 Hisa Hui Ling Tseng1
Hisa Hui Ling Tseng1 Yiu Wa Kwan
Yiu Wa Kwan Simon Ming-Yuen Lee
Simon Ming-Yuen Lee Maggie Pui Man Hoi
Maggie Pui Man Hoi- 1State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau
- 2Faculty of Medicine, School of Biomedical Sciences, The Chinese University of Hong Kong, Shatin, Hong Kong
Aberrant activation of the innate immune system, including NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome-dependent interleukin-1β (IL-1β) secretion, has been implicated in the pathogenesis of type 2 diabetes mellitus (T2DM) and its complication. Our previous study demonstrated that hyperglycemia, a hallmark characteristic of T2DM, induced NLRP3 inflammasome-dependent caspase-1 activation and IL-1β maturation in human monocytic cells. In this study, we examined the underlying mechanisms of secreting IL-1β during hyperglycemia, with a focus on the alteration of Ca2+ homeostasis and lysosomal exocytosis. We found that high glucose (HG; 30 mM glucose for 48 h) altered Ca2+ homeostasis by reducing lysosomal Ca2+ concentration that appeared to be resulted from Ca2+ moving out of lysosomes into cytosol in human monocytic cell lines, U937 and THP-1 cells. Moreover, HG-induced lysosomal Ca2+-dependent mature IL-1β release was strongly correlated with the activation and upregulation of two lysosomal marker proteins, cathepsin D and lysosomal-associated membrane protein-1 (LAMP-1). This involved calcineurin/transcription factor EB (TFEB) pathway and its target genes, cathepsin B, cathepsin D, and LAMP-1, to mediate lysosomal exocytosis. Therefore in this study, we revealed a novel mechanism of HG-induced lysosomal exocytosis which was regulated by lysosomal Ca2+ signals through calcineurin/TFEB pathway, thus contributing to IL-1β secretion in human monocytic cells.
Introduction
Interleukin-1β (IL-1β) is one of the pro-inflammatory cytokines that is involved in the pathogenesis of type 1 diabetes, type 2 diabetes mellitus (T2DM), and diabetic vascular complication, such as atherosclerosis (1–3). IL-1β mediates inflammatory responses contributing to impaired insulin secretion and sensitivity in insulin-sensitive cells (2, 4). Indeed, IL-1β maturation was tightly controlled by the inflammasome, a multiprotein complex that consists of an inflammasome sensor molecule, the adaptor protein apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC) and caspase-1 (5). NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome is now the best studied inflammasome and has been implicated in the progression of T2DM (6, 7). Recent studies suggested that the activation of NLRP3 inflammasome was a key mechanism in obesity- and high-fat diet-induced insulin resistance and inflammation (8, 9). Moreover, our previous study demonstrated that hyperglycemia, a hallmark of T2DM, could induce reactive oxygen species (ROS)-sensitive NLRP3 inflammasome activation in human monocytes (10), suggesting that high glucose (HG) is a key factor of activated innate immunity in T2DM, which could be sensed by NLRP3 inflammasome and mediate the processing of IL-1β under diabetic condition.
It has been recognized that there are three steps involved in IL-1β secretion, first step is to stimulate the synthesis of pro-IL-1β, then pro-IL-1β is cleaved into mature IL-1β by caspase-1, which is followed by IL-1β secretion via non-classical secretory pathway into the extracellular milieu (11). In most phagocytic cells, such as monocytes, macrophages, and dendritic cells, IL-1β secretion was associated with the exocytosis of secretory lysosomes (11), which suggested the importance of lysosomes in IL-1β secretory pathways. Indeed, conventional lysosome is defined by the common function of degrading or recycling processes of intracellular materials (12). In monocytes or macrophages, lysosomes also serve as a secretory compartment for sorting and secretory pathways (13). There are two key features of secretory lysosomal exocytosis. First, signals stimulate the recruitment of lysosomes trafficking to the plasma membrane (PM). Second, intracellular Ca2+ concentration ([Ca2+]i) rise triggers secretory lysosomes to fuse with the PM and release secretory proteins (13, 14). Ca2+ influx was known to be a critical regulator of lysosomal exocytosis to mediate IL-1β secretion (15, 16), and prolonged hyperglycemia was known to be resulted in Ca2+ influx and an increase in [Ca2+]i in different cell types (10, 17–19). Moreover, our previous study has demonstrated that HG could enhance [Ca2+]i and induced caspase-1-dependent IL-1β secretion via transient receptor potential melastatin-2 (TRPM2) in human monocytic cells (10). However, the mechanism of secreting IL-1β into extracellular milieu by HG remains to be clarified.
Transcription factor EB (TFEB) is an essential transcriptional regulator for lysosomal function (20, 21), which was regulated by lysosomal Ca2+ signals that could promote cellular processes, including autophagy and lysosomal exocytosis (22–24). Furthermore, lysosomal Ca2+ release by glycyl-l-phenylalaninebeta-naphthylamide (GPN) could cooperate with endoplasmic reticulum (ER) Ca2+ store and resulted in lysosomal exocytosis and IL-1β secretion in human monocytic cells (15, 25). These observations indicated a close relationship between the alteration of Ca2+ homeostasis and lysosomal exocytosis. Although many stimuli were shown to activate TFEB and mediate lysosome-dependent cellular processes (26), it is unclear how TFEB mediates these processes at the transcriptional level.
Here, we used hyperglycemic environment to mimic the diabetic condition in vitro. Treatment with 30 mM glucose for 48 h was regarded as the HG model in U937 and THP-1 monocytic cells. In this study, we demonstrated that HG could induce change in [Ca2+]i and affect lysosomal Ca2+ homeostasis, and mediate lysosomal exocytosis. We also found that this lysosomal Ca2+ signaling by HG could trigger calcineurin/TFEB pathway and its target genes cathepsin D and lysosomal-associated membrane protein-1 (LAMP-1), and then subsequently release IL-1β in human monocytic cells.
Materials and Methods
Reagents and Chemicals
Carbonyl cyanide 3-chlorophenylhydrazone (CCCP), ethylene glycol tetra acetic acid (EGTA), hydrogen peroxide solution (H2O2), d-mannitol, and lipopolysaccharides were purchased from Sigma-Aldrich, USA. Bafilomycin A1 and GPN were from Santa Cruz Biotechnology, while BAPTA-AM, cyclosporin A, FK506, ionomycin, nicotinic acid adenine dinucleotide phosphate (NAADP), trans-Ned-19 (Ned-19), and U18666A were from Tocris Biosciences, USA. Thapsigargin (TG) was bought from Almone Labs, USA, while LysoTracker Red DND-99 Dye and Rhod dextran were from Invitrogen, USA. Antibodies used for immunoblotting and immunostaining were as follows: anti-mouse lysosome-associated membrane protein-1 (LAMP-1; sc-20011, Santa Cruz Biotechnology, USA), anti-rabbit cathepsin D (2284S, Cell Signaling, USA), anti-rabbit caspase-1 (2225S, Cell Signaling, USA), anti-rabbit TFEB (37785S, Cell Signaling, USA), anti-rabbit histone H3 (D1H2) (4499S, Cell Signaling, USA), anti-rabbit Integrin β1 (4706S, Cell Signaling, USA), anti-rabbit GAPDH (2118S, Cell Signaling, USA), and anti-rabbit α/β-tubulin (2148S, Cell Signaling, USA).
Cell Culture, Treatments, and ELISA
U937 (ATCC, USA) and THP-1 (InvivoGen, USA) monocytic cell lines were grown in RPMI 1640 (Gibco, USA) supplemented with 10% FBS, 2 mM l-glutamine, and 100 U/mL of penicillin and streptomycin. In HG experiments, before HG stimulation, the cells were cultured in RPMI 1640 with 5.5 mM glucose for 48 h, and then were changed to 10, 20, or 30 mM glucose RPMI 1640 for indicated time points. 30 mM mannitol was used as an osmotic control. For the experiments using chemical inhibitors, Cs A, FK506, and U18666A were pre-treated for 24 h, while TG was pre-treated for 45 min. EGTA and BAPTA-AM were treated in the presence of HG stimulation. For the immunoblotting experiments measuring TFEB translocation by calcium inducers, GPN, H2O2, ionomycin, NAADP, and TG were stimulated for 20 min. The supernatants from U937 and THP-1 cells were collected for the detection of human IL-1β levels by ELISA (eBioscience, USA).
Specific Small Interfering RNA (siRNA) Experiments
Cells were transiently transfected with TFEB siRNA (100 nmol/L; Ambion, USA), by using Lipofectamine® RNAiMAX Transfection reagent (Gibco, USA). The protocol was synthesized according to the manufacturer’s protocol. GAPDH siRNA was used as a control (40 nmol/L; Ambion, USA). Transfection efficiency was >70% assessed by BLOCK-iT™ Alexa Fluor® Red Fluorescent Control (Ambion, USA) and western blotting. Cells were transfected with siRNA for 24 h before experiments.
[Ca2+]i Measurements
The intracellular Ca2+ concentration ([Ca2+]i) was measured in single cells as previously described (27). Cells were loaded with Fluo-4 AM (2 µM; Molecular Probes, USA) in Tyrode solution containing 136.5 mM NaCl, 5.4 mM KCl, 0.53 mM MgCl2, 1.8 mM CaCl2, 0.33 mM NaH2PO4, 5.5 mM glucose, and 5.5 mM HEPES (pH 7.4, adjusted with NaOH) for 30 min at 37°C. Fluo-4 fluorescence intensity (494 nm excitation; 506 nm emission) was sampled at 5 s intervals using a Cell® system (MT20, Olympus, USA). To enable comparisons between cells, the maximal change in fluorescence intensity was measured before and after GPN (400 µM), NAADP (1 µM), Baf A1 (500 nM), or TG (1 µM) was added.
