 Rosa Muñoz-Cano1,2*
Rosa Muñoz-Cano1,2* Mariona Pascal2,3Giovanna Araujo1,2M. J. Goikoetxea4Antonio L. Valero1,2Cesar Picado1,2
Mariona Pascal2,3Giovanna Araujo1,2M. J. Goikoetxea4Antonio L. Valero1,2Cesar Picado1,2 Joan Bartra1,2
Joan Bartra1,2
- 1Unitat d’Allergia, Servei de Pneumologia, Hospital Clinic, Universitat de Barcelona, Barcelona, Spain
- 2Institut d’Investigacions Biomediques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 3Servei d’Immunologia, Centre de Diagnòstic Biomèdic (CDB), Hospital Clinic, Universitat de Barcelona, Barcelona, Spain
- 4Allergy and Immunology Department, Universidad de Navarra, Navarra, Spain
Anaphylaxis is an acute and life-threatening systemic reaction. Many triggers have been described, including food, drug, and hymenoptera allergens, which are the most frequently involved. The mechanisms described in anaphylactic reactions are complex and implicate a diversity of pathways. Some of these mechanisms may be key to the development of the anaphylactic reaction, while others may only modify its severity. Although specific IgE, mast cells, and basophils are considered the principal players in anaphylaxis, alternative mechanisms have been proposed in non-IgE anaphylactic reactions. Neutrophils, macrophages, as well as basophils, have been involved, as have IgG-dependent, complement and contact system activation. A range of cationic substances can induce antibody-independent mast cells activation through MRGPRX2 receptor. Cofactors and augmenting factors may explain why, in some patients, food allergen exposure can cause anaphylaxis, while in other clinical scenario it can be tolerated or elicits a mild reaction. With the influence of these factors, food allergic reactions may be induced at lower doses of allergen and/or become more severe. Exercise, alcohol, estrogens, and some drugs such as Non-steroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors, β-blockers, and lipid-lowering drugs are the main factors described, though their mechanisms and signaling pathways are poorly understood.
Introduction
Anaphylaxis is an acute, life-threatening, systemic reaction caused by the mediators released from different cells (1). Although the underlying mechanism is frequently IgE-dependent, some other mechanisms there are also involved (2–4). Complement activation, neuropeptide release, T-cell activation, immune complex formation, cytotoxicity, IgG-dependent activation, induction of purinergic metabolism, and activation of the receptor MRGPRX2, are some of those alternative pathways (2, 5). Mast cells are considered the pivotal cells in IgE-mediated anaphylaxis (6), and the role of macrophages and neutrophils has been described in IgE-independent reactions (6, 7). Interestingly, basophil implication has been suggested in both IgE-dependent and -independent reactions, although its role in allergic reactions has been considered as redundant with mast cells for some time (8). In humans, CD203c and CD63, basophil activation surface markers, are used to confirm allergen sensitization (9). Recently, a decrease on circulating basophil and an increase in serum concentration of the major basophil chemotactic factor (CCL2) during food and hymenoptera-induced anaphylaxis has been observed, suggesting the role of basophil in human IgE-dependent anaphylaxis (10).
Immune-Mediated Anaphylaxis
IgE-Mediated Reactions
The more common mechanism involved in an anaphylactic reaction is promoted by an allergen recognized by an allergen-specific IgE bound to the FcεRI receptor on the surface of mast cells and basophils. When the signal is sufficiently powerful, mast cell and basophil activation make progress, releasing mediators (11). Those mediators also lead to the amplification of the allergic reaction through the recruitment and activation of other cells involved in the IgE immunological response (12–14). However, this explanation is too simple to understand what take place in an in vivo reaction, and sundry factors could influence allergen-dependent mast cell and basophil activation under specific conditions (15).
IgG-Mediated Reactions
There are six different Fcgamma receptors (FcγRI, FcγRIIA, FcγRIIB, FcγRIIC, FcγRIIIA, and FcγRIIIB), and all of them bind IgG. Among them, FcγRI is considered a high-affinity receptor (16). Most of these receptors induce cell activation, except for FcγRIIB, which induces an inhibitory signal and has been proposed as a key player in IgG subclass-dependent anaphylaxis in a recent study (17).
Mouse models have been used to demonstrate the relevance of IgG in anaphylaxis. A passive systemic anaphylaxis model has suggested that FcγRIII on cells as macrophages and basophils mediates these reactions (3, 4, 18), and platelet-activating factor (PAF) but not histamine (3, 4) is the main mediator involved.
An IgG-dependent mechanism has been also suggested in human anaphylaxis. PAF, mostly associated with an IgG mechanism, has a key role in human anaphylaxis as several authors have suggested. Vadas et al. (19) found increased circulating PAF levels and decreased PAF acetylhydrolase (PAF-AH) activity in proportion to the severity of the anaphylactic reaction (20). Indeed, the lowest levels of PAF-AH activity were related with a 27 times more risk of severe or fatal anaphylaxis compared to normal activity (19, 21).
Several authors have suggested that both IgG and neutrophils may be involved in human anaphylaxis. Muñoz-Cano et al. (5) studied patients with anaphylaxis induced by lipid transfer proteins (LTP) and mediated by IgE. They found an increase of specific anti-LTP IgG1 and IgG3 levels and increased expression of the three genes coding for FcγRI (CD64), an activating receptor (5). It has been shown that FcγRI mediates mast cell and neutrophil activation via IgG (22, 23), by both IgG1 and IgG3 (16) also in humans. Interestingly, Muñoz-Cano et al. (5) found specific IgG and anti-LTP IgE in those patients, suggesting that both IgG and IgE pathways may contribute substantially to anaphylaxis. Rispens et al. (24) also found both specific IgE and IgG1 anti-α-gal in patients with galactose-alpha-1,3-alpha-galactose (α-gal) allergy.
