 Amy Houghtelin
Amy Houghtelin Catherine M. Bollard*
Catherine M. Bollard*
- Program for Cell Enhancement and Technologies for Immunotherapy, Children’s National Health System, The George Washington University, Washington, DC, United States
While progress has been made in the treatment of both hematologic cancers and solid tumors, chemorefractory or relapsed disease often portends a dismal prognosis, and salvage chemotherapy or radiation expose patients to intolerable toxicities and may not be effective. Hematopoietic stem cell transplant offers the promise of cure for many patients, and while mismatched, unrelated or haploidentical donors are increasingly available, the recipients are at higher risk of severe immunosuppression and immune dysregulation due to graft versus host disease. Viral infections remain a primary cause of severe morbidity and mortality in this patient population. Again, many therapeutic options for viral disease are toxic, may be ineffective or generate resistance, or fail to convey long-term protection. Adoptive cell therapy with virus-specific T cells (VSTs) is a targeted therapy that is efficacious and has minimal toxicity in immunocompromised patients with CMV and EBV infections in particular. Products have since been generated specific for multiple viral antigens (multi-VST), which are not only effective but also confer protection in 70–90% of recipients when used as prophylaxis. Notably, these products can be generated from either virus-naive or virus-experienced autologous or allogeneic sources, including partially matched HLA-matched third-party donors. Obstacles to effective VST treatment are donor availability and product generation time. Banking of third-party VST is an attractive way to overcome these constraints and provide products on an as-needed basis. Other developments include epitope discovery to broaden the number of viral antigens targets in a single product, the optimization of VST generation from naive donor sources, and the modification of VSTs to enhance persistence and efficacy in vivo.
Introduction
While hematopoietic stem cell transplant (HSCT) offers a chance of cure for patients with many high risk cancers or primary immunodeficiency syndromes, transplant recipients remain vulnerable to infectious complications due to prolonged and profound immunosuppression (1–4). These risks are modified by preparative regimen, transplant type, and duration of myelosuppression (1–4). With advances in conditioning regimens and improved posttransplant management, an increasing number of patients are eligible to receive mismatched, unrelated, or haploidentical donor HSCT. While there have been great improvements in outcome for patients with severe or otherwise untreatable disease, the immunosuppression required for engraftment and, when indicated, to treat graft versus host disease (GVHD), opens the door for infection. In particular, viral infections cause significant morbidity and mortality, and the risk increases when T cell immune reconstitution is delayed (1–3). The relationship between immunosuppression, immune reconstitution, and the effects of GVHD, and infection are complicated and intertwined (5). Pharmacologic treatment and prophylactic options for viral infections remain limited and often ineffective, with associated morbidities notably from acute kidney injury and myelosuppression. Treatment may also generate resistance, and does not confer extended protection leaving patients at risk for viral reactivation (4). Given the correlation between delay in T cell immune recovery and viral disease, adoptive cell therapy is a logical alternative to pharmacologic therapy. Unmanipulated lymphocyte infusions from seropositive donors have been infused in patients with life-threatening disease such as EBV-associated lymphoma, demonstrating clinical efficacy with risks primarily associated with GVHD (6). This strategy has evolved over the past two decades, and donor lymphocyte products have been successful in reconstituting viral immunity in the host as a treatment for viral disease (including reactivation, new exposure, and lymphoma) and as prophylaxis (7). Following these initial studies, virus-specific T cell (VST) selection and/or expansion has been refined to maximize viral cytotoxicity and minimize alloreactivity to reduce and largely eliminate the risk of GVHD. In the current studies, VSTs offer targeted therapy and have demonstrated a very good safety profile to date (8–11). This review will detail developments in the manufacturing process, describe clinical success of VSTs and discuss future directions, including the use of naive donor sources and third-party banks.
Materials and Methods
Antigen Selection
To successfully generate and expand VSTs, specific immunogenic epitopes need to be defined for each pathogen. It is well established that some viruses, notably CMV and EBV, are known to have certain antigens expressed at various stages of disease (12–14). Using available tools, epitope mapping has allowed identification of immunogenic antigens for other viruses, including adenovirus, human herpes virus 6 (HHV6), and BK virus (15–18). For many of these viruses, the immunodominant and subdominant antigens have been characterized, as well as antigens which promote enhanced T cell proliferation and immune protection in vivo (19). Several methods have been used to expand and select VSTs. Most recently, antigen-presenting cells (APCs) exposed to peptide mixtures consisting of overlapping, 15mer peptide libraries have proved highly successful for direct stimulation of CD4+ and CD8+ T cells (17–19). Alternative approaches use APC exposed to whole virus, viral lysates, whole proteins, or viral vectors (7, 9, 15, 20–24).
Antigen Presentation
Once an appropriate antigen has been identified as an immune target, it must be effectively presented by APC to T cells in conjunction with costimulatory signals to promote T cell activation and proliferation. The APC type impacts production time, cell numbers, and product phenotype. Examples include dendritic cells, monocytes, B cells, and various artificial APCs. Table 1 summarizes antigen and APC options. While dendritic cells are very effective APCs, they are limited by low numbers; thus, repeat stimulations require increasing amounts of donor cells. Whole virus has also been used to create potent APCs. For example, EBV lymphoblastoid cell lines (LCLs) infected with the B95-8 EBV strain, are effective and safe APC for generation of clinically useful products (9). However, this strategy is limited by lengthy incubation time with a potential risk of infection. Donor PHA blasts are potent, polyclonal stimulators of T cells and require only low cell numbers for generation (19). Artificial K-562 cells are another potential option for APC, and may be especially helpful to provide costimulation for generating VSTs from seronegative donors. This complementary costimulation in the presence of artificial APCs creates an effective antigen-presenting complex to promote VST stimulation and expansion (25).
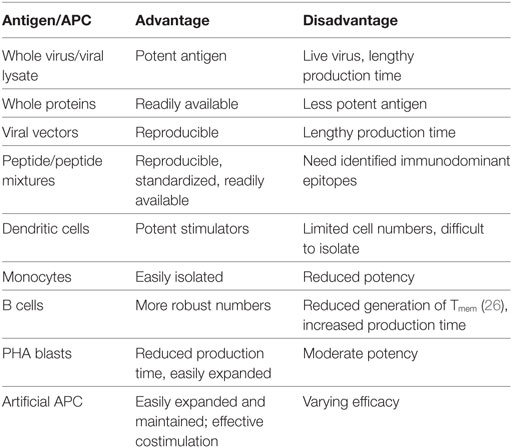
Table 1. Antigen selection and presentation.
