 Aline Cristina Abreu Moreira-Souza1,2†
Aline Cristina Abreu Moreira-Souza1,2† Cássio Luiz Coutinho Almeida-da-Silva1†
Cássio Luiz Coutinho Almeida-da-Silva1† Thuany Prado Rangel1,2
Thuany Prado Rangel1,2 Gabrielle da Costa Rocha1
Gabrielle da Costa Rocha1 Maria Bellio3
Maria Bellio3 Dario Simões Zamboni4
Dario Simões Zamboni4 Rossiane Claudia Vommaro2
Rossiane Claudia Vommaro2 Robson Coutinho-Silva1*
Robson Coutinho-Silva1*
- 1Immunobiology Program, Biophysics Institute Carlos Chagas Filho, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 2Parasitology and Cell Biology Program, Biophysics Institute Carlos Chagas Filho, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 3Department of Immunology, Institute of Microbiology Paulo de Goes, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 4Department of Cell Biology, Ribeirão Preto Medical School, University of São Paulo, São Paulo, Brazil
Toxoplasma gondii (T. gondii) is the protozoan parasite that causes toxoplasmosis, a potentially fatal disease to immunocompromised patients, and which affects approximately 30% of the world’s population. Previously, we showed that purinergic signaling via the P2X7 receptor contributes to T. gondii elimination in macrophages, through reactive oxygen species (ROS) production and lysosome fusion with the parasitophorous vacuole. Moreover, we demonstrated that P2X7 receptor activation promotes the production of anti-parasitic pro-inflammatory cytokines during early T. gondii infection in vivo. However, the cascade of signaling events that leads to parasite elimination via P2X7 receptor activation remained to be elucidated. Here, we investigated the cellular pathways involved in T. gondii elimination triggered by P2X7 receptor signaling, during early infection in macrophages. We focused on the potential role of the inflammasome, a protein complex that can be co-activated by the P2X7 receptor, and which is involved in the host immune defense against T. gondii infection. Using peritoneal and bone marrow-derived macrophages from knockout mice deficient for inflammasome components (NLRP3−/−, Caspase-1/11−/−, Caspase-11−/−), we show that the control of T. gondii infection via P2X7 receptor activation by extracellular ATP (eATP) depends on the canonical inflammasome effector caspase-1, but not on caspase-11 (a non-canonical inflammasome effector). Parasite elimination via P2X7 receptor and inflammasome activation was also dependent on ROS generation and pannexin-1 channel. Treatment with eATP increased IL-1β secretion from infected macrophages, and this effect was dependent on the canonical NLRP3 inflammasome. Finally, treatment with recombinant IL-1β promoted parasite elimination via mitochondrial ROS generation (as assessed using Mito-TEMPO). Together, our results support a model where P2X7 receptor activation by eATP inhibits T. gondii growth in macrophages by triggering NADPH-oxidase-dependent ROS production, and also by activating a canonical NLRP3 inflammasome, which increases IL-1β production (via caspase-1 activity), leading to mitochondrial ROS generation.
Introduction
Toxoplasma gondii is a parasitic protozoan from the phylum Apicomplexa and the etiologic agent of toxoplasmosis, an important disease that affects 30% of the world’s population (1, 2). Toxoplasmosis can be transmitted by organ transplantation, blood transfusion or congenital infection, due to the parasite’s ability to cross biological barriers (3, 4). In most cases, T. gondii causes asymptomatic disease, but toxoplasmosis can be fatal in immunocompromised patients, such as those with HIV/AIDS, cancer, and organ transplants (2). Also, severe ocular disease and multiple organ failure in immunocompetent individuals have been observed in endemic countries, due to the circulation of atypical parasite strains (5, 6). The current therapy for toxoplasmosis is based on drug combinations such as sulfadiazine/pyrimethamine, which reduce parasite replication, controlling host damage and symptoms; however, combination therapy causes important side effects and does not eliminate the resistant T. gondii cysts (7, 8).
T. gondii is an obligate intracellular parasite, able to infect virtually all nucleated cell types in a wide variety of hosts (2). It has been proposed that T. gondii utilizes innate immune cells, like macrophages and dendritic cells, to migrate to preferential infection sites, such as the immune “shielded” environment of the central nervous system, establishing lifelong chronic infection (9, 10). The parasite survives inside host cells by overcoming many host antimicrobial mechanisms, including reactive oxygen species (ROS) and nitric oxide (NO) production, lysosome fusion (to the parasitophorous vacuole), host cell death induction, and the secretion of pro-inflammatory cytokines and chemokines (11). Concomitantly, T. gondii triggers the activation of host cell anti-inflammatory transcription factors, inducing the over expression of receptors involved in host cell migration to the brain, which characterizes the chronic phase of toxoplasmosis (12). The ability to subvert immune system mechanisms and the fast establishment of the latent (chronic) phase of the disease directly jeopardize treatment for toxoplasmosis.
Inflammasomes are multimeric protein complexes that produce active caspase-1 by cleavage of procaspase-1 (13), and whose activation triggers maturation of IL-1β/IL-18 and pyroptosis, a type of inflammatory cell death (14). The inflammasome NLRP3 are formed by nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins, an apoptosis-associated spec-like protein containing a caspase-recruitment domain (ASC) protein adaptor and an inactive zymogen, procaspase-1 (Casp-1) (14). In mice, activation of the NLRP3 inflammasome requires two signals. First, recognition of a pathogen-associated molecular pattern by cell surface molecules known as pattern recognition receptors (PRRs) leads to the transcription of pro-inflammatory cytokines (15). Then, activation of the cytosolic PRRs results in the oligomerization of inflammasome components and procaspase-1 cleavage into active caspase-1 (13). The active caspase-1 subsequently cleaves the pro-inflammatory IL-1-family cytokines into their bioactive forms IL-1β/IL-18 and induces pyroptosis (14). Inflammasomes involved in caspase-1 activation are known as canonical inflammasomes, while those related to caspase-11 (in mice) or caspase-4/5 (in humans) are known as non-canonical inflammasomes (16, 17). Activation of the canonical NLRP3 inflammasome is trigged by cytosolic stress conditions such as K+ efflux, leakage of lysosome components, mitochondrial damage, and ROS production (14, 18). Interestingly, the P2X7 nucleotide receptor activation involved in the elimination of different intracellular parasites, such as Leishmania amazonensis (19), Porphyromonas gingivalis (20), Mycobacterium tuberculosis (21–24), and T. gondii (25–27), induces most of these stress conditions and, thus, is capable of activating the NLRP3 inflammasome (28–30). Polymorphisms in the human P2X7 receptor are directly associated with host susceptibility to congenital or acquired toxoplasmosis, in immunocompetent patients (25, 31). Also, P2X7 absence or disruption of P2X7 receptor function increases toxoplasmosis severity in murine infection with virulent (RH, type I) or non-virulent (Me-49, type II) strains of T. gondii (27, 32).
Our group demonstrated that P2X7 receptor activation by extracellular ATP (eATP) in T. gondii-infected macrophages contributes to parasite elimination by ROS production and lysosome fusion with the parasitophorous vacuole (26). We also showed that the P2X7 receptor is an important activator of the anti-parasitic pro-inflammatory response that occurs in early T. gondii infection in vivo (32). However, the cascade of intracellular events trigged by purinergic signaling—which results in parasite control in early infection—remains to be elucidated.
In this work, we used macrophages from knockout mice lacking inflammasome components to examine the hypothesis that the inflammasome is involved in T. gondii infection control via eATP and the P2X7 receptor. We also studied the roles of ROS production and IL-1β secretion induced by P2X7 receptor in infected cell. Our combined data provide an overall model of the cellular pathways involved in T. gondii infection control mediated by the P2X7 receptor, in murine macrophages.
Materials and Methods
Reagents
Adenosine-5’-triphosphate (ATP), N-acetyl cysteine (NAC), penicillin, streptomycin, HEPES, Mito-TEMPO, apocynin, carbenoxolone, paraformaldehyde, and bovine serum albumin were purchased from Sigma Aldrich (USA). Fetal bovine serum (FBS) was from Gibco/Life Technologies (USA). Z-YVAD and recombinant IL-1β were from R&D Systems (USA).
