 Brice Laffleur
Brice Laffleur Orianne Debeaupuis2
Orianne Debeaupuis2 Zeinab Dalloul
Zeinab Dalloul Michel Cogné
Michel Cogné- 1Department of Microbiology and Immunology, College of Physicians and Surgeons, Columbia University, New York, NY, United States
- 2Faculté de Pharmacie, Université Paris-Descartes, Paris, France
- 3UMR 7276 Centre National de la Recherche Scientifique: Contrôle de la Réponse Immune B et des Lymphoproliférations, Université de Limoges, Limoges, France
- 4Institut Universitaire de France, Paris, France
Memory B cells and long-lived plasma cells are key elements of adaptive humoral immunity. Regardless of the immunoglobulin class produced, these cells can ensure long-lasting protection but also long-lasting immunopathology, thus requiring tight regulation of their generation and survival. Among all antibody classes, this is especially true for IgE, which stands as the most potent, and can trigger dramatic inflammatory reactions even when present in minute amounts. IgE responses and memory crucially protect against parasites and toxic components of venoms, conferring selective advantages and explaining their conservation in all mammalian species despite a parallel broad spectrum of IgE-mediated immunopathology. Long-term memory of sensitization and anaphylactic responses to allergens constitute the dark side of IgE responses, which can trigger multiple acute or chronic pathologic manifestations, some punctuated with life-threatening events. This Janus face of the IgE response and memory, both necessary and potentially dangerous, thus obviously deserves the most elaborated self-control schemes.
Introduction
B cells are specialized in immunoglobulin (Ig) selection and production, the development of which implies several phases. The first is antigen (Ag) independent, with V(D)J assembly creating functional B cell receptor (BCR) genes. In a second phase after cells have reached the periphery, Ag activation can induce expression of activation-induced deaminase (AID) (1, 2), then allowing a second round of Ig gene diversification by somatic hypermutation (SHM) and/or class switch recombination (CSR), which strongly influences their fate (3). In parallel, activated lymphocytes can further differentiate into memory B cells or into plasma cells (PCs) secreting antibodies. PCs can themselves be split into either short- or long-lived cells, the latter surviving and secreting Ig for years in human. CSR switches Ig production from IgM (+/−IgD) to IgG, IgA, and IgE. IgE was the last discovered Ig class (4, 5), notably due to its low amount in body fluids and the scarcity of IgE+ cells in vivo.
Both IgE and IgG emerged in proto-mammals from the ancestral reptilian IgY molecule and further diverged (6). IgG and IgE now stand as the predominant vs. less abundant Ig class in mammals. IgE structure (i.e., constant domains, positions of disulfide bonds, and a CH3 domain N-glycosylation site) is well conserved, with homology culminating at the Fc receptor high-affinity (FcεRI) binding site (7), in agreement with its major functional role. Tiny production is another IgE conserved feature, indicative of a stringently controlled process, consistent with IgE potentially hazardous properties during anaphylactic reactions (8).
IgE antibodies are notably active against helminths, in responses featuring high total and specific IgE levels, hyper-eosinophilia and Th2 polarization (9). IgE have crucial FcεRI-dependent anti-venom properties (10, 11). They also show an adjuvant effect for IgG responses, dependent on the low-affinity FcεRII receptor (CD23) (12–14), which could participate in Ag endocytosis and presentation to T cells. IgE can finally act against tumors by recruiting eosinophils, mastocytes, macrophages, and CD8 T-cells, in a FcεRI-dependent manner (15–17).
Unfortunately, these benefits come with multiple deleterious effects in allergies, and even also autoimmunity (18, 19). Accordingly, the immune system evolved with multiple regulations tempering IgE responses. IgE CSR is stimulated by Th2 cytokines IL4 and IL13, and by IL9 producing T-cells and mast cells [for review (20, 21)], whereas other cytokines (IFNγ, TGFβ, IL10, etc.), T REG cells, and dendritic cells dampen IgE responses (22, 23). These extrinsic controls are consolidated by B cell intrinsic brakes ensuring tight regulation of IgE CSR, mostly short-lived IgE PC differentiation and an extremely short-lived fate of lymphocytes expressing membrane IgE (mIgE). We review recent findings documenting how IgE production is intrinsically self-controlled in B cells.
IgE CSR Regulation
Class switch recombination occurs between switch (S) donor and acceptor regions (Figure 1). S regions are preceded with non-translatable I exons and I promoters, regulated both by the cytokine and cellular environment and in a B cell intrinsic manner. The IgH super-enhancer 3′ regulatory region (3′RR), a hub for multiple factors important for B-cell identity and/or activation is a major regulator of CSR (24, 25). Following B cell activation, germinal transcription of I-S regions is induced, recruiting AID, which initiates DNA double strand breaks under the control of the 3′RR [for review (26)]. Synapsis between broken S regions is also 3′RR dependent (27).
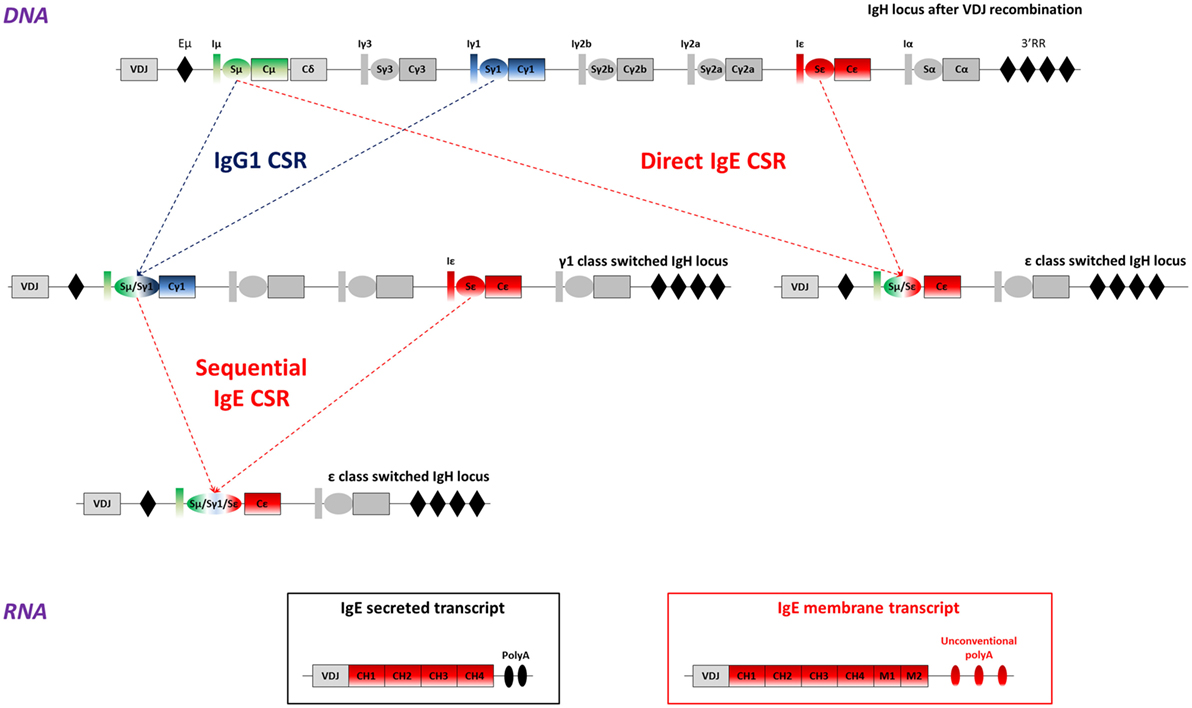
Figure 1. IgE class switch recombination, secreted, and membrane IgE (mIgE) transcripts. Top: mouse WT IgH locus is represented after VDJ recombination (not to scale). The recombined VDJ gene and constant (C) genes are represented as outlined boxes. I exons (I) and switch (S) regions preceding each constant gene (excepted Cδ) are represented as boxes and ovals, respectively. Black diamonds portray important regulatory elements: the intronic enhancer μ (Eμ), and the super-enhancer 3′ regulatory region (3′RR). Upper-middle: after B-cell activation, activation-induced deaminase (AID) is expressed and exerts its mutagenic activity on the VDJ gene, allowing production of antibodies with increased affinity for the Ag. AID is also targeted to S regions to initiate DNA double strand breaks, thereby inducing class switch recombination (CSR) between two S regions, here Sμ and Sγ1, or Sμ and Sε. This process allows the production of high-affinity IgG1 antibodies, or low-affinity IgE. Lower-middle: cells can undergo more rounds of cell cycle or re-express AID after re-exposure to antigen and undergo a sequential CSR event, from γ1 to ε here, to produce high-affinity IgE antibodies. Bottom: IgE class-switched cells produce two main transcripts: mIgE transcripts including the four constant exons (CH1 to CH4), the two membrane (M1 and M2) anchor domains and unconventional polyadenylation (polyA) signals and/or secreted IgE transcripts, containing the constant genes and two conventional polyA signals. Alternate IgE transcripts have been described and could contribute to IgE response regulation.
