 Karen M. Garcia-Rodriguez1,2
Karen M. Garcia-Rodriguez1,2 Anu Goenka1
Anu Goenka1 Maria T. Alonso-Rasgado2
Maria T. Alonso-Rasgado2 Rogelio Hernández-Pando3
Rogelio Hernández-Pando3 Silvia Bulfone-Paus1,4*
Silvia Bulfone-Paus1,4*
- 1Manchester Collaborative Centre for Inflammation Research, Faculty of Biology, Medicine and Health, School of Biological Sciences, Manchester, United Kingdom
- 2Faculty of Science and Engineering, School of Materials, University of Manchester, Manchester, United Kingdom
- 3Departamento de Patología Experimental, Instituto Nacional de Ciencias Médicas y Nutrición “Salvador Zubiran”, Mexico City, Mexico
- 4Division of Musculoskeletal and Dermatological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom
Tuberculosis causes more annual deaths globally than any other infectious disease. However, progress in developing novel vaccines, diagnostics, and therapies has been hampered by an incomplete understanding of the immune response to Mycobacterium tuberculosis (Mtb). While the role of many immune cells has been extensively explored, mast cells (MCs) have been relatively ignored. MCs are tissue resident cells involved in defense against bacterial infections playing an important role mediating immune cell crosstalk. This review discusses specific interactions between MCs and Mtb, their contribution to both immunity and disease pathogenesis, and explores their role in orchestrating other immune cells against infections.
Introduction
Tuberculosis (TB) is the world’s major infectious disease killer, accounting for 1.4 million deaths in 2015 (1). Progress in developing vaccines, diagnostics, and therapies has been hampered by an incomplete understanding of the immune response to the causative pathogen, Mycobacterium tuberculosis (Mtb).
Following entry of Mtb-containing droplets into the airways, bacilli are initially phagocytosed by alveolar macrophages (AMφ), providing a comfortable niche in which Mtb can reside, replicate, and evade immune cell detection (2). Mycobacterial pathogen-associated molecular patterns engage pattern recognition receptors (PRRs) to trigger signaling pathways, resulting in the release of various chemokines and cytokines and the recruitment and activation of immune cells (3). This process results in the internalization of mycobacteria by dendritic cells (DCs), which migrate to lymph nodes where they polarize naïve T cells to antigen-specific Th1 effector cells in an IL-12-dependent manner (4, 5). IFN-γ produced by Th1-polarized T cells activates mycobactericidal mechanisms in AMφ (6). Various immune cells are sequentially recruited to the sites of infection; neutrophils in the earliest stages as well as T cells, NK cells, and fibroblasts. These surround the infected AMφ to form a mycobacterial granuloma (2, 7), which acts as a “physical barrier” limiting bacillary dissemination. However, chronic granulomas also promote Mtb’s intracellular survival and impair elimination, resulting clinically, in latent TB disease (2). One-third of the world’s population is latently infected with Mtb and between 1 and 10% will develop progressive TB disease following “reactivation” of infection in later life (8). The mechanism of TB reactivation is unclear as yet, however, it is suggested that a failure of granuloma maintenance may be the cause (9). Thus, the granuloma is the result of a non-efficient immune control that will eventually progress to a chronic infection, rather than mycobacterial clearance.
Mast cells (MCs) are tissue resident cells strategically located in mucosal tissues (10) and are among the first cells to come in contact with pathogens (11). MCs contribute to bacterial immunity through multiple mechanisms such as bacterial recognition, activation, recruitment of immune cells to the site of infection, release of inflammatory mediators, and direct bacterial killing by extracellular traps (ETs). However, their main role may be orchestrating other immune cells against infections (11–14).
Since little is known about the MC contribution to TB pathogenesis, this review summarizes the MC strategies used in bacterial defense as well as potential and reported interactions occurring between Mtb and MCs (Figure 1).
