 Shalini Chakraborty
Shalini Chakraborty Sonti Roy
Sonti Roy Hiral Uday Mistry
Hiral Uday Mistry Shweta Murthy
Shweta Murthy Neena George
Neena George Vasundhra Bhandari†
Vasundhra Bhandari† Paresh Sharma*†
Paresh Sharma*†
- National Institute of Animal Biotechnology (NIAB-DBT), Hyderabad, India
Plasmodium, Toxoplasma, Cryptosporidium, Babesia, and Theileria are the major apicomplexan parasites affecting humans or animals worldwide. These pathogens represent an excellent example of host manipulators who can overturn host signaling pathways for their survival. They infect different types of host cells and take charge of the host machinery to gain nutrients and prevent itself from host attack. The mechanisms by which these pathogens modulate the host signaling pathways are well studied for Plasmodium, Toxoplasma, Cryptosporidium, and Theileria, except for limited studies on Babesia. Theileria is a unique pathogen taking into account the way it modulates host cell transformation, resulting in its clonal expansion. These parasites majorly modulate similar host signaling pathways, however, the disease outcome and effect is different among them. In this review, we discuss the approaches of these apicomplexan to manipulate the host–parasite clearance pathways during infection, invasion, survival, and egress.
Introduction
The Apicomplexan parasites represent a major class of pathogens with a wide host range. They have emerged as one of the most successful intracellular parasites, which efficiently modulate the host for their survival benefits. In this review, we focus on the potential sabotage mechanisms adopted by the five well-studied pathogens of human and veterinary importance: Plasmodium falciparum (malaria), Babesia bovis (babesiosis), Theileria annulata (theileriosis), Toxoplasma gondii (toxoplasmosis), and Cryptosporidium parvum (cryptosporidiosis). These parasites are morphologically similar; however, variations exist in the context of host range, mode of infection, invasion, and replication inside the host (Table 1).
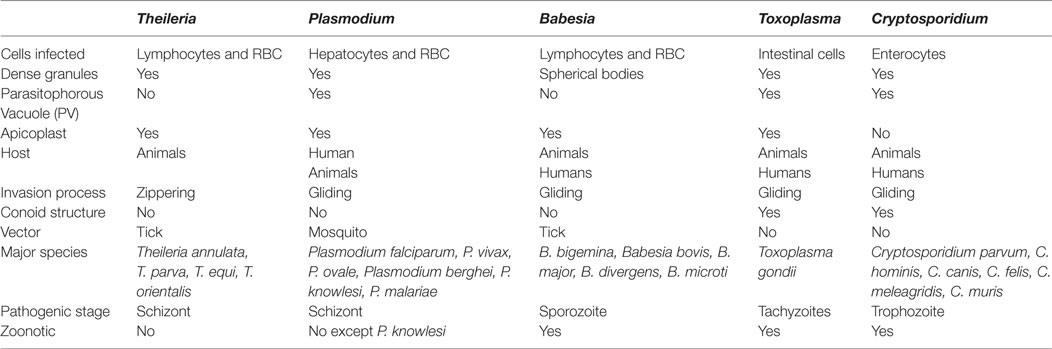
Table 1. A generalized comparative account among Theileria, Plasmodium, Babesia, Toxoplasma, and Cryptosporidium parasites.
Beginning with transmission, P. falciparum, T. annulata, and B. bovis are vector borne; however, C. parvum and T. gondii do not require a vector and the host is infected by oocyst-ingestion (Table 1). T. annulata solely infects animals impacting their health and causing huge economic loss, whereas other parasites have broader host preference range. P. falciparum and T. gondii infections affect human health and cause mortality worldwide. On the other hand, B. bovis and C. parvum are comparatively less pathogenic with fewer reported cases of mortality and morbidity.
In this review, we epitomize the major blueprint of the pathways targeted by these parasites to sabotage the host defense mechanism for their survival and consequent disease progression.
Plasmodium: The Malaria Parasite
Plasmodium falciparum is considered the most lethal among the Plasmodium species, as it accounts for serious illness and high mortality (1–5). Two hundred fourteen million new cases of malaria are reported worldwide with a 35% mortality rate reported for children below 5 years of age (6).
Malaria transmission cycle starts with the female Anopheles feeding on a mammalian host. Thereafter, Plasmodium smartly exploits host cell machinery in numerous ways discussed hereafter to complete its life cycle (7–9). The sporozoites harbored in the salivary gland enter the host blood stream and passes on to the hepatic sinusoid (10–12). The presence of antihistamines and immunomodulators in the salivary gland secretion protects Plasmodium from the initial host immune response (10, 11, 13). The endothelial cell lining the liver sinusoid, guarded by kupffer cells (liver macrophages) prevents sporozoite entry into the hepatocytes (12, 14, 15). The circumsporozoite protein (CSP) of the parasite interacts with LRP-1 (low-density lipoprotein receptor-related protein) present on the kupffer cells thereby upregulating cAMP. Thereafter, cAMP mediates EPAC (exchange protein activated by cAMP) inhibition of reactive oxygen species (ROS) production ultimately suppressing the macrophage defense (15–17) (Figure 1). Simultaneously, the expression of TNFα, IL-6, and monocyte chemoattractant protein-1 (MCP-1) is downregulated and there is an increased production of anti-inflammatory IL-10 cytokine (15). The sporozoite also downregulates expression of kupffer cells MHC-1 and IL-12 to overturn their antigen presenting ability and ease infiltration of sporozoites into hepatocytes (15, 17). All these events result in the successful invasion.
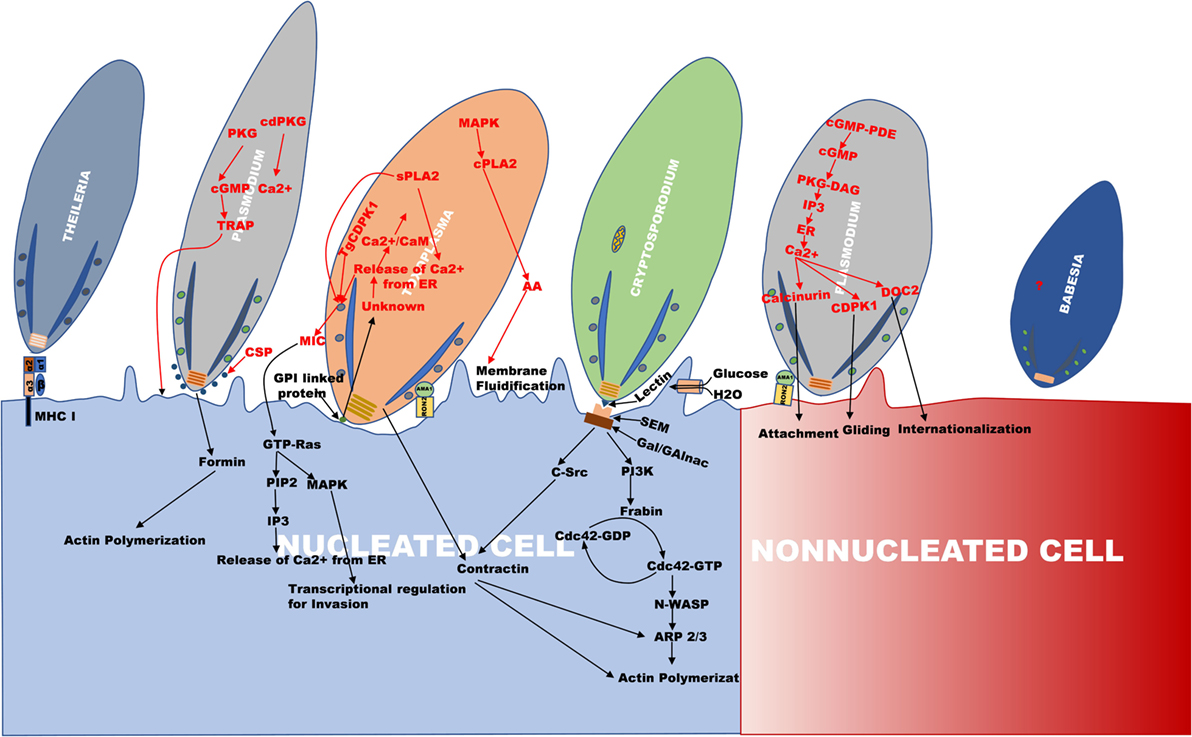
Figure 1. An outline of the invasion mechanism used by Theileria, Plasmodium, Toxoplasma, Cryptosporidium, and Babesia. The figure is the representation of the invasion process, which happens during internalization of Theileria, Plasmodium, Toxoplasma, Cryptosporidium, and Babesia. The apical region of all the parasites faces toward the cell surface for their entry. In Plasmodium, invasion occurs in two types of cells, erythrocyte (non-nucleated cell) and hepatocyte (nucleated cell) as compared to other parasites where invasion occurs in the nucleated cell.
Furthermore, the role of calcium (Ca2+) in activating various parasite proteins involved in the process of invasion, egress, motility, and cell cycle regulation has been observed (18–21). In Plasmodium, endoplasmic reticulum and acidocalcisomes are the major Ca2+ reservoirs which are also observed in Toxoplasma. The activation of protein kinase G (PKG) by an unknown parasitic signal during invasion or egress releases Ca2+ from the parasite endoplasmic reticulum mediated by cyclic guanosine monophosphate (cGMP) (20). Furthermore, phosphoinositide phospholipase C (PI-PLC) is activated by cGMP-dependent PKG which results in hydrolysis of phosphatidylinositol 4, 5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). The translocation of IP3 on to the ER surface causes efflux of Ca2+ to the cytoplasm by the formation of IP3-Ca2+ channel (20, 22) (Figure 1). The increase in the cytoplasmic Ca2+ levels activates various calcium-dependent proteases and kinases, like calcium-dependent protein kinases (PfCDPK), double C2 domain protein (PfDOC2) which induce the secretion of microneme and rhoptry proteins for cell adherence and invasion (20, 23, 24). The hepatocyte invasion of the sporozoites occurs via Ca2+-mediated activation and secretion of microneme proteins, CSP and thrombospondin-related adhesion protein (TRAP) such as Trap-like protein (TLP) (12, 15, 25) (Figure 1). The CSP secreted to the apex in association with actin covers the surface of the sporozoites and its glycosyl phosphatidyl inositol (GPI) anchored C terminus helps in the invasion of sporozoites (12, 26, 27). PfTRAP (TLP) protein interaction with actomyosin motor complex helps in gliding movement of the parasite (15, 25, 28).
The transcellular migration by sporozoites is mediated by the secretion of perforin protein SPECT or perforin like protein1 (PfPLP1), which is also demonstrated to be important in cell traversal, to perforate the hepatocytes (12, 29). Hepatocytic growth factor (HGF) is released by the perforated hepatocyte (30, 31), which activates c-MET receptor tyrosine kinase (c-MET RTK) on them resulting in the activation of tyrosine residues at the cytoplasmic domain of the c-MET receptor (32, 33). This recruits phosphoinositide 3-kinase (PI3-K) which phosphorylates and sequesters proapoptotic proteins of the BCL-2 family (Bad, Bim, PUMA) through AKT (32–35). AKT, which activates anti-apoptotic proteins (BCl-2, BCL-XL, A1), inhibits Bax on the outer mitochondrial membrane and hinders the permeabilization of the mitochondrial membrane and the subsequent release of proapoptotic signaling molecule such as cytochrome-c (Cyt-c) and eventually blocks apoptosis (Figure 2) (12, 27, 32, 33).