I promoters are crucial CSR regulators for each isotype by carrying sites for specific transcription factors. The Iε promoter binds E2A, AP1, C/EBP, STAT6, PU.1, Pax5, and NF-κB (28, 29). Classically, Iε-Sε transcription before IgE CSR is induced by phosphorylation and dimerization of STAT6, which is stabilized by SWAP-70 (30). While IL4 induces STAT6 and NFIL3/E4BP4, CD40 ligation synergistically activates NF-κB (31). In contrast, several negative regulators inhibit Sε transcription and CSR. Bcl6, strongly expressed in GC, competes with STAT6 for the same binding sites, then restricting IgE CSR. Id2 sequesters E2A and Pax5 and inhibits Iε transcription (32) [for review (33)]. TGF-β, IFNγ, and IL10 likely contribute to these negative regulations, and in human by concomitant binding of HoxC4 and Oct-1 to the Iε promoter (34).
Sε structure itself contributes to IgE CSR regulation, as the shortest and least repetitive S region in both mouse and human (2 kb only, whereas Sγ1 is around 10 kb) (35). Consistently, either Sε replacement by Sμ or insertion of Cε downstream of Sγ1 enhances IgE production (36, 37). Epigenetic changes also affect Sε: H3 acetylation and K4 tri-methylation (38) and IL4-dependent demethylation (39). Immature B cells, with potentially different epigenetic marks, thus preferentially switch to IgE rather than IgG1 (40).
IgE CSR can be direct (Sμ to Sε) or sequential (Sμ to Sγ1 first and then to Sε) with an IgG1 intermediate (41) (Figure 1). Direct CSR produces low-affinity IgE that competes with high-affinity IgE generated by sequential CSR (42). A probabilistic model suggests that low activation induces IgE CSR while high level favors IgG1 CSR with eventual secondary CSR to IgE (43). This could impact in vivo IgE responses which can be either inside or outside the GC, respectively, providing stronger or weaker co-receptor signaling. Human IgE+ cells are also produced by either direct or sequential CSR (44). Taken together, these features limit Sε accessibility during GC reactions, ensuring a first control for IgE production in cells rather biased toward preferential IgG1 CSR (45).
Multiple IgE Membrane and Secreted IgE Transcripts
After IgE CSR, B-lymphocytes express mIgE heavy chain transcripts, containing four constant (CH) and two membrane exons (M1 and M2), while PCs produce secreted transcripts lacking membrane exons (Figure 1). Alternatively spliced human IgE transcripts are found both for secreted and membrane forms, and can notably include a supplemental sequence of 156 nucleotides using an acceptor splicing site upstream of the M1 exon (46–50). Human mIgE BCR variants thus feature short mSIgE and long mLIgE, expressed after B cell activation (51). Alternately spliced IgE transcripts were also found in the mouse (52).
Noticeably and contrary to other Ig genes, membrane-type IgE transcripts lack a canonical AATAAA polyadenylation (polyA) site downstream of membrane exons. Maturation of these mRNAs then relies on suboptimal variant polyA sites likely lowers mIgE expression. The resulting ratio of secreted-type/membrane-type ε mRNA is indeed higher than for IgG in stimulated B cells, but partially normalized upon insertion of a classical polyA site downstream the IgE membrane exons in vitro (53). However, the insertion of a classical polyA sequence downstream the IgE gene does not really increase IgE level in vivo (54). Unconventional polyA sites are found in many sequenced organisms, notably 26 primate and 12 non-primate species (55).
Altogether, this alternative and non-canonical maturation of IgE transcripts could dampen IgE production, notably with low mIgE expression conferring specific properties to mIgE+ cells. In other models, low BCR expression was shown to boost tonic BCR signaling and plasma cell differentiation (56, 57).
IgE+ Lymphocytes are Prone to Differentiation into Short-Lived PCs
Following Ag activation, some cells differentiate into memory B cells (58), while others evolve into short- or long-lived PCs, then ensuring short-or long-term infusion of specific Ig in body fluids.
Studying IgE+ cell fate in vivo in WT mice suggested that these cells transiently appeared outside GCs and then rapidly yielded PCs (59). This differentiation was also observed in a transgenic model, where IgE+ cells become GFP+, with IgE+ cells overexpressing Blimp-1 and prompt to become short-lived PCs (60). This effect is also observed in vitro, using feeder cells and cytokines for B cell stimulation, exclusively generating IgM+ and IgG1+ B cells, while IgE+ cells again rapidly become PCs (61). Other in vitro B cell stimulation protocols also suggest PC predisposition for IgE+ cells expressing a WT or an exogenous mIgE (62, 63).
We also found exacerbated PC differentiation in vivo within the LATY136F mouse model, with a ratio of IgE+ plasmablasts vs. B-lymphocytes 10- to 100-fold higher than for IgM and IgG1 (64). Transfer experiments into immunodeficient mice showed a rapid collapse of these IgE+ cells, both lymphocytes, and PCs, arguing for a short-lived fate.
Normal B cell differentiation and activation are known to involve mobility, notably into GC. In vitro migration assays, by contrast, revealed that cells expressing mIgE poorly migrate toward chemokines (64). This likely compromises their entry into optimal zones of long-term survival as memory B cells or even as long-lived PCs in physiological conditions.
That IgE plasmablasts have an intrinsic lower chance to migrate to long-lived PC niches was indeed demonstrated in vivo, using chimeric BCR IgE/IgG1 [Cγ1 membrane exons knock-in (KI) into Cε, Figure 2] (65). However, long-term allergy obviously involves the continuous infusion of specific IgE into the blood by long-lived PCs, which may then constitute the only cell compartment surviving after IgE CSR. Such PCs can indeed be demonstrated in vivo in spleen and bone marrow (66). Also in a model of forced IgE CSR, mIgE-expressing cells disappeared within a few days following induction, while IgE secretion persisted for months (64). In human, bone marrow transplantation can induce the transfer of allergen-specific IgE production, likely by transferring long-lived PCs (67). Of note, IgE serum half-life is less than 2 days, ensuring their rapid disappearance if secretion ceases. Although this half-life is higher for cell-bound IgE carried by FcεRI-expressing cells (68), only the permanent production of IgE by persisting long-lived IgE PCs can account for lifelong allergic conditions. Beside immediate allergy related to persistent IgE secretion but with some delay after an antigenic boost, IgG memory B cells, can also generate new IgE-secreting cells after sequential CSR.
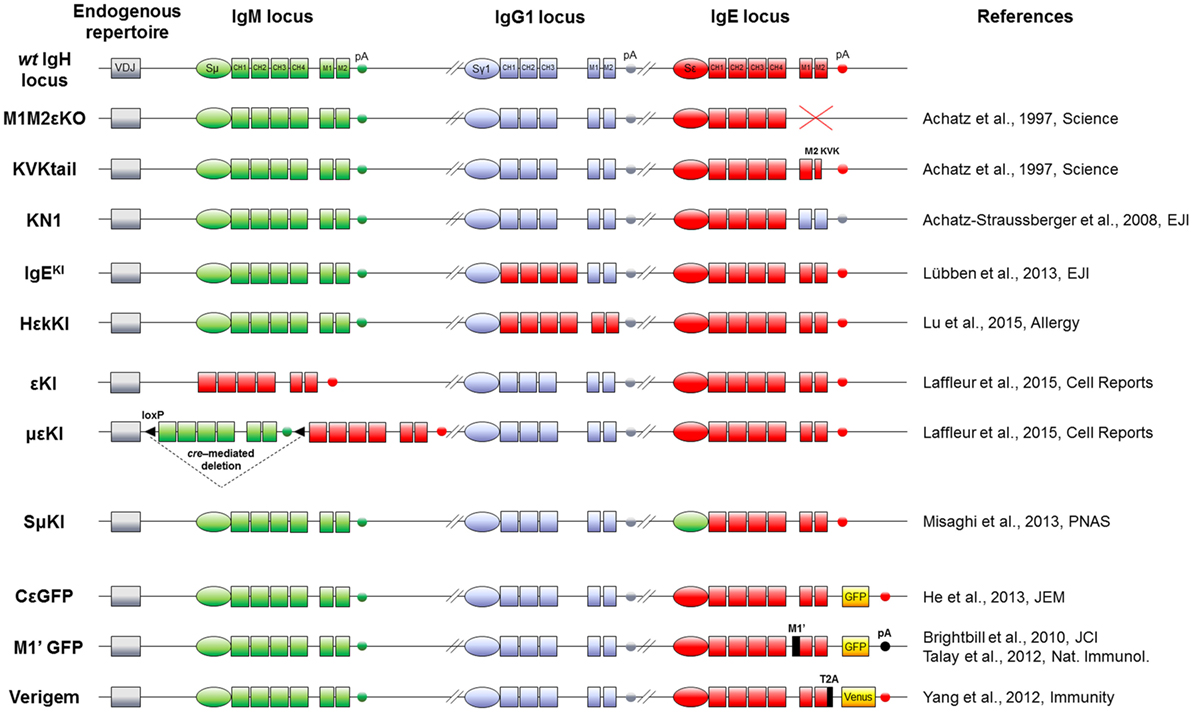
Figure 2. IgH mouse models to study IgE responses in vivo. Top: mouse WT IgH locus is represented after VDJ recombination (not to scale). IgM (green), IgG1 (blue), and IgE (red) loci including their switch regions (S), constant exons (CH), membrane genes (M), and membrane polyadenylation sites (pA) are shown. Middle: knock-out and knock-in alleles are represented, using the color code (IgM elements in green, IgG1 in blue and IgE in red) for modifications of switch regions, exons and polyadenylation sites. Bottom: the three last models feature insertion of fluorescent reporter genes (yellow boxes) and exogenous sequences (black). Name and references of each mouse model are specified on sides.
