 S. M. Shahjahan Miah1†
S. M. Shahjahan Miah1† Chathuraka T. Jayasuriya2†
Chathuraka T. Jayasuriya2† Alexander I. Salter1
Alexander I. Salter1 Emma C. Reilly1Céline Fugere1Wentian Yang2Qian Chen2
Emma C. Reilly1Céline Fugere1Wentian Yang2Qian Chen2 Laurent Brossay1*
Laurent Brossay1*
- 1Department of Molecular Microbiology and Immunology, Graduate Program in Pathobiology, Division of Biology and Medicine, Brown University Alpert Medical School, Providence, RI, United States
- 2Department of Orthopaedics, Rhode Island Hospital and Brown University Alpert Medical School, Providence, RI, United States
The ubiquitously expressed tyrosine phosphatase Src homology region 2 domain-containing phosphatase-2 (SHP-2, encoded by Ptpn11) is required for constitutive cellular processes including proliferation, differentiation, and the regulation of immune responses. During development and maturation, subsets of T cells express a variety of inhibitory receptors known to associate with phosphatases, which in turn, dephosphorylate key players of activating receptor signaling pathways. We hypothesized that SHP-2 deletion would have major effects on T cell development by altering the thresholds for activation, as well as positive and negative selection. Surprisingly, using mice conditionally deficient for SHP-2 in the T cell lineage, we show that the development of these lymphocytes is globally intact. In addition, our data demonstrate that SHP-2 absence does not compromise T cell effector functions, suggesting that SHP-2 is dispensable in these cells. Unexpectedly, in aging mice, Ptpn11 gene deletion driven by CD4 Cre recombinase leads to cartilage tumors in wrist bones in a T cell-independent manner. These tumors resemble miniature cartilaginous growth plates and contain CD4-lineage positive chondrocyte-like cells. Altogether these results indicate that SHP-2 is a cartilage tumor suppressor during aging.
Introduction
Src homology region 2 domain-containing phosphatase-2 (SHP-2) is ubiquitously expressed and is required for gastrulation, hematopoiesis, and embryonic development (1, 2). SHP-2’s widespread expression positions it to affect the outcome of many cellular processes and human diseases. In humans, gain-of-function mutations in SHP-2 result in Noonan syndrome, a severe disease marked by dysmorphic facial features, short stature, heart disease, skeletal malformation, webbed neck, and mental retardation (3, 4). SHP-2 has also been postulated to function as a tumor suppressor, whereby diminished protein expression increases the risk for hepatocellular carcinoma (5). Additionally, mutation in Ptpn11 has been shown to cause metachondromatosis, a rare syndrome leading to cartilage tumors (6).
Because SHP-2 is required for gastrulation, knockout mice die during gestation and conditional knockout animals must be used to study its effects in specific cell subsets. SHP-2 has been shown to both positively and negatively regulate cellular responses, depending on its interactions with other proteins; however, most reports suggest that SHP-2 plays a positive role in hematopoiesis (3, 7–10). SHP-2 has also been shown to act through the Ras pathway to promote cell proliferation (3). Nevertheless, SHP-2 has inhibitory functions in other contexts. For example, SHP-2 can inhibit the mTOR pathway and cell-cycle entry of activated T cells (11). SHP-2 is also believed to negatively regulate multiple JAK/STAT pathways, although the mechanism through which it acts remains unclear (3, 12). SHP-2 contains two tandem SH2 domains and a protein tyrosine phosphatase domain. The N-terminal SH2 domain mediates the binding of SHP-2 to other signaling proteins. At the steady state, SHP-2 activity is repressed due to the intra-molecular association of the N-terminal SH2 domain with the tyrosine phosphatase domain (13). Importantly, SHP-2 associates with immunoreceptor tyrosine-based inhibitory motifs on natural killer cell receptors (NKRs), further suggesting an inhibitory role (14).