T Cell Expansion
Initial attempts at adoptive immunotherapy used unmanipulated donor lymphocyte infusions to transfer cytotoxic and memory T cells specific for certain viral infections. While an effective antiviral strategy, a major complication was GVHD, a natural consequence from infusing alloreactive T cells (6, 27). Furthermore this strategy was essentially limited to EBV and CMV where high frequencies of VSTs circulate in the donor (28). Modifications in the generation and ex vivo expansion of these T cell products have minimized GVHD to an almost negligible risk (28, 29). These processes have evolved over time to select and expand VSTs while minimizing alloreactive or naive T cells in the final product, with the emphasis currently on reducing production times and maximizing product functionality.
Reduction of Alloreactive Cells
Several strategies have been aimed at inactivating or removing alloreactive T cells from donor products. One strategy evaluated blockade via monoclonal antibodies to the B7:CD28 costimulatory complex to produce an anergic response to recipient cells (30). This was successful, although time-consuming and not completely effective at preventing GVHD. Other strategies have employed selective depletion of alloreactive cells, either ex vivo or in vivo. Ex vivo methods use cells stimulated by recipient APCs followed by targeting alloreactive T cells through CD25, CD69, or CD95 and eliminating them by magnetic-coupled monoclonal antibodies or immununotoxins, photodynamic depletion, or apoptosis activation (31–34). These methods have seen some success in vivo, but results have been unreliable in terms of conveying viral protection and preventing GVHD. In vivo strategies employ a safety or suicide switch to deplete alloreactive T cells, to induce apoptosis in response to a specific signal. A thymidine kinase gene from herpes simplex virus I, acting as a trigger for cell elimination via ganciclovir exposure, was found to be effective but hampered by increased immunogenicity and a delay of several days to clinical effect after ganciclovir administration (35). Newer studies have transduced cells with the suicide gene-inducible caspase 9 (iC9), which triggers apoptosis after exposure to a dimerizing drug (36, 37). In this case, VSTs conveyed viral protection, and patients showed clinical improvement of GVHD symptoms soon after administration of the “safety switch” dimerizing drug.
Approaches for Selection and Expansion of VSTs
Isolation of VSTs with or without ex vivo expansion offers an alternative means of eliminating alloreactive cells. More recent methods have simplified this process to reduce production times and simplify manufacturing strategies.
Selection of VSTs
Virus-specific T cells may be isolated directly from donor peripheral blood with the use of peptide-HLA multimers to facilitate the identification and purification of antigen-specific T cells. This process was originally hindered by irreversible binding and significant changes in T cell phenotype. The use of streptamers greatly improved this method, acting as multimers that use an HLA-peptide complex to reversibly bind the desired T cells without altering T cell phenotype or functional status. While a major benefit of this method is the rapid availability of an antigen-specific T cell product, the selected T cells are limited by HLA-restriction imposed by the streptamer (38–40). This process also requires knowledge of defined Class I HLA-restricted viral epitopes for effective isolation, and it selects for a limited repertoire of CD8+ cells rather than the entire polyclonal, polyfunctional population of CD4+ and CD8+ T cells recognizing the full spectrum of available viral antigens. Despite noted limitations, investigators have been successful using VSTs isolated in this manner in the adoptive therapy for diseases with higher numbers of circulating VSTs, such as CMV and EBV. Such infused VSTs have also demonstrated expansion in vivo following transfer of these selected cells (41).
Interferon-γ (IFN-γ) capture also directly selects circulating VSTs from peripheral blood. Peripheral blood mononuclear cells (PBMCs) are stimulated with antigens specific for targeted virus and incubated over 4–16 h, inducing IFN-γ production in stimulated cells. A monoclonal antibody to IFN-γ coupled to a leukocyte-specific antibody (anti-CD45) then captures the IFN-γ producing cells, which are then selected via magnetic beads. This also allows rapid selection of VSTs free of HLA-restriction with the added benefit of stimulating and capturing a polyclonal population of CD4+ and CD8+ cells. This is clinically relevant, as the presence of CD4+ cells enhances the memory and effector response and supports persistence and expansion of the cytotoxic T cells (42, 43). It also allows for selection of VSTs responding to multiple viral epitopes and has been successful in generating clinically functional VSTs targeted to various viruses (44–47).
Both these capture methods allow for rapid and precise selection of circulating VSTs, with obvious benefits for timely treatment of patients with active disease. However, they require VSTs to be circulating at a detectable level and leukapheresis is often needed to collect clinically relevant cell numbers. These methods are thus not useful for naive donors and ineffective if numbers of circulating VSTs are too low to generate a useful product.
Expansion of VSTs
The process of ex vivo T cell selection and expansion has been refined over the past two decades, with an emphasis on decreasing production times and complexity and optimizing in vivo function. The first techniques using EBV-LCL lines required at least 3 months to generate the APC and make a product. Despite still taking at least 10 days, current VST culture expansion systems generate polyclonal and polyfunctional products, properties which enhance in vivo expansion, function, and persistence. Clinical trials using ex vivo stimulated and expanded VSTs show that infused T cells persist long-term, detectible by gene-marking studies for as long as 9 years (48, 49). Ex vivo stimulation and expansion requires only a small volume of blood to establish the culture, eliminating the need for costly, time-consuming, and invasive leukapheresis. Lastly, expansion cultures make possible the generation of VSTs from low levels of circulating VSTs and naive donor sources (19–21, 50). Expanded VSTs infused in post-HSCT recipients carries a potential risk of causing GVHD. While some studies have shown cross reactivity of these VSTs with recipient targets in vitro, no increase in either acute or chronic GVHD has been reported (51). In fact, even when mild cross-reactivity of expanded VSTs with HLA-mismatched targets has been demonstrated in vitro, it has not correlated with increased risk of GVHD in vivo (52). Further refinements in this process continue to evolve as these procedures become standardized, including the use of overlapping peptide pools and alternative APCs to improve reliability and reproducibility of products.