Neutrophils, activated through FcγRIV-IgG2, are proposed to play a major role in a mouse model of anaphylaxis (7). They are important PAF producers, and a differential PAF release has been observed in neutrophil-dependent reactions in mice (7, 25). However, PAF is also observed in IgE-mediated reactions in animal models (11). Muñoz-Cano et al. (5) showed that several markers of neutrophil activation and trafficking were highly expressed in patients with IgE-dependent anaphylaxis allergic to LTP. Moreover, the authors found increased levels of reactive oxygen species/reactive nitrogen species, known as a measure of oxidative outburst, suggesting an enhancement of neutrophilic activity. Francis et al. (26) also found increased neutrophil activation markers (myeloperoxidase and CD62L) during an acute anaphylactic reaction.
In the light of these findings, the paradigm of anaphylaxis mediated only by IgE and mast cell/basophil seems not totally accurate. In the LTP particular case, anaphylaxis may be elicited via IgE, IgG, or both, with the involvement of neutrophils and not only of mast cells and basophils, although other allergens may act similarly.
Complement Activation in Anaphylaxis
Monomeric IgG and IgG immune complexes can bind FcγRI receptors (27, 28) and are key in the novel paradigm in human anaphylaxis (IgG anaphylaxis). Furthermore, the complement system can also be activated by immune complexes, resulting in the generation of anaphylatoxins such as C3a (23, 29). Interestingly, C3a has demonstrated a direct effect on mast cell and also a synergistic effect (twofold increase) with FcγRI receptor activation (23). Therefore, the combination of IgG and C3a activation results in greater mast cell activation or activation under circumstances in which neither of the stimuli would elicit maximal release on its own.
Large amounts of the anaphylatoxin C3a have been found in peanut severe allergic reactions by Khodoun et al. (30), in both mouse and human plasma. However, allergens such as milk and egg white did not have the ability to activate complement in humans (30). Therefore, several factor as patient susceptibility (5), cofactors (31) and characteristics of a particular allergen may determine the severity of an allergic reaction.
Reactions with drugs solubilized in therapeutic liposomes and lipid-based excipients have been related with the activation of complement in the absence of immune complex. It is the case of Cremophor EL, a diluent used in the older preparations of propofol and paclitaxel, which has been found to induce complement activation (32).
Finally, it has been also demonstrated that lypopolysaccharides (LPS) can induce a strong activation of the complement and trigger an anaphylactic reaction in a mouse model (33). Recently, Rodriguez et al. (34) demonstrated the role of LPS as a co-stimulus triggering anaphylaxis in a mouse model; specific Pru p 3-induced anaphylaxis was generated after nasal sensitization to Pru p 3 in combination with LPS.
Non-Immune-Mediated Anaphylaxis
Contact System Activation in Anaphylaxis
It has been identified as direct or indirect activation of the blood coagulation pathway in allergic reactions mediated by IgE (35). During acute anaphylaxis, an increase of the heparin levels and an activation of the factor XII-driven contact system has been observed, which results in the production of bradykinin (36). In fact, after the analysis of more than 150 deaths associated to anaphylaxis induced by oversulfated chondroitin sulfate-contaminated heparin, the possible role of heparin as a trigger of bradykinin formation through contact activation was suggested (37, 38). Therefore, targeting its generation may be a promising strategy for treatment of severe allergic reactions, importantly those with hypotension (39).
New Mast Cell Receptors in Anaphylaxis: MRGPRX2
Mast cells are classically activated by IgE antibodies, although a range of cationic substances, called basic secretagogues, can induce antibody-independent activation. Among those secretagogues, there are peptides with pro-inflammatory effects and several drugs. Recently, Mrgprb2, the ortholog of the human G-protein-coupled receptor MRGPRX2, has been described to mediate this activation in a mouse model. This receptor seems to be the target of many small-molecule drugs involved in non-IgE anaphylactic reactions, such as non-steroidal neuromuscular blocking drugs (tubocurarine, atracurium, or ciprofloxacin). This work identified a chemical motif that is common to several of these molecules and may be linked to some of the observed side effects. In conclusion, MRGPRX2 may be considered a potential therapeutic target to reduce some adverse effects induced by some drugs (40).
Cofactors and Augmenting Factors in Anaphylaxis
The so-called accompanying factors may explain why an allergen can either be tolerated or trigger a mild reaction or, in the same patients, induce a severe anaphylaxis. In the presence of cofactors, reactions become more severe and/or the amount of allergen eliciting the allergic reaction can be lower. According to published data, the presence of those accompanying factors occurs in up to 30% of episodes of anaphylaxis (31, 41). Niggemann and Beyer (42) postulated three categories of risk factors for anaphylactic reactions: first, the augmenting factors, which influence the immunological mechanism, such as physical exercise, acute infections, drugs [non-steroidal anti-inflammatory drugs (NSAIDs), proton pump inhibitors], alcohol, and menstruation; second, concomitant diseases, such as asthma, mastocytosis, and cardiovascular disease, which are associated with more severe reactions and/or increased mortality; and third, cofactors, which do not have any influence on the immunological mechanism, as psychological factors (e.g., emotional stress) or specific allergens. Nevertheless, the lack of knowledge about the mechanisms underlying these risk factors limits a strict categorization. Therefore, for the purposes of this review, the terms cofactor and augmenting factor are used indistinctively.