Lysosomal Ca2+ Measurements
The lysosomal Ca2+ concentration was measured as previously described (28). For lysosomal Ca2+ measurements, the cells were incubated with Rhod dextran (25 mg/ml) for 12 h after treatment as indicated in results, while for all cytosolic Ca2+ measurements, the cells were incubated with Fluo-4 (2 µM) for 30 min. The median fluorescence intensity (MFI) was determined using a FACS Canto flow cytometer (BD Biosciences, USA), and the data were analyzed using FlowJo software (Tree Star, USA).
Western Blot Analysis
Total protein was extracted with ice-cold lysis buffer, the nuclear/cytosolic proteins were extracted by using the Nuclear and Cytoplasmic Extraction Kit (Pierce, USA), and the PM/cytosolic proteins were extracted by using the Mem-PER™ Plus Membrane Protein Extraction Kit (Pierce, USA). The protein concentrations of the lysates were measured by the bicinchoninic acid kit (Pierce, USA). 40 µg proteins were used and separated by 10% SDS-PAGE gels and were transferred onto the nitrocellulose membranes. Membranes were incubated with primary antibodies (1/1,000 dilution) overnight at 4°C, and secondary antibodies (1/1,000 dilution) for 1 h at room temperature, and the immunoblots were developed by enhanced chemiluminescence (GE Healthcare Life Sciences, USA) with a ChemiDoc™ MP System (Bio-Rad Laboratories, USA). GAPDH, β-actin, α/β-Tubulin, Histone H3, and Integrin β1 were used as housekeeping controls.
Real-time PCR Analysis
Total RNA was extracted using RNeasy Mini Kit (Qiagen, USA), and cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA). cDNA was quantified using Taqman assays by ViiA 7 Real-Time PCR System (Applied Biosystems, USA). The Taqman probes (Applied Biosystems, USA) used were as follows: TFEB (Hs00292981_m1), Cathepsin B (Hs00947433_m1), Cathepsin D (Hs00157205_m1), LAMP-1 (Hs00174766_m1), IL-1β (Hs00174097_m1), and β-actin (4326315E). β-Actin was used as an endogenous control. All gene expressions were calculated using the ΔΔCt method and were normalized to control.
Flow Cytometry
For cells labeling with lysotracker, the cells were incubated with LysoTracker DND-99 Dye (250 nM) for 45 min at 37°C after treatment as indicated in results. The MFI was determined using a FACS Canto flow cytometer (BD Biosciences, USA), and the data were analyzed using FlowJo software (Tree Star, USA).
The LAMP-1 level on the PM was measured as previously described (29). After treatment as indicated in results, the intact cells were incubated with LAMP-1 antibody overnight at 4°C and then fixed with 4% paraformaldehyde solution (PFA; Santa Cruz Biotechnology, USA). After fixation, the cells were incubated with secondary antibody (1/400 dilution). The MFI was determined using a FACS Canto flow cytometer (BD Biosciences, USA), and the data were analyzed using FlowJo software (Tree Star, USA).
β-Hexosaminidase Secretion Assay
β-Hexosaminidase secretion was measured as previously described (25). After treatment, 200 ml supernatants of the cells were equilibrated in 1 mM EGTA-Ca2+-free buffer for 3 h and then mixed with 200 ml of 1 mM 4-methylumbelliferyl N-acetyl-β-d-glucosaminide (Sigma-Aldrich, USA) in 0.1 M citrate buffer (0.05 M citric acid, 0.05 M sodium citrate, pH 4.5, Sigma-Aldrich, USA) for 1 h at 37°C. The reaction was stopped with 400 ml 0.1 M sodium carbonate buffer (Sigma-Aldrich, USA), and the absorbance was measured at 405 nm. To determine the total cellular content of β-hexosaminidase, the cells were lysed with 1% (v/v) Triton X-100, and 10 µl of the cell extracts were used for the enzyme activity reaction. The percentage of β-hexosaminidase release was calculated from the enzyme activity of the supernatants and lysates.
Cathepsin D Activity Assay
Cells were extracted with 200 µl of chilled Cell Lysis Buffer following the manufacturer’s instruction. Cathepsin D activity was measured by using a flourimetric assay kit (Abcam, USA) and was normalized to control.
TFEB Nuclear Translocation Assay
After treatment as indicated in results, the cells were fixed with 4% PFA for 15 min, followed by permeabilization with 0.1% Triton X-100 for 5 min, and were blocked in 20% goat serum (Cell Signaling, USA) for 30 min. Next, the cells were incubated with TFEB antibody (1/50 dilution) overnight at 4°C, and stained with secondary antibody (1/400 dilution) for 1 h and DAPI for 10 min. For the acquisition of the images, at least six images were taken per well of the 96-well plate by IN Cell Analyzer 2000 (GE Healthcare, USA), and quantitative analysis was performed with ImageJ software.
Immunofluorescence Staining
The cells were seeded onto confocal dishes (SPL Life Sciences, Korea) and were treated with indicated conditions as described. The cells were fixed with 4% PFA for 15 min, blocked in 20% goat serum (Cell Signaling, USA) for 30 min, and incubated with primary antibodies (1/50 dilution) overnight at 4°C, and then secondary antibodies (1/400 dilution) for 1 h. Images were captured with a confocal microscope (LEICA TCS SP8, Leica Microsystems, Germany), and quantitative analysis was performed with the ImageJ software.
Statistical Analysis
All data were expressed as mean ± SEM and were analyzed by GraphPad Prism 5.0 (GraphPad, USA). Significant differences were determined by one-way ANOVA followed by a Dunnett’s test. P < 0.05 was considered as significant. Sample size (n) represented the number of independent experiments.
Results
HG Alters Lysosomal Ca2+ Homeostasis in Human Monocytic Cells
Impaired lysosomal Ca2+ homeostasis could lead to lysosomal dysfunction (30), and chronic exposure of HG to macrophages was demonstrated to induce the inhibition of lysosomal function (31); however, whether lysosomal Ca2+ homeostasis was altered under HG condition is still unclear. To examine the role of lysosomes in hyperglycemic environment in human monocytic cells, we first measured Ca2+ release from the lysosomes. GPN is a cathepsin C substrate that was reported to induce lysosomal Ca2+ release in monocytes (25). In Fluo-4-loaded U937 cells, treatment with HG (10, 20, 30 mM glucose for 48 h) or 30 mM glucose for 24, 48, or 72 h significantly reduced GPN-evoked Ca2+ release (Figures 1A–C), compared to low glucose (LG; 5.5 mM glucose) and 30 mM mannitol (Ma). Ma was used to as an osmotic control. Since 30 mM glucose treatment for 48 h, but not Ma, induced approximately 85% reduction of GPN-evoked Ca2+ release in U937 cells; therefore, it was regarded as our HG model in this study. Moreover, we also used another human monocytic cell line, THP-1, to confirm this observation. Similarly, we also observed that there was a reduction of GPN-evoked Ca2+ release under HG condition in THP-1 cells (Figure 1D), suggesting that HG might influence lysosomal Ca2+ homeostasis in human monocytic cells. In THP-1 cells, pre-treatment with U18666A, a drug that was used to deplete lysosomal Ca2+ store, significantly blocked GPN-evoked Ca2+ release (Figure 1D), this confirmed that GPN-evoked Ca2+ release was from the lysosomes in human monocytic cells.
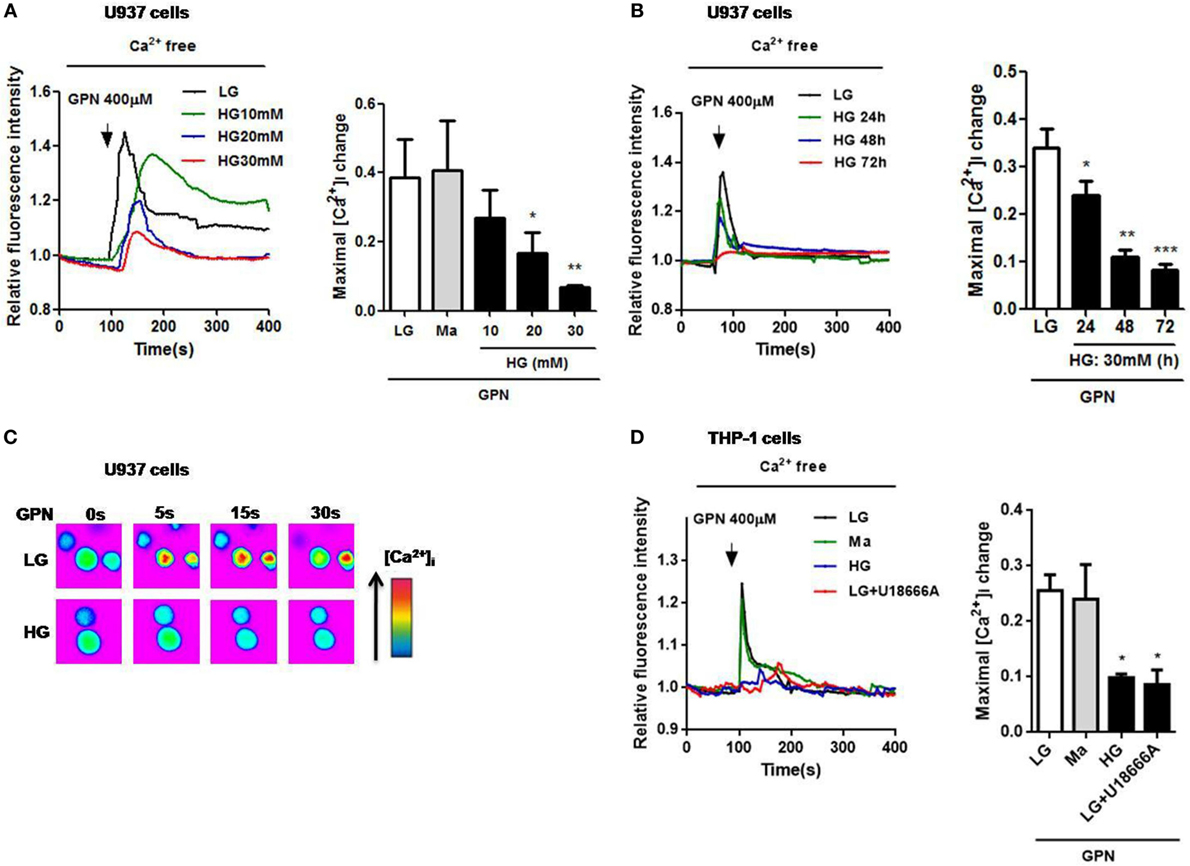
Figure 1. High glucose (HG) reduced GPN-evoked lysosomal Ca2+ release in U937 and THP-1 cells. The cells were loaded with Fluo-4-AM and were treated with glycyl-l-phenylalanine-beta-naphthylamide (GPN) to evoke Ca2+ responses. Representative and relative changes in intracellular Ca2+ concentration ([Ca2+]i) evoked by GPN (400 µM) under low glucose (LG; 5.5 mM glucose), mannitol (Ma; 30 mM mannitol) or (A) HG (10, 20, 30 mM glucose for 48 h), or (B) HG (30 mM glucose) for 24, 48, 72 h (n = 4–5), or (C) HG (30 mM glucose for 48 h) in U937 cells. (D) Representative and relative changes in [Ca2+]i evoked by GPN (400 µM), with or without pre-treatment of U18666A (2 µg/ml) under HG (30 mM glucose for 48 h) in THP-1 cells (n = 4). Data were shown as mean ± SEM. (A,B,D) *P < 0.05, **P < 0.01, and ***P < 0.001 vs. LG.