Results of VSTs in Clinical Use
CMV
Human cytomegalovirus is a pervasive β-herpes virus with prevalence rates of 50–100% in the general population. While it may manifest as mild self-limiting disease in the immunocompetent host, CMV can cause severe life-threatening disease in the immunocompromised host. Because CMV persists in the latent form after acute infection, CMV-specific CD4+ and CD8+ T cells are necessary to maintain viral quiescence. In post-HSCT patients, in the absence of donor immunity and in other immunodeficient states, CMV may reactivate in the form of retinitis, pneumonitis, hepatitis, or enterocolitis (53). The adoptive transfer of CMV-specific T cells is a logical strategy for treating and preventing CMV reactivation in such individuals, and numerous clinical trials confirm the overall excellent efficacy of VST (10, 41, 46, 54–60). CMV-specific VSTs generated from naive T cells in umbilical cord blood (UCB) have also proved effective. These VSTs show specificity for atypical epitopes while maintaining functionality (21). Naive donor sources such as UCB are being explored as a source for other VSTs for generating third-party banks for on-demand use as well (see Other Viruses and Third-Party VST Products) (21).
EBV
EBV is a ubiquitous, highly immunogenic γ-herpesvirus that can cause unique complications following transplant. Over 90% of the general population have been infected and retain lifelong seropositivity. Manifestations of primary EBV infection vary widely from asymptomatic infection to a debilitating viral illness (61). Thereafter in most cases, EBV remains latent lifelong in a B cell and mucosal epithelial reservoir under continuous T cell immune surveillance. In these healthy individuals, up to 2% of circulating T cells are EBV specific. In the period of immune deficiency after HSCT, EBV reactivation may cause viremia and life-threatening posttransplant lymphoproliferative disease (PTLD). While the monoclonal antibody rituximab successfully treats severe EBV disease in many patients by eliminating B cells in which the EBV virus resides, it results in long-term reduction in antibody production and is not always successful at controlling PTLD (61). Adoptive T cell therapy for PTLD is facilitated by the high probability of finding healthy EBV-exposed donors with measurable frequencies of circulating EBV-specific T cells. First attempts using donor lymphocyte infusions to treat EBV-PTLD were complicated by high rates of GVHD (6). Subsequent ex vivo strategies to select and expand EBV-specific T cells show broad efficacy and safety of EBV VST in numerous clinical trials for prevention and treatment of viremia and PTLD (47–49, 62–68).
Adenovirus
Adenovirus infection can range from mild upper respiratory tract infections to a spectrum of life-threatening pneumonia, gastrointestinal, hepatic, renal, and neurologic complications. Following infection, latency is maintained in the lymphoid tissues, but the virus can reactivate during periods of prolonged absence of T cell immunity (69). Adenovirus causes potentially lethal viral complication in post-HSCT recipients. Antiviral drugs such as ribavirin are largely ineffective. However, adenovirus-specific T cells generated from healthy donors have proven effective at treating even advanced disease (45, 70, 71). For this reason adenovirus antigens are often incorporated in the generation of multivirus-specific T cell products (see below).
Other Viruses
The BK and JC polyomaviruses, normally latent in healthy tissues of most adult individuals, reactivate after HSCT and in immunodeficient individuals (72). BK virus may manifest as nephropathy and life-threatening hemorrhagic cystitis (HC). Rarely, the closely associated JC virus causes fatal brain damage from progressive multifocal leukoencephalopathy (73). Polyoma-specific VST are being developed to combat these viruses. A single case report describes the successful use of BK VSTs, after which the patient had complete resolution of HC without bystander organ toxicity, GVHD, or graft rejection (74). It is now clear that the platforms developed for ex vivo selected and expanded VSTs are readily adaptable to many other viruses that complicate immune deficient states, and future developments include developing VST to target an array of viruses including VZV, HHV, and even HIV (16, 75–79).
VSTs Targeting Multiple Viruses
Given the success in prophylaxis and treatment of individual viral infections with single-virus-specific VST, the targeting of multiple viruses in a single product is a logical extension for managing the post-HSCT patient at risk from multiple viral infections. Several groups have successfully manufactured multivirus-specific T cells for the more common viruses (11, 76, 80). Challenges, as with single virus-specific products, include production time, labor, and cost. Several groups have increased manufacturing efficiency through use of viral plasmids, standardized pepmixes, alternative APCs, and alternative donor sources such as UCB to produce polyclonal, clinically efficacious VSTs (19, 81, 82). A potential obstacle for multivirus pepmixes is the risk that the most immunodominant antigens will outcompete other T cell expansions and dilute the final product of clonal diversity. Various ways to maintain multiviral specificity are being explored (80, 83). To broaden the applicability of multivirus VST, Hanley et al used virus naive donors and UCB sources to generate tri-virus-specific T cells with success (20, 84). The viral repertoire of multi VST products is continually being extended and there is no apparent limit to the number of viruses that could be targeted in a single product. As an example, Gerdemann et al established a Good Manufacturing Practices (GMP) grade method for generating VST targeting seven different viruses: CMV, EBV, adenovirus, BK virus, HHV6, RSV, influenza (19). More recently, the group at Children’s National has established a rapid, reproducible method in GMP-compliance for generating VSTs to CMV, EBV, Adenovirus, and BK virus from naive (cord blood) donor sources (82), paving the way for establishing third-party VST banks for “off the shelf” distribution.
Third-Party VST Products
One of the more exciting developments in VST therapy is the generation of third-party VST banks. The development of a bank of efficacious, clinical-grade cell therapy products which pass all release testing requires an initial outlay in time, labor, and cost. However, immediate product availability avoids any risky delay in treatment of life threatening viral disease. Several groups have created third party VST banks for “off the shelf” administration. Since these products are derived from unmatched donors and not autologous or HLA-matched sources, they carry an increased risk of GVHD. Nevertheless, with attempts to match at least one HLA molecule with the recipient, third-party VST products have been successful in several clinical trials (Table 2) (85, 86). Predictably higher number of HLA matches between the VST product and the recipient (particularly HLA class I) correlate with better in vivo proliferation and superior efficacy (86). However, even less closely matched products can be effective despite limited persistence of these mismatched T cell products. In one study, third-party cells were identified up to 12 weeks after infusion and approximately 70% of VST recipients benefited (22). Reassuringly there is no indication of enhanced alloreactivity from the VST as measured by GVHD or graft rejection. Third-party banks are thus emerging as a promising option for treating refractory post-transplant viral infections.