Several subsets of T cells express inhibitory receptors known to associate with SHP-2. For instance, activated classical effector CD8+ T lymphocytes express members of the Ly49 receptor family, KLRG1, and PD-1 in addition to an αβ T cell receptor (TCR) (15). Invariant natural killer T (iNKT) cells also express certain inhibitory receptors (CD94/NKG2A, and members of the Ly49 family) in addition to a semi-invariant αβ TCR, which recognizes glycolipid antigens presented by the non-classical MHC class I molecule CD1d (16). Given that SHP-2 is implicated in TCR signaling and is known to associate with inhibitory NKRs expressed on CD8+ T cells and iNKT cells, we hypothesized that SHP-2 deletion would have major effects on development by altering the thresholds for activation, as well as positive and negative selection.
Both conventional T cells and iNKT cells develop in the thymus from shared T cell precursors. During maturation, T cell precursors pass through four double-negative (DN) stages, marked by an absence of CD4 or CD8 surface expression. As these immature cells proliferate, they begin to express CD4 and CD8 and enter the double-positive (DP, CD4+CD8+) stage of conventional T cell development. iNKT cells branch off from the DP stage following TCR α chain rearrangement and positive selection by CD1d-expressing CD4+CD8+ cortical thymocytes (17). Because conventional T cells and iNKT cells derive from DP thymocytes, we crossed mice with loxP-flanked alleles of Ptpn11 with CD4-Cre transgenic and Lck transgenic lines to delete SHP-2 in both subsets. Unexpectedly, we found that SHP-2 is dispensable for the development, differentiation, and functions of both effector CD8+ T cells and iNKT cells.
Surprisingly, in aging mice, Ptpn11 gene deletion driven by CD4 Cre recombinase (but not LckCre) led to cartilage tumors presenting large chondrocyte-like cells and fibrochondrocyte-like cells. Importantly, SHP-2fl/fl-CD4-Cre mice on a RAG null background also developed cartilage tumors, ruling out contributions from T cells. In support of this conclusion, we found that CD4-Cre was not restricted to T cells and was active in cartilage as well as several non-T cell subsets. Therefore, SHP-2 regulates cartilage homeostasis through a CD4-lineage positive subset.
Materials and Methods
Mice
Inbred C57BL/6 mice and B6.Cg-Tg(CD4-cre)1Cwi mice were purchased from Taconic Farms (Hudson, NY, USA). Inbred C57BL/6 mice, B6.SJL-Ptprca Pepcb/BoyJ, B6.Cg-Tg(Lck-cre)3779Nik/J, B6.129S7-Rag1Tm1Mom/J and B6.129P2-Ptpn6tm1Rsky/J (SHP-1fl/fl) were purchased from The Jackson Laboratory. Ptpn11fl/fl (SHP-2fl/fl) mice with LoxP recombination sites flanking Ptpn11 were previously described (18) and bred with CD4-Cre mice and Lck-Cre mice. SHP-2fl/fl-CD4-Cre mice were crossed to R26R-EFYP mice to produce SHP-2fl/fl-CD4-Cre-ROSAEYFP mice. Jα18−/− mice were bred, crossed to B6 (>12 generations). SHP-1fl/fl mice were bred with CD4-Cre recombinase mice. All mice were bred in pathogen-free breeding facilities at Brown University. All experiments were conducted in accordance with institutional guidelines for animal care.
Murine Lymphocyte Isolation
Mice were sacrificed by isofluorane treatment. Cardiac puncture was performed prior to the harvesting of the organs. Liver was perfused before harvesting. Spleens were dissociated and lymphocytes were enriched using Lympholyte Cell Separation Media (Cederlane). Livers were dissociated using a gentleMACS Dissociator (Miltenyi Biotec) and lymphocytes were enriched using a 40–70% discontinuous Percoll gradient (GE Healthcare) as previously described (19). Thymi were dissociated and thymocytes were washed prior to analysis.