To further examine the role of Ca2+ homeostasis under HG condition in human monocytic cells, we used NAADP, a Ca2+-mobilizing secondary messenger that was known to release Ca2+ from the acidic endo-lysosomal vesicles (32), and bafilomycin A1, an inhibitor of the vacuolar-ATPase to induce lysosomal Ca2+ release. Figures 2A,B showed that NAADP- and bafilomycin A1-evoked Ca2+ release were significantly reduced under HG in U937 cells. By contrast, we also measured Ca2+ release from the ER and mitochondria under HG condition. The cells were treated with TG to release ER Ca2+, or with CCCP, a mitochondrial uncoupler to release mitochondria Ca2+. No differences in Ca2+ release from the ER or mitochondria were observed between LG-, Ma-, and HG-treated U937 cells (Figures 2C,D). Taken together, this suggested that HG induced a disruption of Ca2+ homeostasis within lysosomes, but was dispensable for ER and mitochondria Ca2+ in human monocytic cells.
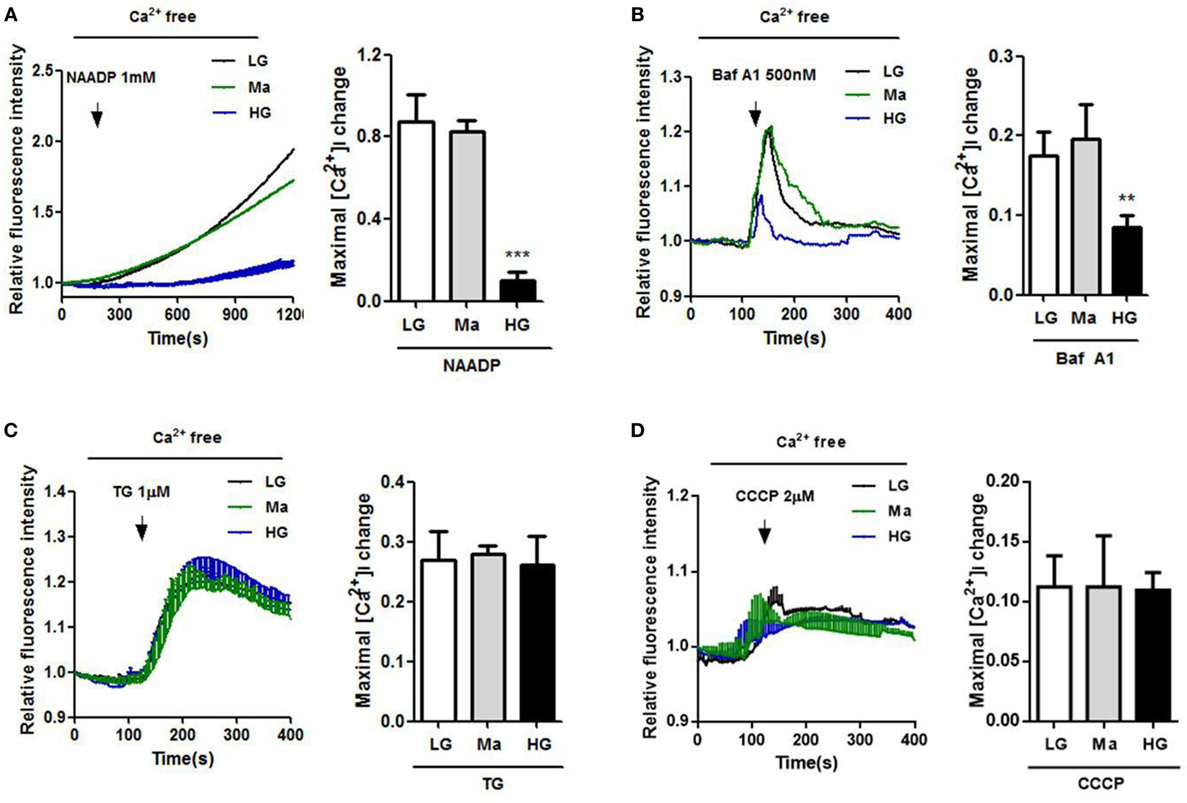
Figure 2. High glucose (HG) reduced Ca2+ release by nicotinic acid adenine dinucleotide phosphate (NAADP) and bafilomycin A1, but not by thapsigargin (TG) and carbonyl cyanide 3-chlorophenylhydrazone (CCCP) in U937 cells. (A–D) U937 cells were loaded with Fluo-4-AM, and were treated with indicated intracellular Ca2+ activators to evoke Ca2+ responses. Representative and relative changes in intracellular Ca2+ concentration ([Ca2+]i) evoked by (A) NAADP (1 mM), or (B) bafilomycin A1 (Baf A1; 500 nM), or (C) TG (1 µM), or (D) CCCP (2 µM) under low glucose (LG; 5.5 mM glucose), mannitol (Ma; 30 mM mannitol), and HG (30 mM glucose for 48 h) in U937 cells (n = 4–5). Data were shown as mean ± SEM. (A–D) **P < 0.01 and ***P < 0.001 vs. LG.
HG Increases Cytosolic Ca2+ Concentration by Reducing Lysosomal Ca2+ Concentration in Monocytic Cells
To determine the relationship between Ca2+ homeostasis and lysosomes, we measured cytosolic and lysosomal Ca2+ levels directly with Fluo-4 and Rhod-dextran, respectively, as previously described (28). Bafilomycin A1 was reported to increase the pH level of lysosomes that increased cytosolic Ca2+ concentration by reducing lysosomal Ca2+ level (33). In agreement with that, after 60 min treatment with bafilomycin A1, an increase in Fluo-4 MFI and a decrease in Rhod-dextran MFI were observed in U937 cells (Figure 3A). This further confirmed the change in cytosolic and lysosomal Ca2+ levels with Fluo-4 and Rhod-dextran by bafilomycin A1. Next, we examined whether HG affected Ca2+ homeostasis in monocytic cells, we observed a decrease in lysosomal Ca2+ level with Rhod-dextran and elevation in cytosolic Ca2+ level with Fluo-4 under HG (30 mM; 24, 48, 72 h) in U937 cells, this strongly suggested that HG decreased lysosomal Ca2+ concentration and affected cytosolic Ca2+ homeostasis (Figure 3B). Besides, we also determined whether HG influenced lysosomal function in human monocytic cells. LysoTracker dye was used to label lysosomes in live cells (34). By using flow cytometry, we observed a significant decrease in LysoTracker staining under HG for 72 h, but not for 24 h and 48 h in U937 cells (Figure 3C), suggesting that HG for 48 h caused a defect in lysosomal Ca2+ store, and HG for up to 72 h could inhibit lysosomal function in human monocytic cells. Taken together, our results suggested that HG induced the loss of lysosomes, affected lysosomal and cytosolic Ca2+ homeostasis in human monocytic cells.