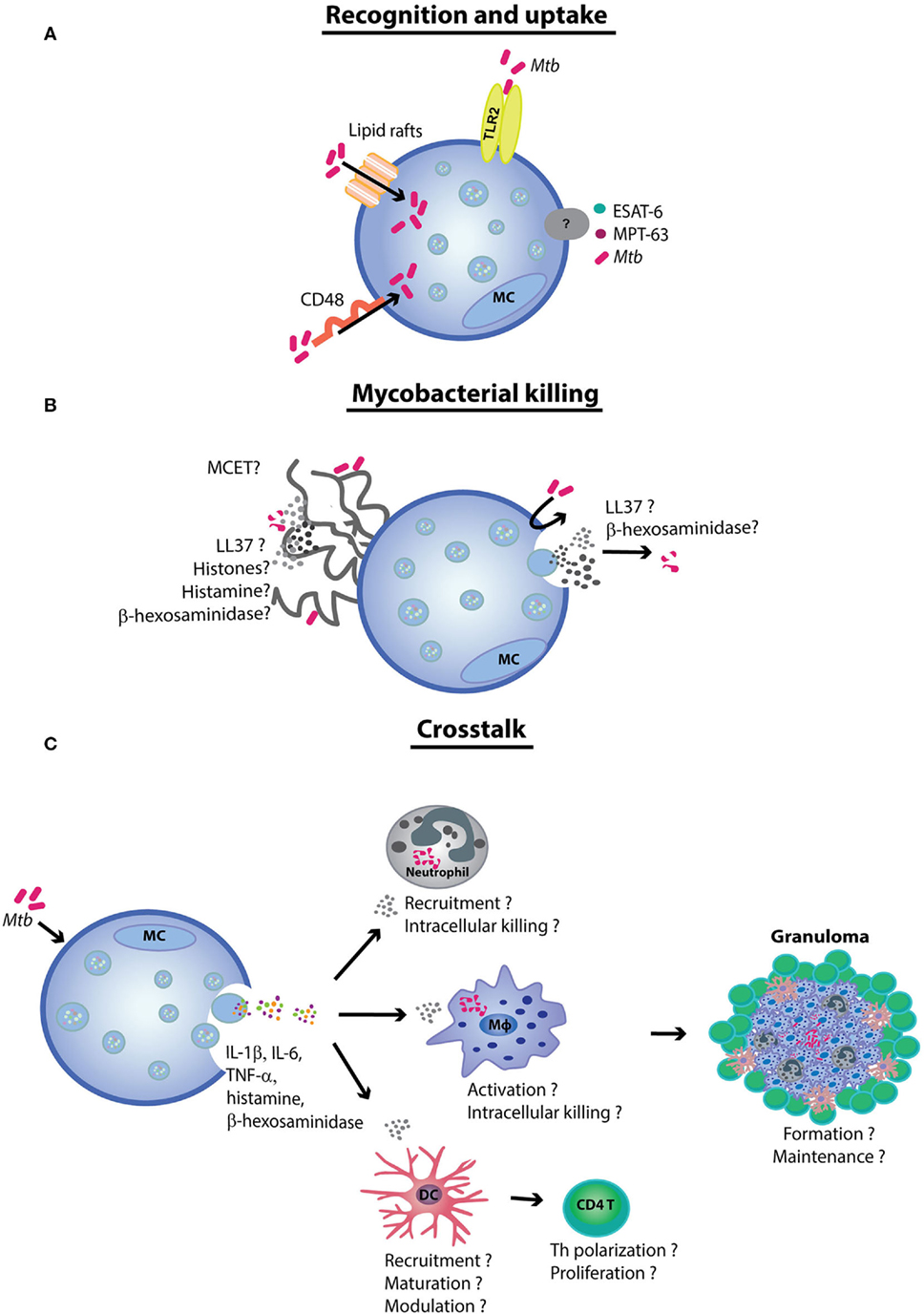
Figure 1. The role of mast cells (MCs) in tuberculosis. (A) MCs recognize Mycobacterium tuberculosis (Mtb) via the TLR2 and CD48 receptors. The latter also contributes to Mtb uptake. Although the uptake process remains yet unclear, mycobacteria have been demonstrated to be internalized by lipid rafts. (B) Mtb and the mycobacterial antigens early secretory antigenic target 6 (ESAT-6) and MPT-63 induce MC degranulation and cytokine release. It is likely that Mtb exposure induces antimicrobial peptide secretion and mast cell extracellular trap (MCET) formation. MCETs possibly contain antimycobacterial mediators, such as β-hexosaminidase and LL-37. (C) Finally, the MCs crosstalk with other immune cells [e.g., neutrophils, dendritic cells (DCs), and macrophages (Mφ)] contribute to antimycobacterial immunity. Although further experimental evidence is needed to prove the hypothesis, MCs seem to play a role in orchestrating tuberculosis granuloma formation and maintenance.
MC Ontogeny and Functions
Mast cells originate from pluripotent CD34+ and CD117+ hematopoietic stem cells and migrate as progenitors to various tissues where they mature influenced by the local microenvironment (15, 16). MCs are mainly located in the skin (~12,000/mm3) and mucosa including the lungs (~500–4,000/mm3) (10).
Mast cells express various PRRs (e.g., TLRs and CD48), complement receptors, and Fc receptors which upon engagement induce cell activation, degranulation, or both (17). Thereby, MCs release a great variety of pro- and anti-inflammatory pre-stored and de novo synthesized mediators such as histamine, heparin, tryptase, chymase, PGD2, LTC4/D4, chemokines as CCL1, CCL2, CCL4, CCL7, CCL12, CCL17, CXCL5, and CXCL8, and cytokines including IL-4, IL-3, IL-5, GM-CSF, IL-6, IL-13, IL-12, IFN-γ, TGFβ1, and TNF-α (18). This wide variety of products and their rapid release (in min/s) make MCs important modulators of inflammatory responses.
MC Involvement in Early Mycobacterial Infection
Although interactions between MCs and Mtb have been reported (Figure 1), the role of MCs in TB pathogenesis remains unclear. MC involvement in mycobacterial immunity was first observed in guinea pigs using electron microscopy. Shortly, after intratracheal infection with Mtb, a significant increase in MCs was detected in guinea pig lungs (19). A later study demonstrated that the number of MCs in mice lungs increases by ~23% after 15 days of Mtb exposure (20).
Bacillary Recognition
Rat peritoneal MCs (rPMCs) recognize Mtb via CD48 that is a glycosyl phosphoinositol-anchored cell surface protein (Figure 1A). Incubation with increasing concentrations of anti-CD48 antibodies together with Mtb correlate with a proportional decrease in histamine release (21). Previous work has shown that CD48 recognizes FimH protein expressed by fimbriated bacteria (such as Escherichia coli and Staphylococcus aureus) resulting in MC degranulation (12, 22, 23), raising the question of how precisely CD48 recognizes Mtb, which is not known to be fimbriated (24).