Estrogens
Gender differences have been reported in the incidence of anaphylactic reactions, demonstrating that anaphylaxis is more frequent in women than men (43, 44), but only during the reproductive years, suggesting that sexual hormones might play a role. Additionally, like the episodes of asthma or urticaria associated with the menstrual cycle (45, 46), recurrent episodes of anaphylactic reactions around menstruation have been described, pointing at the estrogens or progesterone as the augmenting factors involved (47).
The susceptibility of women to develop anaphylactic reactions observed in clinical studies was also demonstrated in a murine model (48). Female mice were ovariectomized to eliminate the major source of estrogens and the result was the decrease in the severity of anaphylaxis. Moreover, the implant of subcutaneous estradiol-releasing pellets in the ovariectomized mice resulted in an increase in the severity of anaphylaxis. The mechanism involved was not related with the increase in mast cell degranulation, but with an augmentation of the vascular permeability. A higher production of nitric oxide as a result of major expression of endothelial nitric oxide synthase was in fact the cause (48).
Exercise
Exercise is involved up to 10% of anaphylactic reactions, being one of the more frequent augmenting factors (49). However, the knowledge of its pathogenic mechanism still continues to be poorly understood, and some theories have being proposed. Although, according to the literature, the foods involved in food-dependent exercise-induced anaphylaxis are very diverse, wheat is the most frequent one, being ω-5 gliadin the culprit protein in most cases (50). As a result, most of the mechanistic studies have been performed in patients with wheat-dependent exercise-induced anaphylaxis (WDEIA).
One of the theories hypothesizes that exercise induces an activation of tissue transglutaminase (tTG), resulting in a formation of large ω-5 gliadin/tTG complexes that would facilitate the ω-5gliadin–IgE binding. Nonetheless, no direct evidence of this phenomenon has been found in patients with WDEIA (51).
A second hypothesis establishes that exercise would induce an increase in the intestinal allergen absorption, and as a consequence, an increase of blood allergen concentration and the likelihood to develop an anaphylactic reaction (52, 53). Some murine models of food allergy have demonstrated how physical exercise increases the absorption of allergen from the gastrointestinal tract due to mucosa injury (54, 55). An increase of the core temperature in the gastrointestinal tract due to intense exercise would result in epithelial cell damage owing to the phosphorylation state of tight junction proteins. In addition, another mechanism involved in the mucosal damage may be related with the deviation of blood flow away from the splanchnic arteries to the working muscle, resulting in an ischemia/reperfusion cycle that causes the epithelial damage (56, 57).
Another hypothesis establishes that exercise, due to a direct effect on mast cells, would modify the threshold dose of allergen in patients with WDEIA. It has been described that physical exercise induces an increment of the plasma osmolarity (58), and this increase results in mast cells activation and release of inflammatory mediators (59). Additionally, a previous in vitro study showed that IgE activation and hyperosmolar stimuli at the same time have a synergistic effect on IgE-induced mast cell release (60). However, the increase of plasma osmolarity due to physical exercise is only significant when the exercise is strenuous and, in patients with WDEIA reactions, the intensity of the physical exercise is frequently moderate.
Lipid-Lowering Drugs (Statins)
Lipid-lowering drugs can be considered as a risk factor in anaphylaxis since some studies posited that low plasma levels of low-density lipoprotein (LDL) may augment the risk of severe or fatal anaphylaxis. These drugs increase plasma concentration of PAF by lowering PAF-AH activity (19, 61). In this way, Perelman et al. (62) demonstrated a significant direct correlation between PAF-AH activity and LDL levels in patients with peanut allergy. Moreover, a significant correlation between PAF plasma levels and the severity of anaphylaxis has also been demonstrated (33).
Non-Steroidal Anti-inflammatory Drugs
Non-steroidal anti-inflammatory drugs are other well-known augmentation factors in anaphylaxis. They have been reported to be present in up to 22% of cases of food-induced severe anaphylaxis, constituting a risk factor with an odds ratio >11 (63). In the Mediterranean area, NSAIDs are involved up to 58% of cofactor-induced food-related anaphylaxis episodes (64) and in up to 33% of cases of anaphylactic reaction induced by LTP (65). Two hypotheses have been proposed to explain the underlying mechanisms involved in food-dependent NSAID-induced anaphylaxis (FDNIA).
The first hypothesis suggests that the increase of gastrointestinal permeability and allergen absorption may account for the augmentation effect of NSAIDs (53). It is well known that prostaglandins play an important role in gastrointestinal mucosa defense and repair. NSAIDs, through prostaglandin inhibition, leave gastrointestinal tissues more susceptible to the injury caused by gastric acid and bile and with less capacity to retrieve the mucosa function (38). Additionally, NSAIDs induce mitochondrial damage that leads to the malfunction of the intestinal epithelial cells and increase of the intestinal permeability (66, 67).
A second hypothesis suggests that NSAIDs have a direct impact on mast cells and basophils IgE activation, amplifying their activation and degranulation (68, 69). However, the underlying mechanisms involved remain still unknown.
Bartra et al. (70) suggested that the enhancing effect of NSAIDs in food allergic reactions might be related with the cyclooxygenase (COX) pathway. Several authors (71–73) have also shown that this effect is a class effect, therefore COX-dependent. Moreover, it has been demonstrated that selective COX-2 inhibitors (nimesulide and etodolac) (68, 69) did not increase the severity of food allergic reactions. Additionally, prostaglandin E1, an important prostanoid derived from the COX pathway (74), has been demonstrated to be protective in patients with FDNIA (75).