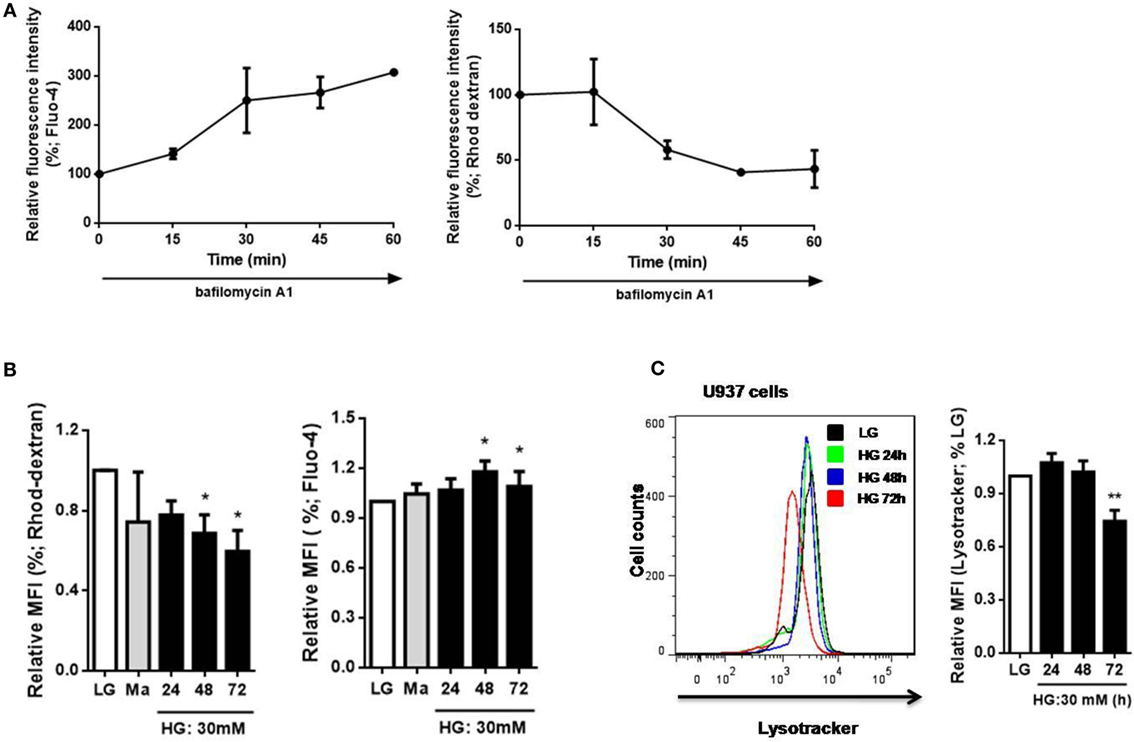
Figure 3. High glucose (HG) induced changes in cytosolic Ca2+ level by affecting lysosomal Ca2+ level and lysosomal function in U937 cells. (A) The percentages of relative median fluorescence intensity (MFI) by Fluo-4-AM or Rhod-dextran staining after bafilomycin A1 (500 nM) stimulation for 15–60 min in U937 cells. (B,C) The MFI by (B) Fluo-4 or Rhod-dextran staining, or (C) Lysotracker staining under low glucose (LG; 5.5 mM glucose), mannitol (Ma; 30 mM mannitol), and HG (30 mM glucose for 48 h) in U937 cells (n = 5). Data were shown as mean ± SEM. (B,C) *P < 0.05 and **P < 0.01 vs. LG.
HG Alters Intracellular Ca2+ Homeostasis to Mediate Lysosomal Exocytosis, Cathepsin D Activity, and IL-1β Secretion in Monocytic Cells
Previous studies have suggested that Ca2+ signals was involved in lysosomal exocytosis-mediated IL-1β secretion in response to multiple stimuli (15, 25, 35, 36), whether HG disturbed Ca2+ homeostasis to promote lysosomal exocytosis is still unknown. To examine lysosomal exocytosis, we stained surface LAMP-1, a marker of the lysosomal exocytosis process (37). Figure 4A showed that HG (10, 20, and 30 mM glucose for 48 h) induced LAMP-1 translocation from cytosol to the PM in a dose-dependent manner in U937 cells. Similarly, we observed that treatment with HG (30 mM glucose) for 24, 48, and 72 h significantly increased surface LAMP-1 level by flow cytometry, where it reached maximum at 48 h in U937 cells (Figure 4B), suggesting that HG induced an active movement of lysosomes toward the PM. Moreover, we also examined the effects of different Ca2+ chelators and blockers on the surface LAMP-1 level under HG. In U937 cells, buffering of [Ca2+]i by BAPTA significantly inhibited HG-induced surface LAMP-1 level (Figure 4C). Besides, we observed that the depletion of lysosomal Ca2+ store by U18666A also blocked this effect (Figure 4C). Similar results were obtained in THP-1 cells (Figure 4C). By contrast, EGTA did not affect the LAMP-1 level (Figure 4C), suggesting that HG rapidly triggered intracellular Ca2+ signals, which contributed to lysosomal exocytosis in human monocytic cells. Furthermore, HG-triggered translocation of LAMP-1 was accompanied by the lysosomal hydrolase, including cathepsin D (Figure 4D). We found that HG induced the maturation and activity of cathepsin D with maximal effects occurring at 48 h in U937 cells, whereas pre-treatment with U18666A could block these effects (Figures 4D–G). Similarly, Ned-19, an inhibitor of NAADP that blocks NAADP-induced Ca2+ mobilization from the lysosomes, also inhibited HG-induced cathepsin D activity in U937 cells (Figure 4G). This indicated that lysosomal Ca2+ signals was involved in HG-induced lysosomal exocytosis.
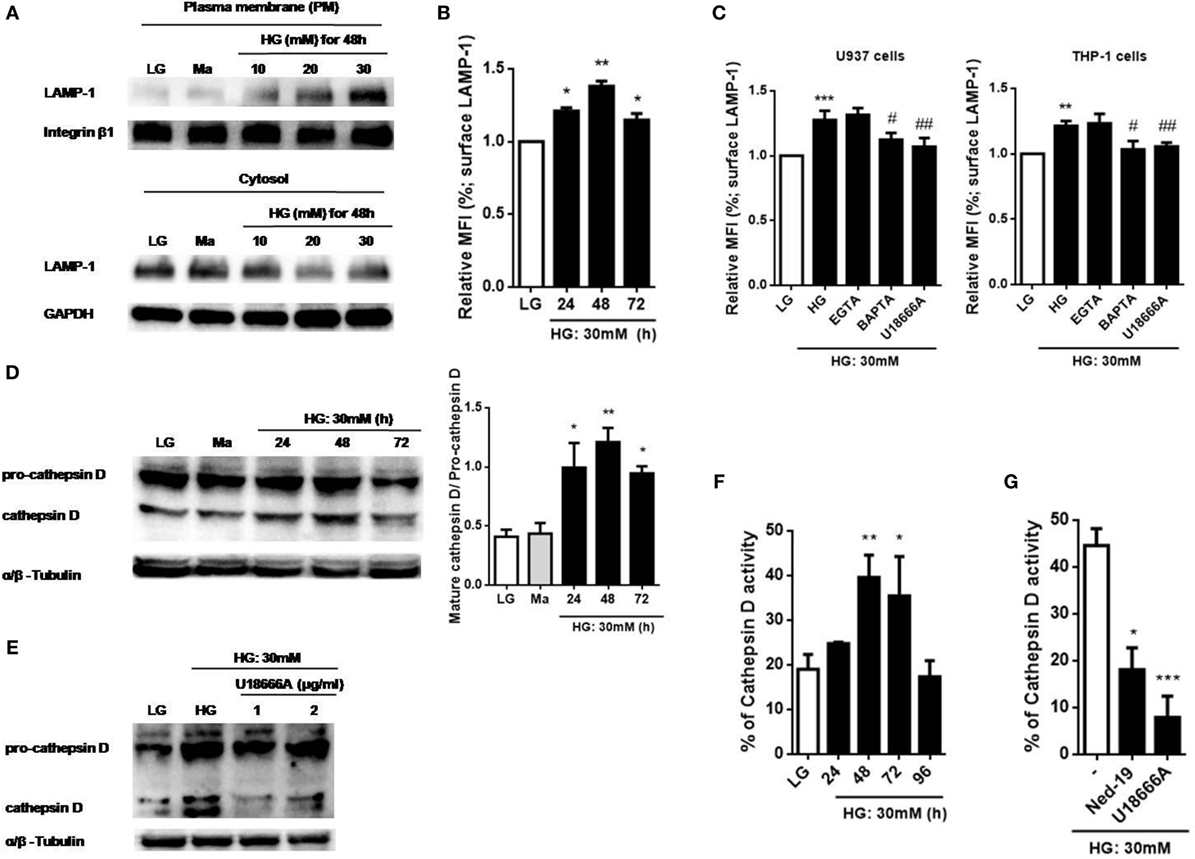
Figure 4. High glucose (HG) increased intracellular Ca2+-dependent surface lysosomal-associated membrane protein-1 (LAMP-1) level and cathepsin D maturation and activity in U937 and THP-1 cells. (A) Representative immunoblots for LAMP-1 and integrin β1 levels on the plasma membrane (PM), and LAMP-1 and GAPDH levels in the cytoplasm under low glucose (LG; 5.5 mM glucose), mannitol (Ma; 30 mM mannitol) and HG (10, 20, 30 mM glucose for 48 h) in U937 cells. (B,C) The relative median fluorescence intensity (MFI) of surface LAMP-1 staining, (B) under LG and HG (30 mM glucose for 24–72 h) in U937 cells, or (C) in the presence of ethylene glycol tetra acetic acid (EGTA) (5 mM), BAPTA (10 µM), or with the pre-treatment of U18666A (2 µg/ml) under HG (30 mM glucose for 48 h) in U937 and THP-1 cells (n = 4). (D,E) Representative immunoblots and graphs for pro- and mature cathepsin D, and α/β-Tubulin under LG or HG (30 mM glucose for 24–72 h), or (E) with the pre-treatment of U18666A (1, 2 µg/ml) under HG (30 mM glucose for 48 h) in U937 cells. (F,G) Cathepsin D activity was measured by cathepsin D activity kit. U937 cells were (F) stimulated with HG (30 mM glucose for 24–96 h), or (F) with the pre-treatment of U18666A (2 µg/ml) or trans-Ned-19 (Ned-19; 100 µM) under HG. The percentage of cathepsin D activity was normalized to (F) LG or (G) HG (n = 3). Data were shown as mean ± SEM. (B–F) *P < 0.05, **P < 0.01, and ***P < 0.001 vs. LG; #P < 0.05 and ##P < 0.01 vs. HG. (G) *P < 0.05 and ***P < 0.001 vs. HG.