TLRs are a key receptor family implicated in pathogen recognition (22). Carlos and colleagues found MC TLR2 to be relevant in Mtb recognition (Figure 1A) (9). The transfer of TLR2+/+ MCs into TLR−/− Mtb-infected mice showed an increase of cytokine release and cell recruitment, suggesting MC TLR2 as key receptor upon mycobacterial challenge (9). MCs also express TLR4 that serves as a mannose receptor in complex with soluble CD14. In the absence of CD14, high concentrations of TLR4 ligands are required for MC–TLR4 activation (25–27). Presently, it is unclear if TLR4 (or other TLRs) are involved in Mtb recognition and if the TLR4–CD14 complex is necessary to trigger MC functions. Besides the TLRs and CD48, the CR3, CR4, C3aR, and C5aR complement receptors and the FcεRI, FcγRI, FcγRII, and FcγRIII receptors mediate MC responses to other bacteria, but it is as yet unknown if they are involved in MC–Mtb interactions (12, 22, 28).
Bacillary Binding and Uptake
Muñoz and colleagues suggested that Mtb is internalized by MCs via lipid rafts (Figure 1A). Cholesterol depletion of the rat basophilic leukemia cells (RBL-2H3) reduced internalization of mycobacteria. Interestingly, once internalized, mycobacteria survived intracellularly for 4 days after which most of the infected MCs (70%) had undergone lysis. These findings implicate MCs as a reservoir for Mtb (29). However, still unclear are the requirements and dynamics of Mtb internalization by MCs since few studies have demonstrated it experimentally and all were performed in animal models (21, 29). It is therefore important that the Mtb internalization is demonstrated in human MCs, together with the underlying mechanisms of internalization (aside from lipid rafts) and the intracellular compartments in which Mtb may reside. Published data describing MC interactions with other bacteria may guide the design of these investigations. For example, Salmonella typhi is taken up by complement receptors while E. coli is attached and internalized by FimH to the MC surface (30). Furthermore, preincubation of human MCs with IFN-γ increases the membrane attachment of S. aureus (31). Recently, the MC phagosome was demonstrated to interact with NOD-like receptors thus modulating cytokine production (32), and indicating that MC phagocytosis acts as an inducer of other MC activities.
Bacterial-Induced Mediator Release
Few studies have characterized the Mtb-induced MC production of soluble mediators of inflammation. Muñoz et al. reported that after stimulation with Mtb, rPMCs released de novo synthesized TNF-α and IL-6, followed by secretion of histamine and β-hexosaminidase. In addition, specific Mtb antigens [MPT-63 and early secretory antigenic target 6 (ESAT-6)] have also been shown to induce rPMCs to release TNF-α, IL-6, histamine, and β-hexosaminidase (Figure 1) (21).
Cytokines and Chemokines
Cytokines and chemokines released by MCs contribute to protective immunity in the context of bacterial infections. For example, it has been shown that MC depletion reduces TNF-α (important in mycobacterial granuloma maintenance) concentrations in bronchoalveolar lavage (BAL) of Bordetella pertussis-infected mice (33). Furthermore, following infection with Streptococcus pneumoniae, increased TNF-α concentrations in the BAL and MC numbers in the lung correlated with protection (34). Similarly, MC-derived IL-6 produced during Klebsiella pneumoniae challenge was observed to improve mouse survival by promoting neutrophil recruitment and intra-neutrophil killing (35), the importance of which in mycobacterial immunity is being increasingly recognized (36). MCs also produce a wide variety of soluble mediators potentially relevant to mycobacterial immunity, including IL-13, IL-12, IL-6, IL-4, TNF-α, CCL5, CXCL2, CCL7, and CCL2, following infection with Streptococcus equi (37). Since the cocktail of soluble mediators produced by MCs appears dependent on the specific pathogen, a comprehensive proteomic assessment of mediators produced by MCs in response to Mtb is needed.
Antimicrobial Peptides (AMPs)
Antimicrobial peptides kill pathogens by forming pores in cytoplasmic membranes; defensins and cathelicidins are the most studied (38). Early in infection, cathelicidins promote phagocytosis, upregulate the expression of costimulatory molecules in DCs and stimulate Th1 cytokine production, while later in the course of the disease they inhibit the production of pro-inflammatory molecules (39). Although little is known regarding the AMP repertoire that MCs may release, MC supernatants reduce bacterial burden (40, 41). Cathelicidin LL-37 is expressed in human dermal skin MCs while the respective murine homolog cathelicidin-related AMP (CRAMP) is produced by bone marrow-derived MCs (BMMCs). Upregulation of CRAMP expression by LPS reduces group A streptococcus extracellular titers (42), while MC-derived LL-37 promotes clearance of Enterococcus faecalis (43). Finally, β-hexosaminidase, which is released by MCs after degranulation, was observed to exhibit antimicrobial activities upon intracellular Listeria monocytogenes infection (39) and mMCP-6, a mouse tryptase, was essential for K. pneumoniae clearance in mMCP-6−/− mice (44). Thus, antimycobacterial molecules are likely to be secreted by MCs upon Mtb exposure (Figure 1B).
