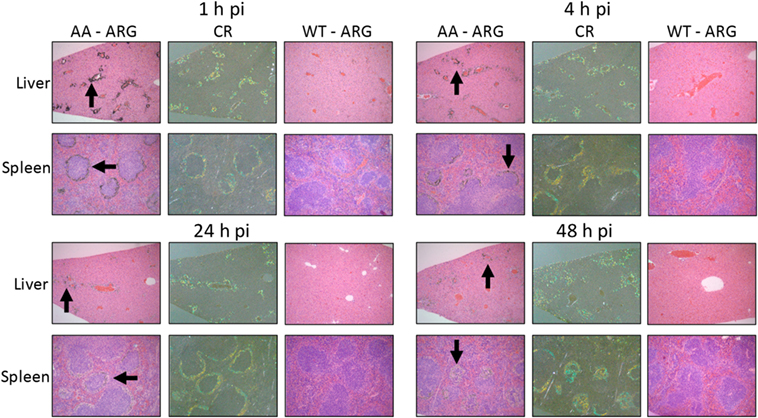
Figure 7. 125I-Fcp5 binds AA amyloid deposits in vivo. The presence of 125I-Fcp5 in the liver and spleen at 1, 4, 24, and 48 h postinjection (pi) was evidenced by the presence of black silver deposits (arrows) in autoradiographs (ARG). The tissue distribution of 125I-Fcp5 correlated with the presence of amyloid seen as green-gold birefringent material in Congo red-stained (CR) consecutive tissue sections. No evidence of specific binding of 125I-Fcp5 to wild-type (WT) liver and spleen was observed.
The distribution of Iba-1-positive macrophages was studied microscopically in these mice (Figure 8). For this analysis, only the liver was evaluated since this organ yielded evidence of the greatest accumulation of 125I-Fcp5 in amyloid deposits (Figure 7). Formalin-fixed paraffin-embedded tissue sections were immunostained using an Iba-1-reactive pAb and the number of stained cells quantified by image segmentation from digital images (Figure 8A). Positively stained macrophages were more prevalent in the Fcp5-treated mice at 48 h postinjection as compared to 1 h postinjection (Figure 8A). The number of macrophages was calculated from tissue sections of three AA mice (Figure 8B) and three WT mice (Figure 8C) at 1 and 48 h postinjection. There was a significant increase in the Iba-1-positive cells in the AA liver, but not the WT liver of Fcp5-treated mice.
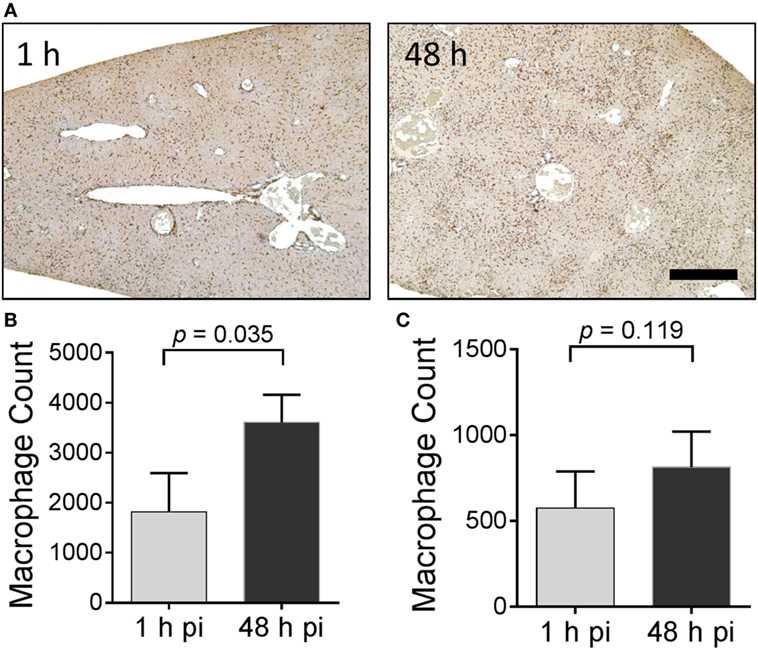
Figure 8. Administration of Fcp5 enhances hepatic macrophage number over 48 h in AA mice but not wild-type (WT) mice. (A) Formalin-fixed liver tissue from mice at 1 and 48 h postinjection of Fcp5 was immunostained with Iba-1-reactive monoclonal antibody and the number of immunopositive cells quantified by image segmentation. The bar is 300 pixels in length. Comparison (two-tailed t-test) of hepatic macrophage number in AA (B) and WT (C) mice at 1 and 48 h postinjection of Fcp5. Mean ± SD, n = 3.