Surface Staining, Antibodies, and Flow Cytometry
Cells were stained with fluorochrome-conjugated monoclonal antibodies and 2.4G2 blocking antibody and incubated in the dark for 30 min on ice. For CD1d tetramer staining, cells were incubated in the dark for 15 min at room temperature followed by 15 min on ice. Flow cytometric analysis was performed on a FACSAria (BD Biosciences) or a MACSQuant (Miltenyi Biotec). Data were analyzed using FlowJo (Tree Star).
TCRβ-FITC, HSA-FITC, CD4-FITC, TCRβ-PE, Ly49C/I/F/H-PE, TCRβ-PerCPCy5.5, Ly49G2-PerCPeFluor710, CD62L-PE-Cy6, NK1.1-PE-Cy7, KLRG1-APC, CD25-APC, CD19-PE, CD44-APCeFluor780, CD8α-eFluor450, and CD3-eFluor450 were purchased from ebioscience. Ly49G2-FITC, Ly49C/I-FITC, Ly49A/D-PE, and CD4-PerCP were purchased from BD Pharmingen. BV421-CD127, BV510-TCRβ, BV570-CD45, BV605-CD3, BV605-CCR6, and BV785-NK1.1 were purchased from Biolegend. PE- and APC-conjugated α-GalCer loaded CD1d tetramer was prepared in our laboratory. CD1d, M45 and M57 tetramers were kindly provided by the National Institute of Allergy and Infectious Disease MHC Tetramer Core Facility at Emory University (Atlanta, GA, USA).
Western Blot
Splenocytes from SHP-2 heterozygous (fl/+) and knockout (fl/fl) animals were sorted using a FACSAria to greater than 95% efficiency. TCRβ+ and TCRβ− populations were collected and lysed in Lysis buffer. Cellular lysates were added to SDS/sample buffer, boiled, and run on a Ready Gel 4–20% Tris-HCl (Bio-Rad, Hercules, CA, USA) with 1× SDS-PAGE running buffer. The gel was blotted against a nitrocellulose membrane (Bio-Rad) using 1× transfer buffer with 10% methanol. After blocking in a 5% milk solution for 1 h at room temperature, rabbit α-mouse SHP-2 (Abcam) and β-actin (Ambion) antibodies were incubated with the membrane overnight at 4°C. HRP-conjugated-donkey α-rabbit IgG secondary antibody (Jackson Immunoresearch, Westgrove, PA, USA) was incubated with the membrane for 1 h at room temperature. Membranes were developed with Super signal Chemiluminescent Substrate (Pierce) and photographed using a ChemiDoc XRS + System (Bio-Rad).
Microcomputed Tomography (μ-CT) and X-Ray Analysis
X-ray images of the entire skeleton were obtained immediately after euthanasia using a Faxitron X-ray system (Wheeling). After fixation in 4% paraformaldehyde, μ-CT images of skeletal tissues were scanned with a desktop microcomputer graphic imaging system (μ-CT40, Scanco Medical AG). For μ-CT, 3D image acquisitions took 36 min with a beam parameter of 70 kVp.
Histology
Mice were euthanized at the indicated ages, and femurs, tibiae, and paws were removed and fixed in 4% paraformaldehyde overnight at 4°C. Postnatal skeletal tissues were decalcified in 0.5 M EDTA before embedding. Tissue sections (5 µm) were stained with hematoxylin and eosin. Some sections were separately stained with Safranin O and Fast Green.
Murine Cytomegalovirus (MCMV) Infection
American Type Culture Collection Strain #VR1399 and RVG-102 clone of MCMV recombinant for GFP under the promoter of the immediate early gene-1 (i.e., 1) (a gift of Dr. Hamilton, Duke University) were prepared from salivary glands as previously described (20, 21). On day 0, mice were infected intraperitoneally with 5 × 104 plaque-forming units (pfu) of MCMV. Mice were sacrificed for analysis at 7 days and 8–12 weeks postinfection.