It thus appears that the actors of long-term IgE responses are mostly PCs. Despite their scarcity, their persistent production of a highly pro-inflammatory Ig class endows them with a major role in both physiology and pathology. Since IgE CSR is very efficient in vitro and since many reports showed that IgE-switched cells more efficiently differentiate into PCs than into persisting IgE+ lymphocytes, it remains to be understood why IgE PCs are so rare in vivo and IgE production so low (about 1,000-fold below that of specific IgG). Convergent data now show that the rate-limiting step of IgE PC generation is a specifically fragile and short-lived status of mIgE+ lymphocytes. Poorly moving and briefly surviving, these mandatory precursors (69) constitute a bottleneck in IgE PC generation.
mIgE Expression Strongly Impacts Lymphocyte Cell Fate
In physiological conditions, mIgE+ lymphocytes are barely detectable in vivo either in mouse or human. They are, however, mandatory for building IgE responses, knock-out (KO) of membrane exons (Figures 2 and 3) decreasing total serum IgE by 95%, and preventing the production of specific IgE after immunization (69). Replacement of the IgE intracellular tail with a KVK tail (similar to IgM BCR) (Figures 2 and 3) also decreased IgE production, showing that this intracellular tail positively contributed to IgE secretion (69). That antibody targeting the mIgE extracellular portion decrease IgE production in vitro as well as in vivo, further underlines the importance of mIgE+ cells as IgE PC precursors. A first study showed in vitro induction of apoptosis of mIgE+ cells and inhibition of specific IgE responses in allergic mice, using an anti-EMPD (extracellular membrane-proximal domain) monoclonal antibody (MAb) (70). Anti-human extra-membrane proximal domain (EMPD) antibodies proved efficient in vivo, targeting humanized mouse (Figure 2), or human PBMC, by promoting apoptosis, decreasing IgE PC number, and lowering IgE production in models of asthma induction and helminth infection (71). Soluble IgE neutralization and decrease of IgE+ cells was also obtained using a single-chain antibody against total IgE (72). Another antibody targeting total IgE and including a FcγRIIb binding Fc region is 40-fold more efficient than the classical omalizumab MAb anti-IgE in HuSCID mice, diminishing PCs and IgE production, likely due to co-engagement of mIgE and the inhibitory receptor FcγRIIb at the lymphocyte surface (73). It remains to be explored whether the IgE BCR is only expressed on B-lymphocytes, as for IgG, or also on some bone marrow long-lived PCs, as for some IgA and IgM-PCs (74, 75). Altogether, mIgE+ cells are a mandatory step of IgE responses, but since they poorly show up in vivo, their study is challenging and has needed specific biochemical and cellular in vitro and in vivo strategies.
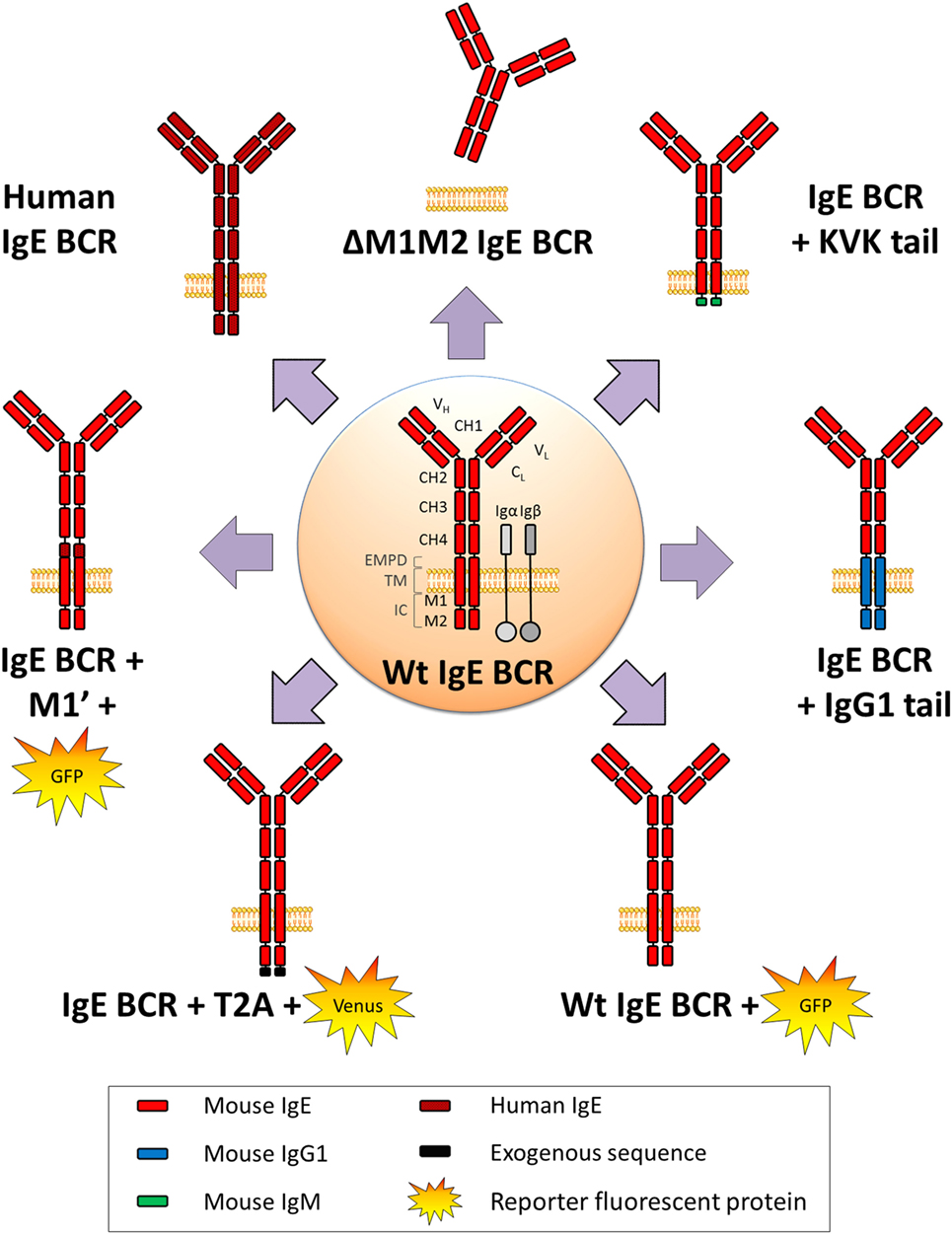
Figure 3. WT and variant IgE B cell receptor (BCR) expressed in mouse models. Mouse WT IgE BCR structure is schematized in the center. Heavy and light chain variable (VH and VL) domains form the antigen-binding site, constant domains (CH), transmembrane (TM), and intracellular domains (IC) are shown, and anchored in the lipid membrane. IgE BCR includes an extra-membrane proximal domain (EMPD). Co-signaling molecules Igα and Igβ are represented in gray. Variant and chimeric IgE BCR resulting from the different knock-in and knock-out alleles (described in Figure 2) are represented, and reporter fluorescent proteins are illustrated.
Phage display helped identify two mIgE intra-cytosolic binding proteins: HPK1 binds mIgE and other isotype tails (76) while HAX1 only binds IgE (77). IgE production, however, remained unaffected in corresponding KO models (78–80).
As for transfected IgE BCRs expressed in vitro in cell lines, both human mIgE forms expressed into the immature WEHI cell line, associated with mouse signaling co-receptors Igα/Igβ, and inhibited cell growth after mSIgE BCR cross-linking (51). Chimeric receptors (missing CH1 to CH3 exons) expressed in A20 mature B cell line induce caspase-dependent apoptosis upon cross-linking when lacking the EMPD domain (81). Short chimeric constructions expressed into Ramos human mature cells showed Grb2 adaptor recruitment to intra-cytoplasmic mIgG or mIgE tails, enhancing BCR signals and proliferation (82). Membrane IgE expression alone induced some apoptosis of the mature Daudi cell line, which was increased by mIgE cross-linking and reversed by caspase inhibitors (71).