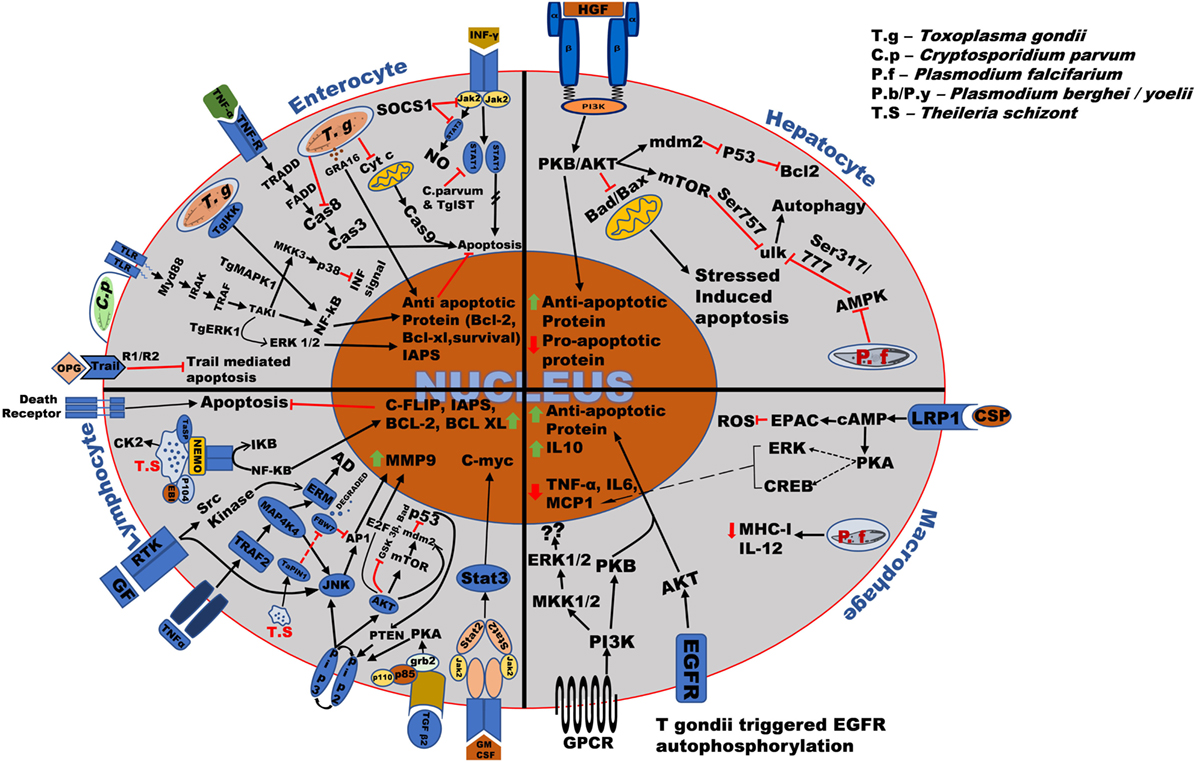
Figure 2. Overall survival mechanism used by the Apicomplexan parasites in different host cells. Toxoplasma and Cryptosporidium bind to the surface receptor of host cells through the ligands such as EGF, TNF-α, and parasitic surface proteins such as circumsporozoite protein (CSP). After invasion into the host cells such as enterocytes, macrophages, hepatocytes, etc., the parasite modify the host signaling pathway such as TRADD, NF-kB, PKB/AKT resulting in production and upregulation of anti-apoptotic proteins such as Bcl-2, Bcl-xl, and anti-inflammatory cytokines such as IL-10 thereby stopping cytochrome-c (Cyt-c), TNF-alpha-related-apoptosis-inducing ligand (TRAIL) and BAD, BAX production, and ensuring its survival in the host. Plasmodium parasite mainly modifies host PKB/AKT signaling pathway causing upregulation of anti-apoptotic protein and downregulation of pro-apoptotic proteins such as BAD/BAX. Theileria schizont proliferates uncontrollably within the host macrophages and lymphocytes. Right after invasion, it upregulates anti-apoptotic proteins such as c-FLIP, IAPs, Bcl-2, Bcl-XL, and proto-oncogenic proteins such as C-myc, antiapoptotic genes such as C-FLIP, Bcl-2, and matrix metallo-protein (MMP9) by majorly targeting host signaling pathways such as NF-κB, JNK/AKT, JAK/STAT, phosphoinositide 3-kinase (PI3-K)/MAPK, and TGF-β2. The regulation of these host signaling pathways causes continuous survival and proliferations of the parasite infected cells which are also common in some cases.
Once the parasite has already invaded the hepatocyte, host cell apoptosis block is independent of the PI3-K pathway. It seems that the direct intervention of parasite proteins is necessary for modulating the host survival signal (36). One such example is hypoxia mediated by host-dependent HIF-α through AMPK activation which promotes proliferation and parasite survival in the liver (37). The presence of the autophagy marker Atg8 on P. falciparum might suggest the parasite’s involving degradative functions, but it instead majorly contributes toward biogenic process (38, 39). After the exoerythrocytic merogony, they trigger apoptosis, although this does not seem to occur by activating the caspase-dependent pathway nor via the expression of phosphatidylserine (36, 40). However, a serine-repeated antigen (SERA), a cysteine protease identified in Plasmodium berghei, is upregulated and is reported to be playing a role in parasite-induced cell death, parasitophorous vacuole (PV) disruption and merosome formation at the time of exoerythrocytic merozoite egress (12). It suggests that the parasite secretory proteins are mediators of host cell apoptosis in the late liver stage. Among seven calcium-dependent protein kinases (CDPK1–7) known in Plasmodium, inhibition of PfCDPK5 leads to schizont stage arrest (20).
Erythrocyte invasion of Plasmodium occurs in two stages. First, the interaction of merozoite with the erythrocyte causes host cytoskeletal distortion with the help of high Ca2+ level, increasing the contact area between the two favoring merozoite entry and the alignment of its apical pole (14, 19, 20). The role of calcineurin (CnA, CnB) has been implicated in merozoite attachment to erythrocytes, which when knocked down results in impaired invasion (20, 41). Furthermore, DOC2 activation induces microneme secretion of erythrocyte binding antigen (EBA175) and AMA1 (microneme apical antigen1), which are involved in the attachment (20, 42) (Figure 1). At the second stage of invasion, AMA1, in association with RON2 (rhoptry neck protein), binds to the erythrocyte ligand resulting in the formation of tight junction via TRAP (43). It has also been observed that the localization of formin at the apical pole nucleates the parasite F-actin with its FH2 domain and helps in parasite motility (44). The forward propulsion of the actin-myosin filament helps in the invasion of merozoite and encapsulation into the PV in the host cytoplasm (14, 45–47). Plasmodium invasion entails increased erythrocyte membrane permeability in order to gain nutrients from the extracellular fluid for its survival (45, 48) and further, utilizes the NF-kB-dependent pathway to inhibit host cell apoptosis (49). Following the invasion, the parasite secretes proteins essential for survival, cell adhesion, and pathogenicity. These are transported from the cytosol to the plasma membrane through vesicular transport. The interplay of protein export elements (PEXEL) and Plasmodium translocon of exported proteins (PTEX), cause the exported proteins to be transported to the plasma membrane of the RBC (50–52).
Cell cytolysis and infecting new cells are an essential strategy associated with disease progression in Plasmodium. Parasite egress is a tightly regulated proteolytic activity of parasite proteases PfSUB1 and dipeptidyl peptidase 3 (DPAP3) in response to increased Ca2+ level in the cytosol of the parasite. The PfSUB1, an exoneme secreted protease acts on the PV and the plasma membrane of RBC causing cell rupture and egress (53, 54). Millholland et al. 2013 showed the role of host calpains in cytolysis. Krebs cycle intermediates, formed during replication, act through host GPCR, diffused from host plasma membrane to the PV, also help in cytolysis. The ligand activation of GPCR signal through Gαq activates PKC via PLC activation. PKC phosphorylates and liberates adducin (a protein that maintains cytoskeletal integrity) resulting in host cell cytoskeletal deformation, which in turn opens the TRPC6 cation channel on the plasma membrane of RBC. The influx of Ca2+ from the extracellular fluid activates CaMK (Ca2+/calmodulin dependent kinase) via calcium-dependent calmodulin, which phosphorylates host cytoskeletal substrates also resulting in a rapid influx of Ca2+. This Ca2+ activates and releases host calpain, which causes lysis and dissolution of host cytoskeleton facilitating parasite release (55, 56). The modulation of host survival signaling by Plasmodium allows them to successfully establish a specific environment where they can proliferate and differentiate leading to pathogenesis.
Despite substantial progress in the malaria research, restraining the disease still remains a challenge. P falciparum parasites owing to their multiple forms/stages, antigenic polymorphisms and AT-rich genome have further impended the problem. Current chemotherapy is based on using artemisinin and artemisinin-based combination therapies (ACTs), however, reports of drug resistance have already emerged. An effective vaccine should be the ultimate goal for long-term control of the disease. To date only the RTS, S/AS01 vaccine, targeting the CSP 178 of P. falciparum has reached phase three trials, but has not shown much efficacy (57). There are still many gaps in the understanding of the invasion process of P. falciparum, such as which molecules signal the release of Ca2+, which leads to adherence and invasion by activating many pathways. These pathways playing role in Ca2+ release can be targeted for identifying the novel antigens for developing future vaccines and therapeutics.
Babesia: Malaria Like Parasite
Babesiosis is a hemolytic disease prevalent in tropical and subtropical parts of the world with a broad host range. B. microti and B. divergens infect humans and have emerged as a public health concern predominantly in the United States and Europe, respectively (58). Human cases of babesiosis have surfaced recently in Asian countries, including India and Korea (59). B. bovis and B. bigemina both infect cattle but, however, higher morbidity and mortality are associated with B. bovis resulting in a huge economic loss (60). There are very limited studies investigating the parasite and its host interactions as compared to the other apicomplexan parasites. Owing to the striking similarity between Babesia and Plasmodium (61), the disease pathogenesis is considered to be similar to malaria during infection in cattle (60).
Babesia multiplies in the host erythrocyte in a similar fashion to Plasmodium, and they are transovarially transmitted in the vector except for B. microti (Table 1) (62). The life cycle of the parasite begins with tick feeding on host blood and simultaneously releasing sporozoites into the host bloodstream (63). Invasion occurs in a similar fashion as observed for Plasmodium, Toxoplasma, and Cryptosporidium via gliding mechanism using microneme and rhoptry secretions (61, 64). Parasite loosely attaches to the surface of the host RBC by its surface GPI anchored proteins and within the apical secretory organelles. Variant erythrocyte surface antigen 1 (VESA1), a heterodimeric protein of Babesia is known to play role in cytoadherence to the host erythrocyte surface (62, 65). After entry of the sporozoites into the red blood cells, they divide by binary fission and produce merozoites. Erythrocyte lysis further allows each merozoite to invade a new RBC and successive merogonies follow (62, 64, 66, 67). Merozoites interact with the RBC surface receptors and play a major role in invasion.
In Babesia, the role of Ca2+ has been primarily described in invasion and egress mechanism of the parasite, however, the modulation of the host signaling pathways are not thoroughly understood. In B. bovis, Ca2+-dependent protein kinase inhibitor showed growth limiting effects (68), though, in B. divergens, it impacted egress of the merozoites from erythrocytes (69, 70). Since there is no PV formation in Babesia, less Ca2+ is released during egress as compared to P. falciparum parasites (Table 2) (70, 71).
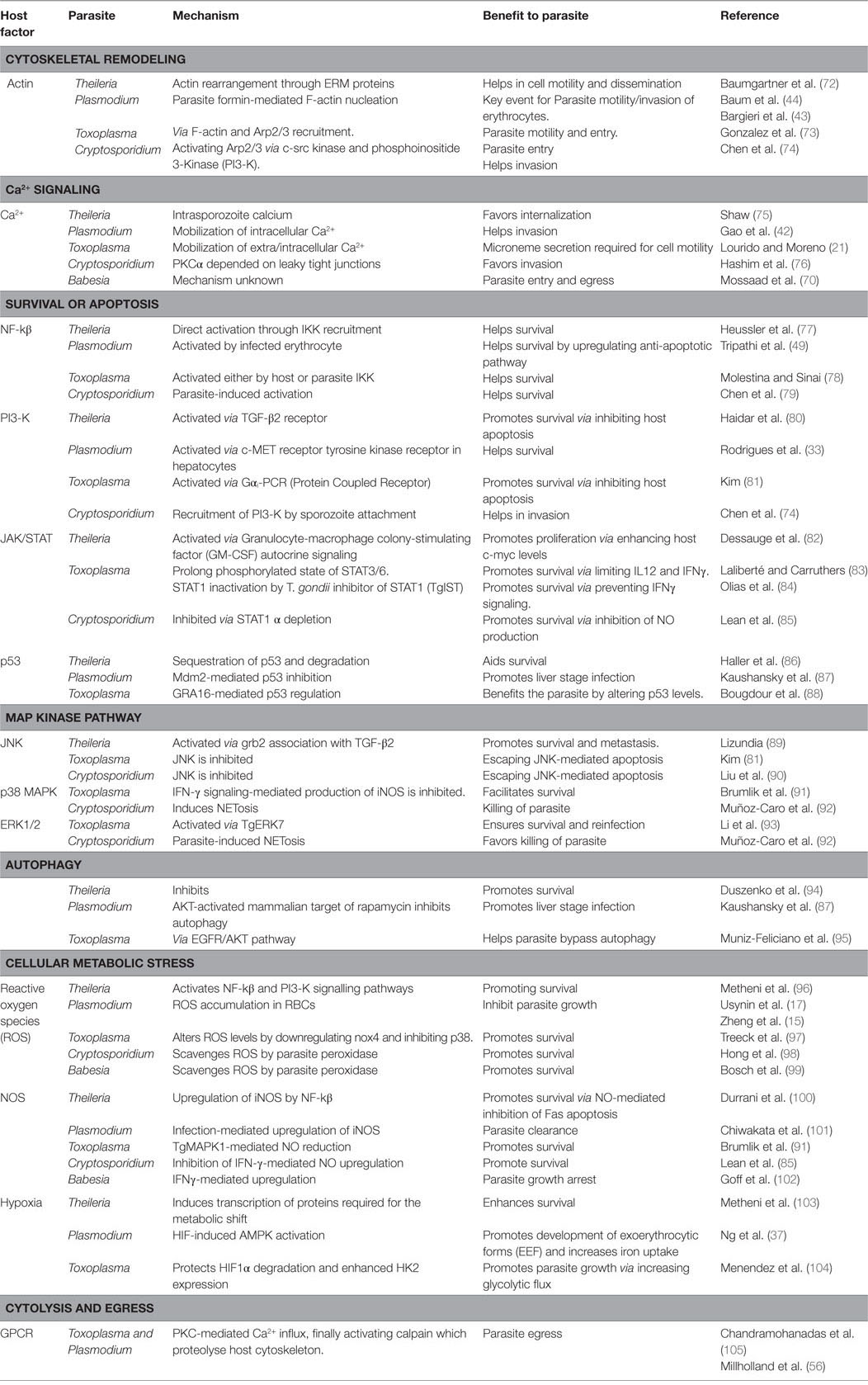
Table 2. A number of host signaling pathways modulated by Theileria, Plasmodium, Babesia, Toxoplasma, and Cryptosporidium during its invasion, survival, expansion, and egress in the host cell.