In Vitro iNKT Cell Stimulation and Cytokine Analysis
A method described by Stankovic et al was used (22). Briefly, CD1d tetramer-positive cells were sorted using a FACSAria to greater than 95% efficiency. A 96-well plate was coated with 0.5 μg/well purified α-mouse CD3ε and CD28 antibodies (eBioscience) and iNKT cells were plated at a concentration of 10,000 cells per well. Supernatant was collected 24 h poststimulation. Cytokine production was quantified by Cytometric Bead Array (BD Biosciences). Samples were run on a FACSAria and data were analyzed using FCAP Array Software (BD Biosciences).
Proliferation Analysis
Recipient mice were non-lethally irradiated with 7.5 Gy and placed on oral Sulfamethoxazole and Trimethoprim (Hi-tech Pharmacal). On day 3 postirradiation, donor mice were sacrificed, thymi were harvested, and thymocytes were extracted and counted. CD8+ T cells were depleted using anti-CD8 magnetic beads via an AutoMACS. Remaining cells were stained for 10 min at 37°C in the dark with Cell Proliferation Dye eFluor450 (eBioscience) in PBS at a labeling concentration of 10 µM. 8–10 million cells were injected into each irradiated recipient. Six days postinjection, mice were sacrificed and the spleen and liver were harvested. Lymphocytes were extracted, stained, run on a MACSquant, and analyzed using FlowJo.
Statistical Analysis
Data sets were analyzed for statistical significance using unpaired, two-tailed, Student’s t-tests. Values of p < 0.05 were considered significant. Graphs and statistics were generated and calculated using Prism (GraphPad).
Results
SHP-2 Is Dispensable for T Cell Development
SHP-2 floxed/floxed (fl/fl) mice (18) were crossed to mice carrying a transgene for Cre recombinase driven by the cd4 enhancer/promoter/silencer (23, 24). As a result, the Ptpn11 gene encoding SHP-2 is deleted at the DN4/DP stages of T cell development when CD4 is first expressed. For simplicity, we will refer to SHP-2fl/fl-CD4-Cre as SHP-2−/− and their littermate controls, SHP-2fl/+-CD4-Cre, as wild type (WT). To verify that SHP-2 was efficiently deleted in T cells, splenic T cells and non-T cell populations from WT and SHP-2−/− animals were sorted for protein analysis by Western blot using α-mouse SHP-2 primary antibody. Expression of SHP-2 in T cells was strongly reduced to undetectable levels SHP-2−/− mice (Figure 1A).
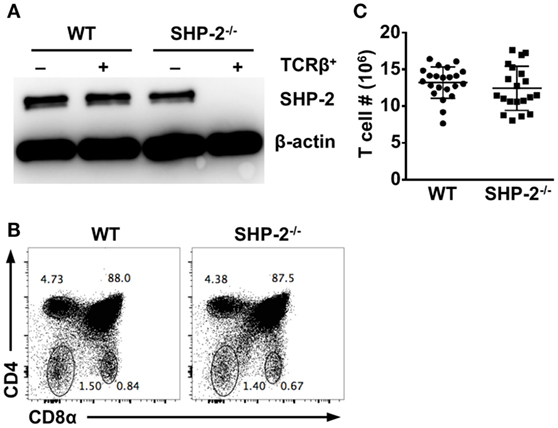
Figure 1. Src homology region 2 domain-containing phosphatase-2 (SHP-2) deficiency does not affect T cell development. (A) SHP-2 expression in splenocytes was evaluated by Western blot. YFP+CD3+TCRβ+ and YFP−CD3−TCRβ− splenocytes were sorted from wild-type (WT) and SHP-2−/− mice. Cellular lysates were blotted with α-SHP-2 and anti-β-actin antibodies. (B) Representative FACS plots show thymic T cell development in WT and SHP-2−/− mice. (C) Number of T cells in the spleen of SHP-2 deficient and littermate controls.