Mice
The following mouse strains were used in this work: Swiss CF1, C57BL/6 (wild-type strain), P2X7−/− (originally from the Jackson Laboratory), Caspase-11−/−, Caspase1/11−/− (Genentech, USA), and NLRP3−/− mice generated on the C57BL/6 background (8–12 weeks old) were obtained from the Isogenic Breeding Unit at Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil. Mice were maintained at the Animal House for Transgenic Mice of the Federal University of Rio de Janeiro (UFRJ), at 22°C in a 12-h light/dark cycle. Mice aged between 8 and 12 weeks were used in all experiments. The procedures for the care and use of animals were according to the guidelines of the Brazilian College of Animal Experimentation (COBEA). All efforts were made to minimize animal suffering and to reduce the number of animals used in this study. This study was approved and followed all the guidelines established by the Ethics Committee on the Use of Animals (CEUA) of the Biophysic Institute Carlos Chagas Filho (IBCCF, UFRJ, no. 082/15).
Parasites
T. gondii tachyzoites from the RH strain were maintained in Swiss CF-1 mice as previously described in Ref. (33). Briefly, 5-week-old mice were infected intraperitoneally with 106 tachyzoites. After 72 h of infection, parasites were harvested from the peritoneal cavity by PBS washing and then collected by centrifugation at 1,000 × g for 10 min. Harvested parasites were counted in a hemocytometer for experimental infections or for the next passage in mice.
Bone Marrow-Derived Macrophages (BMDMs) and Peritoneal Macrophages
Bone marrow-derived macrophages were generated using L929 cell conditioned media (LCCM) as a source of macrophage colony-stimulating factor, as previously described (34, 35). RPMI containing 100 U/mL penicillin, 100 µg/mL streptomycin, and 1 mM sodium pyruvate was used throughout the procedure. In brief, fresh bone marrow from C57BL/6, NLRP3−/−, P2X7−/−, Caspase-11−/−, and Caspase-1/11−/− mice were obtained and 5 × 106 cells were plated in the petri dishes in RPMI with 20% FBS and 30% LCCM. On day 3 after plating, fresh medium was added to the cultures. On day 7 after plating, cells were harvested, washed with PBS, counted and plated in RPMI with 10% FBS and 5% LCCM, for cytokine detection assays (in 6-well plates, at 2 × 106 cells/well) or light microscopy analysis (in 24-well plates with 13 mm coverslips, at 2 × 105 cells/well). Cells were then incubated at 37°C for at least 18 h before the start of experiments. All experimental infections and treatments were performed in RPMI with 10% FBS (but without LCCM). Macrophage differentiation to >95% purity was confirmed by flow cytometry as described previously (35), before the start of each experiment.
Peritoneal macrophages were collected as previously described (33). Briefly, peritoneal exudates from C57BL/6 mice were obtained by washing the peritoneal cavity with 10 mL of sterile PBS. Peritoneal cells were counted by light microscopy, resuspended in DMEM medium, and 2 × 105 cells were plated in 24-well plates with 13-mm coverslips for light microscopy analyzes. Cells were left to adhere for 1 h, at 37°C (and 5% CO2), and then non-adherent cells were removed by PBS washes before “supplemented DMEM” (DMEM with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, and 10 mM HEPES) was added to cultures.
Infection Assays
Cells were infected in supplemented DMEM medium, at 37°C (and 5% CO2). BMDMs or peritoneal macrophages were infected at a ratio of 3:1 tachyzoites to host cells for 2 h at 37°C (and 5% CO2), and non-internalized parasites were removed by PBS washes. Where required, cells were then incubated for 30 min in medium containing 3 mM eATP and/or 1 ng/mL IL-1β. In some experiments, cells were pretreated for 40 min with 10 mM NAC, 2 µM Z-YVAD, 1 µM Apocynin, 50 µM carbenoxolone, or 10 nM Mito-TEMPO, before ATP or IL-1β treatment. Then, cells were maintained at 37°C (and 5% CO2). After 18 h post-infection, aliquots of culture supernatants were collected and kept at −20°C, for IL-1β quantification (see “ELISA”) and cultures were processed for light microscopy, as described below.
Light Microscopy
Infected cells were fixed in 4% paraformaldehyde and stained with Panoptic (“Panotico Rapido kit,” LaborClin, Brazil) following the manufacturer’s instructions. A minimum of 300 cells/sample were evaluated by light microscopy, in a Zeiss microscope (manufacturer). As previously described (33), the percentage of infection and infection index, which represents the overall infection load, were determined using the formula:
where iC is number of infected cells; totalC is the total number of cells; and IntP is the number of intracellular parasites.
Cytokine Assay—ELISA
IL-1β levels were quantified in supernatant samples from infected BMDM cultures (see Infection Assays), using the Mouse IL-1 beta/IL-1F2 DuoSet ELISA #DY401 kit, according to the manufacturer’s instructions (R&D Systems).
ROS Assay
To estimate ROS production, cells were plated (at a density of 2 × 105 cells/well) in opaque-black 96-well plates with a transparent and flat bottom. After 2 h of infection with T. gondii, cells were kept untreated or were subjected to one or both of the following treatments, in the presence of 2 µM of H2DCFDA: 10 mM NAC for 30 min, followed by 3 mM ATP for up to 60 min. Fluorescence was measured at 40, 50, and 60 min of exposure to ATP, using a Spectra Max3 spectrophotometer (Molecular Devices), at 37°C, at excitation and emission wavelengths of 490 and 520 nm, respectively.
Statistical Analyses
A two-tailed t-test was used for comparisons of two groups, while multiple comparisons were performed by one-way ANOVA followed by Tukey’s post-test. All statistical analyses were performed using Graph Pad Prism 5 (La Jolla, CA, USA).
Results
P2X7 Receptor Activation Promotes T. gondii Control in Macrophages, via Inflammasome Activation, Caspase-1 and ROS Production
In a previous study, our group demonstrated that P2X7 receptor activation via eATP reduces T. gondii infection burden in peritoneal macrophages and J774.G8 cells (26). However, the mechanism of P2X7-induced T. gondii infection control in these cells remained unclear. On the other hand, eATP is capable of activating the inflammasome in infected cells (30) and promotes pathogen elimination in different models of infection (36). Therefore, we investigated whether T. gondii elimination in infected cells via eATP treatment was dependent on inflammasome activation.
To address this question, we infected BMDMs from wild-type C57BL/6 mice, as well as from NLRP3−/− and Caspase1/11−/− mice with T. gondii, and treated infected cells with eATP (3 mM), which activates the P2X7 receptor (26). Light microscopy-based quantification of the percentage of infected cells and the infection index (which combines the proportion of infected cells and the number of parasites per cell) showed that treatment with eATP decreased the parasite load in wild-type macrophages, while the parasite load remained high in cells from NLRP3−/− and Caspase1/11−/− mice (Figure 1). These data suggest that, in macrophages, the control of T. gondii infection mediated by eATP depends on the assembly of the NLRP3 inflammasome.