We then investigated whether IL-1β was accompanied by lysosomal exocytosis. The intracellular distribution of IL-1β and cathepsin D was examined under HG in U937 cells by confocal microscopy. We found out that IL-1β was co-localized with cathepsin D under HG, whereas this effect was abolished by the removal of Ca2+ with BAPTA plus EGTA (Figure 5A). Moreover, during HG (30 mM glucose) stimulation, IL-1β maturation and release were also abolished by buffering of [Ca2+]i with BAPTA and the removal of extracellular Ca2+ with EGTA in U937 cells (Figure 5B). Meanwhile, we also examined the effect of Ca2+ chelators and agents on IL-1β secretion under HG condition in U937 and THP-1 cells. As expected, BAPTA significantly reduced IL-1β secretion by HG (Figure 5C). To further investigate whether lysosomal Ca2+ release participated in IL-1β secretion by HG, we used three antagonists, U18666A, Ned-19, and bafilomycin A1. Figure 5C showed that U18666A, Ned-19, and bafilomycin A1 markedly blocked HG-induced IL-1β secretion in U937 and THP-1 cells. Taken together, these results indicated that HG altered lysosomal Ca2+ homeostasis, which resulted in an increase in [Ca2+]i and surface LAMP-1 level, facilitation in lysosomal exocytosis, lysosomal cathepsin D maturation and activity, and IL-1β release in human monocytic cells.
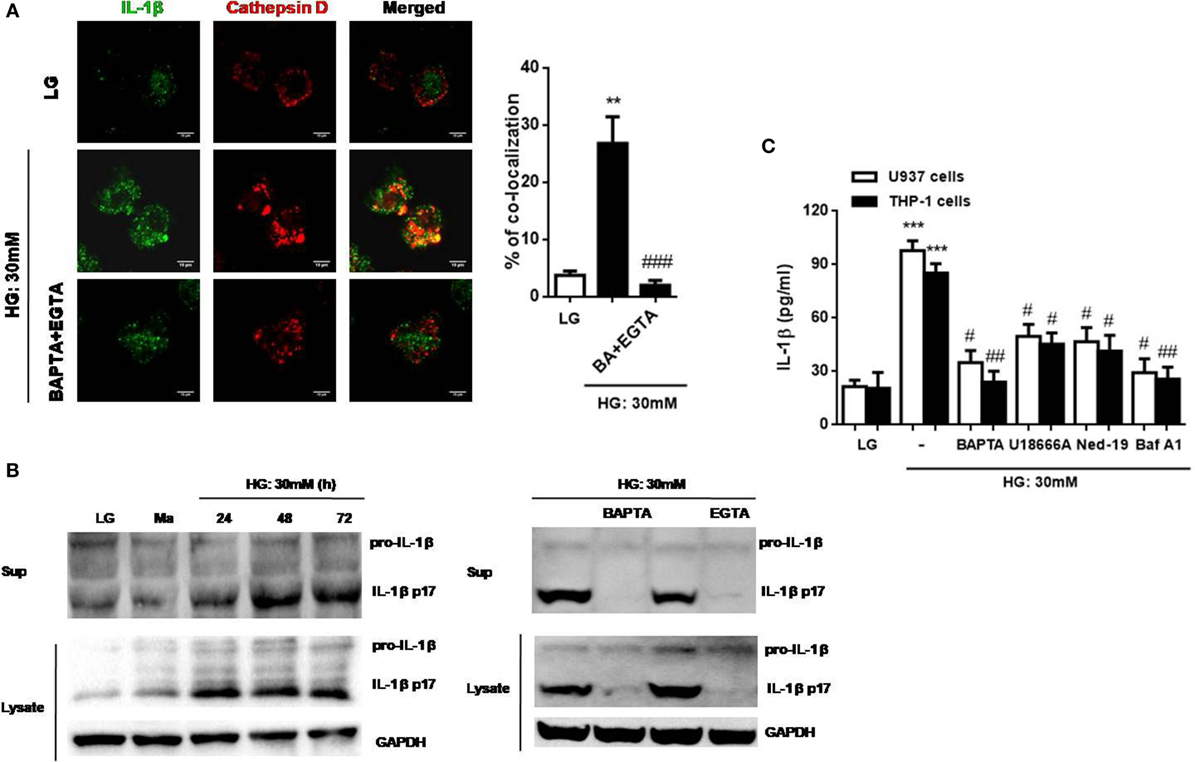
Figure 5. High glucose (HG) induced cathepsin d-dependent interleukin-1β (IL-1β) secretion, which was dependent on lysosomal Ca2+ release in U937 and THP-1 cells. (A) Immunofluorescence images showing the location of IL-1β and cathepsin D in fixed U937 cells under HG (30 mM glucose for 48 h) by confocal microscopy. The U937 cells were pre-treated with BAPTA (10 µM) plus ethylene glycol tetra acetic acid (EGTA) (5 mM). The percentages of co-localization were calculated as the average volume of the overlapping areas (n = 4). (B) Representative immunoblots for pro-IL-1β, IL-1β (p17), and GAPDH protein expressions under low glucose (LG; 5.5 mM glucose for 48 h) or HG (30 mM glucose for 24–72 h), or in the presence of BAPTA (BA; 10 µM) or EGTA (5 mM) under HG (30 mM glucose for 48 h) in U937 cells. (C) ELISA for IL-1β secretion from the supernatants of treated cells. U937 cells were stimulated with HG (30 mM glucose for 48 h) in the presence of BAPTA (10 µM), or with the pre-treatment of U18666A (2 µg/ml), trans-Ned-19 (Ned-19; 100 µM), or bafilomycin A1 (Baf A1; 500 nM). Data were shown as mean ± SEM. (A,C) **P < 0.01 and ***P < 0.001 vs. LG; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. HG.
HG Induces Lysosomal Ca2+ Release-Dependent TFEB Translocation in Monocytic Cells
The activation of TFEB was reported to regulate lysosomal exocytosis by raising [Ca2+]i (22); therefore, we examined whether it was also involved in HG stimulation. Immunoblotting results showed that HG increased TFEB translocation to the nucleus in a dose-dependent manner in U937 cells (Figure 6A). In addition to the nuclear translocation of TFEB, we also observed that HG upregulated TFEB mRNA in U937 cells (Figure 6B), indicating that HG did not only induce TFEB activation, but could also increase its mRNA expression. Notably, we found that the depletion of internal Ca2+ stores by ionomycin (28), or U18666A significantly reduced HG-induced nuclear translocation of TFEB (Figure 6C). By contrast, the depletion of ER Ca2+ store by TG had no effect on it (Figure 6C), suggesting that Ca2+ release from the lysosomes, but not from the ER, mediated the activation of TFEB under HG. Conversely, short and acute exposure to ionomycin, GPN, NAADP, or TG, that triggered internal Ca2+ release, could significantly induce nuclear translocation of TFEB in U937 cells (Figure 6D). However, H2O2 stimulation, which was reported to regulate monocytic function via extracellular Ca2+ influx (38), did not induce nuclear translocation of TFEB (Figure 6D). Therefore, our results supported that lysosmal Ca2+ signals played a key role in the regulation of TFEB translocation during HG condition in human monocytic cells.
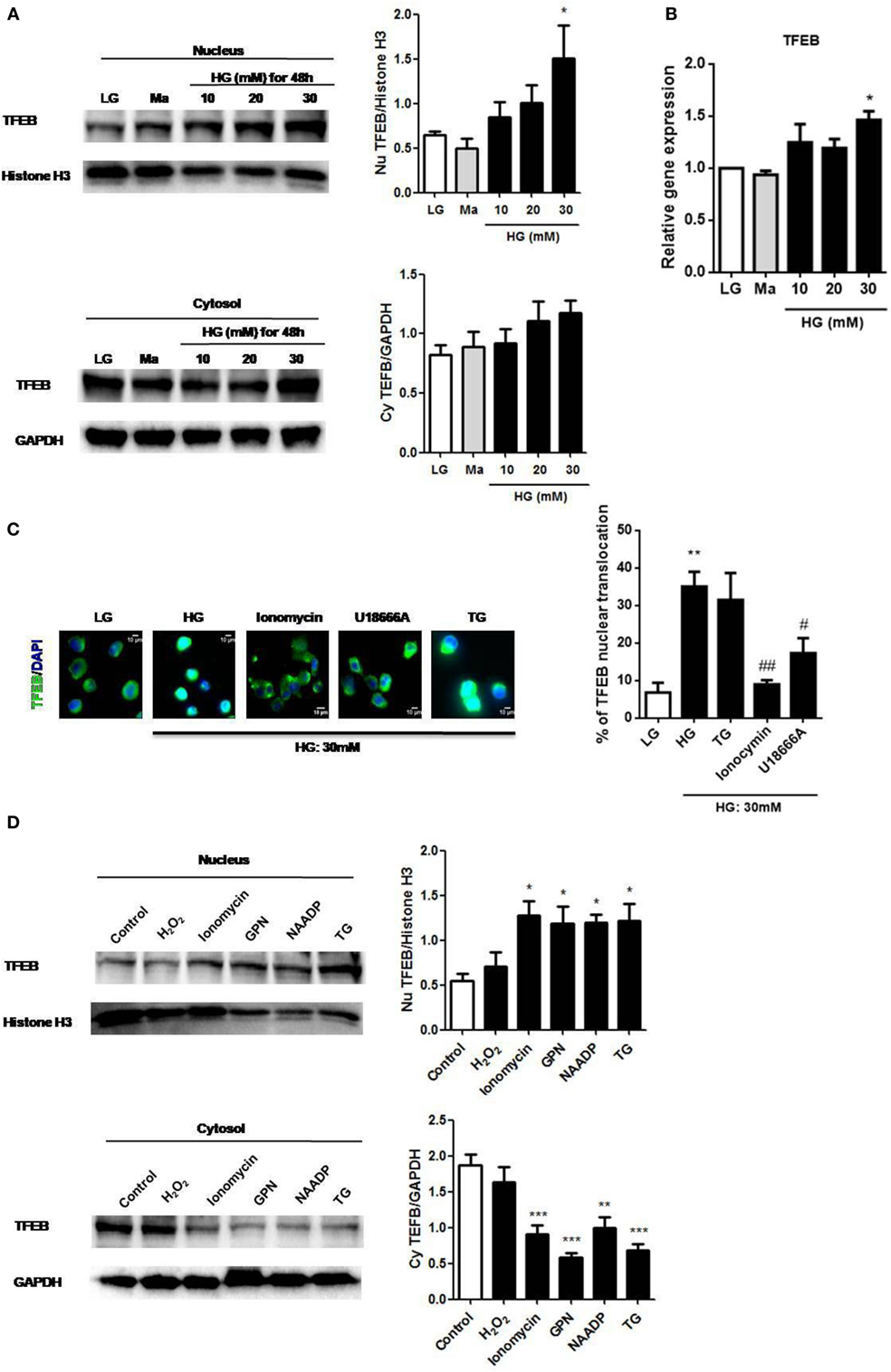
Figure 6. High glucose (HG) upregulated transcription factor EB (TFEB) expression and lysosomal Ca2+-dependent TFEB nuclear translocation in U937 cells. (A) Representative immunoblots and graphs for TFEB and Histone H3 expressions in the nucleus, and TFEB and GAPDH expressions in the cytoplasm under low glucose (LG; 5.5 mM glucose), mannitol (Ma; 30 mM), or HG (10, 20, 30 mM glucose for 48 h) in U937 cells. The relative expression of TFEB was normalized to representative controls (Histone H3/GAPDH) (n = 4). (B) Relative gene expression of TFEB under LG, Ma, or HG (10, 20, and 30 mM glucose for 48 h) in U937 cells (n = 5). (C) Immunofluorescence images and representative graph showing the nuclear translocation of TFEB in U937 cells that were pre-treated with ionomycin (10 µM), U18666A (2 µg/ml), or thapsigargin (TG; 1 µM) under HG (30 mM glucose for 48 h). The graph represented the percentage of the cells with nuclear translocation of TFEB (n = 4). (D) Representative immunoblots and graphs for TFEB and Histone H3 expressions in the nucleus, and TFEB and GAPDH expressions in the cytoplasm after stimulation with H2O2 (400 µM), ionomycin (1 µM), glycyl-l-phenylalanine-beta-naphthylamide (GPN; 400 µM), nicotinic acid adenine dinucleotide phosphate (NAADP; 1 mM), or TG (400 nM) in U937 cells. The relative protein expression of TFEB was normalized to representative controls (histone H3/GAPDH) (n = 4). Data were shown as mean ± SEM. (A–C) *P < 0.05 and **P < 0.01 vs. LG; #P < 0.05 and ##P < 0.01 vs. HG. (D) *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control.