We then analyzed thymic T cell development in our conditional knockout mice. We found that WT and SHP-2−/− mice had comparable frequencies of DP (CD4+CD8+), single-positive (CD4+ or CD8+), and DN (CD4−CD8−) thymic lymphocytes (Figure 1B). Furthermore, no obvious T cell defects were observed in peripheral compartments. The number of splenic T cells (Figure 1C), splenic CD8+ T cells (Figures S1A,B in Supplementary Material) and CD4+ T cells (data not shown) were comparable between SHP-2−/− mice and WT controls. However, not all T cells express inhibitory receptors, so the absence of SHP-2 could potentially affect the development of small subsets of T cells known to express these receptors. These include iNKT cells, Ly49+CD8+ T cells with a memory phenotype (CD44hiCD122+Ly6C+) (25), and exhausted T cells during chronic infection (26). To this end, we first examined CD8+ T cells with a memory phenotype by staining for CD44 and CD62L. No significant difference in the frequency of effector memory (CD44hiCD62Llo) CD8+ T cells between WT and SHP-2−/− mice was observed (Figure S1C in Supplementary Material). SHP-2-deficiency also did not affect the frequency of KLRG1+ effector CD8+ T cells in all organs we tested (Figure S1D in Supplementary Material). Ly49 antibody cocktail staining revealed that Ly49 (Ly49C/I/F/H) expression did not substantially differ in the spleen, blood, or liver (Figure S1E in Supplementary Material). When analyzed separately, only in the case of the Ly49G2 receptor did we find a modest decrease of hepatic Ly49G2+ CD8+ T cells in SHP-2−/− mice (Figure S1F in Supplementary Material).
As previously mentioned, iNKT cells also express a variety of inhibitory receptors such as Ly49 (27). While the role of inhibitory receptors on iNKT cells has not been defined, it has been suggested that they control iNKT cell autoreactivity and tolerance (28, 29). Due to the association of these receptors with SH2 domain-containing phosphatases, we hypothesized that the absence of SHP-2 might affect iNKT cell development. Because iNKT cells are positively selected by CD1d-expressing CD4+CD8+ cortical thymocytes (30), SHP-2 is absent in both iNKT cell precursors and DP cells mediating positive selection in our conditional knockout mice. However, CD1d tetramer staining showed no significant difference in the absolute number of iNKT cells (Figures S2A,B in Supplementary Material). Additionally, no defects were observed in iNKT cell frequency among each stage of thymic maturation, where the majority of cells progressed to mature Stage III iNKT cells (Figure S2C in Supplementary Material). The TCR Vβ chain repertoire and Ly49 expression profile of iNKT cells from WT and SHP-2−/− mice were also comparable (data not shown). Collectively, these results indicate that SHP-2 is dispensable during iNKT cell development and maturation.
It has been previously reported that conditional deletion of SHP-2 by placing Cre recombinase under the control of Lck promoter results in defective conventional T cell development (8). As we did not find a T cell defect when SHP-2 was deleted by CD4-Cre, we also crossed SHP-2fl/fl mice to Lck-Cre transgenic mice, another T cell-specific Cre recombinase mouse line. Similarly to the SHP-2fl/fl-CD4-Cre mice, no developmental defect was observed in T cells in the thymus (Figure 2A) or in the periphery (Figure 2B). Altogether the data using both SHP-2fl/fl-CD4-Cre mice and SHP-2fl/fl-Lck-Cre mice demonstrate that SHP-2 is globally dispensable for T cell development.
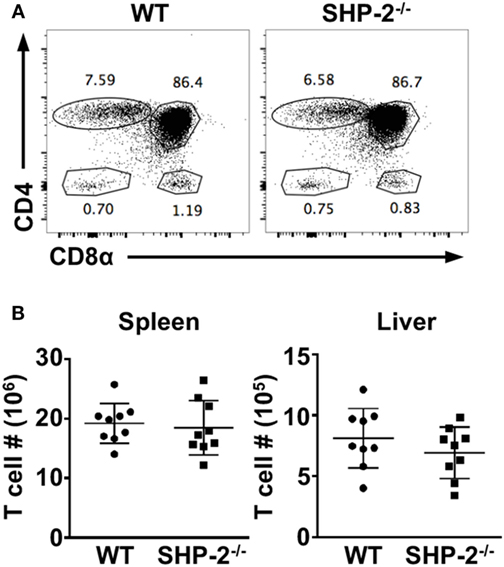
Figure 2. T cell development is mostly normal in SHP-2−/−LCK-Cre mice. (A) Representative FACS plots show thymic T cell development in wild-type (WT) and SHP-2−/− mice. (B) Number of T cells in the spleen and liver of SHP-2−/− mice and littermate controls. Data are pooled from three independent experiments, n = 9. Error bars indicate SD.