Adenosine metabolism and A3 receptor (A3R) have been linked with the underlying mechanisms of some diseases exacerbated by NSAIDs, such as NSAID-dependent urticaria (76) and aspirin-induced asthma (77). Interestingly, it has also been demonstrated that the activation of A3R enhances FcεRI-induced granule release in human mast cells (78–80). Moreover, a study that evaluated the transcriptome of patients with FDNIA showed an overexpression of genes related to adenosine metabolism, particularly A3R gene (5). NSAIDs are able to inhibit oxidative phosphorylation of ATP and promote its hydrolysis which entails the release of adenosine (81, 82); therefore, a link between NSAIDs, adenosine, adenosine receptors, and allergic reaction has been suggested.
Angiotensin-Converting Enzyme Inhibitors and β-Blockers
Angiotensin-converting enzyme inhibitors (ACE inhibitors) and β-blockers have been described as augmenting factors in anaphylactic reactions according to several studies (63, 83, 84). The odds ratio established for β-blockers was 6.8 and 13 for ACE inhibitors. However, other studies concluded that the risk of develop anaphylaxis related to ACE inhibitors and β-blockers does not exist unless both treatments are combined (21, 85, 86). Recently, mast cells were recognized as targets of ACE inhibitors and β-blockers in a murine model, augmenting their activation through FcεRI (86). In spite of the fact of this important data, further epidemiological and in vivo and in vitro studies in humans are necessary to determinate the real impact of these drugs as a risk factors in anaphylaxis.
Alcohol
Alcohol is involved in up to 15% of cases of anaphylactic reaction according to some series (49, 87), independently of their severity (86). Although, the underlying mechanisms are not well established, alcohol may increase allergen absorption. It has been described that alcohol induces a modification in the expression of the tight junction-associated proteins ZO-1 and claudin-1 of the intestinal epithelium, thereby augmenting the permeability of the intestinal epithelial barrier (88).
An adenosine-related mechanism has also been suggested in IgE-mediated anaphylaxis when alcohol is involved. Alcohol inhibits the adenosine uptake, inducing an increase of the extracellular adenosine, thus enhancing FcεRI-induced mast cells and basophil activation (89).
Another mechanism postulated is based on the capacity of alcohol to boost the serum IgE concentration (90). In a murine model, alcohol intake was linked with a raise in IgE serum levels and a decrease in IgG (90). Nevertheless, this acute alcohol intake has also been linked with lower release of mast cell mediators, such tryptase (91).
Conclusion
IgE, mast cells and basophils have been considered the main key players in human anaphylaxis for a long time, although alternative mechanisms have been suggested. Neutrophils and macrophages, IgG-mediated, complement, and contact system activation are some of them. A range of cationic substances can induce antibody-independent activation through the recently described receptor MRGPRX2. The presence of the so-called cofactors (accompanying or augmenting factors) may explain why the intake of some food sometimes lead to anaphylaxis, while in other cases the same allergen induces a milder reaction or is even tolerated. An understanding of the mechanisms underlying the anaphylactic reactions as well as of the predisposing and augmenting factors could help in the development of new prophylactic and therapeutic approaches. These strategies should target the specific pathways involved in anaphylaxis which, in the light of this review, may be more than one (Figure 1).
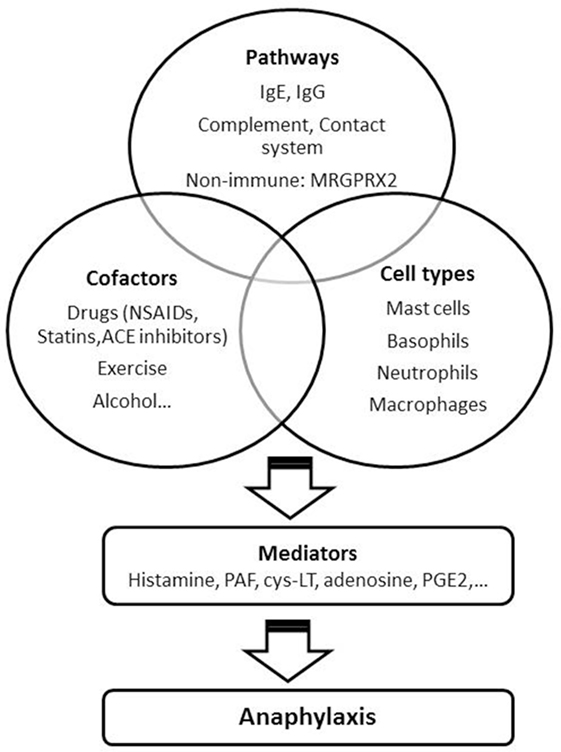
Figure 1. Factors involved in anaphylaxis. NSAIDs, non-steroidal anti-inflammatory drugs; ACE, angiotensin-converting enzyme; PAF, platelet-activating factor; Cys-LT, cysteinyl leukotrienes; PGE2, prostaglandin E2.
Author Contributions
All authors have contributed equally to this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RM-C, MP, GA, MG, and JB belong to the Spanish Research Network ARADyAL RD16/0006/0007 of the Carlos III Health Institute. RM-C is a recipient of a Juan Rodes fellowship (Carlos II Health Institute JR16/00016).