Transovarial transmissions in tick vector and straight entry of sporozoites into erythrocytes are some key features, which make Babesia parasites distinct from Plasmodium or Theileria parasites. Few studies to understand the mechanism of disease pathogenesis during Babesia infections have been published. Also, the mechanism of entry and transmission of the parasites are poorly defined. It will be important to investigate the parasite invasion and evasion strategies along with parasite vector interactions for identifying key genes that might play an important role in immune evasion or disease pathogenesis.
Theileria: A Livestock Pathogen
Theileria annulata and T. parva cause tropical theileriosis and east coast fever, respectively, in ruminants predominantly in cattle causing enormous economic loss to the livestock industry (106, 107). Tick vector transmits the parasite upon feeding on animal through the saliva (108–110). After entering the blood stream, it infects WBC of different lineage, T. parva infects B cells and T cells whereas T. annulata infects B cells and cells of monocyte lineage. The sporozoites, i.e., the infective stage of the parasite passively invade the host cell by zippering mechanism, which is unlike other apicomplexan discussed in which a tight continuous junction is formed between the host cell surface and the parasite sporozoites (108). In addition, the role of MHC class I molecule (Figure 1) (111), intrasporozoite Ca2+ and protein kinases of host and parasite and the G-protein linked signaling has been shown in invasion (75, 112). After entry into the host cell, parasite rhoptries and its microsphere discharges dissolve the enveloping PV membrane (108), and move to the host cell cytoplasm rather than to PV in comparison to other apicomplexan parasites and provides it with an advantage of escaping lysosomal degradation (Table 1). Additional advantage of staying in the host cytoplasm allows the parasite to modulate several signaling pathways, such as TGF-β, JNK, PI3-K, NF-κB, src kinase, and casein kinase 2 (CK2) (80, 100, 113–115).
Theileria transforms their host cell into a cell with a cancerous phenotype by modulating several host cell kinases and activating transcription factors (116). Several studies have been done to identify parasite protein instigating epigenetic changes that may lead to successful transformation. T. annulata protein, TaPIN (secretory prolyl isomerase Pin) has been reported to promote transformation by degrading FBW7, a host ubiquitin ligase via stabilizing c-JUN (117). p104 and TaSP are surface proteins which have been reported to be phosphorylated in a host cell cycle-dependent manner and might be involved in transformation (118). Two more proteins are TashAT group of protein, which contains AT hook DNA-binding motif and nuclear localization signal and found to be localized in host nucleus (119). SuAT1, a parasite gene, contains AT hook DNA-binding polypeptide and predicted signal peptide, PEST motifs and nuclear localization signals, which may interact with the host cell and play a role in transformation (120). Studies to identify epigenetics changes are scarce, only one study has shown the role of oncomiR mir155 in repressing DET1 protein (involve in c-Jun ubiquitination) and stabilizing c-Jun (121). The parasite schizont hijacks the host mitotic assembly resulting in its clonal expansion (122–124).
Theileria transformed cells can be reversed, unlike tumor cells upon treatment with BW720c (122). The transformation occurs by modulating several signaling pathways which ultimately inhibits apoptosis, increases proliferation, and encourages metastatis (116). NF-κB is constitutively expressed in Theileria infected cells, which in turn upregulates many anti-apoptotic proteins, such as c-FLIP, IAPs, Bcl-2, and Bcl-XL, and induces Gadd45β that blocks the pro-JNKK2-mediated apoptotic JNK pathway. NF-κB is activated by recruitment and phosphorylation of IKK signalosome α and β subunits, which further phosphorylates inhibitory κB (iκB) setting NF-κB free to translocate to the nucleus (125). Infected cells release a plethora of cytokines and growth factors that activate TGF-β receptor (I and II) and TNF-α receptor (126). TGF-β2 activates smad2/3 and subsequently smad4, which over-expresses COX-2 resulting in increased levels of prostaglandins and downregulates PKIG, a potent inhibitor of PKA pathway. Simultaneously, TGF-β2 is accounted for parasite motility and invasiveness by activating Rho–ROCK kinase and recruiting an adaptor protein growth factor receptor-bound protein 2 (Grb2) to TGF-RII receptor. The signaling descends by Grb2, activating downstream PI3-K/AKT and JNK pathway (80, 127). Activator protein 1 (AP1), a JNK activated transcription factor drives B cell integration cluster (BIC) transcription upregulating miRNA 155 which inhibits DET1 resulting in accumulation of c-Jun and increased proliferation (89, 121, 128–131). B-1 a bovine analog of MMP9 (ECM degrading proteinase) containing AP1 binding sites also helps in detachment and metastasis of the infected cell to other organs (89, 132, 133).
Theileria modulates the host PI3-K/AKT pathway to be regulated by granulocyte-macrophage colony-stimulating factor (GM-CSF), depending on an autocrine loop and, hence, sharing a major role in cell proliferation (134, 135). Phosphorylation of the AKT protein by class I PI3-K facilitates the release of Rb bound E2F transcription factor, activating MDM2 (E3 protein ubiquitin ligase and negative regulator of p53 tumor suppressor gene) directly or through mammalian target of rapamycin (mTOR). AKT-mediated inhibition of several proapoptotic genes (bad, foxo) and GSK-3β help the infected cells to combat the stress-induced mitochondrial-mediated apoptosis and to maintain elevated c-Myc levels, respectively (Table 2; Figure 2) (136). Hypoxia-inducing factor (HIF-1α) is activated by mTOR pathway and by constitutive NF-κB and AP1 production (137). Increased levels of ROS during infection and HIF-1 expression induce the Warburg effect allowing the parasite to establish uncontrolled proliferation (96, 103, 138, 139). PI3-K/AKT pathway, therefore, plays a very important role in survival and proliferation as well as in metastasis of Theileria. PTEN, an inhibitor of the PI3-K pathway and activator of p53 are suppressed majorly by NF-κB and CK2 (82, 140, 141). CK2 also dampens TNF/Fas-mediated apoptosis and accelerates iκB degradation augmenting NF-κB activation. Activation of JAK/STAT pathway via GM-CSF autocrine loop enhances c-myc expression, whereas phosphorylation by CK2 stabilizes this potent oncogene primarily upregulated in Theileria infected cells (Figure 2) (82, 115).
The proliferation of infected cells is followed by evasion and metastasis, which involves cytoskeleton alteration. TNF-α binding to its receptor recruits TNF receptor-associated factor 2 (TRAF2), which may either activates NF-κB or a mitogen-activated protein kinase, MAP4K4 (134). MAP4K4 bifurcates into JNK activation and ERM (ezrin/radixin/moesin) cytoskeletal protein phosphorylation, helping in actin dynamics through Rho kinase. ERM may also be activated through src kinases (72). Interfering with autophagic mechanisms also augments survival of the parasite within the cell. Dampening peptide presentation by CD4+ and CD8+ infected cells is a way opted by the parasite to increase its chances of survival enhancing the establishment of infection (94). In contrast to other apicomplexan parasites, Theileria usually does not egress and remains attached to the host mitotic assembly. Although under unfavorable circumstances the schizont ruptures releasing merozoites, which invade RBCs, forming piroplasm, which is taken up by the tick and the cycle resumes. Theileria parasites have thereby evolved a wide range of strategies to help them survive and proliferate inside the host cells.
Theileria parasites are considered as the smartest among the apicomplexan group for their ability to manipulate the host cells. However, the parasite proteins and molecular mechanisms behind the host cell manipulation are still not clear. Very few proteins are identified till now, which are involved in the host–parasite interaction. The presence of a heterogeneous population of T. annulata parasite strains in the field is making it difficult for a currently used attenuated vaccine (schizont stage) to be effective against T. annulata parasites. Resistance against burpaquone, a drug currently being used for the treatment of theileriosis, and acaricide, used for controlling the tick vector, has hampered the control of the disease. In this post genomic era with the availability of advanced genomic and proteomic tools, better studies are needed to dissect the pathways modulated by Theileria in detail and select new targets for the development of second-generation drugs and vaccines for disease control.
Enteric and Zoonotic Apicomplexan Pathogen
Toxoplasma
Toxoplasmosis, caused by Toxoplasma gondii, is one of the most well-studied zoonoses (142). Toxoplasma appears to be one of the most feared apicomplexan parasites due to a considerable number of congenital transmission incidents and subsequent fetal damage in animals and humans (143, 144). It also causes neurologic deficits (145) and chorioretinitis (146). The life cycle of the parasite circulates between definitive (feline) and intermediate hosts (mammals/birds, etc.). Sexual phase occurs in the small intestine of the feline host from which the oocysts/tissue cysts are excreted along with the feces and ingested by the intermediate host through multiple routes. The oocysts release sporozoites, which invade the intestinal lining. On the other hand, tissue cysts release bradyzoites which differentiate into tachyzoites (147–149). The tachyzoites further replicate in the host and may again differentiate into bradyzoites in the brain, liver, and muscle tissue forming cysts (147, 150, 151). This inter-conversion between tachyzoites and bradyzoites appears essential to the life cycle and infective potential of the parasite (Table 1) (152).
Host–parasite interactions are mostly via secreted parasite proteins from their rhoptries, micronemes, and dense granules, which help parasite in cell invasion, survival, and egress. Invasion by T. gondii involves gliding mobility prompted by an actin-myosin motor based complex (153) and interrelated signaling cascades as well. The parasite attaches loosely with the host cell surface via GPI-linked proteins, surface antigens (SAGs), SAG-related sequences (SRSs), and SAG unrelated surface antigens (SUSAs) (154, 155). After the secretion of Toxoplasma microneme adhesion proteins (MICs) into the host, there is an increased activity of Calcium/Calmodulin (Ca/CAM)-dependent processes leading to the secretion of Phospholipases (sPLA2 and PLA2) (Figure 1). T. gondii calcium-dependent protein kinase1 (TgCDPK1) has been reported to be involved in microneme secretion and can thereby regulate cell motility which is essential for invasion (156). sPLA2 secretion causes the release of microneme proteins MIC3/MIC2, which induces Ca2+ release from host ER via Protein Kinase C-Inositol 1,4,5 triphosphate (PKC–IP3) pathway. cPLA2 activated by parasite MAPK causes membrane fluidification by hydrolyzing host membrane phospholipids (157). Therefore, Ca2+signaling induce a lot of complex cascades facilitating parasite invasion (Figure 1; Table 2). Apicomplexan have been reported to have several Ca2+ATPases and CA2+/H+ exchangers, which help in invasion (21).
Toxoplasma invades via gliding movement, which results in actin remodeling by F-actin ring formation at the point of entry with the subsequent recruitment of Arp2/3 complex (Figure 1) (73, 158). Parasite rhoptries are secreted followed by microneme secretion, which consists of RON and traditional rhoptries proteins (ROP). RON2 and AMA1 associate together to form a tight junction between the host and the parasite referred to as moving junction (MJ) facilitating the formation of PV (159–161). Sporozoites also invade using paralogs of AMA1 and RON2 conveniently named as sporoAMA1 and sporoRON2 (162). Traditional rhoptry proteins such as Rop 17, Rop 18 (kinase), and Rop 5 (pseudokinase) reside on the PV membrane inhibiting the accumulation of immunity-related GTPases (IRGs). This complex also has a dense granule protein, namely GRA 7 which has a definite impact on IRG turnover (163, 164).