SHP-2 Is Not Required for iNKT Cell Effector Functions and Effector CD8+ T Cell Differentiation
We next sought to determine whether deletion of SHP-2 affected iNKT cell and T cells functions. Because activated iNKT cells are known to rapidly secrete large amounts of cytokines upon activation, we quantified cytokine production following in vitro TCR cross-linking as previously described (22). Normalization of three independent experiments revealed that iNKT cell cytokine production was unaffected by SHP-2 absence (Figure S2D in Supplementary Material). More specifically, IL-2, IL-4, IFN-γ, and TNF-α production by SHP-2-deficient iNKT cells did not differ significantly from WT controls. To assess TCR-mediated cytokine production in an in vivo setting, we injected WT and SHP-2−/− mice intraperitoneally with 2 µg of α-GalCer. α-GalCer is the prototypical ligand for iNKT cells and leads to vigorous cytokine production and proliferation (20). Because iNKT cell TCR cell surface expression is reduced after activation, we analyzed iNKT cells at early time points. At 2 h poststimulation, intracellular cytokine staining showed similar frequencies of IL-4−IFN-γ−, IL-4+, IFN-γ+, and IL-4+IFN-γ+ iNKT cells between WT and SHP-2−/− mice (Figure S2E in Supplementary Material). To evaluate whether the absence of SHP-2 causes an iNKT cell proliferative defect, we intravenously injected CD8-depleted SHP-2−/− (CD45.2+) and congenic WT (CD45.1+) thymocytes into sublethally irradiated Jα18−/− mice. Proliferation cycles were quantified by Cell Proliferation Dye eFluor450. In these experiments, SHP-2−/− and congenic WT iNKT cells proliferated to an equal extent in the leukopenic environment (Figure S2F in Supplementary Material). These results suggest that SHP-2 does not affect iNKT cell ability to proliferate.
We next asked whether SHP-2 affected the formation of effector CD8+ T cells during infection. Using the MCMV infection model, we examined CD8+ T responses at early and late time points (7 days and 8–12 weeks postinfection, respectively). We found the frequencies of CD8+ T cells, CD44hiCD62Llo effector memory CD8+ T cells, and KLRG1+CD8+ T cells to be nearly identical between WT and SHP-2−/− mice at both time points (Figure 3). Several MCMV epitopes have been described in C57BL/6 mice including M38, M139, M45, and M57 (21). CD8+ T cells specific to M38 and M139 are classified as inflationary cells (31), while CD8+ T cells specific for M45 and M57 epitopes have been shown to differentiate into a classical central memory phenotype (31, 32). We simultaneously examined CD8+ T cells specific for M45 and M57 epitopes, which expand during acute infection and have been associated with long-term protection against MCMV (21). On day 7 postinfection, no significant difference in the number of effector Ag-specific CD8+ T cells was observed (Figure 3B). Furthermore, at 10 weeks postinfection, we found that SHP-2 deficiency did not affect the formation of Ag-specific memory CD8+ T cells (Figure 3C,D). Overall, the data suggest that SHP-2 is dispensable for Ag-specific CD8+ T cell formation. Interestingly, preliminary analysis of SHP-1fl/fl-CD4-Cre conditional mice indicates that there is no gross defect in T cell and iNKT cell development (Figure S3 in Supplementary Material), suggesting a possible redundancy between SHP-1 and SHP-2 in T cell development. Generation of mice conditionally deficient for both SHP-1 and SHP-2 in the T cell lineage will clarify this issue.