References
1. Sampson HA, Muñoz-Furlong A, Campbell RL, Adkinson NF, Bock SA, Branum A, et al. Second symposium on the definition and management of anaphylaxis: summary report – Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network Symposium. J Allergy Clin Immunol (2006) 117:391–7. doi:10.1016/j.jaci.2005.12.1303
2. Metcalfe DD, Peavy RD, Gilfillan AM. Mechanisms of mast cell signaling in anaphylaxis. J Allergy Clin Immunol (2009) 124:639–48. doi:10.1016/j.jaci.2009.08.035
3. Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol (2007) 120:506–7. doi:10.1016/j.jaci.2007.07.033
4. Strait RT, Morris SC, Yang M, Qu XW, Finkelman FD. Pathways of anaphylaxis in the mouse. J Allergy Clin Immunol (2002) 109:658–68. doi:10.1067/mai.2002.123302
5. Muñoz-Cano R, Pascal P, Bartra J, Picado C, Valero A, Kim D, et al. Distinct transcriptome profiles differentiate nonsteroidal anti-inflammatory drug-dependent from nonsteroidal anti-inflammatory drug-independent food-induced anaphylaxis. J Allergy Clin Immunol (2016) 137:137–46. doi:10.1016/j.jaci.2015.05.042
6. Jonsson F, Daeron M. Mast cells and company. Front Immunol (2012) 3:16. doi:10.3389/fimmu.2012.00016
7. Jönsson F, Mancardi DA, Kita Y, Karasuyama H, Iannascoli B, Van Rooijen N, et al. Mouse and human neutrophils induce anaphylaxis. J Clin Invest (2011) 121:1484–96. doi:10.1172/JCI45232
8. Schroeder JT. Basophils: emerging roles in the pathogenesis of allergic disease. Immunol Rev (2011) 242:144–60. doi:10.1111/j.1600-065X.2011.01023.x
9. Santos AF, Du Toit G, Douiri A, Radulovic S, Stephens A, Turcanu V, et al. Distinct parameters of the basophil activation test reflect the severity and threshold of allergic reactions to peanut. J Allergy Clin Immunol (2015) 135:179–86. doi:10.1016/j.jaci.2014.09.001
10. Korosec P, Turner PJ, Silar M, Kopac P, Kosnik M, Gibbs BF, et al. Basophils, high-affinity IgE receptors, and CCL2 in human anaphylaxis. J Allergy Clin Immunol (2017) 140:750–8. doi:10.1016/j.jaci.2016.12.989
11. Rivera J, Gilfillan A. Molecular regulation of mast cell activation. J Allergy Clin Immunol (2006) 117:1214–25. doi:10.1016/j.jaci.2006.04.015
12. Kajiwara N, Sasaki T, Bradding P, Cruse G, Sagara H, Ohmori K, et al. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunol (2010) 125:1137–45.e6. doi:10.1016/j.jaci.2010.01.056
13. Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, et al. Mast cells and inflammation. Biochim Biophys Acta (2012) 1822:21–33. doi:10.1016/j.bbadis.2010.12.014
14. Kemp SF, Lockey RF. Anaphylaxis: a review of causes and mechanisms. J Allergy Clin Immunol (2002) 110:341–8. doi:10.1067/mai.2002.126811
15. Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol (2006) 6:218–30. doi:10.1038/nri1782
16. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood (2012) 119:5640–9. doi:10.1182/blood-2012-01-380121
17. Beutier H, Gillis CM, Iannascoli B, Godon O, England P, Sibilano R, et al. IgG subclasses determine pathways of anaphylaxis in mice. J Allergy Clin Immunol (2017) 139:269–80.e7. doi:10.1016/j.jaci.2016.03.028
18. Tsujimura Y, Obata K, Mukai K, Shindou H, Yoshida M, Nishikado H, et al. Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity (2008) 28:581–9. doi:10.1016/j.immuni.2008.02.008
19. Vadas P, Gold M, Perelman B, Liss GM, Lack G, Blyth T, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med (2008) 358:28–35. doi:10.1056/NEJMoa070030
20. Stafforini DM. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2). Cardiovasc Drugs Ther (2009) 23:73–83. doi:10.1007/s10557-008-6133-8
21. Brown SGA, Stone SF, Fatovich DM, Burrows SA, Holdgate A, Celenza A, et al. Anaphylaxis: clinical patterns, mediator release, and severity. J Allergy Clin Immunol (2013) 132:1141–9.e5. doi:10.1016/j.jaci.2013.06.015
22. Okayama Y, Hagaman DD, Metcalfe DD. A comparison of mediators released or generated by IFN-γ-treated human mast cells following aggregation of FcγRI or FcεRI1. J Immunol (2001) 166:4705–12. doi:10.4049/jimmunol.166.7.4705
23. Woolhiser MR, Brockow K, Metcalfe DD. Activation of human mast cells by aggregated IgG through FcgammaRI: additive effects of C3a. Clin Immunol (2004) 110:172–80. doi:10.1016/j.clim.2003.11.007
24. Rispens T, Derksen NIL, Commins SP, Platts-Mills TA, Aalberse RC. IgE production to α-gal is accompanied by elevated levels of specific IgG1 antibodies and low amounts of IgE to blood group B. PLoS One (2013) 8:e55566. doi:10.1371/journal.pone.0055566
25. DeNichilo MO, Stewart AG, Vadas MA, Lopez AF. Granulocyte-macrophage colony-stimulating factor is a stimulant of platelet-activating factor and superoxide anion generation by human neutrophils. J Biol Chem (1991) 266:4896–902.