Discussion
Potential Clinical Application
Effective treatment of patients with systemic amyloidosis has, until recently, been challenging. Most therapeutic approaches have focused on reducing the concentration of the amyloid-forming precursor protein by using anti-plasma cell chemotherapy, anti-inflammatory drugs, antisense oligonucleotides, or with protein-stabilizing small molecules for AL-, AA-, and ATTR-associated amyloidoses, respectively (33–35). Patients with AL amyloidosis have benefited from stem cell transplantation and those with ATTR amyloidosis from liver organ transplantation; however, many patients present with advanced disease and are not eligible for transplantation regimens, and even following transplant, disease often recurs.
Recently, three clinical trials have begun that evaluate the therapeutic efficacy of amyloid-reactive mAbs that specifically bind and opsonize the tissue amyloid deposits. These treatments facilitate recruitment of phagocytic macrophages that mediate amyloid removal through, presumably, ADCP; moreover, evidence of mAb binding to potentially toxic oligomeric light chain species may afford an additional measure of protection (36). Interim reports from these trials have indicated improvement in patients based on positive changes in surrogate biomarkers of cardiac and renal function, N-terminal pro B-type natriuretic peptide (NT-proBNP) and proteinuria, respectively (9, 10). In one trial, which incorporates an imaging component, a reduction in hepatic amyloid load has been documented in a subset of patients by using planar gamma scintigraphic imaging of amyloid (8). Thus, passive immunotherapy with amyloid-reactive opsonizing mAbs in patients with systemic amyloidosis can yield clinical benefit.
At present, only one of the current mAbs in clinical trial has the potential to treat many forms of systemic amyloidosis due to reactivity of the mAb with the serum amyloid P component (SAP), which is ubiquitously present in all amyloid deposits (37). Similarly, Fcp5, by virtue of the ability of peptide p5 to bind universal electrostatic motifs in amyloid deposits, is capable of binding many varied amyloid deposits. However, in contrast to Fcp5, anti-SAP passive immunotherapy is a two-step process wherein circulating SAP must first be depleted by pre-treatment using a small molecule (38) while leaving the amyloid-bound SAP intact. There are no data that suggest the presence of a circulating ligand, in mice, that binds Fcp5 and competes for amyloid binding.
Furthermore, there are currently no clinical trial data indicating these mAbs will effectively opsonize leukocyte chemotactic factor 2 (ALect2)-associated amyloid, which is now the third most common form of systemic amyloidosis in the US (39). In contrast, we have shown, albeit immunohistochemically, that Fcp5 binds ALect2 amyloid deposits in formalin-fixed tissues (Figure 3).
Pan-Amyloid Reactivity of Fcp5
To address the need for novel paradigms to treat patients with systemic amyloidosis, we have developed Fcp5, a bifunctional reagent that combines a pan-amyloid-reactive agent, peptide p5, fused with an IgG2a Fc domain. This construct was designed to enhance the circulating half-life of the peptide and generate an amyloid opsonin to mediate ADCP. This reagent has the potential to target many, if not all, forms of the systemic amyloidoses and thereby effect removal of tissue amyloid deposits (Figures 2–4). Immunoglobulin Fc-fusion proteins, sometimes termed peptibodies, have been developed as FDA-approved-therapeutics for numerous indications. The Fc domain, by virtue of its interaction with the FcRn and subsequent recycling, is used to extend the half-life of the associated bioactive protein, which otherwise would be rapidly catabolized. One Fc reagent, fused with the general amyloid interaction motif protein from a bacteriophage capsid protein, has been shown to bind Aβ amyloid and, when used to treat transgenic mice with cerebral Aβ amyloid deposits, showed positive therapeutic benefit (40, 41). We have previously demonstrated that peptide p5, and related polybasic synthetic peptides, bind Aβ amyloid in tissue sections as well as synthetic Aβ(1–40) amyloid-like fibrils in vitro (12, 13, 26). This suggests that Fcp5, delivered intravenously, may, in addition to the systemic amyloidoses, effectively target pathologic cerebral amyloid-related disorders, such as those found in patients with Alzheimer’s disease or spongiform encephalopathies (prion-related disorders).