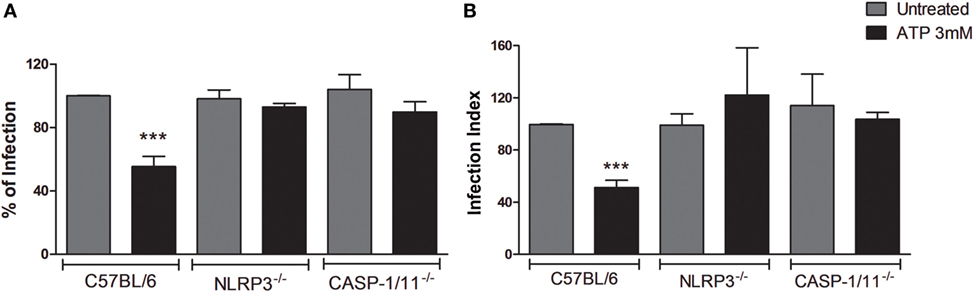
Figure 1. Extracellular ATP (eATP)-induced T. gondii control is dependent on NLRP3 and caspase-1/11 molecules. Bone marrow-derived macrophages from C57BL/6 (WT), NLRP3−/− or Caspase-1/11−/− mice were infected with T. gondii tachyzoites for 2 h and kept untreated, or were treated with 3 mM eATP for 30 min. Then, cells were incubated at 37°C for a total of 18 h, and stained with Panoptic, for light microscopy quantification of the percentage of infection (A) and the infection index (B). Treatment with eATP reduced the percentage of infected cells and the infection index in wild-type (C57BL/6) macrophages, but not in the knockout cells. Normalized data represent mean ± SEM of three independent experiments performed in triplicates. ***p ≤ 0.001 vs. untreated (by two-tailed t-test).
We demonstrated previously that P2X7 receptor activation reduces T. gondii load in macrophages, while also generating ROS (26). Given that, in macrophages, P2X7 receptor activation is associated with activation of the NLRP3 inflammasome (15), we sought to determine if ROS and caspase-1—the hallmark of the canonical NLRP3 inflammasome—are required for the reduction of T. gondii burden in BMDMs, via P2X7 receptor signaling.
To address this issue, we infected BMDMs from C57BL/6 (WT) mice with T. gondii tachyzoites followed by pretreatment with NAC or Z-YVAD (which inhibit ROS and caspase-1 activities, respectively) and then treated infected cells with eATP. We confirmed our previous observations that eATP reduced the percentage of infected cells (Figure 2A) and the infection index (Figure 2B), in wild-type macrophage cultures. This effect was abolished by treatment with either NAC or Z-YVAD (Figure 2). Thus, the reduction in T. gondii infection load via eATP and the P2X7 receptor depends on activation of the canonical NLRP3 inflammasome, and also on ROS production and caspase-1.
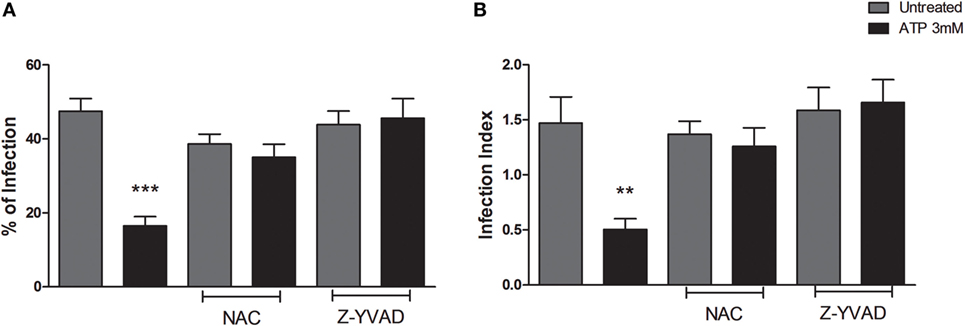
Figure 2. The decrease in T. gondii infection upon extracellular ATP (eATP) treatment depends on reactive oxygen species (ROS) generation and caspase-1 activation. Wild-type (C57BL/6) bone marrow-derived macrophages were infected with T. gondii tachyzoites for 2 h and then left untreated, or subjected to one of the following treatments: 10 mM N-acetyl cysteine (NAC) or 2 µM Z-YVAD for 30 min followed by 3 mM eATP, for 30 min. After treatments, cells were incubated for a further 18 h at 37°C, and stained with Panoptic, for light microscopy quantification of the percentage of infection (A) and the infection index (B). Treatment with NAD or Z-YVAD before exposure to eATP prevented the eATP-dependent reduction in T. gondii infection. Data represent mean ± SEM of three independent experiments performed in triplicates. **p ≤ 0.01 and ***p ≤ 0.001 vs. untreated (by one-way ANOVA followed by Tukey’s test).
A Canonical, but Not a Non-Canonical, Inflammasome Mediates T. gondii Elimination via P2X7 Receptor Signaling
Given that P2X7 receptor activation reduces T. gondii infection load in a caspase1/11-dependent manner (Figures 1 and 2), we investigated whether canonical and/or non-canonical inflammasome were involved in parasite elimination. To differentiate between canonical and non-canonical inflammasome contributions, we quantified the parasite load after eATP treatment in BMDMs from C57BL/6 (wild-type), Caspase11−/−, and Caspase1/11−/− mice. Treatment with eATP reduced the parasite load in wild-type and Caspase-11−/− macrophages, while no reduction in parasite load was observed after nucleotide treatment in Caspase1/11−/−cells (Figure 3). Thus caspase-1, but not caspase-11, is required for eATP-mediated T. gondii infection control. Combined with the data shown in Figures 1 and 2, these results confirm that the activation of the canonical NLRP3 inflammasome is crucial for the control of T. gondii infection in macrophages via P2X7 receptor activation by eATP. These data also show that caspase-11 is dispensable for pathogen elimination via eATP, while caspase-1 is required for this effect, defining a role for the canonical inflammasome only (and not for the non-canonical inflammasome) in T. gondii infection control mediated by purinergic signaling.
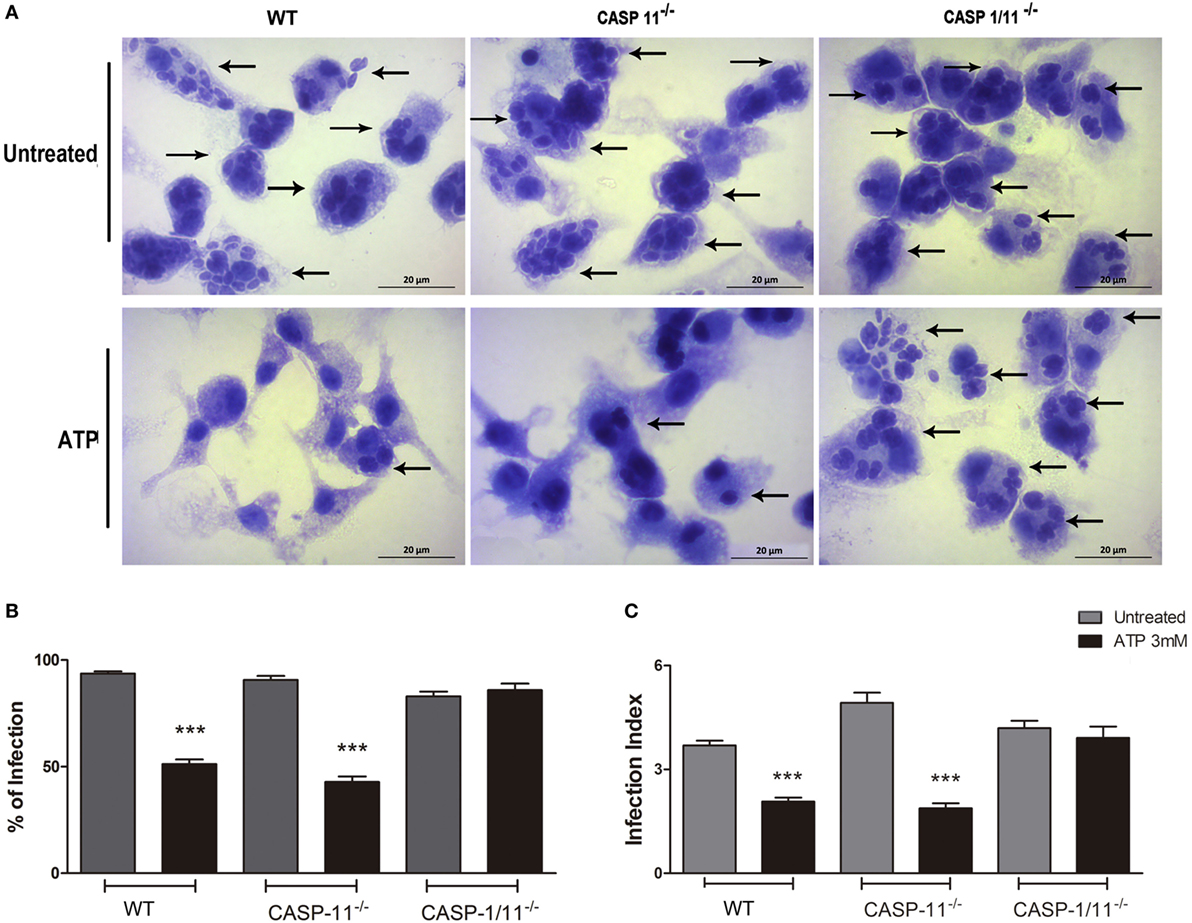
Figure 3. P2X7 receptor activation triggers T. gondii elimination via a canonical (but not via a non-canonical) inflammasome. Bone marrow-derived macrophages (BMDMs) from C57BL/6 (wild-type), and from Caspase-11−/− and Caspase1/11−/−knockout mice (lacking non-canonical and canonical inflammasome effectors, respectively) were infected with T. gondii tachyzoites for 2 h and then kept untreated, or were treated 3 mM extracellular ATP for 30 min. Then, cells were incubated at 37°C for a total of 18 h, and stained with Panoptic, for light microscopy observation [(A); representative images of three independent experiments] and for the quantification of the % of infection (B) and the infection index (C). We observed reduced parasite load in wild-type and Caspase-11−/− BMDMs, but not in Caspase-1/11−/− cells. Black arrows indicate parasitophorous vacuoles containing T. gondii tachyzoites. Micrographs are representative of three independent experiments. Data represent mean ± SEM of three independent experiments performed in triplicates. ***p ≤ 0.001 vs. untreated (by two-tailed t-test).