MC Degranulation and Histamine Release
Mast cell degranulation in Mtb-infected mice is associated with a decrease in leukocytes, neutrophils, mononuclear cells, IL-1β, TNF-α, MIP-2, IL-12, IFN-γ, and MCP-1 (20). Histamine is released during MC degranulation. Carlos and colleagues used histamine-deficient C57BL/6 mice to investigate the role of histamine in Mtb infection, which is detectable in high concentrations 28 days after Mtb infection. Mice lacking histamine showed decreased neutrophils numbers, as well as TNF-α and IL-6 levels in lung tissue, while IL-12 and IFN-γ concentrations were increased. Furthermore, the histamine-deficient lungs showed lymphocytic infiltration with an increase in the number of CD4+ T cells that correlated with reduced bacterial growth (45). Taken together, these findings suggest that MC degranulation may have a complex role in modulating the inflammatory response to Mtb. It would be important to investigate the redundancy of these pathways in Mtb infection using in vivo models, as well as determine whether MCs are the source of histamine in this context, which may indicate novel therapeutic avenues.
MC Extracellular Traps
The formation of ETs, named ETosis, is a type of cell death characterized by release of DNA (46). ETosis differs from apoptosis and necrosis since it lacks DNA fragmentation, disruption of the nuclear envelope, absence of phosphatidylserine in the outer membrane and caspase-independent activation (47). The formation of MCETs upon cell stimulation relies on ROS production by MCs that in turn promotes nuclear envelope disruption and release of DNA together with granular components with antimicrobial properties. The DNA backbone in combination with histones, proteases, and AMPs (39, 48) forms physical traps that catch and expose pathogens to high concentrations of antimicrobial molecules (Figure 1B) (48, 49).
A human mast cell line (HMC-1), showed ET formation upon L. monocytogenes infection. This Gram-positive bacterium was shown to promote disruption of the nuclear envelope followed by an increase in ROS production. Interestingly, the presence of β-hexosaminidase in the traps was observed to have an antimicrobial activity to intracellular L. monocytogenes (39). E. faecalis was also found to induce MCET after 3 h of incubation with BMMCs. However, the level of MCET observed was lower compared with the one promoted by other bacteria (43). This was possibly due to the low multiplicity of infection (MOI) used in this study (MOI 1:1). Streptococcus pyogenes induced MCET after infection (MOI 25:1) in HMC-1 cells exhibiting cathelicidin LL-37, histones, and tryptase in the traps and in murine BMMCs displaying tryptase and histones (50). This suggests that high bacterial burden promotes MCET activation. Interestingly, besides bacterial stimulation, IL-12 and IL-1β were found to induce MCET containing IL-17 after the stimulation of MCs from skin explants of patients with psoriasis (51).
Mycobacterium–MCET
Mycobacterium tuberculosis induces neutrophil ETs (NETs). However, Ramos-Kichik et al. have reported that although mycobacteria induce the formation of NETs, which include elastase and histones, the AMPs contained in the NETs are unable to kill mycobacteria (52). Three hour incubation of human neutrophils with the virulent Mtb and the less virulent Mycobacterium canettii showed that both mycobacteria were entrapped in NETs; however, neither Mtb nor M. canettii were killed. In fact, mycobacteria were not eliminated even at low bacterial concentrations (MOI 0.1:1), nor did NETs restrict ongoing mycobacterial replication.
Virulent Mtb secretes ESAT-6 and CFP-10 (10-kDa culture filtrate protein) through the ESX-1 secretory system. Both factors are important for the pathogenic intracellular pore-forming activities and phagosomal subversion observed in the early phase of TB (53). Interestingly, ESAT-6 has been shown to induce extracellular NETs by Ca+ influx (54). In addition, Mtb can induce ETs in human macrophages (Mφ) via the ESX-1 system, which is enhanced by IFN-γ (55). By contrast, in highly infected Mφ, it has been reported that after IFN-γ initiates necrosis without NETosis (56). This information suggests that virulent mycobacteria may actively promote NET formation to achieve their own ends of persistence, raising the hypothesis that MCETs may also be involved (Figure 1B). Although mycobacteria–MCETs have not been demonstrated, MCs produce a large repertoire of immunomodulatory mediators that are known to be contained in traps. Therefore, it is important that future studies investigate the mycobacteria induction of MCETs and the inclusion of antimycobacterial mediators.
MC Immune Crosstalk and the Mycobacterial Granuloma
Studies involving a broad array of bacterial pathogens have demonstrated the important role of MCs in promoting recruitment, maturation, and bactericidal activity of Mφ, DCs, and neutrophils (Table 1). However, the potential roles of MCs in modulating the delicate orchestration of immune crosstalk in mycobacterial immunity have not yet been described (Figure 1C). MCs could easily coordinate granuloma formation and maintenance. In support of this notion, Taweevisit and Poumsuk reported a correlation between MC numbers and granuloma formation (57). Briefly, 45 lymph nodes from patients with TB lymphadenitis were analyzed to determine the frequency of MCs in the granulomatous region. The authors observed that the number of MCs positively correlated with the number of granulomas. A similar correlation was found between multinucleated giant cells and MCs in the lymph nodes (57). Similar studies have been performed using skin biopsies of patients suffering with leprosy (Mycobacterium leprae) (58). Lepromatous leprosy (disseminated disease with high bacillary load) was associated with the lowest dermal MC density compared with paucibacillary and localized tuberculous leprosy. This suggests that MC functions may have a role in driving a differential susceptibility to these polar forms of leprosy, an as yet poorly explained clinical phenomenon (59). Furthermore, the higher MC numbers located around granulomas in the tuberculous group were considered to be indirect evidence of the role of MCs in activating the immune response to M. leprae infection. Interestingly, numerous MCs were found in the highly fibrotic dermal area and in the epineurial layer of lepromatous leprosy lesions, suggesting that MCs could be involved in the induction of fibrosis, including fibrotic leprosy neuritis (60).