Herein, we have demonstrated that the Fcp5 protein retains the amyloid reactivity of the parent 31-mer peptide p5 both in vitro and in vivo. Based on the estimated EC50 of Fcp5 for surface-adsorbed low molecular weight heparin and amyloid-like fibrils, as evidenced in EuLISA studies, the binding affinity was greater than that of the p5 peptide. This is likely due, in part, to the dimeric structure of Fcp5, which can mediate bivalent interactions and thus engage in high avidity binding to amyloid. Similar enhanced affinity has been observed for other Fc constructs where, in addition to bivalency, two or more copies of the binding motif have been incorporated into each of the Fc arms (42). In contrast to the EuLISA that favors bivalent interactions due to the high target density in the microplate well, solution-phase pulldown assays are likely governed by univalent interactions (Figures 2 and 4). When human amyloid extracts, containing, in addition to fibrils, heparan sulfate proteoglycans and other accessory molecules, were used as a substrate in pulldown assays, 125I-p5 bound in greater amounts as compared to 125I-Fcp5 (Figure 4). We hypothesize that the size of the Fc moiety may negatively impact the interaction of the Fc-associated p5 peptide in this assay. Alternatively, given that Fcp5 is 20-fold larger than peptide p5, it may reflect a decrease in the availability of binding sites due to the inability of the Fcp5 to penetrate the complex amyloid extract, as compared to the smaller peptide p5. That said, 125I-Fcp5 bound to all AL and ATTR amyloid extracts (with the exception of the ALκ4 amyloid, Cab) more effectively than 125I-11-1F4 mAb, which has been shown to image AL amyloid deposits in patients (43) and afforded clinical benefit in ~70% of enrolled subjects in the Phase 1a/b trial (11).
Effective development of peptides and polypeptides as therapeutics has been plagued by limitations due to their short circulation half-life and rapid catabolism; however, numerous strategies have been developed to overcome this problem (44). Of the potential approaches, Fc-fusion strategies have been successfully employed to address this shortcoming, and numerous reagents have been approved by the FDA for human use (45). Although our goal is to develop Fc-conjugated peptides as novel passive immunotherapeutics in a non-radiolabeled form, to study the behavior of Fcp5 in vivo, it was radiolabeled using 125I. The purified 125I-labeled Fcp5 retained its reactivity with amyloid-like fibrils composed of rVλ6Wil and was stable during storage for up to 30 days—fibril binding declined at only 1.8% of maximum, per day (Figure 6A). The serum half-life of 125I-Fcp5 was measured by following the radioiodinated reagent in the circulation over 24 h postinjection. Given that data collection was not extended to days, the estimated T1/2 of 3.4 h in WT mice likely represents only the fast (α) component of the two-phase decay curve. It is notable that even at 24 h, ~25% of the 125I-Fcp5 remained in the circulation at 24 h postinjection (as compared to the 5 min time point). These observations are consistent with other measurements of Fc circulating half-life where T1/2fast (α) was ~3 h, which is followed by a T1/2slow (β) of >100 h (46). Future studies are required that include earlier and later time points to validate this supposition. Regardless, addition of the Fc moiety extended the serum half-life of the peptide relative to that of 124I-p5, which was shown to have a T1/2slow component of the bi-exponential decay of ~30 min and a peak kidney uptake time of only 7 min postinjection (32). Prolongation of the serum half-life should contribute to improved interactions of Fcp5 and similar variants with the tissue amyloid in patients.