26. Francis A, Bosio E, Stone SF, Fatovich DM, Arendts G, Nagree Y, et al. Neutrophil activation during acute human anaphylaxis: analysis of MPO and sCD62L. Clin Exp Allergy (2017) 47:361–70. doi:10.1111/cea.12868
27. Mancardi DA, Albanesi M, Jönsson F, Iannascoli B, Van Rooijen N, Kang X, et al. The high-affinity human IgG receptor FcγRI (CD64) promotes IgG-mediated inflammation, anaphylaxis, and antitumor immunotherapy. Blood (2013) 121:1563–73. doi:10.1182/blood-2012-07-442541
28. van der Poel CE, Spaapen RM, van de Winkel JGJ, Leusen JHW. Functional characteristics of the high affinity IgG receptor, Fc{gamma}RI. J Immunol (2011) 186:2699–704. doi:10.4049/jimmunol.1003526
29. Zwirner J, Götze O, Sieber A, Kapp A, Begemann G, Zuberbier T, et al. The human mast cell line HMC-1 binds and responds to C3a but not C3a(desArg). Scand J Immunol (1998) 47:19–24. doi:10.1046/j.1365-3083.1998.00250.x
30. Khodoun M, Strait R, Orekov T, Hogan S, Karasuyama H, Herbert DR, et al. Peanuts can contribute to anaphylactic shock by activating complement. J Allergy Clin Immunol (2009) 123:342–51. doi:10.1016/j.jaci.2008.11.004
31. Wölbing F, Fischer J, Köberle M, Kaesler S, Biedermann T. About the role and underlying mechanisms of cofactors in anaphylaxis. Allergy (2013) 68:1085–92. doi:10.1111/all.12193
32. Szebeni J, Muggia FM, Alving CR. Complement activation by Cremophor EL as a possible contributor to hypersensitivity to paclitaxel: an in vitro study. J Natl Cancer Inst (1998) 90:300–6. doi:10.1093/jnci/90.4.300
33. Kawabata Y, Yang TS, Yokochi TT, Matsushita M, Fujita T, Shibazaki M, et al. Complement system is involved in anaphylactoid reaction induced by lipopolysaccharides in muramyldipeptide-treated mice. Shock (2000) 14:572–7. doi:10.1097/00024382-200014050-00013
34. Rodriguez MJ, Aranda A, Fernandez TD, Cubells-Baeza N, Torres MJ, Gomez F, et al. LPS promotes Th2 dependent sensitisation leading to anaphylaxis in a Pru p 3 mouse model. Sci Rep (2017) 7:40449. doi:10.1038/srep40449
35. Pinckard RN, Tanigawa C, Halonen M. IgE-induced blood coagulation alterations in the rabbit: consumption of coagulation factors XII, XI, and IX in vivo. J Immunol (1975) 115:525–32.
36. Sala-Cunill A, Björkqvist J, Senter R, Guilarte M, Cardona V, Labrador M, et al. Plasma contact system activation drives anaphylaxis in severe mast cell-mediated allergic reactions. J Allergy Clin Immunol (2015) 135:1031–43.e6. doi:10.1016/j.jaci.2014.07.057
37. Guerrini M, Beccati D, Shriver Z, Naggi A, Viswanathan K, Bisio A, et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat Biotechnol (2008) 26:669–75. doi:10.1038/nbt1407
38. Kishimoto TK, Viswanathan K, Ganguly T, Elankumaran S, Smith S, Pelzer K, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med (2008) 358:2457–67. doi:10.1056/NEJMoa0803200
39. Siebeck M, Cheronis JC, Fink E, Kohl J, Spies B, Spannagl M, et al. Dextran sulfate activates contact system and mediates arterial hypotension via B2 kinin receptors. J Appl Physiol (1994) 77:2675–80.
40. McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature (2015) 519:237–41. doi:10.1038/nature14022
41. Worm M, Edenharter G, Ruëff F, Scherer K, Pföhler C, Mahler V, et al. Symptom profile and risk factors of anaphylaxis in Central Europe. Allergy (2012) 67:691–8. doi:10.1111/j.1398-9995.2012.02795.x
42. Niggemann B, Beyer K. Factors augmenting allergic reactions. Allergy (2014) 69:1582–7. doi:10.1111/all.12532
43. Webb LM, Lieberman P. Anaphylaxis: a review of 601 cases. Ann Allergy Asthma Immunol (2006) 97(1):39–43. doi:10.1016/S1081-1206(10)61367-1
44. Liew WK, Williamson E, Tang MLK. Anaphylaxis fatalities and admissions in Australia. J Allergy Clin Immunol (2009) 123:434–42. doi:10.1016/j.jaci.2008.10.049
45. Pereira Vega A, Sanchez Ramos JL, Maldonado Perez JA, Alvarez Gutierrez FJ, Ignacio Garcia JM, Vazquez Oliva R, et al. Variability in the prevalence of premenstrual asthma. Eur Respir J (2010) 35:980–6. doi:10.1183/09031936.00045109
46. Vasconcelos C, Xavier P, Vieira AP, Martinho M, Rodrigues J, Bodas A, et al. Autoimmune progesterone urticaria. Gynecol Endocrinol (2000) 14:245–7. doi:10.3109/09513590009167688
47. Bauer CS, Kampitak T, Messieh ML, Kelly KJ, Vadas P. Heterogeneity in presentation and treatment of catamenial anaphylaxis. Ann Allergy Asthma Immunol (2013) 111:107–11. doi:10.1016/j.anai.2013.06.001
48. Hox V, Desai A, Bandara G, Gilfillan AM, Metcalfe DD, Olivera A. Estrogen increases the severity of anaphylaxis in female mice through enhanced endothelial nitric oxide synthase expression and nitric oxide production. J Allergy Clin Immunol (2015) 135:729–36.e5. doi:10.1016/j.jaci.2014.11.003
49. Versluis A, van Os-Medendorp H, Kruizinga AG, Blom WM, Houben GF, Knulst AC. Cofactors in allergic reactions to food: physical exercise and alcohol are the most important. Immun Inflamm Dis (2016) 4:392–400. doi:10.1002/iid3.120
50. Morita E, Kunie K, Matsuo H. Food-dependent exercise-induced anaphylaxis. J Dermatol Sci (2007) 47:109–17. doi:10.1016/j.jdermsci.2007.03.004
51. Palosuo K, Varjonen E, Nurkkala J, Kalkkinen N, Harvima R, Reunala T, et al. Transglutaminase-mediated cross-linking of a peptic fraction of ω-5 gliadin enhances IgE reactivity in wheat-dependent, exercise-induced anaphylaxis. J Allergy Clin Immunol (2003) 111:1386–92. doi:10.1067/mai.2003.1498
52. Lambert GP, Broussard LJ, Mason BL, Mauermann WJ, Gisolfi CV. Gastrointestinal permeability during exercise: effects of aspirin and energy-containing beverages. J Appl Physiol (2001) 90(6):2075–80.