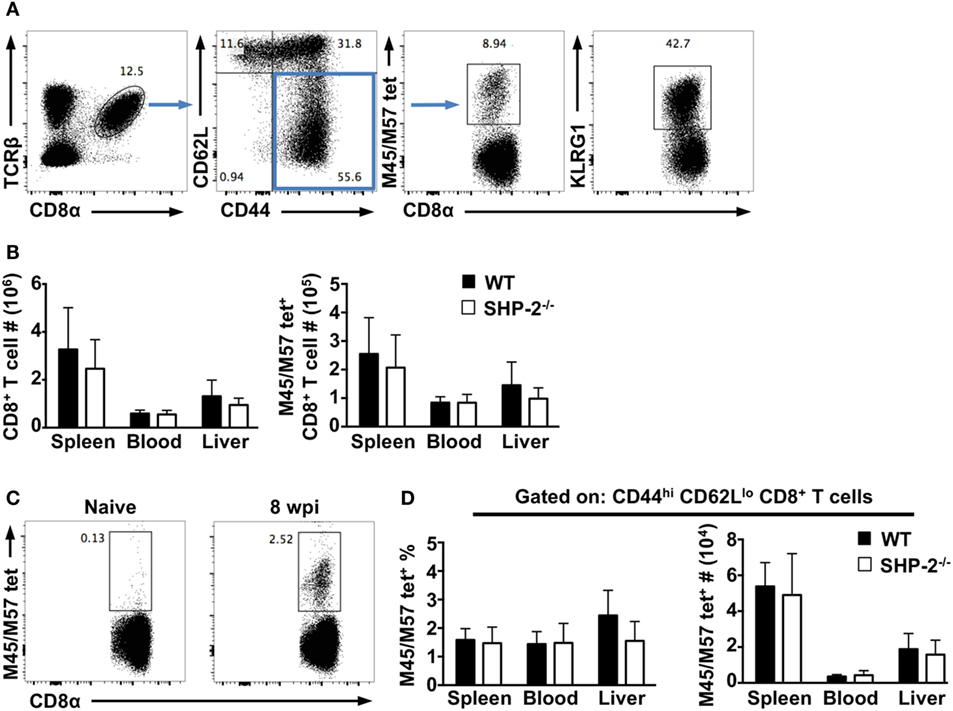
Figure 3. Src homology region 2 domain-containing phosphatase-2 (SHP-2) does not affect early immune responses and memory CD8+ T cell differentiation during murine cytomegalovirus (MCMV) infection. (A) Representative FACS plots show staining of CD8+ T cells and m45/57tet+ CD8+ T cells at day 7 postinfection. (B) The absolute numbers of CD8+ T cells and m45/57tet+ CD8+ T cells at day 7 postinfection. n = 10 wild-type (WT) and 8 SHP-2−/− mice over two independent experiments. Graphs show mean and SD. (C) Representative FACS plots show staining for antigen-specific CD8+ T cells in naïve and infected WT mice. (D) The absolute number and frequency of m45/57tet+ CD44hiCD62Llo CD8+ T cells from the spleen, blood, and liver of mice at 10 weeks post infection. n = 5 WT and 5 SHP-2−/− mice. Graphs show mean and SD.