Opsonization and In Vivo Amyloid Reactivity
A critical requirement of the bifunctional Fcp5 for its use as an amyloid therapeutic, in addition to specific amyloid binding, is its ability to serve as an opsonin. We have demonstrated, using a quantitative fluorescence-based phagocytosis assay, that synthetic amyloid-like fibrils and human ATTR amyloid extracts can be effectively opsonized by pre-incubation with Fcp5, leading to enhanced uptake by surface-bound macrophages (Figure 5). Although the effector cell(s) that mediate amyloid dissolution in patients remain undefined, data suggest that macrophages and giant cells are likely involved (47, 48).
Complement fixation may have been important in the passive immuno-dissolution of amyloid in vivo when the SAP-reactive antibody was used (8). The fixation of complement by Fcp5 has not been evaluated and may not be an absolute requirement for ADCP of amyloidosis. Recent advances in Fc engineering have led to amino acid substituted variants and modified glycoforms that impart altered biological properties such as increased affinity for FcRn, increased complement fixation, enhanced and selective FcγR isoform binding, and decreased immunological activity [reviewed in Park et al. (18)]. Antigen-reactive Fc moieties have also been developed that incorporate paratopes engineered into the heavy constant domain 2 (CH2) to serve as truncated IgG (49). In the case of Fcp5, we have chosen to incorporate peptide p5 to function as the “paratope” and direct highly specific, high-affinity binding of the Fc to the target amyloid deposits. The initial Fcp5 protein produced in HEK cells has not been optimized for any biological function; however, this can be readily explored by using an engineered IgG1 isotype-based Fcp5 and altering the glycoform by production in CHO cells (50–52). Studies to compare the activities of HEK- and CHO-generated Fcp5 are underway.
There are currently no murine models of AL-, ATTR-, or ALect2-associated amyloidosis that effectively recapitulate the pervasive amyloid deposits seen in patients. Therefore, the murine model of AA-associated amyloidosis has become the exemplar experimental model of systemic amyloid disease used in preclinical studies. In susceptible mice, such as those expressing the pro-inflammatory human interleukin-6 transgene, AA amyloidosis can be induced by exogenous stimuli such as a suspension of preformed AA amyloid extract [AEF (31)], synthetic fibrils (53), or even pate de foie gras (54). Within weeks of induction, the mice develop severe systemic AA-associated amyloidosis predominantly in the spleen and liver, but the pancreas, kidney, adrenal, heart, and vasculature are also involved. These extensive deposits can serve as a target for the in vivo quantitative evaluation of amyloid-reactive reagents such as peptides (13, 14, 17) and the Fc-peptide fusion reagents. Using this experimental model, we observed specific co-localization of 125I-Fcp5 was observed in hepatosplenic AA amyloid deposits, as evidenced by the presence of black punctate staining in microautoradiographs that correlated with the anatomic distribution of amyloid (Figure 7). The presence of radiolabeled Fcp5 in the amyloid deposits decreased over 48 h postinjection. This contrasts with similar observations of bound half-life of radiolabeled peptides p5 and the related p5+14, which were retained at high density for more than 72 h (13). The loss of radiolabel from the amyloid-bound Fcp5 may reflect dissociation of 125I-Fcp5 or, alternatively, may indicate dehalogenation of the 125I-Fcp5 in situ, and subsequent loss of radioiodine to the circulation. Although we cannot definitively discern between these two scenarios, we observed a substantial influx of Iba1-positive macrophages in the livers of Fcp5-treated AA mice. Importantly, deiodinases are generally intracellular enzymes (55); therefore, if dehalogenation of amyloid-bound 125I-Fcp5 is occurring, it may reflect Fcp5-mediated ADCP (Figure 8). Further investigation and more direct measurement of the pharmacokinetics of Fcp5 in amyloid-laden mice and of the opsonization function in vivo are necessary to confirm this hypothesis.
If, however, the rapid loss of radioiodine from the amyloid deposits within 48 h postinjection reflects a short amyloid-bound half-life, improved Fc-peptide constructs may be required to enhance the peptide–amyloid interaction. To address this, we are generating reagents that increase the charge density of the amyloidophilic peptide moiety and exploring the use, either as an alternative or in conjunction with p5, of polybasic peptides with β-sheet primary structure [e.g., peptide p5(sheet) (14)].