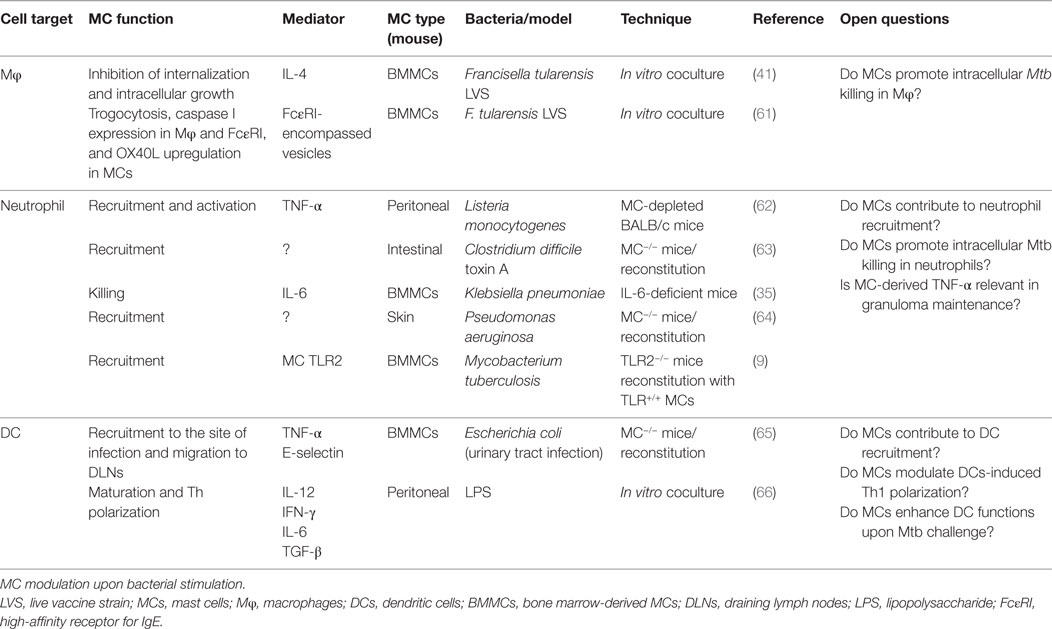
Table 1. MCs: immune cell crosstalk in antibacterial immunity.
MC-Derived Soluble Mediators and Mycobacterial Granuloma Maintenance
Mast cell-derived LL-37 and CRAMP are bactericidal for Mtb (67). Ramos-Espinosa and colleagues reported that the administration of adenovirus encoding the human cathelicidin LL-37 (AdLL37) and TNF-α (AdTNFα) had a protective role in inducing granuloma maintenance, and thus TB disease reactivation (68). These observations suggest that MCs and MC mediators in particular are involved in granuloma maintenance (Figure 1C). Similarly, von Stebut and colleagues reported that upon encounter with pathogens MCs release pre-stored TNF-α that induces neutrophil recruitment to the site of granulomatous inflammation. This was followed by the release of neutrophil-derived MIP-1α/β and MIP-2 chemokines both responsible for Mφ recruitment. Lack of this immediate pre-stored TNF-α release delayed Mφ recruitment and granuloma formation (69).
The study by Carlos et al. discussed earlier also described that during Mtb infection, TLR2 engagement induces cytokine release in the lung; as observed in a reconstitution murine model after 60 days of infection (9). The transfer of TLR2+/+ MCs into TLR−/− Mtb-infected mice showed diminished lung bacterial growth and an increase of TNF, IL-6, IL-1β concentrations, and neutrophil and mononuclear cell recruitment resulting in the restoration of granuloma formation (9). Therefore, MCs may be involved not only in the early but also in the late phase of infection. Furthermore, MC-derived IL-6 and TNF-α in this phase of infection may contribute to granuloma maintenance (9). The precise contribution of MCs in mycobacterial granuloma maintenance remains an important open question, and in vivo Mtb infection models combined with MC reconstitution experiments may yield critical insights into this area.
Conclusion
Tuberculosis is a highly contagious infectious disease caused by Mtb which infects billions, and kills millions of people worldwide. Although many efforts have been made to reduce TB mortality, the infection remains one the most important threats to human health. MCs are key lung resident immune sentinels that contribute to antibacterial immunity and are likely to play a key role in TB pathogenesis. A potential important and unique function of MCs is the crosstalk with other immune cells to orchestrate multiple effector functions, which may contribute to granuloma formation and maintenance. We have highlighted the potential roles MCs may play during TB that once addressed could inform the design of novel therapeutic strategies.
Author Contributions
KG-R and SB-P conceived, wrote, designed, and coordinated the manuscript. AG, MA-R, and RH-P provided helpful discussions and edited the manuscript. All the authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Martin Barron and Peter West for the critical reading of the manuscript.