Once inside the host cell, the parasite thrives on host nutrients by expressing various parasite transporters, enzymes, and following complex cascades (165). T. gondii inhibits apoptosis and dodges autophagy by manipulating PI3-K pathway, the immediate downstream effector protein kinase B (PKB/Akt), JAK/STATs, mTOR, NF-kβ, ERK1/2, C-myc, and microRNAs to promote its survival (165, 166). The parasite avoids lysosomal degradation by cleverly maintaining the non-fusogenic nature of the PV. Studies suggest that T. gondii micronemal proteins (MICs) with epidermal growth factor (EGF) domains activate Epidermal Growth Factor Receptor (EGFR) on endothelial, retinal cells, and microglia keeping the parasite protected in the vacuole (95). It has been reported that the parasite causes mTOR activation in an infected host cell even in the absence of phosphorylation of 4E-BP1 and S6K1 (167). Later, a study confirmed the role of mTORC1 and C2 in host cell invasion and persistence of infection (168).
Toxoplasma gondii disrupts host apoptotic pathways primarily by affecting the release of Cyt-c and thereby preventing activation of caspase 3 (166). The parasite modulates the host NF-kβ pathway in line with Theileria causing increased expression of anti-apoptotic genes, although, there is also a role of Toxoplasma IKK (TgIKK) in maintaining the NF-kβ response which declines after the initial activation by the host IKK (78). A dense granule protein GRA 15 also activates this pathway via TNF receptor-associated factor 6 (TRAF6) thereby inducing the release of pro-inflammatory cytokines (169). Rop 16, on the other hand, might be playing a role in inhibiting cytokine synthesis by host macrophages (170). Hence, opposed the effect of two of these factors determines macrophage polarization in the host (171). Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation (171) Toxoplasma inhibits the proapoptotic genes (BCL-2, Bad, caspase-9) by modulating the host–PI3-K pathway. It also hinders apoptosis by downregulating phosphorylated c-Jun N-terminal kinase levels (81). AKT/PKB pathway is upregulated, which serves the parasite by inhibiting forkhead transcription factor (FKHRI) resulting in decreased levels of proapoptotic factors, such as Bim and FasL (172). Infected cells also display increased expression of anti-apoptotic proteins, such as BCL-2, BFL1, BCL-XI, BCL-W, and MCI-I, and reduced expression of proapoptotic factors Bad and Bax (Figure 2).
Toxoplasma ERK-7 (TgERK7) protein has been recently demonstrated to play an important role in the intracellular proliferation of the parasite in the host (93). T. gondii also protects itself from host interferon-γ (IFN-γ)-mediated pathway by obstructing the expression of IFN-γ activated genes. T. gondii inhibitor of STAT1 (TgIST) has been shown to bind to activated STAT1 in the host cell membrane and recruits host Mi2/NURD complex which keeps the STAT1 in inactivated stage thereby preventing pro-inflammatory gene expression (84, 173). Elevated Ca2+ during T. gondii infection activates the protein kinase C cascade which further activates COX-2 resulting in increased prostaglandin E2 (PGE2) levels helping in the resolution of inflammation (174). Furthermore, a dual role of TGF-β during infection by inducing or suppressing the immune system has been reported (175).
During infection, TRAF6 is activated by the parasite dense granule protein GRA7 which leads to unusually increased levels of ROS in the cells (97). Increased levels of ROS, few cytokines and growth factors, causes elevated HIF-1 levels via dampening prolyl hydroxylase domain containing protein 2 (PHD-2) levels downstream to Type I TGFβ receptor signaling. Influencing the levels of such a crucial host factor as PHD-2 is pivotal for the maintenance of a secure haunt of the parasite (176). A host kinase-HK2 also activates HIF-1 expression resulting in glycolytic flux and Warburg effect, as identified by siRNA screening (104). A microarray-based study reveals that increased HIF-1 level in infected cells lead to activation of EGR1 and AP1 which play roles in inducing resistance against drugs and proliferation, respectively (177, 178). The parasite also seizes the IFN-γ-induced iNOS production by T. gondii expressed MAP kinase (TgMAPK1), which reduces NO production by p38 MAPK (91). But on the other hand, a dense granule protein GRA24 also plays a role in maintaining p38α autophosphorylation, forming a complex, which consequently activates EGR1 and cFOS which induce the release of MCP-1 and IL-12, which can keep the parasite load in check (179).
Toxoplasma also modulates p53 levels for its own benefit by GRA16, another parasite dense granule protein which binds to two host enzymes-HAUSP and PP2A phosphatase in the host nucleus (88). T. gondii reportedly alters dopaminergic and GABA-ergic signaling due to elevated levels of mi-RNA132 which might be the underlying cause for the neuronal abnormalities often found associated with the infection (180). The parasite utilizes GABA to partially satisfy its carbon requirements and also in egress (181). Once it has successfully established infection, egress mainly occurs via GPCR-coupled signaling pathway similar to Plasmodium. TgCDPK1 and TgCDPK3 activated by Ca2+ influx have been reported to play a role in egress. Studies suggest that a parasite pore forming protein TgPLP1 might be responsible for making the PV perforated to make egress easier (182). Recently, cGMP-dependent PKG has been identified to play an important role in controlling egression (156).
As it appears from the above discussion, the cunning parasite can steer a staggering number of host signaling pathways in direction of its own purpose. However, despite the fact that very specific knowledge is available about particular such proteins, it is not clear how they affect host gene expression since such nucleus targeted proteins do not really resemble host transcription factors neither can they bind to the host cell DNA (164). Interestingly, not all of these secreted proteins benefit the parasite. Some actually trigger the host immune system to call up its guards. Now, how might the parasite strike a balance to sustain infection or how might we use such kind of knowledge to limit infection still remains to be worked out.
Cryptosporidium
Cryptosporidium commonly causes gastrointestinal diseases worldwide, which albeit minimally invasive in the immunocompetent host (both human and animals) can be deadly in immunosuppressed patients (183, 184). C. parvum with a broad host range and zoonoses is considered a more important pathogen in comparison to C. hominis, which only infects human. The disease prevalence ranges from 1 to 37% in countries such as Africa, Asia, Australia, South America, and Central America (185, 186).
Its life cycle comprises of a sexual and an asexual stage, which takes place in a single host (187). Similar to Toxoplasma, Cryptosporidium infection occurs by ingestion of oocysts through contaminated water followed by excystation and release of sporozoites. These zoites then invades the enterocytes by gliding movement (Table 1, Figure 1) (188). Cryptosporidium form an intracellular but extra cytoplasmic PV wherein they get developed into spherical trophozoites (184, 189). Invasion of host epithelial cells occurs via aggregating the host actin and actin binding protein, villin at the site of parasite attachment and further inducing host tyrosine kinase signaling cascades (189, 190). Reports of numerous C. parvum proteins have been implicated in attachment, invasion, and intracellular development (191, 192). p30, a galactose-N-acetylgalactosamine (Gal/GalNAc) lectin parasite protein has been identified which forms an adhesion complex along with gp40 and gp900 (193). Furthermore, the cryptosporidial binding leads to the formation of sphingolipid-enriched membrane microdomains which attracts Gal/GalNAc epitope containing glycoproteins on the host membrane parasite interface, activating PI3-K (192). The PI3-K cascade successively activates Cdc42, N-WASP, and Arp2/3 (actin-related protein 2/3) resulting in the formation of actin plaque (74, 124, 194). The parasite recruited src tyrosine kinase subsequently phosphorylates cortactin stimulating the polymerization and rearrangement of the actin cortex in the cell periphery through activation of Arp2/3 complex proteins (Figure 1) (74, 195). Increase in local cell volume by accumulation of host aquaporin AQP1 and Na+/Glucose co-transporter also aid in efficient membrane protrusions (196). Few studies have also shown the role of host calpain in remodeling host cytoskeleton which is essential during parasite invasion (197).
Ca2+-ATPase located at the Cryptosporidium sporozoites apical and perinuclear regions helps it in fulfilling its Ca2+ requirement during the invasion (193). Cryptosporidium also possesses 7 CDPKs, which has a role in invasive and regulatory processes similar to Plasmodium and Toxoplasma. Cryptosporidium invasion is promoted by a Ca2+-dependent PKC signaling pathway, which disrupts the cell–cell junction. PKC causes downstream activation of PKCα which has been associated with tight junctional leakiness in renal epithelial cells (76, 193, 198). Cryptosporidium embodies a novel Ca2+-activated nucleoside diphosphatase (apyrase, CApy), which interfere with extracellular nucleotide and modulates inflammatory pathways delaying the response against parasite clearance (199). The trophozoite stage of the parasite inhibits apoptosis; however, schizont- and merozoite-affected cells are handled by host apoptosis through Fas/FasL signaling (200, 201).
Cryptosporidium activates NF-κB pathway by inducing IL-8 secretion and acting synergistically with AP1 and IL-6 (79) (Figure 2; Table 2). It has also been reported to play a role in activating other survival signals, e.g., over expression of antiapoptotic proteins (bcl-2, IAP, survivin) and inhibition of proapoptotic proteins (bax) (77, 202). Myc, an oncogenic protein plays a role in positive regulation of parasite survival, whereas PTEN, an inhibitor of PI3-K, negatively regulates the anti-apoptotic protein (90). Also, microarray analysis revealed that TNF-superfamily receptor osteoprotegerin (OPG) is upregulated in infected host intestinal mucosa by microarray. The overexpression of OPG helps in evading host defense by inhibiting TNF-alpha-related-apoptosis-inducing ligand (TRAIL)-mediated apoptosis and supporting the parasite to complete its life cycle (184, 203). The host tries to control the propagation of Cryptosporidium by enhancing Th1 response characterized by the production of IFNγ and IL-12. The parasite, too, in turn, erodes the JAK/STAT-mediated IFNγ signaling by depletion of STAT1-α (85, 204). TNF-α and TGF-β play roles in providing the host protective immunity and healing effect against the infection (85, 205, 206). Again, increased mucin levels in the host by COX-2-mediated PGE2 protects the host (183), ERK1/2 and p38 MAPK pathway also assist the host cells to destroy the parasite by inducing NETosis (Formation of the neutrophil extracellular trap) (92).
Absence of Apicoplast in C. parvum parasites and complications in their in vitro propagation has posed problems for researchers involved in drug or vaccine development. Despite many efforts by Cryptosporidium to modulate the host signaling pathways, the parasite loses the battle against the host. At present, only one drug (nitazoxanide) with limited efficacy is approved for treatment of Cryptosporidiosis. Further studies are needed to better understand the egress mechanism of Cryptosporidium (74). Susceptibility to the parasite has shown to be linked with the immune status of the host. Understanding the host–pathogen interaction will be critical in designing new tools for effective control of the disease.
Final Conclusion
A substantial amount of research has been done to gain insights into pathways by which these parasites modulate and undermine the host defense, yet gaps in knowledge still prevail and many questions remain unanswered. In this review, we have attempted to include all the major work carried out in this field. Advancement in gene editing technologies and whole genome sequencing of these pathogens lead us to better understand the manipulation strategies used by the parasites. Emerging problems of either drug resistance or unavailability of an effective vaccine against some of the parasites make the precise comprehension of the sabotage techniques employed by the parasites a primary requisite in order to curb the morbidity rate.
As discussed above, it is apparent that few of the host defense pathways targeted by these parasites to survive and proliferate in the host cell are common among the mentioned pathogens. Host cell invasion by Plasmodium, Babesia, Toxoplasma, and Cryptosporidium occur via gliding movement, whereas in Theileria an overall different process known as zippering takes place. Plasmodium, Toxoplasma, and Cryptosporidium reside and replicate inside the PV in the host, however, Theileria and Babesia survives in the host cytoplasm. Theileria clearly takes an advantage of staying in the host cytoplasm by modulating numerous pathways, though any such information about Babesia has to be still investigated. Several pathways are modulated by majority of these parasites such as host cytoskeleton remodeling, Ca2++ modulated signaling pathways, and apoptotic pathways which helps in their survival. In the review, we have discussed the cross talks happening between the parasite and the host and observed that the multifaceted nature of the parasite gives them an upper hand over the host.
Future studies focusing on
(i) Exploring the parasite proteins and their role in host–parasite interface interaction will provide in-depth understanding of the invasion process. These targets can be further utilized to develop vaccine or drugs.