Summary
The most important biophysical properties required of a passive immunotherapeutic for the removal of tissue amyloid are: in vivo stability; specific amyloid targeting; extended biological half-life; extended amyloid-bound half-life; and the ability to opsonize and recruit macrophages and induce phagocytosis of the amyloid. We have prepared a protein, Fcp5, that is stable, has 3 h serum half-life, exhibits specific, pan-amyloid reactivity, and is functional in in vitro phagocytosis assays. The Fcp5 specifically binds systemic AA amyloid deposits in a mouse model, and preliminary data suggest that, at least for the liver, this interaction enhances recruitment of macrophages. These data support our premise that Fc-peptide fusion proteins that incorporate synthetic amyloid-reactive peptides may serve as effective, immunologically active amyloid-targeting agents. Further optimization, by peptide or Fc engineering and expression in CHO cells, may lead to an Fcp5 variant that can serve as a beneficial, universal immunotherapeutic for patients with amyloidosis.
Ethics Statement
All patient-derived tissue samples were used in accordance with an Institutional Review Board-approved application. Animal studies were carried out in strict accordance with a protocols approved by the University of Tennessee Institutional Animal Care and Use Committee. All procedures were approved by the IACUC and were performed in accordance with the guidelines provided by OLAW and the Guide for the Care and Use of Laboratory Animals. The University of Tennessee Medical Center animal program is AAALAC-i-accredited.
Author Contributions
JW, AW, JF, and SK designed the experiments; JW, EM, SK, and JF analyzed the data, prepared the figures, and wrote the paper; AW, JF, AS, TR, SM, DW, RK-T, and SK performed the experiments.
Conflict of Interest Statement
JW, JF, and SK are inventors on a US provisional patent application that describes the use of Fcp5 as a potential therapeutic opsonizing agent for amyloidosis. JW is inventor on a patent (USPTO #8,105,594) that describes the use of mAb 11-1F4 as a therapeutic agent for AL amyloidosis.
Acknowledgments
The authors would like to thank Jim Wesley for tissue processing, sample preparation, and assistance with microautoradiography. This work was supported by PHS grant R01DK079984 from The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) as well as funds from the Molecular Imaging and Translational Research Program and the Department of Medicine at UTMCK. A limited amount of data regarding this subject was presented at the XVth International Symposium on Amyloidosis and has recently been published in a proceedings supplement (56). This work is dedicated to Dale Schenk, a friend, visionary, and pioneer in amyloid immunotherapy.
References
1. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet (2016) 387:2641–54. doi:10.1016/S0140-6736(15)01274-X
2. Dispenzieri A, Gertz MA, Buadi F. What do I need to know about immunoglobulin light chain (AL) amyloidosis? Blood Rev (2012) 26:137–54. doi:10.1016/j.blre.2012.03.001
3. Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol (2012) 421:185–203. doi:10.1016/j.jmb.2011.12.060
4. Nasr SH, Dogan A, Larsen CP. Leukocyte cell-derived chemotaxin 2-associated amyloidosis: a recently recognized disease with distinct clinicopathologic characteristics. Clin J Am Soc Nephrol (2015) 10:2084–93. doi:10.2215/CJN.12551214
5. Westermark GT, Fandrich M, Westermark P. AA amyloidosis: pathogenesis and targeted therapy. Annu Rev Pathol (2015) 10:321–44. doi:10.1146/annurev-pathol-020712-163913
6. Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol (2012) 302:H544–52. doi:10.1152/ajpheart.00815.2011
7. Dispenzieri A, Merlini G. Immunoglobulin light chain systemic amyloidosis. Cancer Treat Res (2016) 169:273–318. doi:10.1007/978-3-319-40320-5_15
8. Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med (2015) 373:1106–14. doi:10.1056/NEJMoa1504942
9. Gertz MA, Landau H, Comenzo RL, Seldin D, Weiss B, Zonder J, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol (2016) 34:1097–103. doi:10.1200/JCO.2015.63.6530
10. Gertz MA, Landau HJ, Weiss BM. Organ response in patients with AL amyloidosis treated with NEOD001, an amyloid-directed monoclonal antibody. Am J Hematol (2016) 91:E506–8. doi:10.1002/ajh.24563
11. Edwards CV, Gould J, Langer AL, Mapara M, Radhakrishnan J, Maurer MS, et al. Interim analysis of the phase 1a/b study of chimeric fibril-reactive monoclonal antibody 11-1F4 in patients with AL amyloidosis. Amyloid (2017) 24:58–9. doi:10.1080/13506129.2017.1292900
12. Wall JS, Richey T, Stuckey A, Donnell R, Macy S, Martin EB, et al. In vivo molecular imaging of peripheral amyloidosis using heparin-binding peptides. Proc Natl Acad Sci U S A (2011) 108:E586–94. doi:10.1073/pnas.1103247108
13. Wall JS, Martin EB, Richey T, Stuckey AC, Macy S, Wooliver C, et al. Preclinical validation of the heparin-reactive peptide p5+14 as a molecular imaging agent for visceral amyloidosis. Molecules (2015) 20:7657–82. doi:10.3390/molecules20057657
14. Wall JS, Williams A, Richey T, Stuckey A, Wooliver C, Christopher Scott J, et al. Specific amyloid binding of polybasic peptides in vivo is retained by beta-sheet conformers but lost in the disrupted coil and all d-amino acid variants. Mol Imaging Biol (2017). doi:10.1007/s11307-017-1063-0
15. Wall JS, Williams A, Richey T, Stuckey A, Huang Y, Wooliver C, et al. A binding-site barrier affects imaging efficiency of high affinity amyloid-reactive peptide radiotracers in vivo. PLoS One (2013) 8:e66181. doi:10.1371/journal.pone.0066181
16. Martin EB, Williams A, Heidel E, Macy S, Kennel SJ, Wall JS. Peptide p5 binds both heparinase-sensitive glycosaminoglycans and fibrils in patient-derived AL amyloid extracts. Biochem Biophys Res Commun (2013) 436:85–9. doi:10.1016/j.bbrc.2013.05.063
17. Wall JS, Richey T, Williams A, Stuckey A, Osborne D, Martin E, et al. Comparative analysis of peptide p5 and serum amyloid P component for imaging AA amyloid in mice using dual-isotope SPECT. Mol Imaging Biol (2012) 14:402–7. doi:10.1007/s11307-011-0524-0
18. Park HI, Yoon HW, Jung ST. The highly evolvable antibody Fc domain. Trends Biotechnol (2016) 34:895–908. doi:10.1016/j.tibtech.2016.04.005
19. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7:715–25. doi:10.1038/nri2155
20. Wu B, Sun YN. Pharmacokinetics of peptide-Fc fusion proteins. J Pharm Sci (2014) 103:53–64. doi:10.1002/jps.23783
21. Hao X, Zhu Y, Zheng C, Fu X, Feng J, Shen B, et al. A comparison of biological activity of B lymphocyte stimulator (BLyS) antagonist peptibodies and the elucidation of possible BLyS binding sites. Protein Pept Lett (2016) 23:17–23. doi:10.2174/0929866522666151026122347
22. Goffe B, Cather JC. Etanercept: an overview. J Am Acad Dermatol (2003) 49:S105–11. doi:10.1016/mjd.2003.554
23. Glaesner W, Vick AM, Millican R, Ellis B, Tschang SH, Tian Y, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev (2010) 26:287–96. doi:10.1002/dmrr.1080
24. Sanford M. Dulaglutide: first global approval. Drugs (2014) 74:2097–103. doi:10.1007/s40265-014-0320-7