Primary cell studies need tricky sensitive protocols to specifically identify true mIgE+ cells and not passive IgE binding to Fcε receptors. Several protocols limit false positivity: cells can be intracellularly stained after trypsin treatment, to remove CD23-bound IgE (40), or after saturation of surface IgE with non-fluorescent antibodies (60). An acidic wash can also efficiently remove CD23-bound IgE and then allow for direct mIgE staining (59).
In vitro, IL21 induces apoptosis of IgE+-stimulated B cells, but not IgG1+ (83). High concentration of IgE+ cells also promotes caspase-dependent apoptosis (84). Using a more sophisticated in vitro system, “GC like” structures can be created with IgM/IgD and IgG, but not IgE, “memory” B cells, suggesting that mIgE+ cells die (61).
We observed spontaneous apoptosis induced by the human mIgE expression, both in heterologous (mouse mature B cell line) and homologous (human mature B cell line) transfectants. Consistently, we observed increased apoptosis of mIgE+ lymphocytes among primary B cells stimulated in vitro toward CSR and deprived of cytokines. This apoptosis implicated the mitochondrial and caspase pathways and was partially reversed using cyclosporine A, caspase inhibitors, or bcl2 overexpression. In these cells, survival was affected by multiple means: mobility was drastically reduced while the actin cytoskeleton was reorganized and cells took circular shapes, HAX1 was delocalized from mitochondria, IgE BCR was spontaneously incorporated into lipid rafts and endocytosed, transcriptional program controlling cell death, metabolism, signaling, and mobility was affected (64). Another study confirmed spontaneous apoptosis in vitro, upon expression of an exogenous BCR into stimulated primary B cells, and confirmed that the IgE BCR induces cell death actively, and not due to a signaling defect (63).
All these data are informative about the B-cell intrinsic impact of mIgE expression but do not integrate the in vivo complexity of IgE responses. Beyond these intrinsic aspects, extracellular factors such as cell–cell interactions, Ag presentation by B-cells to T-cells and various soluble factors could strongly influence IgE+ B cell fate. Methods and mouse models have been developed by different labs and help further understand IgE responses.
WT and Transgenic Mouse Models to Investigate IgE Responses In Vivo
One first model involved a transgene encoding secreted IgE, thus increasing serum IgE level, but without providing any clue about mIgE+ cells (85). KI insertion of Cε into the Cγ1 locus (Figures 2 and 3) also increased IgE production without generating mIgE+ B cells in vivo, whereas these cells appeared in vitro (37). IgG1 membrane exons (M1–M2) KI downstream of Cε (Figures 2 and 3) also increased IgE basal levels and secondary IgE responses, suggesting that the transmembrane (TM) and intracellular IgE tail contribute to IgE homeostasis in vivo (65). De Lafaille and colleagues developed a hyper-IgE model revealing several aspects of IgE responses: these mice carry a transgenic anti-OVA TCR and a KI anti-HA BCR, inducing strong IgE response after immunization with OVA-HA peptide and revealing the role of TREGS in controlling IgE production (22). In this model and in helminth (Nippostrongylus brasiliensis)-infected WT mice, IgE+ PCs were localized outside GC, whereas IgE CSR occurred in GC IgG1+ cells, i.e., through sequential CSR (Figure 1). Intracellularly stained IgE+ cells have specific characteristics: they are large, express PC genes but not CXCR5, which is implicated into GC localization (59). Ig affinity was studied after OVA-PEP1 immunization (a variant of OVA-HA), and showed rapid specific IgG1 but delayed specific IgE production, suggesting sequential CSR from IgM to IgG1 and then to IgE, where SHM occurs in parallel to IgG1 CSR (59). Consistently, IgG1-deficient mice show a defect in specific IgE production, whereas total IgE level is unaffected. This suggests that sequential IgE CSR generates high-affinity (potentially pathogenic) IgE, whereas direct IgE CSR generates low-affinity IgE (rather dampening allergy by saturating FcεRI receptors) (42). We can note that a “natural” IgE production by B2 cells, independently of MHCII, could eventually provide some of the protective low-affinity IgE antibodies (86), as well as IgE produced by immature B cells (40). The specific role of IgG1 memory cells into IgE memory has been confirmed using a high-throughput sequencing approach and adoptive transfer, suggesting the absence of mIgE+ memory B-lymphocytes (87).
Three fluorescent reporter mouse models have been generated to study the rare IgE+ cells in vivo. The first one (M1’ GFP) includes a human EMPD IgE domain knocked into the mouse Cε (Figure 2) and an IRES-GFP cassette downstream of membrane exons. Upon N. brasiliensis infection, IgE+ cells were then detected into GCs, but without strong PC differentiation (54). These conclusions could be biased by this chimeric BCR, the use of a classical polyA for membrane exons, and the fact that this IRES does not tag the IgE protein but IgE transcripts (with some GFP+ cells expressing functional IgG1+ on one hand and germline Cε transcripts on the other). A second model used the peptide 2A system to really tag IgE+ cells, coupling translation of mIgE and the fluorescent protein “Venus” (Figure 2), so that IgE+ cells are also Venus+. This model showed IgE+ cells into GC and confirmed their PC bias. These PCs were mostly short-lived but their number can be enhanced by Bcl2 overexpression, they express mIgE, opposite to IgG PCs (60). Another IRES-GFP cassette has been inserted into Cε, conserving the natural architecture of mIgE and the unconventional polyA sites (Figure 2). This showed that along N. brasiliensis infection, IgE+ cells generated into GC were less mobile than IgG1 and restricted to the dark zone, limiting their contribution to memory. This also showed more in vivo apoptosis of IgE+ than IgG1+ lymphocytes (88).
All these models converge toward the conclusion that the IgE response highly differs from other isotypes, by generating few class-switched lymphocytes, which are short-lived and might then face the cell fate decision of differentiating into PCs or dying through apoptosis. Since there are good indications that mIgE by itself elicits signals that cut B-cell mobility, shorten survival, and/or induce apoptosis (64), the issue of BCR expression in long-lived PCs downstream of the B-lymphocyte stage is of interest. For IgM+ and IgA+ PCs expressing a functional BCR, cross-linking this receptor with high doses of anti-BCR antibodies decreases PC survival (74). Regarding the mIgE which by itself shortens the half-life of B-lymphocytes (somehow mimicking high-dose cross-linking of other BCR classes), whether it can also be detrimental to PC survival remains an open issue.
Two mouse models uncoupled mIgE expression to normal CSR and microenvironment signals. Cε was inserted into Sμ (εKI, Figure 2) to force mIgE BCR expression in place of mIgM, as previously done successfully with mIgA (89). A strong blockade of early B cell development hereby occurred, suggesting a defect in ε − pre-BCR signaling or a pro-apoptotic signal. Another model followed the same strategy, but now including a floxed human Cμ upstream of Cε (μεKI, Figure 2), human IgM expression then initially supporting B cell development (90). Upon tamoxifen-inducible cre-deletion, these mice secreted IgE and transiently showed IgE+ B-lymphocytes. IgE levels peaked around 1 week after induction and while IgE+ B-lymphocytes rapidly vanished from lymphoid tissues after a few days independently of any BCR cross-linking, secreted IgE levels persisted for months, indicating that long-lived IgE+ PCs survived (64). Another model replaces Cγ1 with human Cε but conserving Sγ1 and its polyA site (Figure 2). Human IgE level was then higher than mouse IgE, but much lower than IgG1. Very infrequent IgE+ hybridomas could be obtained after immunization, suggesting again an underrepresentation of mIgE+ cells (91).
While expression of human IgE in a mouse context could participate to this phenotype, this was consistent with the observation of increased apoptosis in IgE class-switched primary B-cells. A recent study also confirmed spontaneous apoptosis of IgE+ lymphocytes, induced by mIgE expression in a BLNK-dependent manner, while the same cells were also simultaneously induced to leave the B-cell stage and readily become PCs, with a PC bias attributed to the CD19 and BLNK signaling pathways. This was dependent from mIgE extracellular domains (63). FAS-dependent apoptosis (92) might also dampen IgE responses since FAS inactivation yields expanded IgE PCs, and IgG and IgE autoantibodies (18).
Finally, a recent study showed that expression of mIgE promotes PC differentiation rather than survival of mIgE+ lymphocytes even in the absence of Ag, due to constitutive activation of Syk, CD19, BLNK, Btk, and IRF4 signaling pathways. This model did not demonstrate apoptosis but a proliferative defect and showed an exaggerated GC response for IgE+ B cells after disruption of BCR signaling. IgE+ B cells also poorly presented Ag in vivo, which could explain poor participation to immune responses (62).
Altogether, IgE+ B cells are very scarce in vivo, they can be generated for a short-term window but quickly vanish, apparently only surviving as PCs.