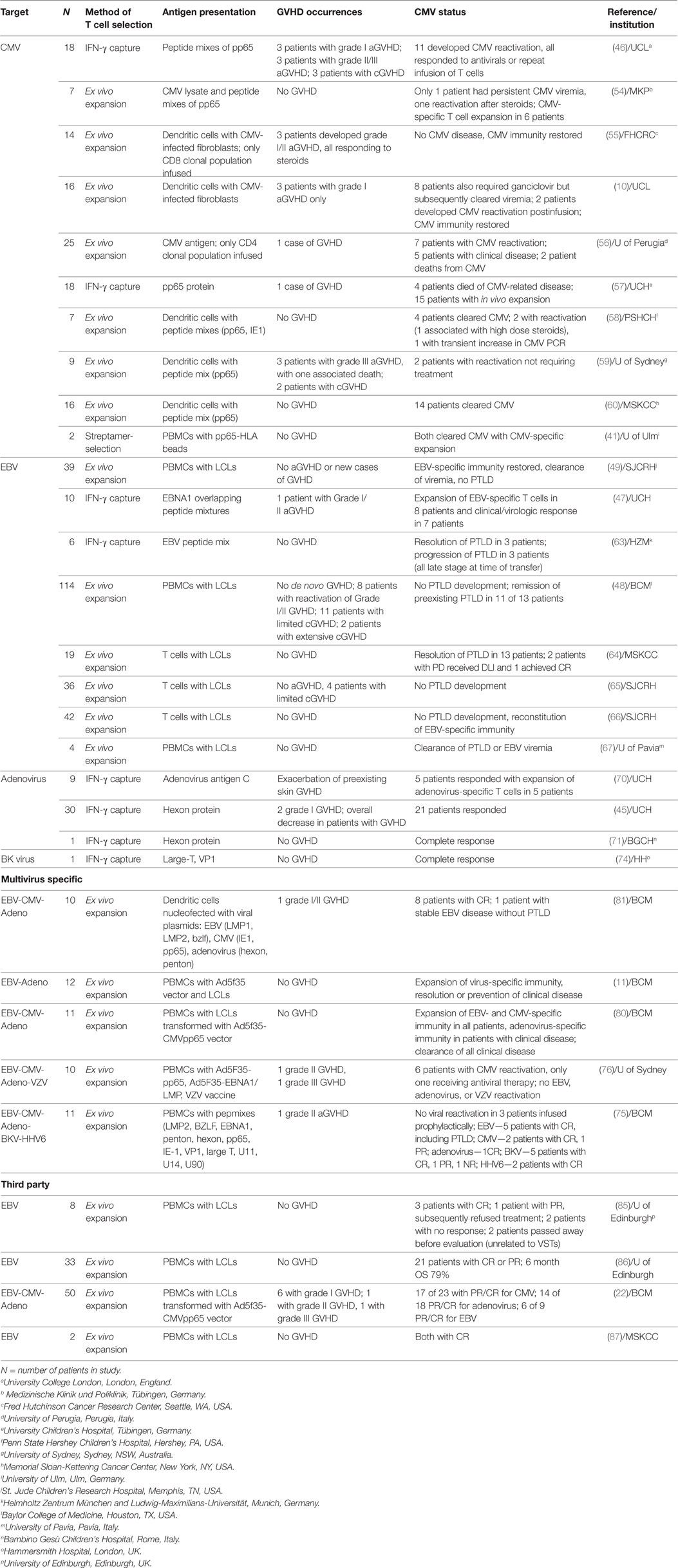
Table 2. VSTs in clinical trials.
Discussion and Future Directions
Over the past two decades, VST treatment has evolved from first proof of principle to a broadening acceptance that these cell products are a valuable, low-risk and effective tool to treat viral infections in immunocompromised individuals. Response rates reach approximately 90% for patients post-HSCT receiving VST from the matched transplant donor, and are around 70% for patients receiving third-party VST products (Table 2). VSTs appear promising as prophylaxis for high risk patients, conferring a high probability of protection against reactivation. A recent review of VSTs given to 36 patients with primary immunodeficiency syndromes over the past 10 years reported excellent responses to VSTs both for treatment (response rates 76–100% depending on the virus) and prophylaxis (81% of patients protected from viral reactivation) (24). Improved technology, including standardized pepmixes and alternative APCs, has improved the speed and efficiency of the manufacturing process. In parallel, the successes with VST from naive donor sources, multivirus-specific products, and generation of third-party banks have widened the scope of VST applicability. Ongoing studies are evaluating the safety and feasibility of increasing the number of viruses in a single product and extending the size of third-party banks for rapid use. Given the success seen in viral infections, the expansion of antigen-specific adoptive cell therapy to other complicated diseases, including HIV, fungal disease, and malignancies is increasingly within reach.
The studies included in Table 2 demonstrate the safety of VSTs in various settings. The risk of GVHD, a primary concern in initial trials using unmanipulated donor products, has been decreased by improved methods of selecting and expanding VSTs. Current GVHD rates after VST do not exceed those expected for patients post-HSCT. Of studies with particularly high rates of GVHD, nearly all of the patients who developed GVHD (both acute and chronic) had prior risk factors that would explain these outcomes, including history of or active GVHD, subtherapeutic immunosuppression, or recipients of T cell-replete grafts (46, 59). Critical, taking into consideration the patient-specific risk factors, no correlation has been identified between GVHD development and the method of VST generation, product phenotype, or duration of in vivo activity of infused VSTs.
Through the multitude of clinical trials utilizing VSTs, we have gained some important information regarding predictors of response. Most methods of generating antigen-specific T cells yield a very heterogenous population of CD4+ (typically about 30%) and CD8+ T cells, unless they are generated against a single CD8+-restricted epitope. While we know this polyclonal phenotype supports persistence of VSTs in vivo (43), it is not clear whether differing proportions of CD4+/CD8+ T cells are associated with increased or decreased clinical efficacy. One special situation is the use of third-party products, where it appears critical to ensure that there is shared antiviral activity through a shared HLA allele when selecting the “right” product. This can be either class I or class II for most cases, although the endogenous immune response is HLA-specific for certain viruses and must be matched accordingly. For example, the response to adenovirus is mediated through HLA class II, thus products for patients with adenoviral disease should be matched through HLA class II, whereas for CMV class I matching is typically preferred.
Immune reconstitution studies in patients following infusion of VSTs have also lead to increased knowledge about the in vivo activity of different VST products. For latent viral infections (e.g., CMV and EBV), enhanced detection of circulating antiviral T cells has been correlated with a better response (10); however, this is not always the case for viruses that are not latent (e.g., adenovirus) (80). While the gene marking studies by Heslop and Rooney suggest that adoptively transferred EBV-specific T cells can persist for a decade, there is also the suggestion that adoptive transfer of VSTs can stimulate endogenous anti-viral immunity. Additionally, epitope spreading is another marker of improved immune response as illustrated in the EBV-associated lymphoma setting (88).