eATP Induces NLRP3 Inflammasome-Dependent IL-1β Secretion and ROS Generation in Infected Macrophages
IL-1β is an important pro-inflammatory cytokine in response to different pathogens (37), and its secretion is induced by inflammasome activation (14). Therefore, we investigated whether activation of the canonical inflammasome via eATP/P2X7 receptor resulted in the secretion of IL-1β by T. gondii-infected cells.
T. gondii infection itself induced discrete (and similar) amounts of IL-1β release by WT (C57BL/6), NLRP3−/− and Caspase1/11−/− BMDMs (Figure 4A). When infected cells were treated with eATP, IL-1β secretion was potentiated in WT macrophages but did not increase in cells from NLRP3−/− or Caspase1/11−/− mice, showing that activation of the canonical NLRP3 inflammasome by eATP (via the P2X7 receptor) triggers IL-1β secretion in T. gondii-infected macrophages.
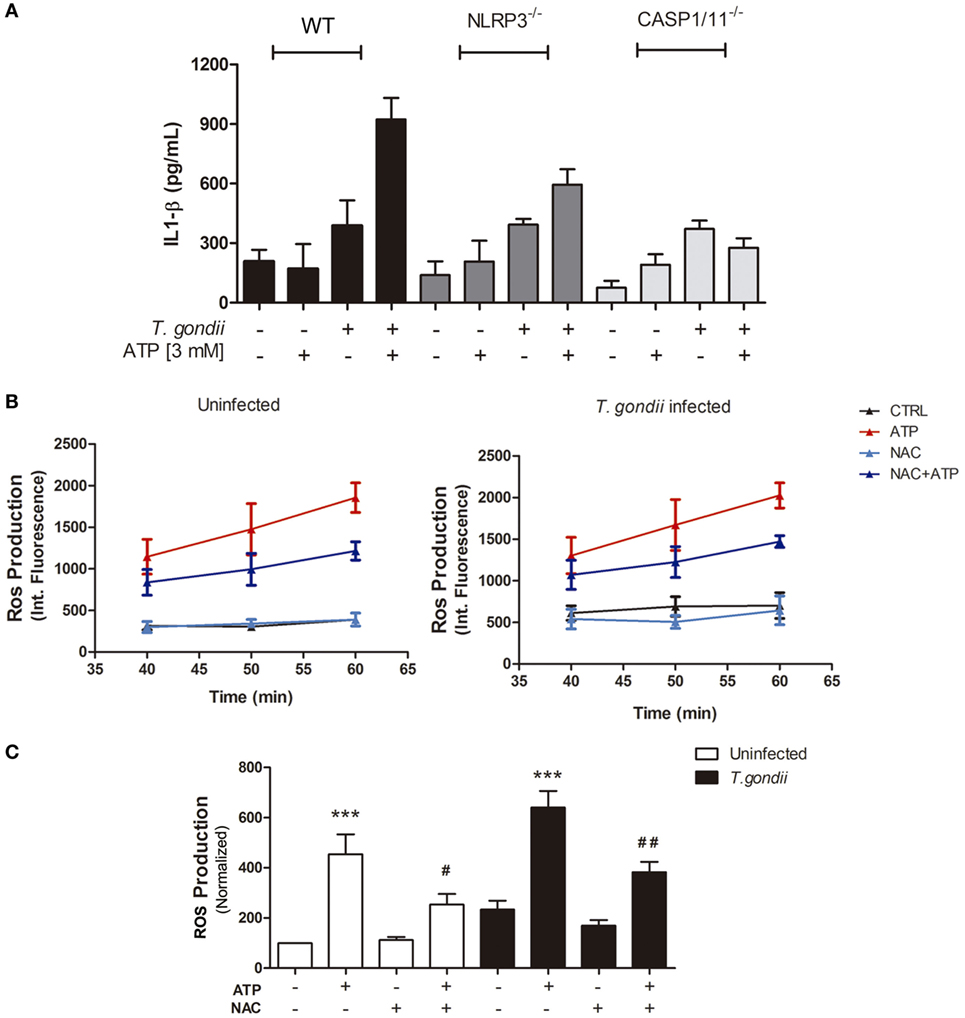
Figure 4. Extracellular ATP (eATP) is important for IL-1β secretion via canonical NLRP3 inflammasome induction and reactive oxygen species (ROS) production, in T. gondii-infected macrophages. Bone marrow-derived macrophages from C57BL/6 (WT), NLRP3−/− or Caspase-1/11−/− mice, either uninfected or infected with T. gondii tachyzoites, were kept untreated or were treated with ATP, and then culture supernatant samples were used to estimate IL-1β secretion (A). Treatment with 3 mM eATP (for 30 min) was performed 2 h post-infection, and all cells were incubated for 24 h prior to IL-1β quantification. Data represent mean ± SEM from triplicates. For ROS detection, before eATP treatment, some cultures were incubated with 10 mM N-acetyl cysteine (NAC) and ROS production was detected. (B,C) ROS levels in cells treated for 30 min, with NAC and then with 3 mM eATP (for up to 60 min), 2 h post-infection. ROS levels were estimated by H2DCFDA fluorescence, at different time-points during treatment with eATP (B), with normalized data from the 50-min time-point in (C). Data represent mean ± SEM of three independent experiments performed in triplicates. #p ≤ 0.05 and ##p ≤ 0.01 vs. corresponding samples treated with eATP only; ***p ≤ 0.001 vs. the corresponding untreated control (by one-way ANOVA followed by Tukey’s test).
ROS production is one of the most potent microbicidal mechanisms against intracellular pathogens, and T. gondii efficiently block ROS production in order to survive and evade host immune mechanisms (38). In a previous study, we showed that treatment with eATP induces ROS production in murine macrophages (26). To test if T. gondii infection subverts eATP-induced ROS generation, we quantified ROS levels in T. gondii-infected peritoneal macrophages at different time-points (40, 50, and 60 min), after eATP. Exposure to eATP increased ROS levels, from 40 to 60 min of treatment (Figure 4B), even in T. gondii-infected cells (Figure 4B). The ROS level increase triggered by eATP after 40 min of treatment was reduced significantly by exposure to the ROS production inhibitor NAC (Figure 4C). Together, these results show that eATP induced the production of key molecules for the control of intracellular parasite infection—ROS and IL-1β—even in the presence of T. gondii and its evasion mechanisms.