Discussion
IgE and Their Receptors
IgE responses are also controlled by IgE receptors, several of which exist under soluble as well as membrane-anchored isoforms. Surface FcεRI expression in mast cells is stabilized by soluble IgE (93) and participates to the clearance of soluble IgE by dendritic cells and monocytes (94). Membrane FcεRI, expressed by a basophilic leukemia cell line, also bound mIgE expressed by lymphocytes (95). The soluble form of FcεRII/CD23 also binds mIgE (96), and interestingly, FcεRII KO increases IgE responses (97). FcγRIV can bind IgE-Ag immune complexes and activate macrophages in mouse (98). Other receptors like soluble galectin 3 and 9 also bind IgE, respectively, with low [for review (99)] or high affinity (100), and might also bind mIgE. We can speculate that this hub of IgE receptors, expressed and secreted by different cell populations or by mIgE+ lymphocytes themselves, and IgE/Ag immune complexes could cross-link mIgE, re-enforcing the intrinsic control mIgE+ lymphocyte homeostasis.
IgE Memory
After primary immunization in the mouse, multiple layers of memory B cells are generated, including IgM+ and IgG+. This ensures secondary responses with rapid PC differentiation of IgG+ memory cells and re-entry into GC center of IgM+ memory cells (101). Long-lived PCs establish soluble Ig memory for long term. Class-switched BCRs strongly influence B cell fate, globally inducing PC differentiation (56, 89, 102) and modulating the memory B cell pool. As discussed above, there are contradictory data about IgE memory. Some studies suggested that mIgE+ memory B cells persisted after IgE responses (54), but convergent reports rather documented that these cells end into short-lived PC and undergo multiple counter-selection processes including apoptosis (36, 37, 60, 64, 87, 88, 91). These contradictory conclusions might result from technical pitfalls, like false mIgE staining, use of leaky IRES, disruption of IgE BCR natural architecture, an introduction of exogenous sequences, heterologous systems, genetic background, etc. Kinetic and spatial issues are also crucial as IgE responses are transient and restricted to specific compartments, which is difficult to investigate in vivo. Differences between mouse and human are also possible.
Numerous indirect pieces of evidence strongly suggest the absence of mIgE+ cells in the memory lymphocyte compartment; thanks to sequential CSR to IgE after an antigenic boost, some IgE memory could in fact indirectly be created through de novo CSR of classical IgG1+ memory lymphocytes then ending with the production of high-affinity IgE. This mechanism could ensure that high-affinity IgE antibodies are not generated as a first defense layer but only in a second stage when Ag reappears in an appropriate cytokine context and that these responses are essentially transient.
Membrane IgE expression strongly affects lymphocyte phenotype and we suggest that evolution selected multiple mechanisms to restrict IgE responses. IgE tail itself perturbs BCR signaling, eventually by sequestrating anti-apoptotic protein (HAX1), recruiting adaptor or signaling proteins (Grb2, Syk), or through still unknown interactions. TM and extra-membrane domains notably contribute to specific interactions with Ig-α, CD19 and probably other molecules. IgE is highly flexible (103, 104) and could spontaneously adopt a closed conformation, even when expressed as a BCR, and be spontaneously cross-linked; this effect could be enhanced by interactions with IgE receptors. Cε domains seem to be implicated in multiple signaling pathways and functions, ensuring both efficient PC differentiation and a selective disadvantage for mIgE+ cells, with reduced proliferation, Ag presentation, and mobility, also affected with constitutive BCR clustering into lipid rafts and endocytosis lowering surface expression, globally promoting poor survival. These effects are now well documented in lymphocytes and might also impact mIgE+ PCs (Figure 4).
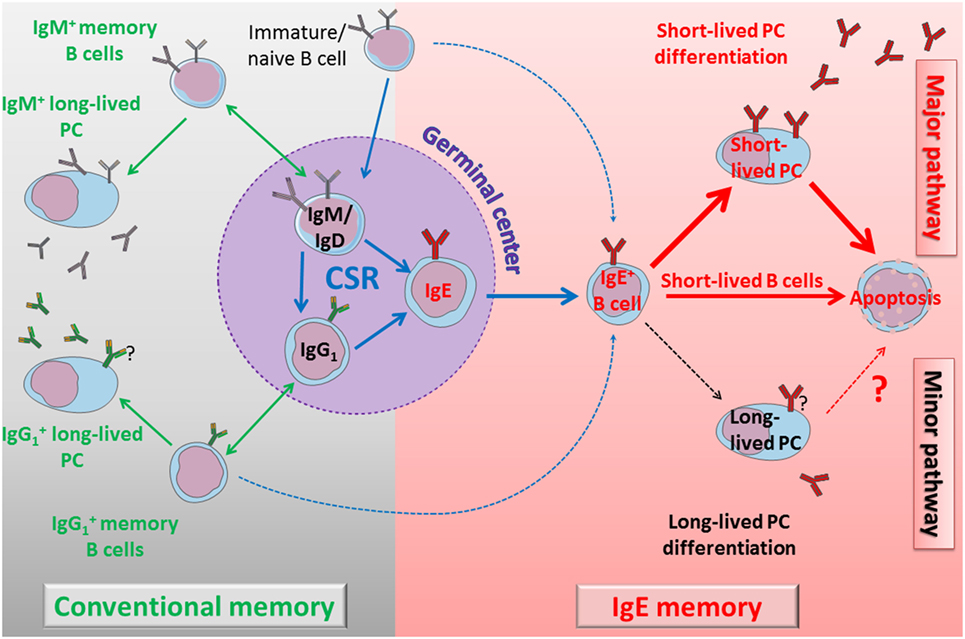
Figure 4. Self-controlled IgE production model. Left part: immature cells differentiate into naïve B cells and could undergo direct class switch recombination (CSR) to IgE or enter into the GC. During the GC reaction, cells can switch directly to IgG1 or IgE, or eventually undergo a sequential switching with an IgG1 intermediate and an extra-follicular CSR to IgE. IgM+ and IgG1+ lymphocytes generate classical humoral memories, with memory B cells and long-lived plasma cells (PCs). These memory B cells can undergo more rounds of GC reaction to increase their antigen (Ag) affinity and to switch to IgG1 or IgE. Direct or sequential CSR produces membrane IgE+ lymphocytes with different Ag affinity, and IgE B cell receptor (BCR) seems to induce a common, short-lived fate. Right part: IgE+ lymphocytes have a bias in short-lived PC differentiation. These short-lived PCs strongly express IgE BCR and undergo massive apoptosis. Membrane IgE+ lymphocytes are also counter-selected by various mechanisms including poor mobility, low membrane IgE expression, and spontaneous apoptosis. Long-lived PCs are very elusive but produce IgE antibodies for a long term, potentially including pathological IgE. A better characterization of these cells and their targeting to eliminate them could lead to new treatments to re-initialize IgE memory and deplete pathological IgE production.
Note Added in Proof
A new mouse model has been generated to force membrane IgE expression from early B cell development (105), similarly to εKI mice (Figure 2), but using a different strategy and a mouse Cε gene. These animals have also a strong blockade at the pro-B cell stage, again showing that Cε gene cannot support B cell development. Interestingly, the authors demonstrate that the strong (secreted vs. membrane) ratio of IgE accounts for this phenotype, which could be partially rescued by inserting an mIgE. Finally, they show that increasing BCR signaling by PTEN deletion increases mIgE cell number and IgE responses.
Conclusion
IgE production is tightly regulated, from CSR to PC differentiation, including IgE half-life and survival of mIgE+ memory B cells. It is interesting to note that upon all different KI strategies deployed to investigate IgE+ cells in vivo (Figure 2), these cells are generated at a very low frequency, only during short intervals, and do not yield mIgE+ memory B cells. IgE CSR is initially controlled by the cytokine microenvironment, the anomalies of which can boost IgE production in allergic individuals (106). The mechanistic of Sε accessibility likely contributes to restricting IgE CSR in mature B cells, as indicated by Sε replacements (36). Unconventional polyA sites could also contribute to IgE regulation (53). Several chimeric IgE BCR models were generated to dissect domain functions (Figures 2 and 3), but it seems that distinct protein parts contribute to disadvantage mIgE+ cells, resulting in overrepresentation of short-lived PCs and almost no accumulation of mIgE+ lymphocytes. The protracted expression of the IgE BCR might also provide counter-selective signals in plasmablasts and contribute to their short-lived fate. These multiple mechanisms induced by mIgE expression even in the absence of Ag could be enhanced in vivo by mIgE cross-linking.
Membrane IgE expression per se is, thus, a powerful gatekeeper of IgE production, both ensuring and restraining a punctual IgE response while minimizing IgE memory. These elements clearly need to be taken into account when dealing with therapy of IgE-mediated disorders. Acting on cytokines and microenvironment of the immune response is important at the initiation of IgE responses and for preventing sequential IgE CSR of memory B-lymphocytes (notably IgG1). Acting on the true memory cells responsible for the long-term infusion of IgE should by contrast rather focus onto IgE PCs.
Author Contributions
BL and MC wrote the manuscript, OD created the figures and participated to the redaction, and ZD helped for references and redaction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from ARC (PGA120150202338), Agence Nationale de la Recherche (ANR 16-CE15-0019-01), and Région Nouvelle Aquitaine.