25. Levin D, Golding B, Strome SE, Sauna ZE. Fc fusion as a platform technology: potential for modulating immunogenicity. Trends Biotechnol (2015) 33:27–34. doi:10.1016/j.tibtech.2014.11.001
26. Wall JS, Williams A, Wooliver C, Martin EB, Cheng X, Heidel RE, et al. Secondary structure propensity and chirality of the amyloidophilic peptide p5 and its analogues impacts ligand binding – in vitro characterization. Biochem Biophys Rep (2016) 8:89–99. doi:10.1016/j.bbrep.2016.08.007
27. Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, Solomon A. Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry (1999) 38:14101–8. doi:10.1021/bi991131j
28. Pras M, Schubert M, Zucker-Franklin D, Rimon A, Franklin EC. The characterization of soluble amyloid prepared in water. J Clin Invest (1968) 47:924–33. doi:10.1172/JCI105784
29. Martin EB, Williams A, Richey T, Stuckey A, Heidel RE, Kennel SJ, et al. Comparative evaluation of p5+14 with SAP and peptide p5 by dual-energy SPECT imaging of mice with AA amyloidosis. Sci Rep (2016) 6:22695. doi:10.1038/srep22695
30. Kennel SJ, Macy S, Wooliver C, Huang Y, Richey T, Heidel E, et al. Phagocyte depletion inhibits AA amyloid accumulation in AEF-induced huIL-6 transgenic mice. Amyloid (2014) 21:45–53. doi:10.3109/13506129.2013.876400
31. Kisilevsky R. Preparation and propagation of amyloid-enhancing factor. Methods Mol Biol (2005) 299:237–41.
32. Martin EB, Kennel SJ, Richey T, Wooliver C, Osborne D, Williams A, et al. Dynamic PET and SPECT imaging with radioiodinated, amyloid-reactive peptide p5 in mice: a positive role for peptide dehalogenation. Peptides (2014) 60:63–70. doi:10.1016/j.peptides.2014.07.024
33. Niemietz C, Chandhok G, Schmidt H. Therapeutic oligonucleotides targeting liver disease: TTR amyloidosis. Molecules (2015) 20:17944–75. doi:10.3390/molecules201017944
34. Adams D, Cauquil C, Labeyrie C, Beaudonnet G, Algalarrondo V, Theaudin M. TTR kinetic stabilizers and TTR gene silencing: a new era in therapy for familial amyloidotic polyneuropathies. Expert Opin Pharmacother (2016) 17:791–802. doi:10.1517/14656566.2016.1145664
35. Zumbo G, Sadeghi-Alavijeh O, Hawkins PN, Fontana M. New and developing therapies for AL amyloidosis. Expert Opin Pharmacother (2017) 18:139–49. doi:10.1080/14656566.2016.1274971
36. Renz M, Torres R, Dolan PJ, Tam SJ, Tapia JR, Li L, et al. 2A4 binds soluble and insoluble light chain aggregates from AL amyloidosis patients and promotes clearance of amyloid deposits by phagocytosis dagger. Amyloid (2016) 23:168–77. doi:10.1080/13506129.2016.1205974
37. Pepys MB. Pathogenesis, diagnosis and treatment of systemic amyloidosis. Philos Trans R Soc Lond B Biol Sci (2001) 356:203–10; discussion 210–1. doi:10.1098/rstb.2000.0766
38. Gillmore JD, Tennent GA, Hutchinson WL, Gallimore JR, Lachmann HJ, Goodman HJ, et al. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br J Haematol (2010) 148:760–7. doi:10.1111/j.1365-2141.2009.08036.x
39. Larsen CP, Beggs ML, Wilson JD, Lathrop SL. Prevalence and organ distribution of leukocyte chemotactic factor 2 amyloidosis (ALECT2) among decedents in New Mexico. Amyloid (2016) 23:119–23. doi:10.3109/13506129.2016.1145110
40. Krishnan R, Tsubery H, Proschitsky MY, Asp E, Lulu M, Gilead S, et al. A bacteriophage capsid protein provides a general amyloid interaction motif (GAIM) that binds and remodels misfolded protein assemblies. J Mol Biol (2014) 426:2500–19. doi:10.1016/j.jmb.2014.04.015
41. Krishnan R, Hefti F, Tsubery H, Lulu M, Proschitsky M, Fisher R. Conformation as the therapeutic target for neurodegenerative diseases. Curr Alzheimer Res (2017) 14:393–402. doi:10.2174/1567205014666170116152622