(ii) What are the alteration in the host cell that leads to the nutrient acquisition after invasion and the host factors contributing to parasite replication?
(iii) Studies are also required to develop inhibitors against known molecules/pathways, which help the in intracellular survival of parasite in the host cell.
Therefore, targeting the common pathways playing crucial role in all parasites survival and dissemination may be a good approach to understand disease pathogenesis and controlling the disease.
In order to deal with these cunning pathogens, we need all the necessary information to be able to target important molecules for a vaccine or drug development. However, a considerable amount of research and thorough screening of presently available literature is still required to better understand how these parasites exploit their hosts for their own survival. Apicomplexan parasites infecting human beings such as Plasmodium is hugely funded and globally studied; however, so is not the case when it comes to parasites such as Babesia and Theileria, which are of veterinary importance. Therefore, for more in-depth understanding of these pathogens, tenacious research is expected which would only be possible through the combined efforts of researchers and support from funding agencies on a global range.
Author Contributions
All authors mentioned have made a significant effort and contributed intellectually to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work was supported by an extramural grant (BT/PR11979/AAQ/1/608/2014) funded by Department of Biotechnology, India and National Institute of Animal Biotechnology (NIAB-DBT).
References
1. Boussaroque A, Fall B, Madamet M, Camara C, Benoit N, Fall M, et al. Emergence of mutations in the K13 Propeller Gene of Plasmodium falciparum Isolates from Dakar, Senegal, in 2013-2014. Antimicrob Agents Chemother (2015) 60:624–7. doi:10.1128/aac.01346-15
2. Cui L, Rosenthal PJ, Rathod PK, Ndiaye D, Mharakurwa S. Antimalarial drug resistance: literature review and activities and findings of the ICEMR network. Am J Trop Med Hyg (2015) 93:57–68. doi:10.4269/ajtmh.15-0007
3. Huang B, Deng C, Yang T, Xue L, Wang Q, Huang S, et al. Polymorphisms of the artemisinin resistant marker (K13) in Plasmodium falciparum parasite populations of Grande Comore Island 10 years after artemisinin combination therapy. Parasit Vectors (2015) 8:634. doi:10.1186/s13071-015-1253-z
4. Isozumi R, Uemura H, Kimata I, Ichinose Y, Logedi J, Omar AH, et al. Novel mutations in K13 propeller gene of artemisinin-resistant Plasmodium falciparum. Emerg Infect Dis (2015) 21:490–2. doi:10.3201/eid2103.140898
5. Sharma RK, Thakor HG, Saha KB, Sonal GS, Dhariwal AC, Singh N. Malaria situation in India with special reference to tribal areas. Indian J Med Res (2015) 141:537–45. doi:10.4103/0971-5916.159510
6. World Health Organization. (WHO fact sheet: world malaria report; 9 December 2015). World Health (2015) 243:10–13.
7. Mota MM, Rodriguez A. Migration through host cells: the first steps of Plasmodium sporozoites in the mammalian host. Cell Microbiol (2004) 6:1113–8. doi:10.1111/j.1462-5822.2004.00460.x
8. Walker DM, Oghumu S, Gupta G, McGwire BS, Drew ME, Satoskar AR. Mechanisms of cellular invasion by intracellular parasites. Cell Mol Life Sci (2014) 71:1245–63. doi:10.1007/s00018-013-1491-1
9. Silva LS, Silva-Filho JL, Caruso-Neves C, Pinheiro AAS. New Concepts in malaria pathogenesis: the role of the renin-angiotensin system. Front Cell Infect Microbiol (2016) 5:103. doi:10.3389/fcimb.2015.00103
10. Baldacci P, Ménard R. The elusive malaria sporozoite in the mammalian host. Mol Microbiol (2004) 54:298–306. doi:10.1111/j.1365-2958.2004.04275.x
11. Ishino T, Yano K, Chinzei Y, Yuda M. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol (2004) 2:e4. doi:10.1371/journal.pbio.0020004
12. Garcia JE, Puentes A, Patarroyo ME. Developmental biology of sporozoite-host interactions in Plasmodium falciparum malaria: implications for vaccine design. Clin Microbiol Rev (2006) 19:686–707. doi:10.1128/cmr.00063-05
14. Dhangadamajhi G, Kar SK, Ranjit M. The survival strategies of malaria parasite in the red blood cell and host cell polymorphisms. Malar Res Treat (2010) 2010:1–9. doi:10.4061/2010/973094
15. Zheng H, Tan Z, Xu W. Immune evasion strategies of pre-erythrocytic malaria parasites. Mediators Inflamm (2014) 2014:1–6. doi:10.1155/2014/362605
16. Coppi A, Tewari R, Bishop JR, Bennett BL, Lawrence R, Esko JD, et al. Heparan sulfate proteoglycans provide a signal to Plasmodium sporozoites to stop migrating and productively invade host cells. Cell Host Microbe (2007) 2:316–27. doi:10.1016/j.chom.2007.10.002
17. Usynin I, Klotz C, Frevert U. Malaria circumsporozoite protein inhibits the respiratory burst in Kupffer cells. Cell Microbiol (2007) 9:2610–28. doi:10.1111/j.1462-5822.2007.00982.x
18. Mota MM, Hafalla JCR, Rodriguez A. Migration through host cells activates Plasmodium sporozoites for infection. Nat Med (2002) 8:1318–22. doi:10.1038/nm785
19. Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, et al. Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol (2014) 12:e1001806. doi:10.1371/journal.pbio.1001806
20. Brochet M, Billker O. Calcium signalling in malaria parasites. Mol Microbiol (2016) 100:397–408. doi:10.1111/mmi.13324
21. Lourido S, Moreno SNJ. The calcium signaling toolkit of the apicomplexan parasites Toxoplasma gondii and Plasmodium spp. Cell Calcium (2015) 57:186–93. doi:10.1016/j.ceca.2014.12.010
22. Hopp CS, Bowyer PW, Baker DA. The role of cGMP signalling in regulating life cycle progression of Plasmodium. Microbes Infect (2012) 14:831–7. doi:10.1016/j.micinf.2012.04.011
23. Cheemadan S, Ramadoss R, Bozdech Z. Role of calcium signaling in the transcriptional regulation of the apicoplast genome of Plasmodium falciparum. Biomed Res Int (2014) 2014:1–12. doi:10.1155/2014/869401
24. Jean S, Zapata-Jenks MA, Farley JM, Tracy E, Mayer DCG. Plasmodium falciparum double C2 domain protein, PfDOC2, binds to calcium when associated with membranes. Exp Parasitol (2014) 144:91–5. doi:10.1016/j.exppara.2014.06.015
25. Moreira CK, Templeton TJ, Lavazec C, Hayward RE, Hobbs CV, Kroeze H, et al. The Plasmodium TRAP/MIC2 family member, TRAP-like protein (TLP), is involved in tissue traversal by sporozoites. Cell Microbiol (2008) 10:1505–16. doi:10.1111/j.1462-5822.2008.01143.x
26. Plassmeyer ML, Reiter K, Shimp RL, Kotova S, Smith PD, Hurt DE, et al. Structure of the Plasmodium falciparum circumsporozoite protein, a leading malaria vaccine candidate. J Biol Chem (2009) 284:26951–63. doi:10.1074/jbc.m109.013706
27. Lüder CGK, Stanway RR, Chaussepied M, Langsley G, Heussler VT. Intracellular survival of apicomplexan parasites and host cell modification. Int J Parasitol (2009) 39:163–73. doi:10.1016/j.ijpara.2008.09.013
28. Ejigiri I, Sinnis P. Plasmodium sporozoite–host interactions from the dermis to the hepatocyte. Curr Opin Microbiol (2009) 12:401–7. doi:10.1016/j.mib.2009.06.006
29. Yang ASP, O’Neill MT, Jennison C, Lopaticki S, Allison CC, Armistead JS, et al. Cell traversal activity is important for Plasmodium falciparum liver infection in humanized mice. Cell Rep (2017) 18:3105–16. doi:10.1016/j.celrep.2017.03.017
30. Mota MM. Migration of Plasmodium sporozoites through cells before infection. Science (2001) 291:141–4. doi:10.1126/science.291.5501.141
31. Carrolo M, Giordano S, Cabrita-Santos L, Corso S, Vigário AM, Silva S, et al. Hepatocyte growth factor and its receptor are required for malaria infection. Nat Med (2003) 9:1363–9. doi:10.1038/nm947
32. Organ SL, Tsao M-S. An overview of the c-MET signaling pathway. Ther Adv Med Oncol (2011) 3:S7–19. doi:10.1177/1758834011422556
33. Rodrigues V, Cordeiro-da-Silva A, Laforge M, Ouaissi A, Silvestre R, Estaquier J. Modulation of mammalian apoptotic pathways by intracellular protozoan parasites. Cell Microbiol (2012) 14:325–33. doi:10.1111/j.1462-5822.2011.01737.x
34. Leirião P, Albuquerque SS, Corso S, Van Gemert G-J, Sauerwein RW, Rodriguez A, et al. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell Microbiol (2005) 7:603–9. doi:10.1111/j.1462-5822.2004.00490.x
35. Regad T. Targeting RTK signaling pathways in cancer. Cancers (Basel) (2015) 7:1758–84. doi:10.3390/cancers7030860
36. Koyama FC, Chakrabarti D, Garcia CRS. Molecular machinery of signal transduction and cell cycle regulation in Plasmodium. Mol Biochem Parasitol (2009) 165:1–7. doi:10.1016/j.molbiopara.2009.01.003
37. Ng S, March S, Galstian A, Hanson K, Carvalho T, Mota MM, et al. Hypoxia promotes liver-stage malaria infection in primary human hepatocytes in vitro. Dis Model Mech (2013) 7:215–24. doi:10.1242/dmm.013490
38. Navale R, Atul, Allanki AD, Sijwali PS. Characterization of the autophagy marker protein Atg8 reveals atypical features of autophagy in Plasmodium falciparum. PLoS One (2014) 9:e113220. doi:10.1371/journal.pone.0113220
39. Thieleke-Matos C, Lopes da Silva M, Cabrita-Santos L, Portal MD, Rodrigues IP, Zuzarte-Luis V, et al. Host cell autophagy contributes to Plasmodium liver development. Cell Microbiol (2015) 18:437–50. doi:10.1111/cmi.12524
40. Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med (2010) 207:1807–17. doi:10.1084/jem.20101157
41. Philip N, Waters AP. Conditional degradation of Plasmodium calcineurin reveals functions in parasite colonization of both host and vector. Cell Host Microbe (2015) 18:122–31. doi:10.1016/j.chom.2015.05.018
42. Gao X, Gunalan K, Yap SSL, Preiser PR. Triggers of key calcium signals during erythrocyte invasion by Plasmodium falciparum. Nat Commun (2013) 4:2862. doi:10.1038/ncomms3862
43. Bargieri D, Lagal V, Andenmatten N, Tardieux I, Meissner M, Ménard R. Host cell invasion by apicomplexan parasites: the junction conundrum. PLoS Pathog (2014) 10:e1004273. doi:10.1371/journal.ppat.1004273
44. Baum J, Tonkin CJ, Paul AS, Rug M, Smith BJ, Gould SB, et al. A malaria parasite formin regulates actin polymerization and localizes to the parasite-erythrocyte moving junction during invasion. Cell Host Microbe (2008) 3:188–98. doi:10.1016/j.chom.2008.02.006
45. Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell (2006) 124:755–66. doi:10.1016/j.cell.2006.02.006
46. Bargieri D, Lagal V, Tardieux I, Ménard R. Host cell invasion by apicomplexans: what do we know? Trends Parasitol (2012) 28:131–5. doi:10.1016/j.pt.2012.01.005
47. Koch M, Baum J. The mechanics of malaria parasite invasion of the human erythrocyte – towards a reassessment of the host cell contribution. Cell Microbiol (2016) 18:319–29. doi:10.1111/cmi.12557
48. Gazarini ML, Thomas AP, Pozzan T, Garcia CRS. Calcium signaling in a low calcium environment. J Cell Biol (2003) 161:103–10. doi:10.1083/jcb.200212130
49. Tripathi AK, Sha W, Shulaev V, Stins MF, Sullivan DJ. Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood (2009) 114:4243–52. doi:10.1182/blood-2009-06-226415
50. Cooke BM, Lingelbach K, Bannister LH, Tilley L. Protein trafficking in Plasmodium falciparum-infected red blood cells. Trends Parasitol (2004) 20:581–9. doi:10.1016/j.pt.2004.09.008
51. Marti M. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science (2004) 306:1930–3. doi:10.1126/science.1102452
52. Boddey JA, Moritz RL, Simpson RJ, Cowman AF. Role of the Plasmodium export element in trafficking parasite proteins to the infected erythrocyte. Traffic (2009) 10:285–99. doi:10.1111/j.1600-0854.2008.00864.x
53. Heussler VT, Stanway RR. Cellular and molecular interactions between the apicomplexan parasites Plasmodium and Theileria and their host cells. Parasite (2008) 15:211–8. doi:10.1051/parasite/2008153211
54. Lee MCS, Fidock DA. Arresting malaria parasite egress from infected red blood cells. Nat Chem Biol (2008) 4:161–2. doi:10.1038/nchembio0308-161
55. Millholland MG, Chandramohanadas R, Pizzarro A, Wehr A, Shi H, Darling C, et al. The malaria parasite progressively dismantles the host erythrocyte cytoskeleton for efficient egress. Mol Cell Proteomics (2011) 10:M111.010678. doi:10.1074/mcp.m111.010678
56. Millholland MG, Mishra S, Dupont CD, Love MS, Patel B, Shilling D, et al. A host GPCR signaling network required for the cytolysis of infected cells facilitates release of apicomplexan parasites. Cell Host Microbe (2013) 13:15–28. doi:10.1016/j.chom.2012.12.001
57. Gosling R, von Seidlein L. The future of the RTS,S/AS01 malaria vaccine: an alternative development plan. PLoS Med (2016) 13:e1001994. doi:10.1371/journal.pmed.1001994
58. Leiby DA. Transfusion-transmitted Babesia spp.: bull’s-eye on Babesia microti. Clin Microbiol Rev (2011) 24:14–28. doi:10.1128/cmr.00022-10