T. gondii Infection Control via eATP and IL-1β Depends on ROS Production and Pannexin-1
Thus far, we showed that eATP treatment inhibits T. gondii infection in macrophages by a mechanism dependent on the canonical NLRP3 inflammasome and on ROS production, and that NLRP3 inflammasome activation in cells treated with eATP leads to the secretion of IL-1β. To examine in more detail the relationships between P2X7 receptor activation (by eATP), IL-1β production and ROS generation, during T. gondii infection control by macrophages, we treated infected peritoneal macrophages with eATP or recombinant IL-1β, alone or in combination with the antioxidant NAC. As expected, treatment with eATP or recombinant IL-1β reduced T. gondii infection in wild-type macrophages (Figure 5A). Interestingly, the antioxidant NAC completely inhibited the control of T. gondii infection by eATP and IL-1β in peritoneal macrophages (Figures 5B,C). These results show that ROS production induced downstream of P2X7 receptor activation or IL-1β secretion is essential for the control of T. gondii infection mediated by purinergic signaling.
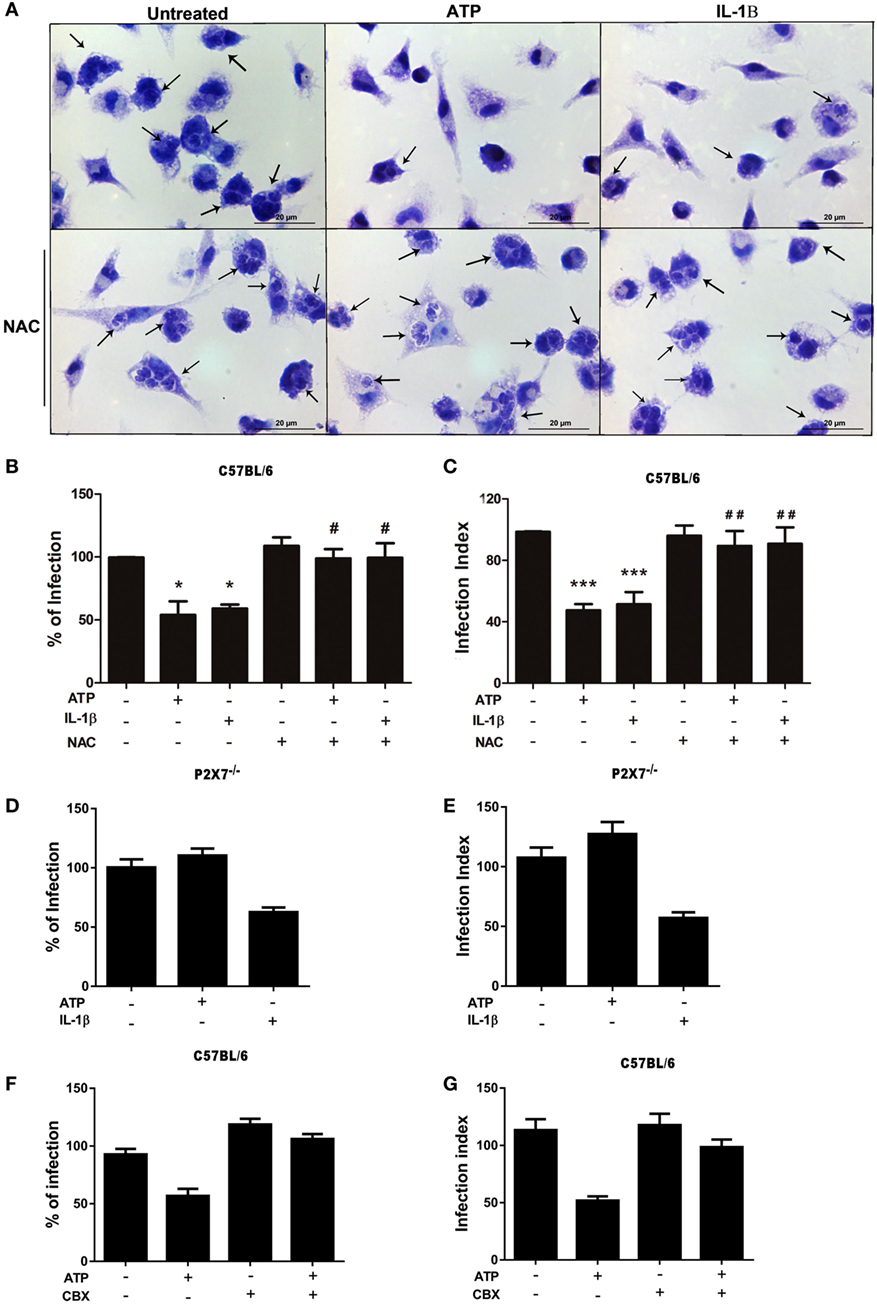
Figure 5. The control of T. gondii infection induced by extracellular ATP (eATP) and IL-1β requires the generation of reactive oxygen species and pannexin-1. Peritoneal macrophages from C57BL/6 (WT) and P2X7−/− mice were infected with T. gondii tachyzoites for 2 h, and then left untreated or subjected to one or both of the following treatments as indicated: 10 mM N-acetyl cysteine (NAC) or 50 µM carbenoxolone for 40 min, followed by 3 mM eATP or 1 ng/mL IL-1β for 30 min. Then, cells were incubated at 37°C for a further 18 h, and stained with Panoptic for light microscopy analysis (A) representative images of three independent experiments, which was used to quantify the percentage of infection (B,D,F) and the infection index (C,E,G) in C57BL/6 (B,C,F,G) and P2X7−/− (D,E) cells. In (A), black arrows indicate parasitophorous vacuoles containing T. gondii tachyzoites in C57BL/6. Normalized data represent mean ± SEM of three independent experiments [in (B–E)] and 2 independent experiments [in (F,G)] performed in triplicates. (F,G) are representative experiments. Treatment with eATP or IL-1β reduced the parasite load in C57BL/6 cells, and this effect was abolished by NAC pretreatment (B,C). Treatment with eATP had no effect in the parasite load in P2X7−/− cells (D,E) or C57BL/6 cells pretreated with carbenoxolone (F,G). #p ≤ 0.05 and ##p ≤ 0.01 vs. sample treated with ATP only; *p ≤ 0.05 and ***p ≤ 0.001 vs. untreated (by one-way ANOVA followed by Tukey’s test).
Importantly, treatment with recombinant IL-1β reduced T. gondii infection load even in macrophages lacking the P2X7 receptor (from P2X7−/−mice) (Figures 5D,E). These data indicate that IL-1β is downstream of P2X7 receptor activation in the signaling network that results in T. gondii infection control by macrophages. In addition, these experiments confirmed that the effects of eATP treatment described here depends on the P2X7 receptor, since eATP treatment did not reduce the percentage of infected cells (Figure 5D) or the infection index (Figure 5E) in P2X7−/− macrophages.
In order to reinforce the role of IL-1β promoting parasite killing, we assessed if the blockade of pannexin-1 channel, which is required for IL-1β secretion in macrophages (39) can inhibit eATP-induced killing of T. gondii. As shown in Figures 5F,G, carbenoxolone pretreatment abolished the eATP-induced control of the parasite load. These data confirm that eATP-induced IL-1β secretion from macrophages is crucial to T. gondii elimination.