42. Mobergslien A, Peng Q, Vasovic V, Sioud M. Cancer cell-binding peptide fused Fc domain activates immune effector cells and blocks tumor growth. Oncotarget (2016) 7:75940–53. doi:10.18632/oncotarget.12445
43. Wall JS, Kennel SJ, Stuckey AC, Long MJ, Townsend DW, Smith GT, et al. Radioimmunodetection of amyloid deposits in patients with AL amyloidosis. Blood (2010) 116:2241–4. doi:10.1182/blood-2010-03-273797
44. van Witteloostuijn SB, Pedersen SL, Jensen KJ. Half-life extension of biopharmaceuticals using chemical methods: alternatives to PEGylation. ChemMedChem (2016) 11:2474–95. doi:10.1002/cmdc.201600374
45. Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW, et al. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol (2015) 35:235–54. doi:10.3109/07388551.2013.834293
46. Unverdorben F, Richter F, Hutt M, Seifert O, Malinge P, Fischer N, et al. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. MAbs (2016) 8:120–8. doi:10.1080/19420862.2015.1113360
47. Nystrom SN, Westermark GT. AA-amyloid is cleared by endogenous immunological mechanisms. Amyloid (2012) 19:138–45. doi:10.3109/13506129.2012.711391
48. Milde R, Ritter J, Tennent GA, Loesch A, Martinez FO, Gordon S, et al. Multinucleated giant cells are specialized for complement-mediated phagocytosis and large target destruction. Cell Rep (2015) 13:1937–48. doi:10.1016/j.celrep.2015.10.065
49. Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschlager M, Antes B, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng Des Sel (2010) 23:289–97. doi:10.1093/protein/gzq005
50. Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov (2009) 8:226–34. doi:10.1038/nrd2804
51. Suen KF, Turner MS, Gao F, Liu B, Althage A, Slavin A, et al. Transient expression of an IL-23R extracellular domain Fc fusion protein in CHO vs. HEK cells results in improved plasma exposure. Protein Expr Purif (2010) 71:96–102. doi:10.1016/j.pep.2009.12.015
52. Monnet C, Jorieux S, Souyris N, Zaki O, Jacquet A, Fournier N, et al. Combined glyco- and protein-Fc engineering simultaneously enhance cytotoxicity and half-life of a therapeutic antibody. MAbs (2014) 6:422–36. doi:10.4161/mabs.27854
53. Johan K, Westermark G, Engstrom U, Gustavsson A, Hultman P, Westermark P. Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci U S A (1998) 95:2558–63. doi:10.1073/pnas.95.5.2558
54. Solomon A, Richey T, Murphy CL, Weiss DT, Wall JS, Westermark GT, et al. Amyloidogenic potential of foie gras. Proc Natl Acad Sci U S A (2007) 104:10998–1001. doi:10.1073/pnas.0700848104
55. Leonard JL, Ekenbarger DM, Frank SJ, Farwell AP, Koehrle J. Localization of type I iodothyronine 5’-deiodinase to the basolateral plasma membrane in renal cortical epithelial cells. J Biol Chem (1991) 266:11262–9.
Keywords: Fc-fusion, peptide p5, amyloidosis, AA amyloid, phagocytosis
Citation: Foster JS, Williams AD, Macy S, Richey T, Stuckey A, Wooliver DC, Koul-Tiwari R, Martin EB, Kennel SJ and Wall JS (2017) A Peptide-Fc Opsonin with Pan-Amyloid Reactivity. Front. Immunol. 8:1082. doi: 10.3389/fimmu.2017.01082
Received: 14 June 2017; Accepted: 21 August 2017;
Published: 04 September 2017
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, United StatesReviewed by:
Ronald B. Corley, Boston University School of Medicine, United StatesMartin Bachmann, Cytos Biotechnology, Switzerland
Copyright: © 2017 Foster, Williams, Macy, Richey, Stuckey, Wooliver, Koul-Tiwari, Martin, Kennel and Wall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan S. Wall, andhbGxAdXRtY2suZWR1