Funding
KG-R is supported by CONACyT funding. AG is supported by fellowships from the MRC (MR/N001427/1) and ESPID.
References
1. World Health Organization. Global Tuberculosis Report 2016. Geneva: World Health Organization (2016).
2. Scriba TJ, Coussens AK, Fletcher HA. Human immunology of tuberculosis. Microbiol Spectr (2016) 4(5):1–24. doi:10.1128/microbiolspec.TBTB2-0016-2016
3. Stamm CE, Collins AC, Shiloh MU. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunol Rev (2015) 264(1):204–19. doi:10.1111/imr.12263
4. Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol (2014) 12(4):289–99. doi:10.1038/nrmicro3230
5. Sia JK, Georgieva M, Rengarajan J. Innate immune defenses in human tuberculosis: an overview of the interactions between Mycobacterium tuberculosis and innate immune cells. J Immunol Res (2015):12. doi:10.1155/2015/747543
6. Sasindran SJ, Torrelles JB. Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front Microbiol (2011) 2:2. doi:10.3389/fmicb.2011.00002
7. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol (2012) 12(5):352–66. doi:10.1038/nri3211
8. Dorhoi A, Kaufmann SH. Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J Immunol (2015) 45(8):2191–202. doi:10.1002/eji.201545493
9. Carlos D, Frantz FG, Souza-Júnior DA, Jamur MC, Oliver C, Ramos SG, et al. TLR2-dependent mast cell activation contributes to the control of Mycobacterium tuberculosis infection. Microbes Infect (2009) 11(8):770–8. doi:10.1016/j.micinf.2009.04.025
10. Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood (2011) 118(20):5383–93. doi:10.1182/blood-2011-07-358432
11. Abraham SN, John ALS. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol (2010) 10(6):440–52. doi:10.1038/nri2782
12. Trivedi NH, Guentzel MN, Rodriguez AR, Yu J-J, Forsthuber TG, Arulanandam BP. Mast cells: multitalented facilitators of protection against bacterial pathogens. Expert Rev Clin Immunol (2013) 9(2):129–38. doi:10.1586/eci.12.95
13. Johnzon C-F, Rönnberg E, Pejler G. The role of mast cells in bacterial infection. Am J Pathol (2016) 186(1):4–14. doi:10.1016/j.ajpath.2015.06.024
14. Urb M, Sheppard DC. The role of mast cells in the defence against pathogens. PLoS Pathog (2012) 8(4):e1002619. doi:10.1371/journal.ppat.1002619
15. Virk H, Arthur G, Bradding P. Mast cells and their activation in lung disease. Transl Res (2016) 174:60–76. doi:10.1016/j.trsl.2016.01.005
16. Bradding P. Human mast cell cytokines. Clin Exp Allergy (1996) 26(1):13–9. doi:10.1111/j.1365-2222.1996.tb00051.x
17. John ALS, Abraham SN. Innate immunity and its regulation by mast cells. J Immunol (2013) 190(9):4458–63. doi:10.4049/jimmunol.1203420
18. Moiseeva EP, Bradding P. Mast cells in lung inflammation. In: Gilfillan AM, Metcalfe DD, editors. Mast Cell Biology: Contemporary and Emerging Topics. Boston, MA: Springer (2011). p. 235–69.
19. Ratnam S, Ratnam S, Puri B, Chandrasekhar S. Mast cell response during the early phase of tuberculosis: an electron-microscopic study. Can J Microbiol (1977) 23(9):1245–51. doi:10.1139/m77-186
20. Carlos D, de Souza Júnior DA, de Paula L, Jamur MC, Oliver C, Ramos SG, et al. Mast cells modulate pulmonary acute inflammation and host defense in a murine model. J Infect Dis (2007) 196(9):1361–8. doi:10.1086/521830
21. Muñoz S, Hernández-Pando R, Abraham SN, Enciso JA. Mast cell activation by Mycobacterium tuberculosis: mediator release and role of CD48. J Immunol (2003) 170(11):5590–6. doi:10.4049/jimmunol.170.11.5590
22. Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol (2004) 4(10):787–99. doi:10.1038/nri1460
23. Malaviya R, Gao Z, Thankavel K, van der Merwe PA, Abraham SN. The mast cell tumor necrosis factor α response to FimH-expressing Escherichia coli is mediated by the glycosylphosphatidylinositol-anchored molecule CD48. Proc Natl Acad Sci U S A (1999) 96(14):8110–5. doi:10.1073/pnas.96.14.8110
24. Ramsugit S, Pillay M. Identification of Mycobacterium tuberculosis adherence-mediating components: a review of key methods to confirm adhesin function. Iran J Basic Med Sci (2016) 19(6):579–84. doi:10.22038/IJBMS.2016.7124
25. Marshall JS, Jawdat DM. Mast cells in innate immunity. J Allergy Clin Immunol (2004) 114(1):21–7. doi:10.1016/j.jaci.2004.04.045
26. McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol (2003) 170(4):1625–9. doi:10.4049/jimmunol.170.4.1625
27. Varadaradjalou S, Féger F, Thieblemont N, Hamouda NB, Pleau JM, Dy M, et al. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. Eur J Immunol (2003) 33(4):899–906. doi:10.1002/eji.200323830
28. Gri G, Frossi B, D’Inca F, Danelli L, Betto E, Mion F, et al. Mast cell: an emerging partner in immune interaction. Front Immunol (2012) 3:120. doi:10.3389/fimmu.2012.00120
29. Munoz S, Rivas-Santiago B, Enciso J. Mycobacterium tuberculosis entry into mast cells through cholesterol-rich membrane microdomains. Scand J Immunol (2009) 70(3):256–63. doi:10.1111/j.1365-3083.2009.02295.x
30. Malaviya R, Abraham SN. Mast cell modulation of immune responses to bacteria. Immunol Rev (2001) 179(1):16–24. doi:10.1034/j.1600-065X.2001.790102.x