53. Matsuo H, Morimoto K, Akaki T, Kaneko S, Kusatake K, Kuroda T, et al. Exercise and aspirin increase levels of circulating gliadin peptides in patients with wheat-dependent exercise-induced anaphylaxis. Clin Exp Allergy (2005) 35:461–6. doi:10.1111/j.1365-2222.2005.02213.x
54. Sakamoto Y, Ohtsuka T, Yoshida H, Ohto K, Onobori M, Matsumoto T, et al. Time course of changes in the intestinal permeability of food-sensitized rats after oral allergen challenge. Pediatr Allergy Immunol (1998) 9:20–4. doi:10.1111/j.1399-3038.1998.tb00295.x
55. Yano H, Kato Y, Matsuda T. Acute exercise induces gastrointestinal leakage of allergen in lysozyme-sensitized mice. Eur J Appl Physiol (2002) 87:358–64. doi:10.1007/s00421-002-0653-x
56. Phillips TLE, Phillips TLE, Neutra MR. Macromolecules can pass through occluding junctions of rat ileal epithelium during cholinergic stimulation. Cell Tissue Res (1987) 247:547–54. doi:10.1007/BF00215748
57. Zuhl M, Schneider S, Lanphere K, Conn C, Dokladny K, Moseley P. Exercise regulation of intestinal tight junction proteins. Br J Sports Med (2014) 48:980–6. doi:10.1136/bjsports-2012-091585
58. KhKhamnei S, Alipour MR, Ahmadiasl N. The combined effects of exercise and post dehydration water drinking on plasma argenine vasopressin, plasma osmolality and body temperature in healthy males. Int J Endocrinol Metab (2005) 2:80–6.
59. Torres-Atencio I, Ainsua-Enrich E, de Mora F, Picado C, Martín M. Prostaglandin E2 prevents hyperosmolar-induced human mast cell activation through prostanoid receptors EP2 and EP4. PLoS One (2014) 9:e110870. doi:10.1371/journal.pone.0110870
60. Eggleston PA, Kagey-Sobotka A, Schleimer RP, Lichtenstein LM. Interaction between hyperosmolar and IgE-mediated histamine release from basophils and mast cells. Am Rev Respir Dis (1984) 130:86–91.
61. Caslake MJ, Packard CJ, Suckling KE, Holmes SD, Chamberlain P, Macphee CH. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase: a potential new risk factor for coronary artery disease. Atherosclerosis (2000) 150:413–9. doi:10.1016/S0021-9150(99)00406-2
62. Perelman B, Adil A, Vadas P. Relationship between platelet activating factor acetylhydrolase activity and apolipoprotein B levels in patients with peanut allergy. Allergy Asthma Clin Immunol (2014) 10:20. doi:10.1186/1710-1492-10-20
63. Moneret-Vautrin D-A, Latarche C. Drugs as risk factors of food anaphylaxis in adults: a case-control study. Bull Acad Natl Med (2009) 193(2):351–62; discussion 362–3.
64. Cardona V, Luengo O, Garriga T, Labrador-Horrillo M, Sala-Cunill A, Izquierdo A, et al. Co-factor-enhanced food allergy. Allergy (2012) 67:1316–8. doi:10.1111/j.1398-9995.2012.02877.x
65. Pascal M, Muñoz-Cano R, Reina Z, Palacín A, Vilella R, Picado C, et al. Lipid transfer protein syndrome: clinical pattern, cofactor effect and profile of molecular sensitization to plant-foods and pollens. Clin Exp Allergy (2012) 42:1529–39. doi:10.1111/j.1365-2222.2012.04071.x
66. Wallace JL, de Nucci G, Sulaieva O. Toward more GI-friendly anti-inflammatory medications. Curr Treat Options Gastroenterol (2015) 13:377–85. doi:10.1007/s11938-015-0064-9
67. Bjarnason I, Takeuchi K. Intestinal permeability in the pathogenesis of NSAID-induced enteropathy. J Gastroenterol (2009) 44:23–9. doi:10.1007/s00535-008-2266-6
68. Matsukura S, Aihara M, Sugawara M, Kunimi Y, Matsuki M, Inoue Y, et al. Two cases of wheat-dependent anaphylaxis induced by aspirin administration but not by exercise. Clin Exp Dermatol (2010) 35:233–7. doi:10.1111/j.1365-2230.2009.03709.x
69. Aihara M, Miyazawa M, Osuna H, Tsubaki K, Ikebe T, Aihara Y, et al. Food-dependent exercise-induced anaphylaxis: influence of concurrent aspirin administration on skin testing and provocation. Br J Dermatol (2002) 146:466–72. doi:10.1046/j.1365-2133.2002.04601.x
70. Pascal M, Muñoz-Cano R, Milà J, Sanz ML, Diaz-Perales A, Sánchez-López J, et al. Nonsteroidal anti-inflammatory drugs enhance IgE-mediated activation of human basophils in patients with food anaphylaxis dependent on and independent of nonsteroidal anti-inflammatory drugs. Clin Exp Allergy (2016) 46:1111–9. doi:10.1111/cea.12735
71. Wojnar RJ, Hearn T, Starkweather S. Augmentation of allergic histamine release from human leukocytes by nonsteroidal anti-inflammatory-analgesic agents. J Allergy Clin Immunol (1980) 66:37–45. doi:10.1016/0091-6749(80)90136-0
72. Kowalski ML, Asero R, Bavbek S, Blanca M, Blanca-Lopez N, Bochenek G, et al. Classification and practical approach to the diagnosis and management of hypersensitivity to nonsteroidal anti-inflammatory drugs. Allergy (2013) 68:1219–32. doi:10.1111/all.12260