59. Marathe A, Tripathi J, Handa V, Date V. Human babesiosis – a case report. Indian J Med Microbiol (2005) 23:267–9.
60. Gohil S, Kats LM, Sturm A, Cooke BM. Recent insights into alteration of red blood cells by Babesia bovis: moovin’ forward. Trends Parasitol (2010) 26:591–9. doi:10.1016/j.pt.2010.06.012
61. Cooke BM, Mohandas N, Cowman AF, Coppel RL. Cellular adhesive phenomena in apicomplexan parasites of red blood cells. Vet Parasitol (2005) 132:273–95. doi:10.1016/j.vetpar.2005.07.002
62. Chauvin A, Moreau E, Bonnet S, Plantard O, Malandrin L. Babesia and its hosts: adaptation to long-lasting interactions as a way to achieve efficient transmission. Vet Res (2009) 40:37. doi:10.1051/vetres/2009020
63. Mehlhorn H, Schein E. The piroplasms: life cycle and sexual stages. Adv Parasitol (1985) 23:37–103. doi:10.1016/s0065-308x(08)60285-7
64. Lobo CA, Rodriguez M, Cursino-Santos JR. Babesia and red cell invasion. Curr Opin Hematol (2012) 19:170–5. doi:10.1097/moh.0b013e328352245a
65. Allred DR. Molecular technology and antigenic variation among intraerythrocytic hemoparasites: do we see reality? Vet Parasitol (2001) 101:261–74. doi:10.1016/s0304-4017(01)00571-4
66. Hines S. Genetic and antigenic characterization of Babesia bovis merozoite spherical body protein Bb-1. Mol Biochem Parasitol (1995) 69:149–59. doi:10.1016/0166-6851(94)00200-7
67. Dowling SC, Perryman LE, Jasmer DP. A Babesia bovis 225-kilodalton spherical-body protein: localization to the cytoplasmic face of infected erythrocytes after merozoite invasion. Infect Immun (1996) 64:2618–26.
68. Bork S, Das S, Okubo K, Yokoyama N, Igarashi I. Effects of protein kinase inhibitors on the in vitro growth of Babesia bovis. Parasitology (2006) 132:775. doi:10.1017/s0031182006009917
69. Okubo K, Wilawan P, Bork S, Okamura M, Yokoyama N, Igarashi I. Calcium-ions are involved in erythrocyte invasion by equine Babesia parasites. Parasitology (2006) 133:289. doi:10.1017/s0031182006000436
70. Mossaad E, Asada M, Nakatani D, Inoue N, Yokoyama N, Kaneko O, et al. Calcium ions are involved in egress of Babesia bovis merozoites from bovine erythrocytes. J Vet Med Sci (2015) 77:53–8. doi:10.1292/jvms.14-0391
71. Agarwal S, Singh MK, Garg S, Chitnis CE, Singh S. Ca2+-mediated exocytosis of subtilisin-like protease 1: a key step in egress of Plasmodium falciparum merozoites. Cell Microbiol (2012) 15:910–21. doi:10.1111/cmi.12086
72. Baumgartner M. Constitutive exclusion of Csk from Hck-positive membrane microdomains permits Src kinase-dependent proliferation of Theileria-transformed B lymphocytes. Blood (2002) 101:1874–81. doi:10.1182/blood-2002-02-0456
73. Gonzalez V, Combe A, David V, Malmquist NA, Delorme V, Leroy C, et al. Host cell entry by apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe (2009) 5:259–72. doi:10.1016/j.chom.2009.01.011
74. Chen XM, Splinter PL, Tietz PS, Huang BQ, Billadeau DD, LaRusso NF. Phosphatidylinositol 3-kinase and frabin mediate Cryptosporidium parvum cellular invasion via activation of Cdc42. J Biol Chem (2004) 279:31671–8. doi:10.1074/jbc.M401592200
75. Shaw MK. Theileria parva sporozoite entry into bovine lymphocytes involves both parasite and host cell signal transduction processes. Exp Parasitol (1996) 84:344–54. doi:10.1006/expr.1996.0123
76. Hashim A, Mulcahy G, Bourke B, Clyne M. Interaction of Cryptosporidium hominis and Cryptosporidium parvum with primary human and bovine intestinal cells. Infect Immun (2005) 74:99–107. doi:10.1128/iai.74.1.99-107.2006
77. Heussler VT, Küenzi P, Rottenberg S. Inhibition of apoptosis by intracellular protozoan parasites. Int J Parasitol (2001) 31:1166–76. doi:10.1016/s0020-7519(01)00271-5
78. Molestina RE, Sinai AP. Detection of a novel parasite kinase activity at the Toxoplasma gondii parasitophorous vacuole membrane capable of phosphorylating host IkappaBalpha. Cell Microbiol (2005) 7:351–62. doi:10.1111/j.1462-5822.2004.00463.x
79. Chen XM, Levine SA, Splinter PL, Tietz PS, Ganong AL, Jobin C, et al. Cryptosporidium parvum activates nuclear factor kappaB in biliary epithelia preventing epithelial cell apoptosis. Gastroenterology (2001) 120:1774–83. doi:10.1053/gast.2001.24850
80. Haidar M, Whitworth J, Noé G, Liu WQ, Vidal M, Langsley G. TGF-β2 induces Grb2 to recruit PI3-K to TGF-RII that activates JNK/AP-1-signaling and augments invasiveness of Theileria-transformed macrophages. Sci Rep (2015) 5:15688. doi:10.1038/srep15688
81. Kim L. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci (2006) 119:2119–26. doi:10.1242/jcs.02934
82. Dessauge F, Lizundia R, Langsley G. Constitutively activated CK2 potentially plays a pivotal role in Theileria-induced lymphocyte transformation. Parasitology (2005) 130:S37. doi:10.1017/s0031182005008140
83. Laliberté J, Carruthers VB. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell Mol Life Sci (2008) 65:1900–15. doi:10.1007/s00018-008-7556-x
84. Olias P, Etheridge RD, Zhang Y, Holtzman MJ, Sibley LD. Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-γ-dependent gene expression. Cell Host Microbe (2016) 20:72–82. doi:10.1016/j.chom.2016.06.006
85. Lean I-S, McDonald SAC, Bajaj-Elliott M, Pollok RCG, Farthing MJG, McDonald V. Interleukin-4 and transforming growth factor have opposing regulatory effects on gamma interferon-mediated inhibition of Cryptosporidium parvum reproduction. Infect Immun (2003) 71:4580–5. doi:10.1128/iai.71.8.4580-4585.2003
86. Haller D, Mackiewicz M, Gerber S, Beyer D, Kullmann B, Schneider I, et al. Cytoplasmic sequestration of p53 promotes survival in leukocytes transformed by Theileria. Oncogene (2010) 29:3079–86. doi:10.1038/onc.2010.61
87. Kaushansky A, Ye AS, Austin LS, Mikolajczak SA, Vaughan AM, Camargo N, et al. Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell Rep (2013) 3:630–7. doi:10.1016/j.celrep.2013.02.010
88. Bougdour A, Durandau E, Brenier-Pinchart M-P, Ortet P, Barakat M, Kieffer S, et al. Host cell subversion by Toxoplasma GRA16, an Exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe (2013) 13:489–500. doi:10.1016/j.chom.2013.03.002
89. Lizundia R. c-Jun NH2-terminal Kinase/c-Jun signaling promotes survival and metastasis of B lymphocytes transformed by Theileria. Cancer Res (2006) 66:6105–10. doi:10.1158/0008-5472.can-05-3861
90. Liu J, Deng M, Lancto CA, Abrahamsen MS, Rutherford MS, Enomoto S. Biphasic modulation of apoptotic pathways in Cryptosporidium parvum-infected human intestinal epithelial cells. Infect Immun (2009) 77:837–49. doi:10.1128/IAI.00955-08
91. Brumlik MJ, Pandeswara S, Ludwig SM, Jeansonne DP, Lacey MR, Murthy K, et al. TgMAPK1 is a Toxoplasma gondii MAP kinase that hijacks host MKK3 signals to regulate virulence and interferon-γ-mediated nitric oxide production. Exp Parasitol (2013) 134:389–99. doi:10.1016/j.exppara.2013.03.016
92. Muñoz-Caro T, Lendner M, Daugschies A, Hermosilla C, Taubert A. NADPH oxidase, MPO, NE, ERK1/2, p38 MAPK and Ca2+ influx are essential for Cryptosporidium parvum-induced NET formation. Dev Comp Immunol (2015) 52:245–54. doi:10.1016/j.dci.2015.05.007
93. Li Z-Y, Wang Z-D, Huang S-Y, Zhu X-Q, Liu Q. TgERK7 is involved in the intracellular proliferation of Toxoplasma gondii. Parasitol Res (2016) 115:3419–24. doi:10.1007/s00436-016-5103-5
94. Duszenko M, Ginger ML, Brennand A, Gualdrón-López M, Colombo MI, Coombs GH, et al. Autophagy in protists. Autophagy (2011) 7:127–58. doi:10.4161/auto.7.2.13310
95. Muniz-Feliciano L, Van Grol J, Portillo J-AC, Liew L, Liu B, Carlin CR, et al. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog (2013) 9:e1003809. doi:10.1371/journal.ppat.1003809
96. Metheni M, Echebli N, Chaussepied M, Ransy C, Chéreau C, Jensen K, et al. The level of H2O2type oxidative stress regulates virulence of Theileria-transformed leukocytes. Cell Microbiol (2013) 16:269–79. doi:10.1111/cmi.12218
97. Treeck M, Sanders JL, Gaji RY, LaFavers KA, Child MA, Arrizabalaga G, et al. The calcium-dependent protein kinase 3 of Toxoplasma influences basal calcium levels and functions beyond egress as revealed by quantitative phosphoproteome analysis. PLoS Pathog (2014) 10:e1004197. doi:10.1371/journal.ppat.1004197
98. Hong S, Kim J-H, Yoon S, Kim K, Sim S, Park WY, et al. Expression of Cryptosporidium parvum thioredoxin peroxidase in COS-7 cells confers radioprotection. Exp Parasitol (2016) 163:8–15. doi:10.1016/j.exppara.2016.01.012
99. Bosch SS, Kronenberger T, Meissner KA, Zimbres FM, Stegehake D, Izui NM, et al. Oxidative stress control by apicomplexan parasites. Biomed Res Int (2015) 2015:351289. doi:10.1155/2015/351289
100. Durrani Z, Weir W, Pillai S, Kinnaird J, Shiels B. Modulation of activation-associated host cell gene expression by the apicomplexan parasite Theileria annulata. Cell Microbiol (2012) 14:1434–54. doi:10.1111/j.1462-5822.2012.01809.x
101. Chiwakata CB, Hemmer CJ, Dietrich M. High levels of inducible nitric oxide synthase mRNA are associated with increased monocyte counts in blood and have a beneficial role in Plasmodium falciparum malaria. Infect Immun (2000) 68:394–9. doi:10.1128/IAI.68.1.394-399.2000
102. Goff WL, Johnson WC, Cluff CW. Babesia bovis immunity: in vitro and in vivo evidence for IL-10 regulation of IFN-γ and iNOS. Ann N Y Acad Sci (1998) 849:161–80. doi:10.1111/j.1749-6632.1998.tb11046.x
103. Metheni M, Lombès A, Bouillaud F, Batteux F, Langsley G. HIF-1α induction, proliferation and glycolysis of Theileria-infected leukocytes. Cell Microbiol (2015) 17:467–72. doi:10.1111/cmi.12421
104. Menendez MT, Teygong C, Wade K, Florimond C, Blader IJ. siRNA screening identifies the host hexokinase 2 (HK2) gene as an important hypoxia-inducible transcription factor 1 (HIF-1) target gene in Toxoplasma gondii-infected cells. MBio (2015) 6:e00462. doi:10.1128/mbio.00462-15
105. Chandramohanadas R, Davis PH, Beiting DP, Harbut MB, Darling C, Velmourougane G, et al. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science (2009) 324:794–7. doi:10.1126/science.1171085
106. Jongejan F, Uilenberg G. The global importance of ticks. Parisitology (2004) 129:3–14. doi:10.1017/S0031182004005967
107. George N, Bhandari V, Reddy DP, Sharma P. Molecular and phylogenetic analysis revealed new genotypes of Theileria annulata parasites from India. Parasit Vectors (2015) 8:468. doi:10.1186/s13071-015-1075-z
108. Shaw MK. Cell invasion by Theileria sporozoites. Trends Parasitol (2003) 19:2–6. doi:10.1016/s1471-4922(02)00015-6
109. Shiels B, Langsley G, Weir W, Pain A, McKellar S, Dobbelaere D. Alteration of host cell phenotype by Theileria annulata and Theileria parva: mining for manipulators in the parasite genomes. Int J Parasitol (2006) 36:9–21. doi:10.1016/j.ijpara.2005.09.002
110. Mans BJ, Pienaar R, Latif AA. A review of Theileria diagnostics and epidemiology. Int J Parasitol (2015) 4:104–18. doi:10.1016/j.ijppaw.2014.12.006
111. Shaw MK, Tilney LG, Musoke AJ. The entry of Theileria parva sporozoites into bovine lymphocytes: evidence for MHC class I involvement. J Cell Biol (1991) 113:87–101. doi:10.1083/Jcb.113.1.87