31. Swindle EJ, Brown JM, Rådinger M, DeLeo FR, Metcalfe DD. Interferon-γ enhances both the anti-bacterial and the pro-inflammatory response of human mast cells to Staphylococcus aureus. Immunology (2015) 146(3):470–85. doi:10.1111/imm.12524
32. Zabucchi G, Trevisan E, Vita F, Soranzo MR, Borelli V. NOD1 and NOD2 interact with the phagosome cargo in mast cells: a detailed morphological evidence. Inflammation (2015) 38(3):1113–25. doi:10.1007/s10753-014-0077-x
33. Mielcarek N, Hörnquist EH, Johansson BR, Locht C, Abraham SN, Holmgren J. Interaction of Bordetella pertussis with mast cells, modulation of cytokine secretion by pertussis toxin. Cell Microbiol (2001) 3(3):181–8. doi:10.1046/j.1462-5822.2001.00106.x
34. Kerr AR, Irvine JJ, Gingles NA, Kadioglu A, Andrew PW, McPheat WL, et al. Role of inflammatory mediators in resistance and susceptibility to pneumococcal infection. Infect Immun (2002) 70(3):1547–57. doi:10.1128/IAI.70.3.1547-1557.2002
35. Sutherland RE, Olsen JS, McKinstry A, Villalta SA, Wolters PJ. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J Immunol (2008) 181(8):5598–605. doi:10.4049/jimmunol.181.8.5598
36. Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol (2012) 33(1):14–25. doi:10.1016/j.it.2011.10.003
37. Rönnberg E, Guss B, Pejler G. Infection of mast cells with live streptococci causes a toll-like receptor 2-and cell-cell contact-dependent cytokine and chemokine response. Infect Immun (2010) 78(2):854–64. doi:10.1128/IAI.01004-09
38. Harder J, Bartels J, Christophers E, Schroder J. A peptide antibiotic from human skin. Nature (1997) 387(6636):861. doi:10.1038/43088
39. Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, González-Jiménez M, Paredes-Vivas Y, Cerbulo-Vázquez A, et al. Listeria monocytogenes induces mast cell extracellular traps. Immunobiology (2017) 222(2):432–9. doi:10.1016/j.imbio.2016.08.006
40. Wei OL, Hilliard A, Kalman D, Sherman M. Mast cells limit systemic bacterial dissemination but not colitis in response to Citrobacter rodentium. Infect Immun (2005) 73(4):1978–85. doi:10.1128/IAI.73.4.1978-1985.2005
41. Ketavarapu JM, Rodriguez AR, Yu J-J, Cong Y, Murthy AK, Forsthuber TG, et al. Mast cells inhibit intramacrophage Francisella tularensis replication via contact and secreted products including IL-4. Proc Natl Acad Sci U S A (2008) 105(27):9313–8. doi:10.1073/pnas.0707636105
42. Di Nardo A, Vitiello A, Gallo RL. Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J Immunol (2003) 170(5):2274–8. doi:10.4049/jimmunol.170.5.2274
43. Scheb-Wetzel M, Rohde M, Bravo A, Goldmann O. New insights into the antimicrobial effect of mast cells against Enterococcus faecalis. Infect Immun (2014) 82(11):4496–507. doi:10.1128/IAI.02114-14
44. Thakurdas SM, Melicoff E, Sansores-Garcia L, Moreira DC, Petrova Y, Stevens RL, et al. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J Biol Chem (2007) 282(29):20809–15. doi:10.1074/jbc.M611842200
45. Carlos D, Fremond C, Samarina A, Vasseur V, Maillet I, Ramos S, et al. Histamine plays an essential regulatory role in lung inflammation and protective immunity in the acute phase of Mycobacterium tuberculosis infection. Infect Immun (2009) 77(12):5359–68. doi:10.1128/IAI.01497-08
46. Wartha F, Henriques-Normark B. ETosis: a novel cell death pathway. Sci Signal (2008) 1(21):e25. doi:10.1126/stke.121pe25
47. Guimaraes-Costa AB, Nascimento MT, Wardini AB, Pinto-da-Silva LH, Saraiva EM. ETosis: a microbicidal mechanism beyond cell death. J Parasitol Res (2012):11. doi:10.1155/2012/929743
48. Möllerherm H, von Köckritz-Blickwede M, Branitzki-Heinemann K. Antimicrobial activity of mast cells: role and relevance of extracellular DNA traps. Front Immunol (2016) 7:265. doi:10.3389/fimmu.2016.00265
49. Goldmann O, Medina E. The expanding world of extracellular traps: not only neutrophils but much more. Front Immunol (2013) 3:420. doi:10.3389/fimmu.2012.00420
50. von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood (2008) 111(6):3070–80. doi:10.1182/blood-2007-07-104018
51. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol (2011) 187(1):490–500. doi:10.4049/jimmunol.1100123
52. Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (2009) 89(1):29–37. doi:10.1016/j.tube.2008.09.009
53. Yu X, Xie J. Roles and underlying mechanisms of ESAT-6 in the context of Mycobacterium tuberculosis–host interaction from a systems biology perspective. Cell Signal (2012) 24(9):1841–6. doi:10.1016/j.cellsig.2012.05.014
54. Francis R, Butler R, Stewart G. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis (2014) 5(10):e1474. doi:10.1038/cddis.2014.394
55. Wong K-W, Jacobs WR. Mycobacterium tuberculosis exploits human interferon γ to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis (2013) 208(1):109–19. doi:10.1093/infdis/jit097
56. Lee J, Kornfeld H. Interferon-γ regulates the death of M. tuberculosis-infected macrophages. J Cell Death (2010) 3:1–11. doi:10.4137/JCD.S2822
57. Taweevisit M, Poumsuk U. High mast cell density associated with granulomatous formation in tuberculous lymphadenitis. Southeast Asian J Trop Med Public Health (2007) 38(1):115–9.