T. gondii Elimination via eATP and IL-1β Depends on Distinct ROS Generation Sources
The precise role and origin of the ROS pool that acts as an anti-parasitic effector during P2X7 receptor activation remain poorly defined. Considering that our data support the notion that P2X7 receptor activation induces ROS generation via eATP treatment, we decided to examine which source of ROS pool is required for T. gondii infection control triggered by eATP, and whether this phenomenon is mediated by the canonical inflammasome effector IL-1β. To examine this question, we pretreated infected peritoneal macrophages with Mito-TEMPO, an inhibitor of mitochondrial ROS production, and then treated infected cells with eATP or recombinant IL-1β. Treatment with Mito-TEMPO effectively inhibited the control of T. gondii infection by IL-1β, while eATP treatment still reduced the parasite load even in the presence of Mito-TEMPO (Figures 6A,B). Then, we pretreated infected peritoneal macrophages with apocynin, an inhibitor of NADPH oxidase ROS production, and treated infected cells with eATP. Treatment with Apocynin completely blocked the eATP-induced reduction of parasite load (Figures 6C,D). Therefore, while both eATP and IL-1β reduce T. gondii infection load via ROS generation (Figure 5), the mitochondrial ROS pool is important only for the effector activity of IL-1β on T. gondii infection control. IL-1β-independent pathways of T. gondii elimination triggered by eATP are likely to depend on ROS production via NADPH oxidase.
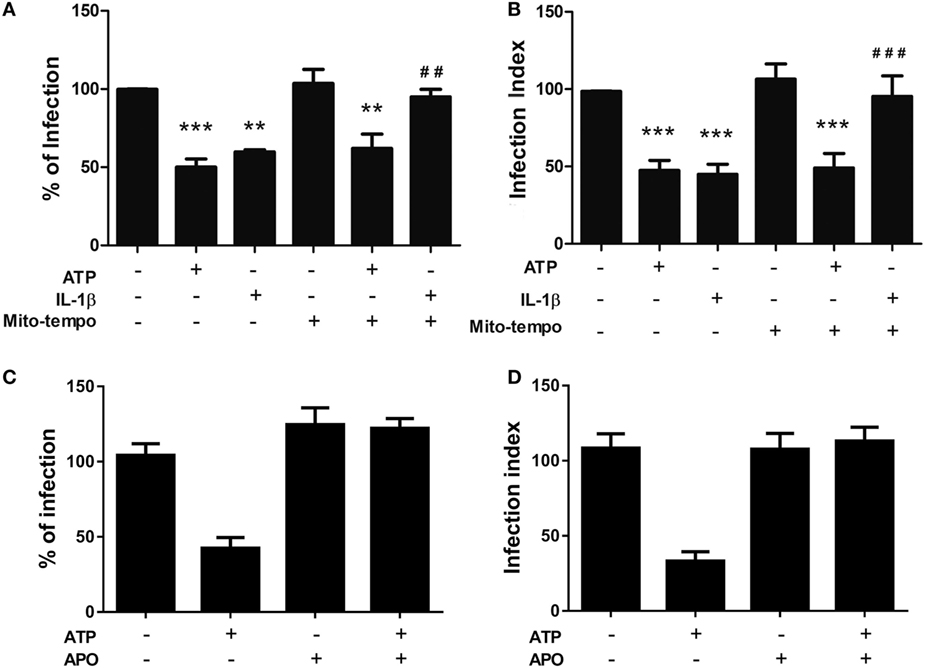
Figure 6. The control of T. gondii infection induced by extracellular ATP (eATP) and IL-1β depends on different sources of reactive oxygen species generation. Peritoneal macrophages from C57BL/6 mice were infected with T. gondii tachyzoites for 2 h and left untreated or subjected to one or more of the following treatments: 100 nM Mito-TEMPO or 1 µM apocynin for 40 min, followed by 3 mM eATP or 1 ng/mL IL-1β for 30 min. Then, cells were incubated at 37°C for a further 18 h, and the percentage of infected cells (A,C), and number of parasites per host cell (B,D) were estimated by light microscopy examination, after staining with Panoptic. Normalized data represent mean ± SEM of three independent experiments performed in triplicates in (A,B) and two independent experiments in (C,D). ##p ≤ 0.01 and ###p ≤ 0.001 vs. sample treated with ATP only; **p ≤ 0.01 and ***p ≤ 0.001 vs. untreated (by one-way ANOVA followed by Tukey’s test).
Discussion
Toxoplasmosis is a major parasitic disease transmitted by food, with a widespread distribution and a global human infection rate of ~30% (1). As toxoplasmosis is an inflammatory disease, the killing of T. gondii requires both innate and adaptive immune responses (40). Several lines of evidence—from gene polymorphism data to mouse knockout analysis—support the notion that P2X7 receptor activity contributes to control toxoplasmosis infection in vivo, by triggering antimicrobial activities in the intracellular environment (such as ROS production and lysosome fusion to the parasitophorous vacuole), and by stimulating pro-inflammatory events, such as the production of IL-12, IL-1β, and IFN-γ (27, 32). However, the downstream pathways that lead to parasite elimination via P2X7 receptor activation had not been dissected previously. Here, we described the cellular pathways that contribute to T. gondii killing induced by P2X7 receptor activation, showing that the control of T. gondii infection triggered by purinergic signaling requires the activity of a canonical NLRP3 inflammasome, and involves the inflammasome-dependent production of ROS and IL-1β (Figure 7).
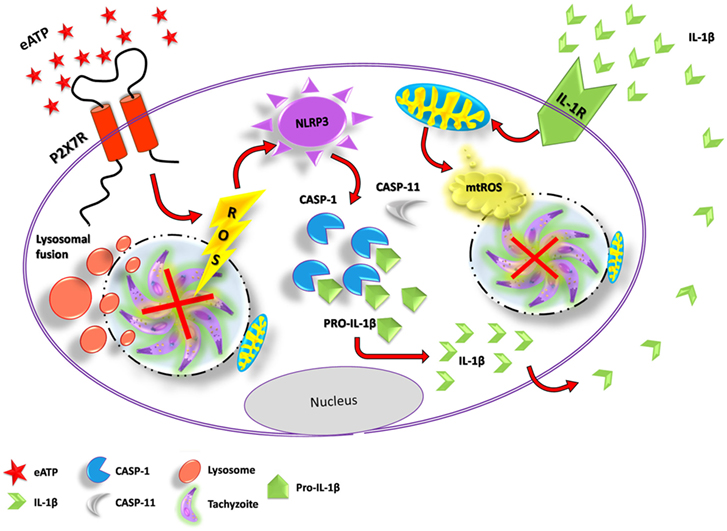
Figure 7. Proposed mechanism of T. gondi infection control mediated by extracellular ATP (eATP)/P2X7 receptor and IL-1β, in macrophages. Binding of eATP to the P2X7 receptor may lead to pathogen elimination via different mechanisms: the fusion of lysosomes to the parasitophorous vacuole (forming a phagolysosome), reactive oxygen species (ROS) generation and the activation of the canonical NLRP3 inflammasome. NLRP3 inflammasome activation by eATP potentiates IL-1β release through caspase-1 activity and, in turn, IL-1β in the extracellular compartment binds to its receptor, triggering T. gondii elimination via mitochondrial ROS generation.
IL-1β secretion, ROS production, and cell death are the most common cellular events linked to P2X7 receptor activation, and also seem to depend on inflammasome activation (20, 29, 35). ATP, a P2X7 receptor ligand, acts as a second signal to activate the NLRP3 inflammasome (41). Both NLRP1 and NLRP3 inflammasomes are involved in the immune responses against T. gondii infection, and mice lacking the inflammasome effectors caspase-1 and caspase-11 have 95% mortality after T. gondii infection (42). In agreement with these data, we show here that the control of T. gondii infection by P2X7 activation in macrophages depends on the NLRP3 inflammasome, and that the inflammasome effector caspase-1 is essential for T. gondii infection control by the P2X7 receptor, because lack of inflammasome components (NLRP3 and caspase-1) or inhibition of caspase-1 activity by Z-YVAD abolished the reduction in parasite load after treatment with ATP. In addition, the release of IL-1β in NLRP3 deficient mice can be explained by activation of NLRP1 inflammasome in macrophages of those mice (42). By examining separately the contributions of caspase-1 and caspase-11 to T. gondii infection control (using mouse knockout cells) we also show that the reduction in infection load caused by ATP treatment requires only caspase-1, and not caspase-11, supporting the hypothesis that a canonical NLRP3 is activated by the P2X7 receptor, to reduce T. gondii infection in macrophages.