For all the successes observed with VST products over the years, some patients still fail to respond to therapy with no identifiable mechanism. Hence, an important area of ongoing research is evaluating the mechanisms underlying VST resistance. For example, tumor (or virus)-secreted TGFβ inactivates antigen-specific T cells. To overcome this obstacle, gene manipulation of the TGFβ receptor on the antigen-specific T cells to render them resistant to the effects of TGFβ is being explored (89, 90). Other groups have found success infusing galunisertib, a small molecule inhibitor of TGFβ, abrogating the anti-inflammatory effect (91). Targets may also evade the immune system by upregulating expression of immunomodulators such as programmed death-1 ligand, which binds PD-1 on T cells in response to IFNγ. Checkpoint inhibitors, such as pembrolizumab targeted to PD-1, have increasing applications to many malignancies or in combination therapies and may also enhance VSTs especially in the HIV setting (92). Such modifications may therefore increase the in vivo efficacy of adoptively transferred VSTs in patients post-HSCT or for virus-associated diseases.
Other challenges of treatment are related to the severe immune dysregulation in the majority of patients at risk of viral disease. Steroid treatment, often used in high doses to treat GVHD, can reactivate dormant viruses and also render VST infusion futile through inactivation of the product. To this end, VST have been gene manipulated to inactivate the glucocorticoid receptor, allowing them to maintain cytotoxicity in the presence of steroids (93). T cells can also be genetically manipulated to be resistant to calcineurin inhibitors (cyclosporine A and tacrolimus, commonly used in post-HSCT setting), which inhibit T cell activation in vivo (94, 95). Such transduced VSTs proliferate, lack alloreactivity, and maintain cytotoxicity.
Planned advanced phase clinical trials will focus on many of these crucial points, including delineating the importance of VST product phenotype for efficacy in vivo, overcoming challenges such as unfavorable microenvironment, and studying immune reconstitution markers following infusion. The application of adoptive cell therapy has become increasingly broad, now extending to other infections as well as hematologic malignancies and solid tumors. As we gain more information about the interplay between the host immune system and the disease, modifications to T cells or combination therapy approach may be increasingly used to maximize efficacy while maintaining safety. Current results and these future developments ensure that VSTs will have an increasingly successful and everwidening role in the management of the immunocompromised patient.
Author Contributions
All authors have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
CB has a licensing agreement with Cell Medica. The other author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NIH grant PO1 CA148600.
References
1. Sahin U, Toprak SK, Atilla PA, Atilla E, Demirer T. An overview of infectious complications after allogeneic hematopoietic stem cell transplantation. J Infect Chemother (2016) 22(8):505–14. doi:10.1016/j.jiac.2016.05.006
2. Wingard JR, Hsu J, Hiemenz JW. Hematopoietic stem cell transplantation: an overview of infection risks and epidemiology. Hematol Oncol Clin North Am (2011) 25(1):101–16. doi:10.1016/j.hoc.2010.11.008
3. Arnaout K, Patel N, Jain M, El-Amm J, Amro F, Tabbara IA. Complications of allogeneic hematopoietic stem cell transplantation. Cancer Invest (2014) 32(7):349–62. doi:10.3109/07357907.2014.919301
4. Tomblyn M, Chiller T, Einsele H, Gress R, Sepkowitz K, Storek J, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant (2009) 15(10):1143–238. doi:10.1016/j.bbmt.2009.06.019
5. Barrett AJ, Bollard CM. The coming of age of adoptive T-cell therapy for viral infection after stem cell transplantation. Ann Transl Med (2015) 3(5):62. doi:10.3978/j.issn.2305-5839.2015.01.18
6. Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med (1994) 330(17):1185–91. doi:10.1056/NEJM199404283301703
7. Bollard CM. Improving T-cell therapy for Epstein-Barr virus lymphoproliferative disorders. J Clin Oncol (2013) 31(1):5–7. doi:10.1200/JCO.2012.43.5784
8. Cruz CR, Hanley PJ, Liu H, Torrano V, Lin YF, Arce JA, et al. Adverse events following infusion of T cells for adoptive immunotherapy: a 10-year experience. Cytotherapy (2010) 12(6):743–9. doi:10.3109/14653241003709686
9. Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet (1995) 345(8941):9–13. doi:10.1016/S0140-6736(95)91150-2
10. Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet (2003) 362(9393):1375–7. doi:10.1016/S0140-6736(03)14634-X
11. Leen AM, Christin A, Myers GD, Liu H, Cruz CR, Hanley PJ, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood (2009) 114(19):4283–92. doi:10.1182/blood-2009-07-232454
12. Niller HH, Wolf H, Minarovits J. Regulation and dysregulation of Epstein-Barr virus latency: implications for the development of autoimmune diseases. Autoimmunity (2008) 41(4):298–328. doi:10.1080/08916930802024772
13. Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev (2009) 22(1):76–98. doi:10.1128/CMR.00034-08
14. Wills MR, Poole E, Lau B, Krishna B, Sinclair JH. The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol (2015) 12(2):128–38. doi:10.1038/cmi.2014.75
15. Feuchtinger T, Richard C, Joachim S, Scheible MH, Schumm M, Hamprecht K, et al. Clinical grade generation of hexon-specific T cells for adoptive T-cell transfer as a treatment of adenovirus infection after allogeneic stem cell transplantation. J Immunother (2008) 31(2):199–206. doi:10.1097/CJI.0b013e31815ef862
16. Gerdemann U, Keukens L, Keirnan JM, Katari UL, Nguyen CT, de Pagter AP, et al. Immunotherapeutic strategies to prevent and treat human herpesvirus 6 reactivation after allogeneic stem cell transplantation. Blood (2013) 121(1):207–18. doi:10.1182/blood-2012-05-430413
17. Binggeli S, Egli A, Schaub S, Binet I, Mayr M, Steiger J, et al. Polyomavirus BK-specific cellular immune response to VP1 and large T-antigen in kidney transplant recipients. Am J Transplant (2007) 7(5):1131–9. doi:10.1111/j.1600-6143.2007.01754.x
18. Blyth E, Clancy L, Simms R, Gaundar S, O’Connell P, Micklethwaite K, et al. BK virus-specific T cells for use in cellular therapy show specificity to multiple antigens and polyfunctional cytokine responses. Transplantation (2011) 92(10):1077–84. doi:10.1097/TP.0b013e31823328c0
19. Gerdemann U, Keirnan JM, Katari UL, Yanagisawa R, Christin AS, Huye LE, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther (2012) 20(8):1622–32. doi:10.1038/mt.2012.130