58. de Oliveira Magalhães G, da Costa Valentim V, dos Santos Pereira MJ, da Costa Nery JA, Illarramendi X, Antunes SLG. A quantitative and morphometric study of tryptase-positive mast cells in cutaneous leprosy lesions. Acta Trop (2008) 105(1):62–6. doi:10.1016/j.actatropica.2007.10.001
59. Talhari C, Talhari S, Penna GO. Clinical aspects of leprosy. Clin Dermatol (2015) 33(1):26–37. doi:10.1016/j.clindermatol.2014.07.002
60. Montagna NA, de Oliveira ML, Mandarim-de-Lacerda CA, Chimelli L. Leprosy: contribution of mast cells to epineurial collagenization. Clin Neuropathol (2005) 24(6):284–90.
61. Rodriguez AR, Yu J-J, Navara C, Chambers JP, Guentzel MN, Arulanandam BP. Contribution of FcɛRI-associated vesicles to mast cell-macrophage communication following Francisella tularensis infection. Innate Immun (2016) 22(7):567. doi:10.1177/1753425916663639
62. Gekara NO, Weiss S. Mast cells initiate early anti-Listeria host defences. Cell Microbiol (2008) 10(1):225–36. doi:10.1111/j.1462-5822.2007.01033.x
63. Wershil BK, Castagliuolo I, Pothoulakis C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology (1998) 114(5):956–64. doi:10.1016/S0016-5085(98)70315-4
64. Siebenhaar F, Syska W, Weller K, Magerl M, Zuberbier T, Metz M, et al. Control of Pseudomonas aeruginosa skin infections in mice is mast cell-dependent. Am J Pathol (2007) 170(6):1910–6. doi:10.2353/ajpath.2007.060770
65. Shelburne CP, Nakano H, John ALS, Chan C, McLachlan JB, Gunn MD, et al. Mast cells augment adaptive immunity by orchestrating dendritic cell trafficking through infected tissues. Cell Host Microbe (2009) 6(4):331–42. doi:10.1016/j.chom.2009.09.004
66. Dudeck A, Suender CA, Kostka SL, von Stebut E, Maurer M. Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur J Immunol (2011) 41(7):1883–93. doi:10.1002/eji.201040994
67. Rivas-Santiago B, Santiago CER, Castañeda-Delgado JE, León-Contreras JC, Hancock RE, Hernandez-Pando R. Activity of LL-37, CRAMP and antimicrobial peptide-derived compounds E2, E6 and CP26 against Mycobacterium tuberculosis. Int J Antimicrob Agents (2013) 41(2):143–8. doi:10.1016/j.ijantimicag.2012.09.015
68. Ramos-Espinosa O, Hernández-Bazán S, Francisco-Cruz A, Mata-Espinosa D, Barrios-Payán J, Marquina-Castillo B, et al. Gene therapy based in antimicrobial peptides and proinflammatory cytokine prevents reactivation of experimental latent tuberculosis. Pathog Dis (2016) 74(7):ftw075. doi:10.1093/femspd/ftw075
Keywords: Mycobacterium tuberculosis, mast cells, degranulation, extracellular traps, granuloma, infection
Citation: Garcia-Rodriguez KM, Goenka A, Alonso-Rasgado MT, Hernández-Pando R and Bulfone-Paus S (2017) The Role of Mast Cells in Tuberculosis: Orchestrating Innate Immune Crosstalk? Front. Immunol. 8:1290. doi: 10.3389/fimmu.2017.01290
Received: 31 July 2017; Accepted: 26 September 2017;
Published: 17 October 2017
Edited by:
Norbert Reiling, Forschungszentrum Borstel (LG), GermanyReviewed by:
Lúcia Helena Faccioli, University of São Paulo, BrazilRoland Lang, University Hospital Erlangen, Germany
Copyright: © 2017 Garcia-Rodriguez, Goenka, Alonso-Rasgado, Hernández-Pando and Bulfone-Paus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Bulfone-Paus, c2lsdmlhLmJ1bGZvbmUtcGF1c0BtYW5jaGVzdGVyLmFjLnVr