TFEB Regulates HG-Induced Lysosomal Exocytosis and Pro-IL-1β Synthesis to Mediate IL-1β Secretion in Monocytic Cells
We next investigated whether TFEB could regulate lysosomal exocytosis in U937 cells. Lysosomal Ca2+ response induced by GPN was proposed to be responsible for lysosomal exocytosis in human monocytes (25). Here, we measured the release of the lysosomal marker enzyme, β-hexosaminidase, to examine lysosome exocytosis. Our results demonstrated that GPN-induced β-hexosaminidase release in a time-dependent manner, and it was inhibited by TFEB siRNA and BAPTA in U937 cells (Figure 7A); this suggested that intracellular Ca2+ signals was involved in GPN-induced lysosomal exocytosis through TFEB pathway. The efficiency of the knockdown was shown by immunoblotting (Figure 7B). Moreover, we also found that HG-induced surface LAMP-1 level was reduced by TFEB siRNA (Figure 7C). Next, to further determine whether TFEB could control lysosomal exocytosis through its target genes, we measured the mRNA levels of TFEB target genes that were previously linked to lysosomal exocytosis, including LAMP-1, cathepsin B, and cathepsin D (15, 29, 39). We demonstrated that the mRNA expressions of LAMP-1, cathepsin B, and cathepsin D were upregulated under HG in U937 cells (Figure 7D). As expected, these effects were abolished by TFEB siRNA (Figure 7D), suggesting that TFEB directly controlled lysosomal exocytosis under HG condition. In addition, previous study demonstrated that calcineurin interacted with TFEB and modulated its activation (24). We observed that calcineurin inhibitors, cyclosporin A and FK506, significantly inhibited HG-induced IL-1β secretion in U937 cells (Figure 7E); so this further confirmed that HG induced lysosomal exocytosis-mediated IL-β secretion via calcineurin/TFEB pathway. Besides, our results also found that HG-mediated upregulation of IL-1β mRNA level and its maturation were suppressed by TFEB siRNA in U937 cells (Figures 7F,G). By contrast, TFEB siRNA did not induce HG-induced caspase-1 cleavage (p20) (Figure 7H). Taken together, this further suggested that TFEB/calcineurin pathway was responsible for HG-induced IL-1β release via regulation of synthesis of pro-IL-1β and lysosomal exocytosis, but independent of caspase-1 activation in human monocytic cells.
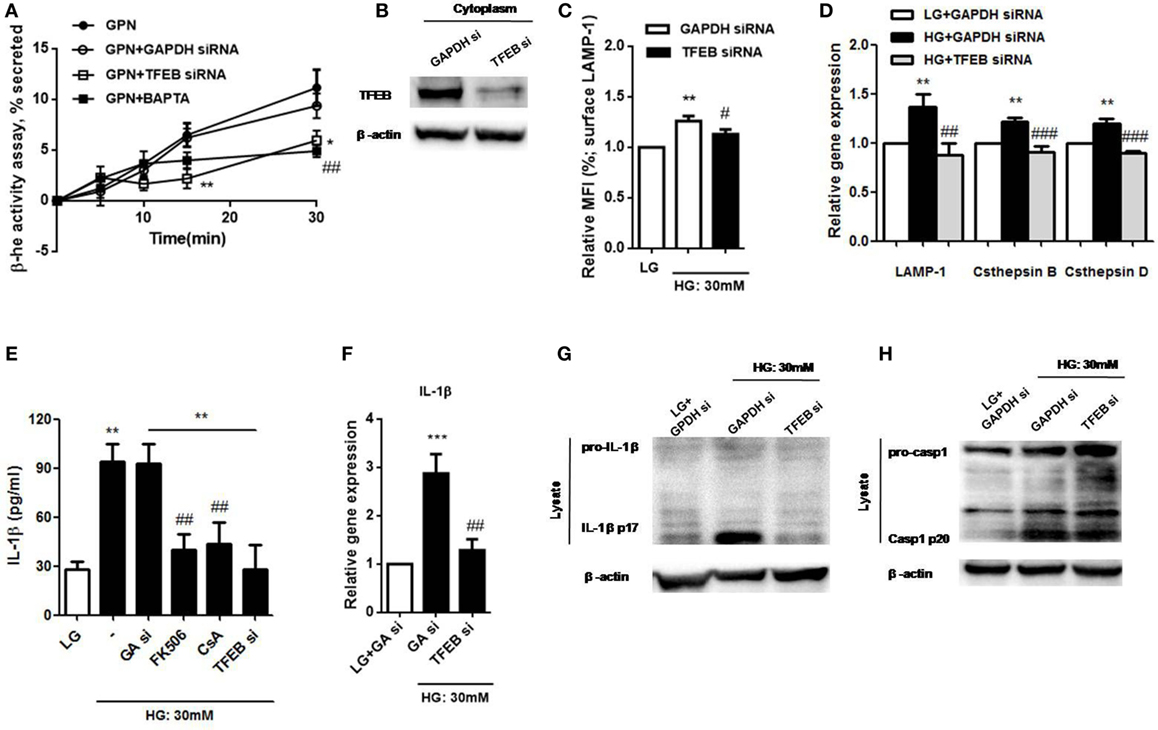
Figure 7. High glucose (HG) induced lysosomal Ca2+-dependent lysosomal exocytosis and interleukin-1β (IL-1β) secretion via calcineurin/transcription factor EB (TFEB) pathway in U937 cells. (A) The percentage of β-hexosaminidase activity was induced by glycyl-l-phenylalanine-b-napthylamide (GPN). U937 cells were treated with GPN (400 µM) in the presence of GAPDH small interfering RNA (siRNA) or TFEB siRNA or BAPTA (10 µM) (n = 4). The results were normalized to control. (B) Representative immunoblots for TFEB and β-actin in GAPDH siRNA (GAPDH si)- or TFEB siRNA-treated cells (TFEB si) (n = 3). (C) The relative median fluorescence intensity (MFI) of surface lysosomal-associated membrane protein-1 (LAMP-1) staining that were in the presence of GAPDH or TFEB siRNA under low glucose (LG; 5.5 mM glucose) or HG (30 mM glucose for 48 h) (n = 4). (D) The relative gene expressions of LAMP-1, cathepsin D, and cathepsin B in the presence of GAPDH or TFEB siRNA under LG or HG (30 mM glucose for 48 h) in U937 cells (n = 5). (E) ELISA for IL-1β secretion from the supernatants of U937 cells that were pre-treated with FK506 (25 µM) or cyclosporin A (Cs A; 10 µM), or in the presence of GAPDH siRNA (GA si) or TFEB siRNA (TFEB si) under LG or HG. (F) The relative gene expressions of IL-1β in the presence of GAPDH or TFEB siRNA under HG in U937 cells (n = 4). (G,H) Representative immunoblots for pro-IL-1β, IL-1β p17, or pro-caspase-1, cleaved caspase-1 (p20) and β-actin in the presence of GAPDH or TFEB siRNA under LG or HG (30 mM glucose for 48 h) in U937 cells (n = 4). Data were shown as mean ± SEM. (A) *P < 0.05 and **P < 0.01 vs. GPN + GAPDH siRNA; ##P < 0.01 vs. GPN. (C,E) **P < 0.01 vs. LG; #P < 0.05 and ##P < 0.01 vs. HG. (D,F) **P < 0.01 and ***P < 0.001 vs. LG + GAPDH siRNA; ##P < 0.01 and ###P < 0.001 vs. HG + GAPDH siRNA.