References
1. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102(5):553–63. doi:10.1016/S0092-8674(00)00078-7
2. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell (2000) 102(5):565–75. doi:10.1016/S0092-8674(00)00079-9
3. Laffleur B, Denis-Lagache N, Péron S, Sirac C, Moreau J, Cogné M. AID-induced remodeling of immunoglobulin genes and B cell fate. Oncotarget (2013) 5(5):1118–31. doi:10.18632/oncotarget.1546
4. Ishizaka K, Ishizaka T. Identification of gamma-E-antibodies as a carrier of reaginic activity. J Immunol (1967) 99(6):1187–98.
5. Stanworth DR, Humphrey JH, Bennich H, Johansson SG. Specific inhibition of the Prausnitz-Küstner reaction by an atypical human myeloma protein. Lancet (1967) 2(7511):330–2. doi:10.1016/S0140-6736(67)90171-7
6. Warr GW, Magor KE, Higgins DA. IgY: clues to the origins of modern antibodies. Immunol Today (1995) 16(8):392–8. doi:10.1016/0167-5699(95)80008-5
7. Vernersson M, Aveskogh M, Hellman L. Cloning of IgE from the echidna (Tachyglossus aculeatus) and a comparative analysis of epsilon chains from all three extant mammalian lineages. Dev Comp Immunol (2004) 28(1):61–75. doi:10.1016/S0145-305X(03)00084-3
8. Wu LC, Zarrin AA. The production and regulation of IgE by the immune system. Nat Rev Immunol (2014) 14(4):247–59. doi:10.1038/nri3632
9. Capron M, Capron A. Immunoglobulin E and effector cells in schistosomiasis. Science (1994) 264(5167):1876–7. doi:10.1126/science.8009216
10. Marichal T, Starkl P, Reber LL, Kalesnikoff J, Oettgen HC, Tsai M, et al. A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity (2013) 39(5):963–75. doi:10.1016/j.immuni.2013.10.005
11. Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, Medzhitov R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective. Immunity (2013) 39(5):976–85. doi:10.1016/j.immuni.2013.10.006
12. Heyman B. Antibodies as natural adjuvants. Curr Top Microbiol Immunol (2014) 382:201–19. doi:10.1007/978-3-319-07911-0_9
13. Xu H, van Mechelen L, Henningsson F, Heyman B. Antigen conjugated to anti-CD23 antibodies is rapidly transported to splenic follicles by recirculating B cells. Scand J Immunol (2015) 81(1):39–45. doi:10.1111/sji.12248
14. Martin RK, Brooks KB, Henningsson F, Heyman B, Conrad DH. Antigen transfer from exosomes to dendritic cells as an explanation for the immune enhancement seen by IgE immune complexes. PLoS One (2014) 9(10):e110609. doi:10.1371/journal.pone.0110609
15. Nigro EA, Brini AT, Soprana E, Ambrosi A, Dombrowicz D, Siccardi AG, et al. Antitumor IgE adjuvanticity: key role of Fc epsilon RI. J Immunol (2009) 183(7):4530–6. doi:10.4049/jimmunol.0900842
16. Jensen-Jarolim E, Turner MC, Karagiannis SN. AllergoOncology: IgE- and IgG4- mediated immune mechanisms linking allergy with cancer and their translational implications. J Allergy Clin Immunol (2017) 140(4):982–84. doi:10.1016/j.jaci.2017.04.034
17. Nigro EA, Brini AT, Yenagi VA, Ferreira LM, Achatz-Straussberger G, Ambrosi A, et al. Cutting edge: IgE plays an active role in tumor immunosurveillance in mice. J Immunol (2016) 197(7):2583–8. doi:10.4049/jimmunol.1601026
18. Butt D, Chan TD, Bourne K, Hermes JR, Nguyen A, Statham A, et al. FAS inactivation releases unconventional germinal center B cells that escape antigen control and drive IgE and autoantibody production. Immunity (2015) 42(5):890–902. doi:10.1016/j.immuni.2015.04.010
19. Sanjuan MA, Sagar D, Kolbeck R. Role of IgE in autoimmunity. J Allergy Clin Immunol (2016) 137(6):1651–61. doi:10.1016/j.jaci.2016.04.007
20. Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol (2003) 3(9):721–32. doi:10.1038/nri1181
21. Kudo M, Ishigatsubo Y, Aoki I. Pathology of asthma. Front Microbiol (2013) 4:263. doi:10.3389/fmicb.2013.00263
22. Curotto de Lafaille MA, Muriglan S, Sunshine MJ, Lei Y, Kutchukhidze N, Furtado GC, et al. Hyper immunoglobulin E response in mice with monoclonal populations of B and T lymphocytes. J Exp Med (2001) 194(9):1349–59. doi:10.1084/jem.194.9.1349
23. Obayashi K, Doi T, Koyasu S. Dendritic cells suppress IgE production in B cells. Int Immunol (2007) 19(2):217–26. doi:10.1093/intimm/dxl138
24. Pinaud E, Marquet M, Fiancette R, Péron S, Vincent-Fabert C, Denizot Y, et al. The IgH locus 3’ regulatory region: pulling the strings from behind. Adv Immunol (2011) 110:27–70. doi:10.1016/B978-0-12-387663-8.00002-8
25. Vincent-Fabert C, Fiancette R, Pinaud E, Truffinet V, Cogné N, Cogné M, et al. Genomic deletion of the whole IgH 3’ regulatory region (hs3a, hs1,2, hs3b, and hs4) dramatically affects class switch recombination and Ig secretion to all isotypes. Blood (2010) 116(11):1895–8. doi:10.1182/blood-2010-01-264689
26. Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. Mutations, kataegis, and translocations in B lymphocytes: towards a mechanistic understanding of AID promiscuous activity. Nat Rev Immunol (2016) 16(3):164–76. doi:10.1038/nri.2016.2
27. Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, et al. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity (2007) 27(5):711–22. doi:10.1016/j.immuni.2007.09.007
28. Monticelli S, Vercelli D. Molecular regulation of class switch recombination to IgE through epsilon germline transcription. Allergy (2001) 56(4):270–8. doi:10.1034/j.1398-9995.2001.00129.x
29. Geha RS. Allergy and hypersensitivity. Nature versus nurture in allergy and hypersensitivity. Curr Opin Immunol (2003) 15(6):603–8. doi:10.1016/j.coi.2003.09.017
30. Audzevich T, Pearce G, Breucha M, Gunal G, Jessberger R. Control of the STAT6-BCL6 antagonism by SWAP-70 determines IgE production. J Immunol (2013) 190(10):4946–55. doi:10.4049/jimmunol.1203014
31. Kashiwada M, Levy DM, McKeag L, Murray K, Schroder AJ, Canfield SM, et al. IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc Natl Acad Sci U S A (2010) 107(2):821–6. doi:10.1073/pnas.0909235107
32. Sugai M, Gonda H, Kusunoki T, Katakai T, Yokota Y, Shimizu A. Essential role of Id2 in negative regulation of IgE class switching. Nat Immunol (2003) 4(1):25–30. doi:10.1038/ni874
33. Tong P, Wesemann DR. Molecular mechanisms of IgE class switch recombination. Curr Top Microbiol Immunol (2015) 388:21–37. doi:10.1007/978-3-319-13725-4_2
34. Schaffer A, Kim EC, Wu X, Zan H, Testoni L, Salamon S, et al. Selective inhibition of class switching to IgG and IgE by recruitment of the HoxC4 and Oct-1 homeodomain proteins and Ku70/Ku86 to newly identified ATTT cis-elements. J Biol Chem (2003) 278(25):23141–50. doi:10.1074/jbc.M212952200
35. Hackney JA, Misaghi S, Senger K, Garris C, Sun Y, Lorenzo MN, et al. DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Adv Immunol (2009) 101:163–89. doi:10.1016/S0065-2776(08)01005-5
36. Misaghi S, Senger K, Sai T, Qu Y, Sun Y, Hamidzadeh K, et al. Polyclonal hyper-IgE mouse model reveals mechanistic insights into antibody class switch recombination. Proc Natl Acad Sci U S A (2013) 110(39):15770–5. doi:10.1073/pnas.1221661110
37. Lübben W, Turqueti-Neves A, Okhrimenko A, Stöberl C, Schmidt V, Pfeffer K, et al. IgE knock-in mice suggest a role for high levels of IgE in basophil-mediated active systemic anaphylaxis. Eur J Immunol (2013) 43(5):1231–42. doi:10.1002/eji.201242675
38. Dayal S, Nedbal J, Hobson P, Cooper AM, Gould HJ, Gellert M, et al. High resolution analysis of the chromatin landscape of the IgE switch region in human B cells. PLoS One (2011) 6(9):e24571. doi:10.1371/journal.pone.0024571
39. Kuwabara N, Kondo N, Fukutomi O, Agata H, Orii T. Relationship between interferon-gamma, interleukin-4 and IgE production of lymphocytes from hen’s egg-sensitive patients. J Investig Allergol Clin Immunol (1995) 5(4):198–204.