Pyroptosis was shown to occur in response to P2X7 activation in macrophages (43), and this cell death mechanism can contribute to the clearance of intracellular pathogens (44). We did not detect significant cell death after 30 min or 18 h of eATP treatment (data not shown). In addition, our data suggest that, as have been shown for several pathogenic bacteria (45), the infection with T gondii may block pyroptosis. Thus, it is possible to conceive that different intracellular pathogens, not only bacteria, may subvert the immune response by operating in the inhibition of pyroptosis.
Even before it interacts with host cells, T. gondii secretes molecules that downregulate PRRs and, consequently, reduce the production of pro-inflammatory mediators (46). PRR activation acts as a first signal for inflammasome activation, inducing the transcription of genes coding for pro-inflammatory molecules. When PRR activation is followed by a second signal, it culminates in the cleavage and secretion of IL-1β (47). Our data show that upon eATP treatment and activation of the P2X7 receptor, T. gondii is not capable of downregulating the inflammasome-induced IL-1β secretion efficiently, since we observed that eATP treatment promotes IL-1β secretion in macrophages from WT mice. In this context, it is already described that P2X7 receptor activation leads to pannexin-1 pore formation and it is important for IL-1β release from macrophages (39). Our data show that inhibiting pannexin-1 channels, eATP-treated macrophages lack the ability to control T. gondii infection, which suggests that eATP-induced IL-1β secretion is crucial for the parasite killing. The results shown here reinforce the notion that IL-1β secretion potentiates the microbicidal effect of P2X7 activation.
Reactive oxygen species production is another key mechanism of intracellular pathogen elimination (48). T. gondii posses a specialized arsenal of antioxidant molecules and can successfully neutralize oxidative stress in the host (38). However, our result show that eATP treatment induced ROS production in murine macrophages even during T. gondii infection. Importantly, treatment with the inflammasome final product IL-1β reduced T. gondii infection load in macrophages, and ROS production was required for T. gondii infection control by eATP and IL-1β, because the pan ROS inhibitor NAC prevented the parasite load reduction caused by ATP and IL-1β. Our data show that, upon eATP treatment, ROS production is maintained during T. gondii infection, suggesting that P2X7 receptor activation overcomes sophisticated antioxidant mechanisms triggered by the pathogen. In phagocytes, NADPH oxidase activity and mitochondria are the two most common sources of ROS involved in immune response events (48), and the mitochondrial-derived ROS also activates the NLRP3 inflammasome (49). Here, we found that ROS derived from mitochondria participates in the control of T. gondii infection downstream of IL-1β signaling only, while IL-1β-independent signaling triggered by eATP is capable of controls T. gondii infection even upon inhibition of mitochondrial ROS production (by Mito-TEMPO). Therefore, we consider that NADPH oxidase-derived ROS are involved in the control of T. gondii infection in macrophages, upon P2X7 signaling activation.
Conclusion
Our data demonstrate that eATP treatment reduces T. gondii infection load in macrophages through activation of the NLRP3 inflammasome, and that eATP promotes IL-1β secretion, and leads to ROS production, in infected cells. By using knockout mice for different inflammasome molecules, such as NLRP3 and caspase1 and 11, we also identified that a canonical (but not a non-canonical) inflammasome is involved in the eATP-induced IL-1β secretion and elimination of the intracellular parasites, in macrophages.
We conclude that P2X7 receptor activation inhibits T. gondii growth via ROS generated, most likely, from NADPH oxidase (rather than mitochondrial ROS), while also activating the canonical NLRP3 inflammasome, which leads to IL-1β secretion and infection control via mitochondrial ROS production.
Ethics Statement
The procedures for the care and use of animals were according to the guidelines of the Brazilian College of Animal Experimentation (COBEA). All efforts were made to minimize animal suffering and to reduce the number of animals used in this study. This study was approved and followed all the guidelines established by the Ethics Committee on the Use of Animals (CEUA) of the Biophysic Institute Carlos Chagas Filho (IBCCF, UFRJ, no. 082/15).
Author Contributions
ACAMS and CLCAS designed and performed the experiments, analyzed the results, and wrote the manuscript. TPR and GCR performed experiments and analyzed the results. MB, DZ, and RCV analyzed the results and revised the manuscript. RCS designed the experiments, analyzed the results, and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DD-R declared a shared affiliation, with no collaboration, with several of the authors, AM-S, TR, and RV, to the handling editor.
Acknowledgments
The authors wish to thank Pryscilla Braga and Kelliane Dias da Silva for technical assistance, Profa. Elvira M. Saraiva for gently providing the apocynin, and Prof. Pedro Muanis Persechini for gently providing the carbenoxolone.
Funding
This work was supported by funds from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil—311362/2014-1, 448152/2014-2), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil; program Science Without Borders, grant no. 038/2012), and Programa de Núcleos de Excelência (PRONEX) from the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ, Brazil—E-26/010.002985/2014). Cássio Luiz Coutinho Almeida-da-Silva received PhD fellowship by CNPq (141715/2014-6) and FAPERJ (200417/2016; 200092/2016) agencies.
References
1. Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet (2004) 363:1965–76. doi:10.1016/S0140-6736(04)16412-X
2. Wang ZD, Liu HH, Ma ZX, Ma HY, Li ZY, Yang ZB, et al. Toxoplasma gondii infection in immunocompromised patients: a systematic review and meta-analysis. Front Microbiol (2017) 8:389. doi:10.3389/fmicb.2017.00389
3. Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect (2002) 8:634–40. doi:10.1046/j.1469-0691.2002.00485.x
4. Barragan A, Sibley LD. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol (2003) 11:426–30. doi:10.1016/S0966-842X(03)00205-1
5. Carme B, Bissuel F, Ajzenberg D, Bouyne R, Aznar C, Demar M, et al. Severe acquired toxoplasmosis in immunocompetent adult patients in French Guiana. J Clin Microbiol (2002) 40:4037–44. doi:10.1128/JCM.40.11.4037-4044.2002
6. Khan A, Jordan C, Muccioli C, Vallochi AL, Rizzo LV, Belfort R Jr, et al. Genetic divergence of Toxoplasma gondii strains associated with ocular toxoplasmosis, Brazil. Emerg Infect Dis (2006) 12:942–9. doi:10.3201/eid1206.060025
7. Lima GS, Saraiva PG, Saraiva FP. Current therapy of acquired ocular toxoplasmosis: a review. J Ocul Pharmacol Ther (2015) 31:511–7. doi:10.1089/jop.2015.0059
8. Dannemann B, McCutchan JA, Israelski D, Antoniskis D, Leport C, Luft B, et al. Treatment of toxoplasmic encephalitis in patients with AIDS. A randomized trial comparing pyrimethamine plus clindamycin to pyrimethamine plus sulfadiazine. The California Collaborative Treatment Group. Ann Intern Med (1992) 116:33–43. doi:10.7326/0003-4819-116-1-33
9. Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Microbiol (2005) 7:837–48. doi:10.1111/j.1462-5822.2005.00517.x
10. Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli AE, Denkers EY. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol (2008) 181:8485–91. doi:10.4049/jimmunol.181.12.8485