Discussion
Interleukin-1β, an inducer of various pro-inflammatory cytokines and chemokines, was implicated in driving tissue inflammation during T2DM (40, 41), and was tightly associated with promoting β-cell death, impaired insulin sensitivity and enhancing the adhesion capacity of circulating monocytes to the vascular endothelium (42–44). Recent studies demonstrated that targeting IL-1β, but not TNF-α antagonism, had beneficial effects for treating T2DM and its complications (3, 45–47). The present study provided mechanistic insights into IL-1β release induced by HG, which was mediated by lysosomal exocytosis via TFEB/calcineurin pathway in human monocytic cell lines, U937 and THP-1 cells. Furthermore, our results demonstrated that HG could cause a defect in lysosomal Ca2+ store and altered cytosolic Ca2+ homeostasis, which was essential for lysosomal exocytosis.
Interleukin-1β is one of the major inflammatory cytokines that is critical for chronic inflammatory response during metabolic disorders, including obesity and T2DM. The secretion of IL-1β is primarily from monocytes and macrophages (11), and HG, a characteristic of T2DM, could upregulate IL-1β mRNA and stimulate its secretion in human monocytes, contributing to impaired insulin secretion and signaling (48, 49). Indeed, there are several steps for IL-1β secretion, first is to produce inactive precursor, pro-IL-1β, which is then cleaved by caspase–1 to produce mature IL-1β, and the maturation of IL-1β should be secreted through non-conventional secreting pathway (50). Our previous study has demonstrated that HG induced NLRP3 inflammasome and caspase-1 activation, which contributed to IL-1β processing and secretion in monocytes (10); however, the mechanisms of secreting IL-1β into extracellular milieu are unclear. In human monocytes, the exocytosis of secretory lysosomes was a key mechanism for IL-1β secretion, and this required the elevation of [Ca2+]i and Ca2+-dependent phospholipases (15, 41). Our results also showed that HG significantly increased [Ca2+]i by reducing lysosomal Ca2+ level, and HG only affected lysosomal Ca2+ homeostasis but not ER and mitochondria Ca2+ homeostasis in human monocytic cells. It has been suggested that lysosomal Ca2+ signals could be linked to regulating endolysosome function, including altering lysosomal morphology, maintaining cytosolic Ca2+ homeostasis and lysosomal exocytosis (24, 25, 33, 51). We found out that lysosomal Ca2+ is a critical determinant of maintaining intracellular Ca2+ homeostasis under HG condition. HG raised [Ca2+]i that was originated from the lysosomes, and this lysosomal Ca2+ signals enhanced lysosomal exocytosis markers, like surface level of LAMP, cathepsin D, and β-hexosaminidase activity, which were critical for lysosome trafficking to the PM (lysosomal exocytosis). Therefore, this lysosomal Ca2+ contributed to secreting IL-1β into extracellular milieu in human monocytic cells.
Transcription factor EB was shown to regulate lysosomal exocytosis (22, 52). Recent study demonstrated that lysosomal stresses, such as Ox-LDL and cholesterol crystals, could induce TFEB nuclear translocation and the activation of lysosomal and autophagy genes in macrophages (53). Here, we showed that HG upregulated TFEB expression and induced TFEB nuclear translocation in U937 monocytic cells, and which was dependent on intracellular Ca2+, particularly lysosomal Ca2+. Interestingly, our results demonstrated that several internal Ca2+ activators, such as ionomycin, GPN, NAADP, and TG, were capable of inducing TFEB nuclear translocation. Therefore, it was likely that HG induced TFEB activation as a consequence of Ca2+ release from the lysosomes. Moreover, in other various cells, such as fibroblasts, neuronal cells, and osteoclasts, it was reported that overexpression of TFEB could mediate lysosomal exocytosis by raising [Ca2+]i (22, 54). Similarly, our results showed that TFEB was critical for HG-induced upregulation of lysosomal gene expressions, such as cathepsin D and LAMP-1, in U937 monocytic cells. Therefore, it was not surprising that TFEB could regulate Ca2+-dependent lysosomal exocytosis via lysosomal genes under HG condition. Although our study with other study showed that lysosomal exocytosis was regulated by TFEB (22), a direct regulation of IL-1β secretion by TFEB was not studied. As expected, we found that HG induced lysosomal exocytosis through calcineurin/TFEB pathway. We further studied the link between TFEB and IL-1β secretion, our results observed that TFEB significantly suppressed mRNA level of IL-1β, but it was dispensable for caspase-1 cleavage under HG. This suggested that TFEB play a critical role for regulating lysosomal exocytosis and pro-IL-1β synthesis, but not participate in caspase-1-dependent processing of pro-IL-1β into mature IL-1β. In addition, the inhibition of calcineurin, a binding partner of TFEB and mediates its activation (24), was reported to reduce IL-1β secretion via the inhibition of pro-IL-1β levels during lipotoxic inflammasome activation (55); this further supported our study, which suggested that calcineurin/TFEB activation was involved in the upregulation of IL-1β level, and subsequently affected its secretion. Taken together, our results suggested that lysosomal Ca2+-mediated TFEB activation could control lysosomal exocytosis through LAMP-1 and cathepsin D, and regulate intracellular pro-IL-1β synthesis by HG in human monocytic cells.
Regarding to the function of lysosomes, prolonged HG treatment was shown to inhibit lysosomal function in different cell types (31, 56–58). We showed that HG for 72 h, but not 48 h, resulted in the loss of lysosomes; however, a defect in lysosomal Ca2+ store was started to occur at 48 h, which suggested that lysosomal Ca2+ depletion was an early event of lysosomal disruption. Since impaired lysosomal Ca2+ store was suggested to induce lysosomal dysfunction (30, 59), we also observed that HG induced a decrease in lysosomal Ca2+ level and an increase in intracellular Ca2+ level. This observation suggested that HG might induce lysosomal Ca2+ release to raise cytosolic Ca2+ concentration and lead to disruption of lysosomal function by preventing Ca2+ refilling back to lysosomes. In particular, ER Ca2+ store and lysosomal pH gradient were responsible for driving Ca2+ refilling of lysosomes (30, 33). Our results demonstrated that HG induced an increase in cytosolic Ca2+ level, a defect in lysosomal Ca2+ level, but did not affect ER Ca2+ store. Therefore, it was likely that HG induced aberrant lysosomal pH, which contributed to the increase in [Ca2+]i and impaired lysosomal Ca2+ store, as supported by two studies (31, 33). Besides, exposure to HG more than 48 h (~72 h) prevented Ca2+ refilling of lysosomes, and lysosomal exocytosis, which was accompanied with enhanced cathepsin D activity, reaching maximum at HG for 48 h and then decreased after 72 h in monocytic cells. These observations suggested that HG induced the processing of exocytosis must be under normal lysosomal function (exposure to HG less than 48 h) to allow lysosomal Ca2+ release under physiological level.
In our previous work, we identified some novel mechanisms involved in the activation of NLRP3 inflammasome under HG in human monocytic cells. We demonstrated that TRPM2-mediated Ca2+ influx could contribute to HG-induced ROS overproduction and NLRP3 inflammasome activation, leading to IL-1β maturation and release (10). Notably, several studies suggested that Ca2+ signals was critical for IL-1β secretion induced by variety of stimulus, which was not only mediated through NLRP3 inflammasome activation, but also by lysosomal exocytosis (25, 35, 60, 61). In this study, we demonstrated two important pathways of HG-induced IL-1β secretion. First, lysosomal Ca2+ release played a vital role in HG-induced secreting IL-1β into extracellular milieu via lysosomal exocytosis/TFEB pathway. Second, TFEB could promote pro-IL-1β synthesis induced by HG. Taken together, our previous and the present study suggested that TRPM2-mediated Ca2+ influx regulate NLRP3 inflammasome activation, whereas internal Ca2+, particularly lysosomal Ca2+ release, was associated with triggering TFEB activation, which contributed to pro-IL-1β synthesis and secretion. Moreover, lysosomal Ca2+ signals was also responsible for secreting IL-1β into extracellular milieu via lysosomal exocytosis in human monocytic cells. The important role of IL-1β in T2DM has been recognized in the recent years (2, 3), and this observation provided more insight into mechanisms of IL-1β secretion in T2DM.
In conclusion, we demonstrated that HG could alter intracellular Ca2+ homeostasis, particularly lysosomal Ca2+ homeostasis, to trigger the activation of calcineurin and TFEB, a master gene for lysosomal function, in monocytic cells. Hence, TFEB could modulate lysosomal exocytosis by enhancing [Ca2+]i and contributed to secreting IL-1β into extracellular milieu under HG. Our results also demonstrated that lysosomal Ca2+ release by GPN or NAADP was sufficient for TFEB activation and induction of lysosomal exocytosis, suggesting that lysosomal Ca2+ signals was crucial for lysosomal exocytosis-dependent IL-1β release in monocytic cells. These findings provided an understanding of the underlying mechanisms of secreting IL-1β into extracellular milieu by HG, with a focus on the involvement of lysosomal Ca2+ signals in lysosomal exocytosis in monocytic cells.
Author Contributions
YK, SL, and MH conceived and designed the study; HT and CV performed the experiments; HT and MH drafted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Professor Alasdair Gibb and Dr. Dean Willis from University College London, UK, for project discussion.
Funding
This work was supported by grants from Science and Technology Development Fund of Macau SAR [127/2014/A3], Research Committee of University of Macau [MYRG124-ICMS12-HPM], [MYRG2015-00161-ICMS-QRCM], and National Natural Science Foundation of China [NSFC-81403139-H2809].