40. Wesemann DR, Magee JM, Boboila C, Calado DP, Gallagher MP, Portuguese AJ, et al. Immature B cells preferentially switch to IgE with increased direct Sµ to Sε recombination. J Exp Med (2011) 208(13):2733–46. doi:10.1084/jem.20111155
41. Yoshida K, Matsuoka M, Usuda S, Mori A, Ishizaka K, Sakano H. Immunoglobulin switch circular DNA in the mouse infected with Nippostrongylus brasiliensis: evidence for successive class switching from mu to epsilon via gamma 1. Proc Natl Acad Sci U S A (1990) 87(20):7829–33. doi:10.1073/pnas.87.20.7829
42. Xiong H, Dolpady J, Wabl M, Curotto de Lafaille MA, Lafaille JJ. Sequential class switching is required for the generation of high affinity IgE antibodies. J Exp Med (2012) 209(2):353–64. doi:10.1084/jem.20111941
43. Wu YL, Stubbington MJT, Daly M, Teichmann SA, Rada C. Intrinsic transcriptional heterogeneity in B cells controls early class switching to IgE. J Exp Med (2017) 214(1):183–96. doi:10.1084/jem.20161056
44. Ramadani F, Bowen H, Upton N, Hobson PS, Chan YC, Chen JB, et al. Ontogeny of human IgE-expressing B cells and plasma cells. Allergy (2017) 72(1):66–76. doi:10.1111/all.12911
45. Siebenkotten G, Esser C, Radbruch A, Wabl M. The murine IgG1/IgE class switch program. Eur J Immunol (1992) 22(7):1827–34. doi:10.1002/eji.1830220723
46. Peng C, Davis FM, Sun LK, Liou RS, Kim YW, Chang TW. A new isoform of human membrane-bound IgE. J Immunol (1992) 148(1):129–36.
47. Zhang K, Saxon A, Max EE. Two unusual forms of human immunoglobulin E encoded by alternative RNA splicing of epsilon heavy chain membrane exons. J Exp Med (1992) 176(1):233–43. doi:10.1084/jem.176.1.233
48. Zhang K, Max EE, Cheah HK, Saxon A. Complex alternative RNA splicing of epsilon-immunoglobulin transcripts produces mRNAs encoding four potential secreted protein isoforms. J Biol Chem (1994) 269(1):456–62.
49. Batista FD, Efremov DG, Tkach T, Burrone OR. Characterization of the human immunoglobulin epsilon mRNAs and their polyadenylation sites. Nucleic Acids Res (1995) 23(23):4805–11. doi:10.1093/nar/23.23.4805
50. Batista FD, Efremov DG, Burrone OR. Characterization of a second secreted IgE isoform and identification of an asymmetric pathway of IgE assembly. Proc Natl Acad Sci U S A (1996) 93(8):3399–404. doi:10.1073/pnas.93.8.3399
51. Batista FD, Anand S, Presani G, Efremov DG, Burrone OR. The two membrane isoforms of human IgE assemble into functionally distinct B cell antigen receptors. J Exp Med (1996) 184(6):2197–205. doi:10.1084/jem.184.6.2197
52. Anand S, Batista FD, Tkach T, Efremov DG, Burrone OR. Multiple transcripts of the murine immunoglobulin epsilon membrane locus are generated by alternative splicing and differential usage of two polyadenylation sites. Mol Immunol (1997) 34(2):175–83. doi:10.1016/S0161-5890(96)00110-1
53. Karnowski A, Achatz-Straussberger G, Klockenbusch C, Achatz G, Lamers MC. Inefficient processing of mRNA for the membraneform of IgE is a genetic mechanism to limit recruitment of IgE-secreting cells. Eur J Immunol (2006) 36(7):1917–25. doi:10.1002/eji.200535495
54. Talay O, Yan D, Brightbill HD, Straney EEM, Zhou M, Ladi E, et al. IgE+ memory B cells and plasma cells generated through a germinal-center pathway. Nat Immunol (2012) 13(4):396–404. doi:10.1038/ni.2256
55. Wu PC, Chen JB, Kawamura S, Roos C, Merker S, Shih CC, et al. The IgE gene in primates exhibits extraordinary evolutionary diversity. Immunogenetics (2012) 64(4):279–87. doi:10.1007/s00251-011-0586-9
56. Martin SW, Goodnow CC. Burst-enhancing role of the IgG membrane tail as a molecular determinant of memory. Nat Immunol (2002) 3(2):182–8. doi:10.1038/ni752
57. Lechouane F, Bonaud A, Delpy L, Casola S, Oruc Z, Chemin G, et al. B-cell receptor signal strength influences terminal differentiation: cellular immune response. Eur J Immunol (2013) 43(3):619–28. doi:10.1002/eji.201242912
58. Phan TG, Tangye SG. Memory B cells: total recall. Curr Opin Immunol (2017) 45:132–40. doi:10.1016/j.coi.2017.03.005
59. Erazo A, Kutchukhidze N, Leung M, Christ AP, Urban JF Jr, Curotto de Lafaille MA, et al. Unique maturation program of the IgE response in vivo. Immunity (2007) 26(2):191–203. doi:10.1016/j.immuni.2006.12.006
60. Yang Z, Sullivan BM, Allen CDC. Fluorescent in vivo detection reveals that IgE(+) B cells are restrained by an intrinsic cell fate predisposition. Immunity (2012) 36(5):857–72. doi:10.1016/j.immuni.2012.02.009
61. Nojima T, Haniuda K, Moutai T, Matsudaira M, Mizokawa S, Shiratori I, et al. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat Commun (2011) 2:465. doi:10.1038/ncomms1475
62. Yang Z, Robinson MJ, Chen X, Smith GA, Taunton J, Liu W, et al. Regulation of B cell fate by chronic activity of the IgE B cell receptor. Elife (2016) 5:e21238. doi:10.7554/eLife.21238
63. Haniuda K, Fukao S, Kodama T, Hasegawa H, Kitamura D. Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol (2016) 17(9):1109–17. doi:10.1038/ni.3508
64. Laffleur B, Duchez S, Tarte K, Denis-Lagache N, Péron S, Carrion C, et al. Self-restrained B cells arise following membrane IgE expression. Cell Rep (2015) 10(6):900–9. doi:10.1016/j.celrep.2015.01.023
65. Achatz-Straussberger G, Zaborsky N, Königsberger S, Luger EO, Lamers M, Crameri R, et al. Migration of antibody secreting cells towards CXCL12 depends on the isotype that forms the BCR. Eur J Immunol (2008) 38(11):3167–77. doi:10.1002/eji.200838456
66. Luger EO, Fokuhl V, Wegmann M, Abram M, Tillack K, Achatz G, et al. Induction of long-lived allergen-specific plasma cells by mucosal allergen challenge. J Allergy Clin Immunol (2009) 124(4):819–26.e4. doi:10.1016/j.jaci.2009.06.047
67. Hallstrand TS, Sprenger JD, Agosti JM, Longton GM, Witherspoon RP, Henderson WR. Long-term acquisition of allergen-specific IgE and asthma following allogeneic bone marrow transplantation from allergic donors. Blood (2004) 104(10):3086–90. doi:10.1182/blood-2004-05-1775
68. Achatz-Straussberger G, Zaborsky N, Königsberger S, Feichtner S, Lenz S, Peckl-Schmid D, et al. Limited humoral immunoglobulin E memory influences serum immunoglobulin E levels in blood. Clin Exp Allergy (2009) 39(9):1307–13. doi:10.1111/j.1365-2222.2009.03278.x
69. Achatz G, Nitschke L, Lamers MC. Effect of transmembrane and cytoplasmic domains of IgE on the IgE response. Science (1997) 276(5311):409–11. doi:10.1126/science.276.5311.409
70. Feichtner S, Inführ D, Achatz-Straussberger G, Schmid D, Karnowski A, Lamers M, et al. Targeting the extracellular membrane-proximal domain of membrane-bound IgE by passive immunization blocks IgE synthesis in vivo. J Immunol (2008) 180(8):5499–505. doi:10.4049/jimmunol.180.8.5499
71. Brightbill HD, Jeet S, Lin Z, Yan D, Zhou M, Tan M, et al. Antibodies specific for a segment of human membrane IgE deplete IgE-producing B cells in humanized mice. J Clin Invest (2010) 120(6):2218–29. doi:10.1172/JCI40141
72. Ota T, Aoki-Ota M, Duong BH, Nemazee D. Suppression of IgE B cells and IgE binding to Fc(epsilon)RI by gene therapy with single-chain anti-IgE. J Immunol (2009) 182(12):8110–7. doi:10.4049/jimmunol.0900300
73. Chu SY, Horton HM, Pong E, Leung IW, Chen H, Nguyen DH, et al. Reduction of total IgE by targeted coengagement of IgE B-cell receptor and FcγRIIb with Fc-engineered antibody. J Allergy Clin Immunol (2012) 129(4):1102–15. doi:10.1016/j.jaci.2011.11.029
74. Pinto D, Montani E, Bolli M, Garavaglia G, Sallusto F, Lanzavecchia A, et al. A functional BCR in human IgA and IgM plasma cells. Blood (2013) 121(20):4110–4. doi:10.1182/blood-2012-09-459289