73. Knights KM, Mangoni AA, Miners JO. Defining the COX inhibitor selectivity of NSAIDs: implications for understanding toxicity. Expert Rev Clin Pharmacol (2010) 3:769–76. doi:10.1586/ecp.10.120
74. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev (2004) 56:387–437. doi:10.1124/pr.56.3.3
75. Takahashi A, Nakajima K, Ikeda M, Sano S, Takahashi A, Nakajima K, et al. Pre-treatment with misoprostol prevents food-dependent exercise-induced anaphylaxis (FDEIA). Int J Dermatol (2011) 50:233–40. doi:10.1111/j.1365-4632.2010.04314.x
76. Kim SH, Nam EJ, Kim YK, Ye YM, Park HS. Functional variability of the adenosine A3 receptor (ADORA3) gene polymorphism in aspirin-induced urticaria. Br J Dermatol (2010) 163:977–85. doi:10.1111/j.1365-2133.2010.09983.x
77. Kim SH, Kim YK, Park HW, Ye YM, Min KU, Park HS. Adenosine deaminase and adenosine receptor polymorphisms in aspirin-intolerant asthma. Respir Med (2009) 103:356–63. doi:10.1016/j.rmed.2008.10.008
78. Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest (1997) 100:2849–57. doi:10.1172/JCI119833
79. Leung CT, Li A, Banerjee J, Gao Z-GG, Kambayashi T, Jacobson KA, et al. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal (2014) 10:1–11. doi:10.1007/s11302-014-9409-4
80. Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem (1993) 268:16887–90.
81. Miyahara JT, Karler R. Effect of salicylate on oxidative phosphorylation and respiration of mitochondrial fragments. Biochem J (1965) 97:194–8. doi:10.1042/bj0970194
82. Cronstein BN, Vandestouwe M, Druska L, Levin RI, Weissmann G. Nonsteroidal antiinflammatory agents inhibit stimulated neutrophil adhesion to endothelium – adenosine-dependent and independent mechanisms. Inflammation (1994) 18:323–35. doi:10.1007/BF01534273
83. Jacobs RL, Rake GW, Fournier DC, Chilton RJ, Culver WG, Beckmann CH. Potentiated anaphylaxis in patients with drug-induced beta-adrenergic blockade. J Allergy Clin Immunol (1981) 68:125–7. doi:10.1016/0091-6749(81)90170-6
84. Ruëff F, Przybilla B, Biló MB, Müller U, Scheipl F, Aberer W, et al. Predictors of severe systemic anaphylactic reactions in patients with Hymenoptera venom allergy: importance of baseline serum tryptase – a study of the European Academy of Allergology and Clinical Immunology Interest Group on insect venom hypersensitivity. J Allergy Clin Immunol (2009) 124(5):1047–54. doi:10.1016/j.jaci.2009.08.027
85. Brown SGA. Clinical features and severity grading of anaphylaxis. J Allergy Clin Immunol (2004) 114:371–6. doi:10.1016/j.jaci.2004.04.029
86. Nassiri M, Babina M, Dölle S, Edenharter G, Ruëff F, Worm M. Ramipril and metoprolol intake aggravate human and murine anaphylaxis: evidence for direct mast cell priming. J Allergy Clin Immunol (2015) 135(2):491–9. doi:10.1016/j.jaci.2014.09.004
87. Zogaj D, Ibranji A, Hoxha M. Exercise-induced anaphylaxis: the role of cofactors. Mater Sociomed (2014) 26:401–4. doi:10.5455/msm.2014.26.401-404
88. Wang Y, Tong J, Chang B, Wang B, Zhang D, Wang B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep (2014) 9:2352–6. doi:10.3892/mmr.2014.2126
89. Nagy LE, Diamond I, Casso DJ, Franklin C, Gordon AS. Ethanol increases extracellular adenosine by inhibiting adenosine uptake via the nucleoside transporter. J Biol Chem (1990) 265:1946–51.
90. Alonso M, Gomez-Rial J, Gude F, Vidal C, Gonzalez-Quintela A. Influence of experimental alcohol administration on serum immunoglobulin levels: contrasting effects on IgE and other immunoglobulin classes. Int J Immunopathol Pharmacol (2012) 25:645–55. doi:10.1177/039463201202500311
Keywords: adenosine, anaphylaxis, cofactor, exercise, IgE, IgG, mast cell, non-steroidal anti-inflammatory drug
Citation: Muñoz-Cano R, Pascal M, Araujo G, Goikoetxea MJ, Valero AL, Picado C and Bartra J (2017) Mechanisms, Cofactors, and Augmenting Factors Involved in Anaphylaxis. Front. Immunol. 8:1193. doi: 10.3389/fimmu.2017.01193
Received: 01 August 2017; Accepted: 08 September 2017;
Published: 26 September 2017
Edited by:
Carlos Pastor Vargas, Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz, SpainReviewed by:
Coen Maas, University Medical Center Utrecht, NetherlandsDavid Dombrowicz, Institut national de la santé et de la recherche médicale, France
Copyright: © 2017 Muñoz-Cano, Pascal, Araujo, Goikoetxea, Valero, Picado and Bartra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Muñoz-Cano, cm11bm96QGNsaW5pYy5jYXQ=