References
1. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (2003) 23(4):656–60. doi:10.1161/01.ATV.0000064374.15232.C3
2. Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of IL-1 beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes (2010) 17(4):314–21. doi:10.1097/MED.0b013e32833bf6dc
3. Herder C, Dalmas E, Boni-Schnetzler M, Donath MY. The IL-1 pathway in type 2 diabetes and cardiovascular complications. Trends Endocrinol Metab (2015) 26(10):551–63. doi:10.1016/j.tem.2015.08.001
4. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1 beta in type 2 diabetes. Nat Immunol (2010) 11(10):897–904. doi:10.1038/ni.1935
5. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi:10.1038/nri3452
6. Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med (2013) 19(9):1132–40. doi:10.1038/nm.3265
7. Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AF, Joosten LA, et al. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes (2011) 60(2):517–24. doi:10.2337/db10-0266
8. Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A (2011) 108(37):15324–9. doi:10.1073/pnas.1100255108
9. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17(2):179–88. doi:10.1038/nm.2279
10. Tseng HH, Vong CT, Kwan YW, Lee SM, Hoi MP. TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci Rep (2016) 6:35016. doi:10.1038/srep35016
11. Eder C. Mechanisms of interleukin-1beta release. Immunobiology (2009) 214(7):543–53. doi:10.1016/j.imbio.2008.11.007
12. Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol (1989) 5:483–525. doi:10.1146/annurev.cb.05.110189.002411
13. Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol (2002) 3(2):122–31. doi:10.1038/nrm732
14. Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol (2007) 8(8):622–32. doi:10.1038/nrm2217
15. Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci U S A (2004) 101(26):9745–50. doi:10.1073/pnas.0308558101
16. Gardella S, Andrei C, Lotti LV, Poggi A, Torrisi MR, Zocchi MR, et al. CD8(+) T lymphocytes induce polarized exocytosis of secretory lysosomes by dendritic cells with release of interleukin-1beta and cathepsin D. Blood (2001) 98(7):2152–9. doi:10.1182/blood.V98.7.2152
17. Bishara NB, Ding H. Glucose enhances expression of TRPC1 and calcium entry in endothelial cells. Am J Physiol Heart Circ Physiol (2010) 298(1):H171–8. doi:10.1152/ajpheart.00699.2009
18. Li J, Wang PP, Yu SP, Zheng Z, Xu X. Calcium entry mediates hyperglycemia-induced apoptosis through Ca2+/calmodulin-dependent kinase II in retinal capillary endothelial cells. Mol Vis (2012) 18(250):2371–9.
19. Yu TZ, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal (2011) 14(3):425–37. doi:10.1089/ars.2010.3284
20. Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science (2009) 325(5939):473–7. doi:10.1126/science.1174447
21. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J (2012) 31(5):1095–108. doi:10.1038/emboj.2012.32
22. Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell (2011) 21(3):421–30. doi:10.1016/j.devcel.2011.07.016
23. Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal (2014) 7(309):ra9. doi:10.1126/scisignal.2004754
24. Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol (2015) 17(3):288. doi:10.1038/ncb3114
25. Sivaramakrishnan V, Bidula S, Campwala H, Katikaneni D, Fountain SJ. Constitutive lysosome exocytosis releases ATP and engages P2Y receptors in human monocytes. J Cell Sci (2012) 125(Pt 19):4567–75. doi:10.1242/jcs.107318
26. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci (2016) 129(13):2475–81. doi:10.1242/jcs.146365
27. Li B, Jie W, Huang L, Wei P, Li S, Luo Z, et al. Nuclear BK channels regulate gene expression via the control of nuclear calcium signaling. Nat Neurosci (2014) 17(8):1055–63. doi:10.1038/nn.3744
28. Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep (2015) 12(9):1430–44. doi:10.1016/j.celrep.2015.07.050
29. Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT. Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol (2011) 187(5):2748–54. doi:10.4049/jimmunol.1100477
30. Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife (2016) 5:1–18. doi:10.7554/eLife.15887
31. Moheimani F, Kim CHJ, Rahmanto AS, van Reyk DM, Davies MJ. Inhibition of lysosomal function in macrophages incubated with elevated glucose concentrations: a potential contributory factor in diabetes-associated atherosclerosis. Atherosclerosis (2012) 223(1):144–51. doi:10.1016/j.atherosclerosis.2012.04.026
32. Galione A, Morgan AJ, Arredouani A, Davis LC, Rietdorf K, Ruas M, et al. NAADP as an intracellular messenger regulating lysosomal calcium-release channels. Biochem Soc Trans (2010) 38:1424–31. doi:10.1042/Bst0381424
33. Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci (2002) 115(Pt 3):599–607.
34. Chazotte B. Labeling lysosomes in live cells with LysoTracker. Cold Spring Harb Protoc (2011) 2011(2):pdb.prot5571. doi:10.1101/pdb.prot5571
35. Qu Y, Franchi L, Nunez G, Dubyak GR. P2X7 receptor-dependent secretion of IL-1beta is mediated by exocytosis of secretory lysosomes. FASEB J (2007) 21(6):A772–A.
36. Carta S, Tassi S, Semino C, Fossati G, Mascagni P, Dinarello CA, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1 beta-containing secretory lysosomes: role of microtubules. Blood (2006) 108(5):1618–26. doi:10.1182/blood-2006-03-014126
37. Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell (2001) 106(2):157–69. doi:10.1016/S0092-8674(01)00421-4
38. Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med (2008) 14(7):738–47. doi:10.1038/nm1758
39. Lopez-Castejon G, Theaker J, Pelegrin P, Clifton AD, Braddock M, Surprenant A. P2X(7) receptor-mediated release of cathepsins from macrophages is a cytokine-independent mechanism potentially involved in joint diseases. J Immunol (2010) 185(4):2611–9. doi:10.4049/jimmunol.1000436
40. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol (2011) 11(2):98–107. doi:10.1038/nri2925
41. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1 beta secretion. Cytokine Growth Factor Rev (2011) 22(4):189–95. doi:10.1016/j.cytogfr.2011.10.001
42. Takahashi M, Ikeda U, Masuyama J, Kitagawa S, Kasahara T, Shimpo M, et al. Monocyte-endothelial cell interaction induces expression of adhesion molecules on human umbilical cord endothelial cells. Cardiovasc Res (1996) 32(2):422–9. doi:10.1016/0008-6363(96)00085-5
43. Masters SL, Latz E, O’Neill LA. The inflammasome in atherosclerosis and type 2 diabetes. Sci Transl Med (2011) 3(81):81ps17. doi:10.1126/scitranslmed.3001902
44. Wen HT, Gris D, Lei Y, Jha S, Zhang L, Huang MTH, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12(5):408–15. doi:10.1038/ni.2022
45. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov (2014) 13(6):465–76. doi:10.1038/nrd4275
46. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356(15):1517–26. doi:10.1056/NEJMoa065213
47. Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein E-deficient mice. Atherosclerosis (2011) 216(2):313–20. doi:10.1016/j.atherosclerosis.2011.02.026
48. Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes (2003) 52(5):1256–64. doi:10.2337/diabetes.52.5.1256
49. Dasu MR, Devaraj S, Jialal I. High glucose induces IL-1beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab (2007) 293(1):E337–46. doi:10.1152/ajpendo.00718.2006
50. Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol (2007) 8(8):622–32. doi:10.1038/nrm2217
51. Cao Q, Yang Y, Zhong XZ, Dong XP. The lysosomal Ca2+ release channel TRPML1 regulates lysosome size by activating calmodulin. J Biol Chem (2017) 292:8424–35. doi:10.1074/jbc.M116.772160
52. Kukic I, Kelleher SL, Kiselyov K. Zn2+ efflux through lysosomal exocytosis prevents Zn2+-induced toxicity. J Cell Sci (2014) 127(14):3094–103. doi:10.1242/jcs.145318
53. Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, et al. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol (2014) 34(9):1942–52. doi:10.1161/Atvbaha.114.303342
54. Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S, Rawlings DJ, et al. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev (2013) 27(8):955–69. doi:10.1101/gad.213827.113
55. Weber K, Schilling JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem (2014) 289(13):9158–71. doi:10.1074/jbc.M113.531202
56. Chaudhari S, Wang YX, Ding M, Ding YF, Yuan J, Ma R. Prolonged high glucose treatment increased STIM1/Orai1 protein expression and enhanced store-operated Ca2+ entry in human glomerular mesangial cells. FASEB J (2013) 27:702–11.
57. Nishimura F, Naruishi K, Yamada H, Kono T, Takashiba S, Murayama Y. High glucose suppresses cathepsin activity in periodontal-ligament-derived fibroblastic cells. J Dent Res (2000) 79(8):1614–7. doi:10.1177/00220345000790081501
58. Vidotti DB, Casarini DE, Cristovam PC, Leite CA, Schor N, Boim MA. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am J Physiol Renal Physiol (2004) 286(6):F1039–45. doi:10.1152/ajprenal.00371.2003
59. Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol (2012) 198(1):23–35. doi:10.1083/jcb.201201076
60. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A (2012) 109(28):11282–7. doi:10.1073/pnas.1117765109
Keywords: high glucose, lysosomal Ca2+, Ca2+ homeostasis, lysosomal exocytosis, interleukin-1β, monocytes
Citation: Tseng HHL, Vong CT, Kwan YW, Lee SM-Y and Hoi MPM (2017) Lysosomal Ca2+ Signaling Regulates High Glucose-Mediated Interleukin-1β Secretion via Transcription Factor EB in Human Monocytic Cells. Front. Immunol. 8:1161. doi: 10.3389/fimmu.2017.01161
Received: 19 April 2017; Accepted: 01 September 2017;
Published: 15 September 2017
Edited by:
Alessandra Mortellaro, Singapore Immunology Network (A*STAR), SingaporeReviewed by:
Gloria Lopez-castejon, University of Manchester, United KingdomPaola Italiani, Consiglio Nazionale Delle Ricerche (CNR), Italy
Copyright: © 2017 Tseng, Vong, Kwan, Lee and Hoi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maggie Pui Man Hoi, bWFnaG9pQHVtYWMubW8=