75. Blanc P, Moro-Sibilot L, Barthly L, Jagot F, This S, de Bernard S, et al. Mature IgM-expressing plasma cells sense antigen and develop competence for cytokine production upon antigenic challenge. Nat Commun (2016) 7:13600. doi:10.1038/ncomms13600
76. Geisberger R, Prlic M, Achatz-Straussberger G, Oberndorfer I, Luger E, Lamers M, et al. Phage display based cloning of proteins interacting with the cytoplasmic tail of membrane immunoglobulins. Dev Immunol (2002) 9(3):127–34. doi:10.1080/1044667031000137584
77. Oberndorfer I, Schmid D, Geisberger R, Achatz-Straussberger G, Crameri R, Lamers M, et al. HS1-associated protein X-1 interacts with membrane-bound IgE: impact on receptor-mediated internalization. J Immunol (2006) 177(2):1139–45. doi:10.4049/jimmunol.177.2.1139
78. Königsberger S, Peckl-Schmid D, Zaborsky N, Patzak I, Kiefer F, Achatz G. HPK1 associates with SKAP-HOM to negatively regulate Rap1-mediated B-lymphocyte adhesion. PLoS One (2010) 5:9. doi:10.1371/journal.pone.0012468
79. Peckl-Schmid D, Wolkerstorfer S, Königsberger S, Achatz-Straussberger G, Feichtner S, Schwaiger E, et al. HAX1 deficiency: impact on lymphopoiesis and B-cell development. Eur J Immunol (2010) 40(11):3161–72. doi:10.1002/eji.200940221
80. Shui JW, Boomer JS, Han J, Xu J, Dement GA, Zhou G, et al. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat Immunol (2007) 8(1):84–91. doi:10.1038/ni1416
81. Poggianella M, Bestagno M, Burrone OR. The extracellular membrane-proximal domain of human membrane IgE controls apoptotic signaling of the B cell receptor in the mature B cell line A20. J Immunol (2006) 177(6):3597–605. doi:10.4049/jimmunol.177.6.3597
82. Engels N, König LM, Heemann C, Lutz J, Tsubata T, Griep S, et al. Recruitment of the cytoplasmic adaptor Grb2 to surface IgG and IgE provides antigen receptor-intrinsic costimulation to class-switched B cells. Nat Immunol (2009) 10(9):1018–25. doi:10.1038/ni.1764
83. Harada M, Magara-Koyanagi K, Watarai H, Nagata Y, Ishii Y, Kojo S, et al. IL-21-induced bepsilon cell apoptosis mediated by natural killer T cells suppresses IgE responses. J Exp Med (2006) 203(13):2929–37. doi:10.1084/jem.20062206
84. Rabah D, Conrad DH. Effect of cell density on in vitro mouse immunoglobulin E production. Immunology (2002) 106(4):503–10. doi:10.1046/j.1365-2567.2002.01435.x
85. Matsuoka K, Taya C, Kubo S, Toyama-Sorimachi N, Kitamura F, Ra C, et al. Establishment of antigen-specific IgE transgenic mice to study pathological and immunobiological roles of IgE in vivo. Int Immunol (1999) 11(6):987–94. doi:10.1093/intimm/11.6.987
86. McCoy KD, Harris NL, Diener P, Hatak S, Odermatt B, Hangartner L, et al. Natural IgE production in the absence of MHC class II cognate help. Immunity (2006) 24(3):329–39. doi:10.1016/j.immuni.2006.01.013
87. Turqueti-Neves A, Otte M, Schwartz C, Schmitt ME, Lindner C, Pabst O, et al. The extracellular domains of IgG1 and T cell-derived IL-4/IL-13 are critical for the polyclonal memory IgE response in vivo. PLoS Biol (2015) 13(11):e1002290. doi:10.1371/journal.pbio.1002290
88. He JS, Meyer-Hermann M, Xiangying D, Zuan LY, Jones LA, Ramakrishna L, et al. The distinctive germinal center phase of IgE+ B lymphocytes limits their contribution to the classical memory response. J Exp Med (2013) 210(12):2755–71. doi:10.1084/jem.20131539
89. Duchez S, Amin R, Cogné N, Delpy L, Sirac C, Pascal V, et al. Premature replacement of mu with alpha immunoglobulin chains impairs lymphopoiesis and mucosal homing but promotes plasma cell maturation. Proc Natl Acad Sci U S A (2010) 107(7):3064–9. doi:10.1073/pnas.0912393107
90. Metzger D, Chambon P. Site- and time-specific gene targeting in the mouse. Methods (2001) 24(1):71–80. doi:10.1006/meth.2001.1159
91. Lu CS, Hung AF, Lin CJ, Chen JB, Chen C, Shiung YY, et al. Generating allergen-specific human IgEs for immunoassays by employing human ε gene knockin mice. Allergy (2015) 70(4):384–90. doi:10.1111/all.12572
92. Volpe E, Sambucci M, Battistini L, Borsellino G. Fas-fas ligand: checkpoint of T cell functions in multiple sclerosis. Front Immunol (2016) 7:382. doi:10.3389/fimmu.2016.00382
93. Yamaguchi M, Lantz CS, Oettgen HC, Katona IM, Fleming T, Miyajima I, et al. IgE enhances mouse mast cell Fc(epsilon)RI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J Exp Med (1997) 185(4):663–72. doi:10.1084/jem.185.4.663
94. Greer AM, Wu N, Putnam AL, Woodruff PG, Wolters P, Kinet JP, et al. Serum IgE clearance is facilitated by human FcεRI internalization. J Clin Invest (2014) 124(3):1187–98. doi:10.1172/JCI68964
95. Vangelista L, Soprana E, Cesco-Gaspere M, Mandiola P, Di Lullo G, Fucci RN, et al. Membrane IgE binds and activates Fc epsilon RI in an antigen-independent manner. J Immunol (2005) 174(9):5602–11. doi:10.4049/jimmunol.174.9.5602
96. Hibbert RG, Teriete P, Grundy GJ, Beavil RL, Reljic R, Holers VM, et al. The structure of human CD23 and its interactions with IgE and CD21. J Exp Med (2005) 202(6):751–60. doi:10.1084/jem.20050811
97. Yu P, Kosco-Vilbois M, Richards M, Köhler G, Lamers MC. Negative feedback regulation of IgE synthesis by murine CD23. Nature (1994) 369(6483):753–6. doi:10.1038/369753a0
98. Hirano M, Davis RS, Fine WD, Nakamura S, Shimizu K, Yagi H, et al. IgEb immune complexes activate macrophages through FcγRIV binding. Nat Immunol (2007) 8(7):762–71. doi:10.1038/ni1477
99. Platzer B, Ruiter F, van der Mee J, Fiebiger E. Soluble IgE receptors—elements of the IgE network. Immunol Lett (2011) 141(1):36–44. doi:10.1016/j.imlet.2011.08.004
100. Niki T, Tsutsui S, Hirose S, Aradono S, Sugimoto Y, Takeshita K, et al. Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. J Biol Chem (2009) 284(47):32344–52. doi:10.1074/jbc.M109.035196
101. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10(12):1292–9. doi:10.1038/ni.1814
102. Gitlin AD, von Boehmer L, Gazumyan A, Shulman Z, Oliveira TY, Nussenzweig MC. Independent roles of switching and hypermutation in the development and persistence of B lymphocyte memory. Immunity (2016) 44(4):769–81. doi:10.1016/j.immuni.2016.01.011
103. Wurzburg BA, Jardetzky TS. Conformational flexibility in immunoglobulin E-Fc 3-4 revealed in multiple crystal forms. J Mol Biol (2009) 393(1):176–90. doi:10.1016/j.jmb.2009.08.012
104. Drinkwater N, Cossins B, Keeble AH, Wright M, Cain K, Hailu H, et al. Human immunoglobulin E flexes between acutely bent and extended conformations. Nat Struct Mol Biol (2014) 21(4):397–404. doi:10.1038/nsmb.2795
105. Tong P, Granato A, Zuo T, Chaudhary N, Zuiani A, Han SS, et al. IgH isotype-specific B cell receptor expression influences B cell fate. Proc Natl Acad Sci U S A (2017) 114(40):E8411–20. doi:10.1073/pnas.1704962114
Keywords: IgE, B cell receptor, B-lymphocytes, IgE regulation, IgE memory
Citation: Laffleur B, Debeaupuis O, Dalloul Z and Cogné M (2017) B Cell Intrinsic Mechanisms Constraining IgE Memory. Front. Immunol. 8:1277. doi: 10.3389/fimmu.2017.01277
Received: 13 August 2017; Accepted: 25 September 2017;
Published: 13 November 2017
Edited by:
Deborah K. Dunn-Walters, University of Surrey, United KingdomReviewed by:
David Holowka, Cornell University, United StatesGregory C. Ippolito, University of Texas at Austin, United States
Copyright: © 2017 Laffleur, Debeaupuis, Dalloul and Cogné. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Cogné, Y29nbmVAdW5pbGltLmZy