112. Shaw MK. The same but different: the biology of Theileria sporozoite entry into bovine cells. Int J Parasitol (1997) 27:457–74. doi:10.1016/s0020-7519(97)00015-5
113. Dobbelaere DAE, Fernandez PC, Heussler VT. Theileria parva: taking control of host cell proliferation and survival mechanisms. Cell Microbiol (2000) 2:91–9. doi:10.1046/j.1462-5822.2000.00045.x
114. Dobbelaere DAE, Küenzi P. The strategies of the Theileria parasite: a new twist in host-pathogen interactions. Curr Opin Immunol (2004) 16:524–30. doi:10.1016/j.coi.2004.05.009
115. Dessauge F, Hilaly S, Baumgartner M, Blumen B, Werling D, Langsley G. c-Myc activation by Theileria parasites promotes survival of infected B-lymphocytes. Oncogene (2005) 24:1075–83. doi:10.1038/sj.onc.1208314
116. Tretina K, Gotia HT, Mann DJ, Silva JC. Theileria-transformed bovine leukocytes have cancer hallmarks. Trends Parasitol (2015) 31:306–14. doi:10.1016/j.pt.2015.04.001
117. Marsolier J, Perichon M, DeBarry JD, Villoutreix BO, Chluba J, Lopez T, et al. Theileria parasites secrete a prolyl isomerase to maintain host leukocyte transformation. Nature (2015) 520:378–82. doi:10.1038/nature14044
118. Wiens O, Xia D, von Schubert C, Wastling JM, Dobbelaere DAE, Heussler VT, et al. Cell cycle-dependent phosphorylation of Theileria annulata schizont surface proteins. PLoS One (2014) 9:e103821. doi:10.1371/journal.pone.0103821
119. Swan DG, Stern R, McKellar S, Phillips K, Oura CA, Karagenc TI, et al. Characterisation of a cluster of genes encoding Theileria annulata AT hook DNA-binding proteins and evidence for localisation to the host cell nucleus. J Cell Sci (2001) 114:2747–54.
120. Shiels BR, McKellar S, Katzer F, Lyons K, Kinnaird J, Ward C, et al. A Theileria annulata DNA binding protein localized to the host cell nucleus alters the phenotype of a bovine macrophage cell line. Eukaryot Cell (2004) 3:495–505. doi:10.1128/ec.3.2.495-505.2004
121. Marsolier J, Pineau S, Medjkane S, Perichon M, Yin Q, Flemington E, et al. OncomiR addiction is generated by a miR-155 feedback loop in Theileria-transformed leukocytes. PLoS Pathog (2013) 9:e1003222. doi:10.1371/journal.ppat.1003222
122. Dobbelaere D, Heussler V. Transformation of leukocytes byTheileria parva and T. annulata. Annu Rev Microbiol (1999) 53:1–42. doi:10.1146/annurev.micro.53.1.1
123. Ahmed JS, Schnittger L, Mehlhorn H. Review: Theileria schizonts induce fundamental alterations in their host cells. Parasitol Res (1999) 85:527–38. doi:10.1007/s004360050592
124. Frénal K, Soldati-Favre D. Role of the parasite and host cytoskeleton in apicomplexa parasitism. Cell Host Microbe (2009) 5:602–11. doi:10.1016/j.chom.2009.05.013
125. De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, et al. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature (2001) 414:308–13. doi:10.1038/35104560
126. Chaussepied M, Janski N, Baumgartner M, Lizundia R, Jensen K, Weir W, et al. TGF-b2 induction regulates invasiveness of Theileria-transformed leukocytes and disease susceptibility. PLoS Pathog (2010) 6:e1001197. doi:10.1371/journal.ppat.1001197
127. Haidar M, Echebli N, Ding Y, Kamau E, Langsley G. Transforming growth factor β2 promotes transcription of COX2 and EP4, leading to a prostaglandin E2-driven autostimulatory loop that enhances virulence of Theileria annulata-transformed macrophages. Infect Immun (2015) 83:1869–80. doi:10.1128/iai.02975-14
128. Botteron C, Dobbelaere D. AP-1 and ATF-2 are constitutively activated via the JNK pathway in Theileria parva-transformed T-cells. Biochem Biophys Res Commun (1998) 246:418–21. doi:10.1006/bbrc.1998.8635
129. Chaussepied M, Lallemand D, Moreau M-F, Adamson R, Hall R, Langsley G. Upregulation of Jun and Fos family members and permanent JNK activity lead to constitutive AP-1 activation in Theileria-transformed leukocytes. Mol Biochem Parasitol (1998) 94:215–26. doi:10.1016/s0166-6851(98)00070-x
130. Lizundia R, Chaussepied M, Naissant B, Masse GX, Quevillon E, Michel F, et al. The JNK/AP-1 pathway upregulates expression of the recycling endosome rab11a gene in B cells transformed by Theileria. Cell Microbiol (2007) 9:1936–45. doi:10.1111/j.1462-5822.2007.00925.x
131. Hakimi M, Cannella D. Apicomplexan parasites and subversion of the host cell microRNA pathway. Trends Parasitol (2011) 27:481–6. doi:10.1016/j.pt.2011.07.001
132. Baylis H. Infection with Theileria annulata induces expression of matrix metalloproteinase 9 and transcription factor AP-1 in bovine leucocytes. Mol Biochem Parasitol (1995) 69:211–22. doi:10.1016/0166-6851(94)00216-a
133. Adamson R, Logan M, Kinnaird J, Langsley G, Hall R. Loss of matrix metalloproteinase 9 activity in Theileria annulata-attenuated cells is at the transcriptional level and is associated with differentially expressed AP-1 species. Mol Biochem Parasitol (2000) 106:51–61. doi:10.1016/s0166-6851(99)00213-3
134. Guergnon J, Chaussepied M, Sopp P, Lizundia R, Moreau M-F, Blumen B, et al. A tumour necrosis factor alpha autocrine loop contributes to proliferation and nuclear factor-kappaB activation of Theileria parva-transformed B cells. Cell Microbiol (2003) 5:709–16. doi:10.1046/j.1462-5822.2003.00314.x
135. Heussler V, Sturm A, Langsley G. Regulation of host cell survival by intracellular Plasmodium and Theileria parasites. Parasitology (2006) 132:S49. doi:10.1017/s0031182006000850
137. Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol (2004) 24:7806–19. doi:10.1128/MCB.24.17.7806-7819.2004
138. Medjkane S, Perichon M, Marsolier J, Dairou J, Weitzman JB. Theileria induces oxidative stress and HIF1α activation that are essential for host leukocyte transformation. Oncogene (2013) 33:1809–17. doi:10.1038/onc.2013.134
139. Medjkane S, Weitzman JB. A reversible Warburg effect is induced by Theileria parasites to transform host leukocytes. Cell Cycle (2013) 12:2167–8. doi:10.4161/cc.25540
140. Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-B prevents apoptosis. Mol Cell Biol (2004) 24:1007–21. doi:10.1128/mcb.24.3.1007-1021.2004
141. Xie Y, Naizabekov S, Chen Z, Tokay T. Power of PTEN/AKT: molecular switch between tumor suppressors and oncogenes (Review). Oncol Lett (2016) 12:375–8. doi:10.3892/ol.2016.4636
142. Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol (2000) 30:1217–58. doi:10.1016/s0020-7519(00)00124-7
143. Vargas-Villavicencio JA, Besné-Mérida A, Correa D. Vertical transmission and fetal damage in animal models of congenital toxoplasmosis: a systematic review. Vet Parasitol (2016) 223:195–204. doi:10.1016/j.vetpar.2016.04.024
144. Berghold C, Herzog SA, Jakse H, Berghold A. Prevalence and incidence of toxoplasmosis: a retrospective analysis of mother-child examinations, Styria, Austria, 1995 to 2012. Euro Surveill (2016) 21:30317. doi:10.2807/1560-7917.es.2016.21.33.30317
145. Elsheikha HM, Büsselberg D, Zhu X-Q. The known and missing links between Toxoplasma gondii and schizophrenia. Metab Brain Dis (2016) 31:749–59. doi:10.1007/s11011-016-9822-1
146. Torgerson PR, Mastroiacovo P. The global burden of congenital toxoplasmosis: a systematic review. Bull World Health Organ (2013) 91:501–8. doi:10.2471/blt.12.111732
147. Furtado J, Smith J, Belfort R, Gattey D, Winthrop K. Toxoplasmosis: a global threat. J Glob Infect Dis (2011) 3:281. doi:10.4103/0974-777x.83536
148. Jones JL, Dubey JP. Foodborne toxoplasmosis. Clin Infect Dis (2012) 55:845–51. doi:10.1093/cid/cis508
149. Dubey JP. History of the discovery of the life cycle of Toxoplasma gondii. Int J Parasitol (2009) 39:877–82. doi:10.1016/j.ijpara.2009.01.005
150. Dubey JP. Toxoplasma gondii. Medical Microbiology (1996). p. 1–11. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21413265
151. Dubey JP. Advances in the life cycle of Toxoplasma gondii. Int J Parasitol (1998) 28:1019–24. doi:10.1016/s0020-7519(98)00023-x
152. Dubey JP. Comparative infectivity of oocysts and bradyzoites of Toxoplasma gondii for intermediate (mice) and definitive (cats) hosts. Vet Parasitol (2006) 140:69–75. doi:10.1016/j.vetpar.2006.03.018
153. Sibley LD. Intracellular parasite invasion strategies. Science (2004) 304:248–53. doi:10.1126/science.1094717
154. Pollard AM, Onatolu KN, Hiller L, Haldar K, Knoll LJ. Highly polymorphic family of glycosylphosphatidylinositol-anchored surface antigens with evidence of developmental regulation in Toxoplasma gondii. Infect Immun (2007) 76:103–10. doi:10.1128/iai.01170-07
155. Blader IJ, Saeij JP. Communication between Toxoplasma gondii and its host: impact on parasite growth, development, immune evasion, and virulence. APMIS (2009) 117:458–76. doi:10.1111/j.1600-0463.2009.02453.x
156. Lourido S, Tang K, Sibley LD. Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J (2012) 31:4524–34. doi:10.1038/emboj.2012.299
157. Bonhomme A, Bouchot A, Pezzella N, Gomez J, Le Moal H, Pinon JM. Signaling during the invasion of host cells by Toxoplasma gondii. FEMS Microbiol Rev (1999) 23:551–61. doi:10.1016/S0168-6445(99)00021-2
158. Heintzelman MB. Gliding motility in apicomplexan parasites. Semin Cell Dev Biol (2015) 46:135–42. doi:10.1016/j.semcdb.2015.09.020
159. Alexander DL, Mital J, Ward GE, Bradley P, Boothroyd JC. Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog (2005) 1:e17. doi:10.1371/journal.ppat.0010017
160. Straub KW, Cheng SJ, Sohn CS, Bradley PJ. Novel components of the apicomplexan moving junction reveal conserved and coccidia-restricted elements. Cell Microbiol (2009) 11:590–603. doi:10.1111/j.1462-5822.2008.01276.x
161. Lamarque M, Besteiro S, Papoin J, Roques M, Vulliez-Le Normand B, Morlon-Guyot J, et al. The RON2-AMA1 interaction is a critical step in moving junction-dependent invasion by apicomplexan parasites. PLoS Pathog (2011) 7:e1001276. doi:10.1371/journal.ppat.1001276
162. Poukchanski A, Fritz HM, Tonkin ML, Treeck M, Boulanger MJ, Boothroyd JC. Toxoplasma gondii sporozoites invade host cells using two novel paralogues of RON2 and AMA1. PLoS One (2013) 8:e70637. doi:10.1371/journal.pone.0070637
163. Etheridge RD, Alaganan A, Tang K, Lou HJ, Turk BE, Sibley LD. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe (2014) 15:537–50. doi:10.1016/j.chom.2014.04.002
164. Hakimi M-A, Olias P, Sibley LD. Toxoplasma effectors targeting host signaling and transcription. Clin Microbiol Rev (2017) 30:615–45. doi:10.1128/CMR.00005-17
165. Blader IJ, Koshy AA. Toxoplasma gondii development of its replicative niche: in its host cell and beyond. Eukaryot Cell (2014) 13:965–76. doi:10.1128/EC.00081-14
166. Hwang I-Y, Quan JH, Ahn M-H, Hassan Ahmed HA, Cha G-H, Shin D-W, et al. Toxoplasma gondii infection inhibits the mitochondrial apoptosis through induction of Bcl-2 and HSP70. Parasitol Res (2010) 107:1313–21. doi:10.1007/s00436-010-1999-3
167. Wang Y, Weiss LM, Orlofsky A. Intracellular parasitism with Toxoplasma gondii stimulates mammalian-target-of-rapamycin-dependent host cell growth despite impaired signalling to S6K1 and 4E-BP1. Cell Microbiol (2009) 11:983–1000. doi:10.1111/j.1462-5822.2009.01305.x
168. Wang Y. Modulation of Host Cell mTOR Signaling by Toxoplasma gondii. New York: Yeshiva University, ProQuest Dissertations Publishing (2010). 3385052 p.
169. Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KDC, et al. Strain-specific activation of the NF-κB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med (2011) 208:195–212. doi:10.1084/jem.20100717
170. Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, et al. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog (2011) 7:e1002236. doi:10.1371/journal.ppat.1002236
171. Jensen KDC, Wang Y, Wojno EDT, Shastri AJ, Hu K, Cornel L, et al. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe (2011) 9:472–83. doi:10.1016/j.chom.2011.04.015
172. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev (1999) 13:2905–27. doi:10.1101/gad.13.22.2905
173. Gay G, Braun L, Brenier-Pinchart M-P, Vollaire J, Josserand V, Bertini R-L, et al. Toxoplasma gondii TgIST co-opts host chromatin repressors dampening STAT1-dependent gene regulation and IFN-γ–mediated host defenses. J Exp Med (2016) 213:1779–98. doi:10.1084/jem.20160340
174. Peng BW, Lin JY, Zhang T. Toxoplasma gondii induces prostaglandin E2 synthesis in macrophages via signal pathways for calcium-dependent arachidonic acid production and PKC-dependent induction of cyclooxygenase-2. Parasitol Res (2008) 102:1043–50. doi:10.1007/s00436-007-0873-4
175. Zare-Bidaki M, Assar S, Hakimi H, Abdollahi SH, Nosratabadi R, Kennedy D, et al. TGF-β in toxoplasmosis: friend or foe? Cytokine (2016) 86:29–35. doi:10.1016/j.cyto.2016.07.002
176. Wiley M, Sweeney KR, Chan DA, Brown KM, McMurtrey C, Howard EW, et al. Toxoplasma gondii activates hypoxia-inducible factor (HIF) by stabilizing the HIF-1α subunit via type I activin-like receptor kinase receptor signaling. J Biol Chem (2010) 285:26852–60. doi:10.1074/jbc.m110.147041
177. Blader IJ, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J Biol Chem (2001) 276:24223–31. doi:10.1074/jbc.M100951200
178. Spear W, Chan D, Coppens I, Johnson RS, Giaccia A, Blader IJ. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol (2006) 8:339–52. doi:10.1111/j.1462-5822.2005.00628.x
179. Braun L, Brenier-Pinchart M-P, Yogavel M, Curt-Varesano A, Curt-Bertini R-L, Hussain T, et al. A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J Exp Med (2013) 210:2071–86. doi:10.1084/jem.20130103
180. Xiao J, Li Y, Prandovszky E, Karuppagounder SS, Talbot CC, Dawson VL, et al. MicroRNA-132 dysregulation in Toxoplasma gondii infection has implications for dopamine signaling pathway. Neuroscience (2014) 268:128–38. doi:10.1016/j.neuroscience.2014.03.015
181. Brooks JM, Carrillo GL, Su J, Lindsay DS, Fox MA, Blader IJ. Toxoplasma gondii infections alter GABAergic synapses and signaling in the central nervous system. MBio (2015) 6:e01428–15. doi:10.1128/mbio.01428-15
182. Roiko MS, Carruthers VB. New roles for perforins and proteases in apicomplexan egress. Cell Microbiol (2009) 11:1444–52. doi:10.1111/j.1462-5822.2009.01357.x
183. Laurent F, McCole D, Eckmann L, Kagnoff MF. Pathogenesis of Cryptosporidium parvum infection. Microbes Infect (1999) 1:141–8. doi:10.1016/s1286-4579(99)80005-7
184. Di Genova BM, Tonelli RR. Infection strategies of intestinal parasite pathogens and host cell responses. Front Microbiol (2016) 7:256. doi:10.3389/fmicb.2016.00256
185. Medema G, Teunis P, Blokker M, Deere D, Charles P, Loret J-F. EHC Cryptosporidium Draft 2. 1. WHO Guidelines for Drinking Water Quality. Geneva: World Health Organization Press (2006).
186. Rossle NF, Latif B. Cryptosporidiosis as threatening health problem: a review. Asian Pac J Trop Biomed (2013) 3:916–24. doi:10.1016/s2221-1691(13)60179-3
187. Ward H, Cevallos A. Cryptosporidium: molecular basis of host-parasite interaction. Adv Parasitol (1998) 40:151–85. doi:10.1016/s0065-308x(08)60120-7
188. Bouzid M, Hunter PR, Chalmers RM, Tyler KM. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev (2013) 26:115–34. doi:10.1128/cmr.00076-12
189. Forney JR, DeWald DB, Yang S, Speer CA, Healey MC. A role for host phosphoinositide 3-kinase and cytoskeletal remodeling during Cryptosporidium parvum infection. Infect Immun (1999) 67:844–52.
190. O’Hara SP, Small AJ, Chen XM, Larusso NF. Host cell actin remodeling in response to Cryptosporidium. Subcell Biochem (2008) 47:92–100. doi:10.1007/978-0-387-78267-6_7
191. Yao L, Yin J, Zhang X, Liu Q, Li J, Chen L, et al. Cryptosporidium parvum: identification of a new surface adhesion protein on sporozoite and oocyst by screening of a phage-display cDNA library. Exp Parasitol (2007) 115:333–8. doi:10.1016/j.exppara.2006.09.018
192. Singh P, Mirdha BR, Srinivasan A, Rukmangadachar LA, Singh S, Sharma P, et al. Identification of invasion proteins of Cryptosporidium parvum. World J Microbiol Biotechnol (2015) 31:1923–34. doi:10.1007/s11274-015-1936-9
193. Lendner M, Daugschies A. Cryptosporidium infections: molecular advances. Parasitology (2014) 141:1511–32. doi:10.1017/s0031182014000237
194. Varughese EA, Kasper S, Anneken EM, Yadav JS. SHP-2 mediates Cryptosporidium parvum infectivity in human intestinal epithelial cells. PLoS One (2015) 10:e0142219. doi:10.1371/journal.pone.0142219
195. Chen X-M, Huang BQ, Splinter PL, Cao H, Zhu G, Mcniven MA, et al. Cryptosporidium parvum invasion of biliary epithelia requires host cell tyrosine phosphorylation of cortactin via c-Src. Gastroenterology (2003) 125:216–28. doi:10.1016/s0016-5085(03)00662-0
196. Chen X-M, O’Hara SP, Huang BQ, Splinter PL, Nelson JB, LaRusso NF. Localized glucose and water influx facilitates Cryptosporidium parvum cellular invasion by means of modulation of host-cell membrane protrusion. Proc Natl Acad Sci U S A (2005) 102:6338–43. doi:10.1073/pnas.0408563102
197. Perez-Cordon G, Nie W, Schmidt D, Tzipori S, Feng H. Involvement of host calpain in the invasion of Cryptosporidium parvum. Microbes Infect (2011) 13:103–7. doi:10.1016/j.micinf.2010.10.007
198. Keyloun KR, Reid MC, Choi R, Song Y, Fox AMW, Hillesland HK, et al. The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology (2014) 141:1499–509. doi:10.1017/s0031182014000857
199. Manque PA, Woehlbier U, Lara AM, Tenjo F, Alves JM, Buck GA. Identification and characterization of a novel calcium-activated apyrase from Cryptosporidium parasites and its potential role in pathogenesis. PLoS One (2012) 7:e31030. doi:10.1371/journal.pone.0031030
200. Schaumburg F, Hippe D, Vutova P, Lüder CGK, Der CGKL. Pro- and anti-apoptotic activities of protozoan parasites pro-and anti-apoptotic activities of protozoan parasites. Parasitology (2006) 132:69–85. doi:10.1017/S0031182006000874
201. Chen XM, Gores GJ, Paya CV, LaRusso NF. Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am J Physiol (1999) 277:G599–608.
202. Liu J, Enomoto S, Lancto CA, Abrahamsen MS, Rutherford MS. Inhibition of apoptosis in Cryptosporidium parvum-infected intestinal epithelial cells is dependent on survivin. Infect Immun (2008) 76(8):3784–92. doi:10.1128/IAI.00308-08
203. Castellanos-Gonzalez A, Yancey LS, Wang H, Pantenburg B, Liscum KR, Lewis DE, et al. Cryptosporidium infection of human intestinal epithelial cells increases expression of osteoprotegerin: a novel mechanism for evasion of host defenses. J Infect Dis (2008) 197:916–23. doi:10.1086/528374
204. Choudhry N, Korbel DS, Edwards LA, Bajaj-Elliott M, McDonald V. Dysregulation of interferon-γ-mediated signalling pathway in intestinal epithelial cells by Cryptosporidium parvum infection. Cell Microbiol (2009) 11:1354–64. doi:10.1111/j.1462-5822.2009.01336.x
205. Robinson P, Okhuysen PC, Chappell CL, Lewis DE, Shahab I, Lahoti S, et al. Transforming growth factor beta 1 is expressed in the jejunum after experimental Cryptosporidium parvum infection in humans. Infect Immun (2000) 68:5405–7. doi:10.1128/iai.68.9.5405-5407.2000
Keywords: Plasmodium, Toxoplasma, Theileria, Babesia, Cryptosporidium, host signaling pathways
Citation: Chakraborty S, Roy S, Mistry HU, Murthy S, George N, Bhandari V and Sharma P (2017) Potential Sabotage of Host Cell Physiology by Apicomplexan Parasites for Their Survival Benefits. Front. Immunol. 8:1261. doi: 10.3389/fimmu.2017.01261
Received: 26 July 2017; Accepted: 21 September 2017;
Published: 13 October 2017
Edited by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilReviewed by:
Avery August, Cornell University, United StatesHem Chandra Jha, University of Pennsylvania, United States
Copyright: © 2017 Chakraborty, Roy, Mistry, Murthy, George, Bhandari and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paresh Sharma, cGFyZXNoQG5pYWIub3JnLmlu, cGFyZXNoc2hhcm1hMjFAZ21haWwuY29t
†Shared Senior Authorship.