11. Denkers EY, Kim L, Butcher BA. In the belly of the beast: subversion of macrophage proinflammatory signalling cascades during Toxoplasma gondii infection. Cell Microbiol (2003) 5:75–83. doi:10.1046/j.1462-5822.2003.00258.x
12. Silva NM, Manzan RM, Carneiro WP, Milanezi CM, Silva JS, Ferro EA, et al. Toxoplasma gondii: the severity of toxoplasmic encephalitis in C57BL/6 mice is associated with increased ALCAM and VCAM-1 expression in the central nervous system and higher blood-brain barrier permeability. Exp Parasitol (2010) 126:167–77. doi:10.1016/j.exppara.2010.04.019
13. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi:10.1016/j.tibs.2016.09.002
14. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi:10.1038/nri.2016.58
15. Prochnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res (2016) 5:1–15. doi:10.12688/f1000research.8614.1
16. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi:10.1038/nature10558
17. Roberts JS, Yilmaz Ö. Dangerous liaisons: caspase-11 and reactive oxygen species crosstalk in pathogen elimination. Int J Mol Sci (2015) 16:23337–54. doi:10.3390/ijms161023337
18. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157:1013–22. doi:10.1016/j.cell.2014.04.007
19. Chaves MM, Marques-da-Silva C, Monteiro AP, Canetti C, Coutinho-Silva R. Leukotriene B4 modulates P2X7 receptor-mediated Leishmania amazonensis elimination in murine macrophages. J Immunol (2014) 192:4765–73. doi:10.4049/jimmunol.1301058
20. Almeida-da-Silva CL, Morandini AC, Ulrich H, Ojcius DM, Coutinho-Silva R. Purinergic signaling during Porphyromonas gingivalis infection. Biomed J (2016) 39:251–60. doi:10.1016/j.bj.2016.08.003
21. Amaral EP, Ribeiro SC, Lanes VR, Almeida FM, de Andrade MR, Bomfim CC, et al. Pulmonary infection with hypervirulent mycobacteria reveals a crucial role for the P2X7 receptor in aggressive forms of tuberculosis. PLoS Pathog (2014) 10:e1004188. doi:10.1371/journal.ppat.1004188
22. Lammas DA, Stober C, Harvey CJ, Kendrick N, Panchalingam S, Kumararatne DS. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity (1997) 7:433–44. doi:10.1016/S1074-7613(00)80364-7
23. Kusner DJ, Adams J. ATP-induced killing of virulent Mycobacterium tuberculosis within human macrophages requires phospholipase D. J Immunol (2000) 164:379–88. doi:10.4049/jimmunol.164.1.379
24. Kusner DJ, Barton JA. ATP stimulates human macrophages to kill intracellular virulent Mycobacterium tuberculosis via calcium-dependent phagosome-lysosome fusion. J Immunol (2001) 167:3308–15. doi:10.4049/jimmunol.167.6.3308
25. Lees MP, Fuller SJ, McLeod R, Boulter NR, Miller CM, Zakrzewski AM, et al. P2X7 receptor-mediated killing of an intracellular parasite, Toxoplasma gondii, by human and murine macrophages. J Immunol (2010) 184:7040–6. doi:10.4049/jimmunol.1000012
26. Correa G, Marques da SC, de Abreu Moreira-Souza AC, Vommaro RC, Coutinho-Silva R. Activation of the P2X(7) receptor triggers the elimination of Toxoplasma gondii tachyzoites from infected macrophages. Microbes Infect (2010) 12:497–504. doi:10.1016/j.micinf.2010.03.004
27. Miller CM, Zakrzewski AM, Robinson DP, Fuller SJ, Walker RA, Ikin RJ, et al. Lack of a functioning P2X7 receptor leads to increased susceptibility to toxoplasmic ileitis. PLoS One (2015) 10:e0129048. doi:10.1371/journal.pone.0129048
28. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi:10.1038/nature04515
29. Coutinho-Silva R, Monteiro da CC, Persechini PM, Ojcius DM. The role of P2 receptors in controlling infections by intracellular pathogens. Purinergic Signal (2007) 3:83–90. doi:10.1007/s11302-006-9039-6
30. Coutinho-Silva R, Kanellopoulos JM, Perfettini JL. Cellular alarms and whispers contribute to the polyphonic melody of danger signals required for immunity. Microbes Infect (2012) 14:1239–40. doi:10.1016/j.micinf.2012.10.002
31. Jamieson SE, Peixoto-Rangel AL, Hargrave AC, Roubaix LA, Mui EJ, Boulter NR, et al. Evidence for associations between the purinergic receptor P2X(7) (P2RX7) and toxoplasmosis. Genes Immun (2010) 11:374–83. doi:10.1038/gene.2010.31
32. Corrêa G, Almeida Lindenberg C, Moreira-Souza AC, Savio LE, Takiya CM, Marques-da-Silva C, et al. Inflammatory early events associated to the role of P2X7 receptor in acute murine toxoplasmosis. Immunobiology (2017) 222:676–83. doi:10.1016/j.imbio.2016.12.007
33. Moreira-Souza AC, Marinho Y, Correa G, Santoro GF, Coutinho CM, Vommaro RC, et al. Pyrimidinergic receptor activation controls Toxoplasma gondii infection in macrophages. PLoS One (2015) 10:e0133502. doi:10.1371/journal.pone.0133502
34. Marim FM, Silveira TN, Lima DS Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One (2010) 5:e15263. doi:10.1371/journal.pone.0015263
35. Morandini AC, Savio LE, Coutinho-Silva R. The role of P2X7 receptor in infectious inflammatory diseases and the influence of ectonucleotidases. Biomed J (2014) 37:169–77. doi:10.4103/2319-4170.127803
36. Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol (2011) 187:597–602. doi:10.4049/jimmunol.1100229
37. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10:89–102. doi:10.1038/nri2691
38. Ding M, Kwok LY, Schluter D, Clayton C, Soldati D. The antioxidant systems in Toxoplasma gondii and the role of cytosolic catalase in defence against oxidative injury. Mol Microbiol (2004) 51:47–61. doi:10.1046/j.1365-2958.2003.03823.x
39. Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J (2006) 25:5071–82. doi:10.1038/sj.emboj.7601378
40. Dupont CD, Christian DA, Hunter CA. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol (2012) 34:793–813. doi:10.1007/s00281-012-0339-3
41. Bording-Jorgensen M, Alipour M, Wine E, Danesh G. Inflammasome activation by ATP enhances Citrobacter rodentium clearance through ROS generation. Cell Physiol Biochem (2017) 41:193–204. doi:10.1159/000455988
42. Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio (2014) 5:1–12. doi:10.1128/mBio.01117-13
43. Le Feuvre RA, Brough D, Iwakura Y, Takeda K, Rothwell NJ. Priming of macrophages with lipopolysaccharide potentiates P2X7-mediated cell death via a caspase-1-dependent mechanism, independently of cytokine production. J Biol Chem (2002) 277:3210–8. doi:10.1074/jbc.M104388200
44. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev (2015) 265:130–42. doi:10.1111/imr.12287
45. Cunha LD, Zamboni DS. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol (2013) 3:76. doi:10.3389/fcimb.2013.00076
46. Morampudi V, Braun MY, D’Souza S. Modulation of early beta-defensin-2 production as a mechanism developed by type I Toxoplasma gondii to evade human intestinal immunity. Infect Immun (2011) 79:2043–50. doi:10.1128/IAI.01086-10
47. Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol (2017) 29(5):201–10. doi:10.1093/intimm/dxx018.:10
48. Dupre-Crochet S, Erard M, Nubetae O. ROS production in phagocytes: why, when, and where? J Leukoc Biol (2013) 94:657–70. doi:10.1189/jlb.1012544
Keywords: P2X7 receptor, Toxoplasma gondii, NLRP3 inflammasome, caspase-1, caspase-11, IL-1β, reactive oxygen species
Citation: Moreira-Souza ACA, Almeida-da-Silva CLC, Rangel TP, Rocha GdC, Bellio M, Zamboni DS, Vommaro RC and Coutinho-Silva R (2017) The P2X7 Receptor Mediates Toxoplasma gondii Control in Macrophages through Canonical NLRP3 Inflammasome Activation and Reactive Oxygen Species Production. Front. Immunol. 8:1257. doi: 10.3389/fimmu.2017.01257
Received: 17 July 2017; Accepted: 21 September 2017;
Published: 12 October 2017
Edited by:
Ian Marriott, University of North Carolina at Charlotte, United StatesReviewed by:
Carlos De Torre, IMIB-Arrixaca, SpainDebora Decote-Ricardo, Universidade Federal Rural do Rio de Janeiro, Brazil
Copyright: © 2017 Moreira-Souza, Almeida-da-Silva, Rangel, Rocha, Bellio, Zamboni, Vommaro and Coutinho-Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robson Coutinho-Silva, cmNzaWx2YUBiaW9mLnVmcmouYnI=
†These authors have contributed equally to this work.