20. Hanley PJ, Lam S, Shpall EJ, Bollard CM. Expanding cytotoxic T lymphocytes from umbilical cord blood that target cytomegalovirus, Epstein-Barr virus, and adenovirus. J Vis Exp (2012) (63):e3627. doi:10.3791/3627
21. Hanley PJ, Melenhorst JJ, Nikiforow S, Scheinberg P, Blaney JW, Demmler-Harrison G, et al. CMV-specific T cells generated from naive T cells recognize atypical epitopes and may be protective in vivo. Sci Transl Med (2015) 7(285):285ra263. doi:10.1126/scitranslmed.aaa2546
22. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood (2013) 121(26):5113–23. doi:10.1182/blood-2013-02-486324
23. Peggs K, Verfuerth S, Mackinnon S. Induction of cytomegalovirus (CMV)-specific T-cell responses using dendritic cells pulsed with CMV antigen: a novel culture system free of live CMV virions. Blood (2001) 97(4):994–1000. doi:10.1182/blood.V97.4.994
24. Naik S, Nicholas SK, Martinez CA, Leen AM, Hanley PJ, Gottschalk SM, et al. Adoptive immunotherapy for primary immunodeficiency disorders with virus-specific T lymphocytes. J Allergy Clin Immunol (2016) 137(5):1498–505.e1. doi:10.1016/j.jaci.2015.12.1311
25. Ngo MC, Ando J, Leen AM, Ennamuri S, Lapteva N, Vera JF, et al. Complementation of antigen-presenting cells to generate T lymphocytes with broad target specificity. J Immunother (2014) 37(4):193–203. doi:10.1097/CJI.0000000000000014
26. Mathieu M, Cotta-Grand N, Daudelin JF, Boulet S, Lapointe R, Labrecque N. CD40-activated B cells can efficiently prime antigen-specific naive CD8+ T cells to generate effector but not memory T cells. PLoS One (2012) 7(1):e30139. doi:10.1371/journal.pone.0030139
27. Heslop HE, Brenner MK, Rooney CM. Donor T cells to treat EBV-associated lymphoma. N Engl J Med (1994) 331(10):679–80. doi:10.1056/NEJM199409083311017
28. Leen AM, Heslop HE, Brenner MK. Antiviral T-cell therapy. Immunol Rev (2014) 258(1):12–29. doi:10.1111/imr.12138
29. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med (2013) 5(174):174ra127. doi:10.1126/scitranslmed.3004916
30. Davies JK, Barbon CM, Voskertchian AR, Nadler LM, Guinan EC. Induction of alloantigen-specific anergy in human peripheral blood mononuclear cells by alloantigen stimulation with co-stimulatory signal blockade. J Vis Exp (2011) (49):e2673. doi:10.3791/2673
31. Solomon SR, Mielke S, Savani BN, Montero A, Wisch L, Childs R, et al. Selective depletion of alloreactive donor lymphocytes: a novel method to reduce the severity of graft-versus-host disease in older patients undergoing matched sibling donor stem cell transplantation. Blood (2005) 106(3):1123–9. doi:10.1182/blood-2005-01-0393
32. Mielke S, McIver ZA, Shenoy A, Fellowes V, Khuu H, Stroncek DF, et al. Selectively T cell-depleted allografts from HLA-matched sibling donors followed by low-dose posttransplantation immunosuppression to improve transplantation outcome in patients with hematologic malignancies. Biol Blood Marrow Transplant (2011) 17(12):1855–61. doi:10.1016/j.bbmt.2011.05.019
33. Hartwig UF, Nonn M, Khan S, Link I, Huber C, Herr W. Depletion of alloreactive donor T lymphocytes by CD95-mediated activation-induced cell death retains antileukemic, antiviral, and immunoregulatory T cell immunity. Biol Blood Marrow Transplant (2008) 14(1):99–109. doi:10.1016/j.bbmt.2007.10.002
34. Amrolia PJ, Muccioli-Casadei G, Yvon E, Huls H, Sili U, Wieder ED, et al. Selective depletion of donor alloreactive T cells without loss of antiviral or antileukemic responses. Blood (2003) 102(6):2292–9. doi:10.1182/blood-2002-11-3516
35. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood (2006) 107(6):2294–302. doi:10.1182/blood-2005-08-3503
36. Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood (2015) 125(26):4103–13. doi:10.1182/blood-2015-02-628354
37. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi:10.1056/NEJMoa1106152
38. Tischer S, Kaireit T, Figueiredo C, Hiller O, Maecker-Kolhoff B, Geyeregger R, et al. Establishment of the reversible peptide-major histocompatibility complex (pMHC) class I Histamer technology: tool for visualization and selection of functionally active antigen-specific CD8(+) T lymphocytes. Int Immunol (2012) 24(9):561–72. doi:10.1093/intimm/dxs059
39. Neudorfer J, Schmidt B, Huster KM, Anderl F, Schiemann M, Holzapfel G, et al. Reversible HLA multimers (Streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods (2007) 320(1–2):119–31. doi:10.1016/j.jim.2007.01.001
40. Knabel M, Franz TJ, Schiemann M, Wulf A, Villmow B, Schmidt B, et al. Reversible MHC multimer staining for functional isolation of T-cell populations and effective adoptive transfer. Nat Med (2002) 8(6):631–7. doi:10.1038/nm0602-631
41. Schmitt A, Tonn T, Busch DH, Grigoleit GU, Einsele H, Odendahl M, et al. Adoptive transfer and selective reconstitution of streptamer-selected cytomegalovirus-specific CD8+ T cells leads to virus clearance in patients after allogeneic peripheral blood stem cell transplantation. Transfusion (2011) 51(3):591–9. doi:10.1111/j.1537-2995.2010.02940.x
42. Pourgheysari B, Piper KP, McLarnon A, Arrazi J, Bruton R, Clark F, et al. Early reconstitution of effector memory CD4+ CMV-specific T cells protects against CMV reactivation following allogeneic SCT. Bone Marrow Transplant (2009) 43(11):853–61. doi:10.1038/bmt.2008.403
43. Novy P, Quigley M, Huang X, Yang Y. CD4 T cells are required for CD8 T cell survival during both primary and memory recall responses. J Immunol (2007) 179(12):8243–51. doi:10.4049/jimmunol.179.12.8243
44. Zandvliet ML, Falkenburg JH, van Liempt E, Veltrop-Duits LA, Lankester AC, Kalpoe JS, et al. Combined CD8+ and CD4+ adenovirus hexon-specific T cells associated with viral clearance after stem cell transplantation as treatment for adenovirus infection. Haematologica (2010) 95(11):1943–51. doi:10.3324/haematol.2010.022947
45. Feucht J, Opherk K, Lang P, Kayser S, Hartl L, Bethge W, et al. Adoptive T-cell therapy with hexon-specific Th1 cells as a treatment of refractory adenovirus infection after HSCT. Blood (2015) 125(12):1986–94. doi:10.1182/blood-2014-06-573725
46. Peggs KS, Thomson K, Samuel E, Dyer G, Armoogum J, Chakraverty R, et al. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis (2011) 52(1):49–57. doi:10.1093/cid/ciq042
47. Icheva V, Kayser S, Wolff D, Tuve S, Kyzirakos C, Bethge W, et al. Adoptive transfer of Epstein-Barr virus (EBV) nuclear antigen 1-specific t cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J Clin Oncol (2013) 31(1):39–48. doi:10.1200/JCO.2011.39.8495
48. Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood (2010) 115(5):925–35. doi:10.1182/blood-2009-08-239186
49. Rooney CM, Smith CA, Ng CY, Loftin SK, Sixbey JW, Gan Y, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood (1998) 92(5):1549–55.
50. Hanley PJ, Bollard CM, Brunstein CG. Adoptive immunotherapy with the use of regulatory T cells and virus-specific T cells derived from cord blood. Cytotherapy (2015) 17(6):749–55. doi:10.1016/j.jcyt.2014.12.007
51. Fuji S, Kapp M, Einsele H. Alloreactivity of virus-specific T cells: possible implication of graft-versus-host disease and graft-versus-leukemia effects. Front Immunol (2013) 4:330. doi:10.3389/fimmu.2013.00330
52. Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, et al. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood (2010) 116(22):4700–2. doi:10.1182/blood-2010-06-289991
53. Smith CJ, Quinn M, Snyder CM. CMV-specific CD8 T cell differentiation and localization: implications for adoptive therapies. Front Immunol (2016) 7:352. doi:10.3389/fimmu.2016.00352
54. Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Löffler J, et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood (2002) 99(11):3916–22. doi:10.1182/blood.V99.11.3916
55. Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med (1995) 333(16):1038–44. doi:10.1056/NEJM199510193331603
56. Perruccio K, Tosti A, Burchielli E, Topini F, Ruggeri L, Carotti A, et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood (2005) 106(13):4397–406. doi:10.1182/blood-2005-05-1775
57. Feuchtinger T, Opherk K, Bethge WA, Topp MS, Schuster FR, Weissinger EM, et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood (2010) 116(20):4360–7. doi:10.1182/blood-2010-01-262089
58. Bao L, Cowan MJ, Dunham K, Horn B, McGuirk J, Gilman A, et al. Adoptive immunotherapy with CMV-specific cytotoxic T lymphocytes for stem cell transplant patients with refractory CMV infections. J Immunother (2012) 35(3):293–8. doi:10.1097/CJI.0b013e31824300a2
59. Micklethwaite K, Hansen A, Foster A, Snape E, Antonenas V, Sartor M, et al. Ex vivo expansion and prophylactic infusion of CMV-pp65 peptide-specific cytotoxic T-lymphocytes following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2007) 13(6):707–14. doi:10.1016/j.bbmt.2007.02.004
60. Koehne G, Hasan A, Doubrovina E, Prockop S, Tyler E, Wasilewski G, et al. Immunotherapy with donor T cells sensitized with overlapping pentadecapeptides for treatment of persistent cytomegalovirus infection or viremia. Biol Blood Marrow Transplant (2015) 21(9):1663–78. doi:10.1016/j.bbmt.2015.05.015
61. Heslop HE. How I treat EBV lymphoproliferation. Blood (2009) 114(19):4002–8. doi:10.1182/blood-2009-07-143545
62. Bollard CM, Rooney CM, Heslop HE. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol (2012) 9(9):510–9. doi:10.1038/nrclinonc.2012.111
63. Moosmann A, Bigalke I, Tischer J, Schirrmann L, Kasten J, Tippmer S, et al. Effective and long-term control of EBV PTLD after transfer of peptide-selected T cells. Blood (2010) 115(14):2960–70. doi:10.1182/blood-2009-08-236356
64. Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood (2012) 119(11):2644–56. doi:10.1182/blood-2011-08-371971
65. Heslop HE, Rooney CM. Adoptive cellular immunotherapy for EBV lymphoproliferative disease. Immunol Rev (1997) 157:217–22. doi:10.1111/j.1600-065X.1997.tb00984.x
66. Rooney CM, Roskrow MA, Smith CA, Brenner MK, Heslop HE. Immunotherapy for Epstein-Barr virus-associated cancers. J Natl Cancer Inst Monogr (1998) 1998(23):89–93. doi:10.1093/oxfordjournals.jncimonographs.a024180
67. Comoli P, Basso S, Zecca M, Pagliara D, Baldanti F, Bernardo ME, et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am J Transplant (2007) 7(6):1648–55. doi:10.1111/j.1600-6143.2007.01823.x
68. Rouce RH, Louis CU, Heslop HE. Epstein-Barr virus lymphoproliferative disease after hematopoietic stem cell transplant. Curr Opin Hematol (2014) 21(6):476–81. doi:10.1097/MOH.0000000000000083
69. Feuchtinger T, Lang P, Handgretinger R. Adenovirus infection after allogeneic stem cell transplantation. Leuk Lymphoma (2007) 48(2):244–55. doi:10.1080/10428190600881157
70. Feuchtinger T, Matthes-Martin S, Richard C, Lion T, Fuhrer M, Hamprecht K, et al. Safe adoptive transfer of virus-specific T-cell immunity for the treatment of systemic adenovirus infection after allogeneic stem cell transplantation. Br J Haematol (2006) 134(1):64–76. doi:10.1111/j.1365-2141.2006.06108.x
71. Di Nardo M, Li Pira G, Amodeo A, Cecchetti C, Giorda E, Ceccarelli S, et al. Adoptive immunotherapy with antigen-specific T cells during extracorporeal membrane oxygenation (ECMO) for adenovirus-related respiratory failure in a child given haploidentical stem cell transplantation. Pediatr Blood Cancer (2014) 61(2):376–9. doi:10.1002/pbc.24753
72. Philippe M, Ranchon F, Gilis L, Schwiertz V, Vantard N, Ader F, et al. Cidofovir in the treatment of BK virus-associated hemorrhagic cystitis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2016) 22(4):723–30. doi:10.1016/j.bbmt.2015.12.009
73. Pinto M, Dobson S. BK and JC virus: a review. J Infect (2014) 68(Suppl 1):S2–8. doi:10.1016/j.jinf.2013.09.009
74. Pello OM, Innes AJ, Bradshaw A, Finn SA, Uddin S, Bray E, et al. BKV-specific T cells in the treatment of severe refractory haemorrhagic cystitis after HLA-haploidentical haematopoietic cell transplantation. Eur J Haematol (2017) 98(6):632–4. doi:10.1111/ejh.12848
75. Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, Martinez C, et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med (2014) 6(242):242ra283. doi:10.1126/scitranslmed.3008825
76. Ma CK, Blyth E, Clancy L, Simms R, Burgess J, Brown R, et al. Addition of varicella zoster virus-specific T cells to cytomegalovirus, Epstein-Barr virus and adenovirus tri-specific T cells as adoptive immunotherapy in patients undergoing allogeneic hematopoietic stem cell transplantation. Cytotherapy (2015) 17(10):1406–20. doi:10.1016/j.jcyt.2015.07.005
77. Blyth E, Gaundar SS, Clancy L, Simms RM, Bilmon I, Micklethwaite KP, et al. Clinical-grade varicella zoster virus-specific T cells produced for adoptive immunotherapy in hemopoietic stem cell transplant recipients. Cytotherapy (2012) 14(6):724–32. doi:10.3109/14653249.2012.663486
78. Patel S, Lam S, Cruz CR, Wright K, Cochran C, Ambinder RF, et al. Functionally active HIV-specific T cells that target gag and Nef can be expanded from virus-naive donors and target a range of viral epitopes: implications for a cure strategy after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2016) 22(3):536–41. doi:10.1016/j.bbmt.2015.12.007
79. Lam S, Sung J, Cruz C, Castillo-Caro P, Ngo M, Garrido C, et al. Broadly-specific cytotoxic T cells targeting multiple HIV antigens are expanded from HIV+ patients: implications for immunotherapy. Mol Ther (2015) 23(2):387–95. doi:10.1038/mt.2014.207
80. Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med (2006) 12(10):1160–6. doi:10.1038/nm1475
81. Gerdemann U, Katari UL, Papadopoulou A, Keirnan JM, Craddock JA, Liu H, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther (2013) 21(11):2113–21. doi:10.1038/mt.2013.151
82. Dave H, Luo M, Blaney JW, Patel S, Barese C, Cruz CR, et al. Toward a rapid production of multivirus-specific T cells targeting BKV, adenovirus, CMV, and EBV from umbilical cord blood. Mol Ther Methods Clin Dev (2017) 5:13–21. doi:10.1016/j.omtm.2017.02.001
83. Karlsson H, Brewin J, Kinnon C, Veys P, Amrolia PJ. Generation of trispecific cytotoxic T cells recognizing cytomegalovirus, adenovirus, and Epstein-Barr virus: an approach for adoptive immunotherapy of multiple pathogens. J Immunother (2007) 30(5):544–56. doi:10.1097/CJI.0b013e3180335b7a
84. Hanley PJ, Cruz CR, Savoldo B, Leen AM, Stanojevic M, Khalil M, et al. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood (2009) 114(9):1958–67. doi:10.1182/blood-2009-03-213256
85. Haque T, Wilkie GM, Taylor C, Amlot PL, Murad P, Iley A, et al. Treatment of Epstein-Barr-virus-positive post-transplantation lymphoproliferative disease with partly HLA-matched allogeneic cytotoxic T cells. Lancet (2002) 360(9331):436–42. doi:10.1016/S0140-6736(02)09672-1
86. Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood (2007) 110(4):1123–31. doi:10.1182/blood-2006-12-063008
87. Barker JN, Doubrovina E, Sauter C, Jaroscak JJ, Perales MA, Doubrovin M, et al. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood (2010) 116(23):5045–9. doi:10.1182/blood-2010-04-281873
88. Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol (2014) 32(8):798–808. doi:10.1200/JCO.2013.51.5304
89. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother (2008) 31(5):500–5. doi:10.1097/CJI.0b013e318177092b
90. Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, et al. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther (2013) 20(5):575–80. doi:10.1038/gt.2012.75
91. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFbetaR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res (2017) 23(3):804–13. doi:10.1158/1078-0432.CCR-16-1743
92. Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol Res (2013) 1(2):85–91. doi:10.1158/2326-6066.CIR-13-0078
93. Menger L, Gouble A, Marzolini MA, Pachnio A, Bergerhoff K, Henry JY, et al. TALEN-mediated genetic inactivation of the glucocorticoid receptor in cytomegalovirus-specific T cells. Blood (2015) 126(26):2781–9. doi:10.1182/blood-2015-08-664755
94. Ricciardelli I, Brewin J, Lugthart G, Albon SJ, Pule M, Amrolia PJ. Rapid generation of EBV-specific cytotoxic T lymphocytes resistant to calcineurin inhibitors for adoptive immunotherapy. Am J Transplant (2013) 13(12):3244–52. doi:10.1111/ajt.12475
Keywords: cell therapy, immunotherapy, adoptive, virus-specific T cells, immunocompromised host, ex vivo expansion, posttransplant complications
Citation: Houghtelin A and Bollard CM (2017) Virus-Specific T Cells for the Immunocompromised Patient. Front. Immunol. 8:1272. doi: 10.3389/fimmu.2017.01272
Received: 08 August 2017; Accepted: 25 September 2017;
Published: 11 October 2017
Edited by:
Katy Rezvani, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Maksim Mamonkin, Baylor College of Medicine, United StatesWilliam K. Decker, Baylor College of Medicine, United States
Copyright: © 2017 Houghtelin and Bollard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine M. Bollard, Y2JvbGxhcmRAY2hpbGRyZW5zbmF0aW9uYWwub3Jn