Adult SHP-2fl/fl-CD4-Cre Mice Develop Cartilage Tumors
Since SHP-2 is a regulator of lymphocyte activation, we hypothesized that aging SHP-2-deficient mice might be predisposed to autoimmune disease, as reported for other T cell tyrosine protein phosphatases (33–35). Although we did not observe any signs of autoimmunity (data not shown), we surprisingly found that 6 month-old SHP-2fl/fl-CD4-Cre mice exhibited markedly decreased mobility, while littermate controls did not. Faxitron X-ray radiographs and μ-CT of SHP-2−/− mice revealed enlarged bony masses at the wrists (Figures 4A,B) and knees (data not shown). Moreover, X-rays showed kinky tails (Figure S4A in Supplementary Material) and spinal abnormalities (Figure S4B in Supplementary Material) in most SHP-2−/− animals, likely due to overgrowth of the bones in the vertebral column (data not shown). We analyzed a larger group of mice (>30 WT and 30 SHP-2−/−) and found this phenotype was gender independent and exhibited 100% penetrance in SHP-2-deficient animals at 5 months of age. Faxitron radiographs indicated that the enlarged bony masses at the wrists were not detectable before 4 months of age (data not shown). H&E staining of the distal radius showed significant differences in bone morphology and revealed that SHP-2−/− mice appear to have cartilage tumors resembling miniature growth plates (Figure 4C). The wrist tumor consists of large areas of cartilage tissue (1–3), indicated by positive Safranin O staining (Figure 4D). Based on their morphology and staining, the cells in these cartilaginous regions appear to be a combination of large chondrocyte-like cells and fibrochondrocyte-like cells, which is not observed in the heterozygote littermate controls. Normal bone growth and development rely on coordination between chondrocyte mass regulation and cartilage structure establishment. Overall, the phenotype observed is reminiscent of metachondromatosis. To this end, both heterozygous Ptpn11 frame shift mutations and deletions in chondroid cells have been shown to cause metachondromatosis (36), suggesting that Ptpn11 is a tumor suppressor gene in cartilage.
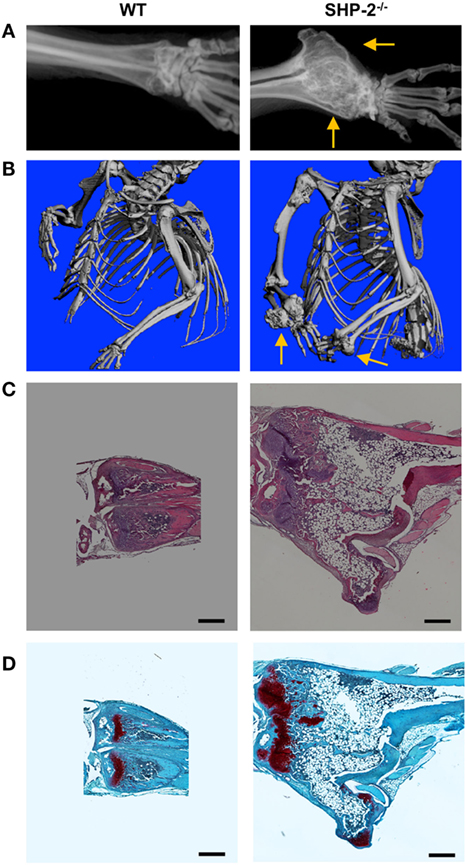
Figure 4. SHP-2−/− mice develop cartilage tumors. (A) Representative Faxitron X-ray of euthanized mice, 9 months of age. Arrows highlight the location of the wrist. (B) Representative microcomputed tomography (μ-CT) analysis shows the wrist tumor in SHP-2−/− mice at 9 months of age. (C) Representative hematoxylin and eosin staining of distal radius of 6-month-old mice. (D) Representative Safranin-O/Fast Green staining of distal radius of 6-month-old mice reveals abnormal cartilaginous regions within the wrist tumor of SHP-2−/− mice. Each analysis was performed on at least three mice per genotype. Scale bar represents 500 µm.
Cartilage Tumor Development Is T Cell Independent
The previous experiments utilized SHP-2−/− mice in which floxed Ptpn11 was deleted by Cre recombinase under the control of the cd4 promoter/enhancer/silencer. However, other mammalian cell lineages, including lymphoid tissue-inducer (LTi) cells, have also been reported to express CD4 during development and the observed phenotype could potentially be explained by CD4 expression outside of the T cell compartment (37, 38). In support of this hypothesis, SHP-2fl/fl-Lck-Cre mice did not develop cartilage tumors suggesting that the phenotype observed was T cell independent (data not shown). We then bred SHP-2fl/fl-CD4-Cre mice on the RAG−/− background. In the resultant SHP-2fl/fl-CD4-Cre-RAG−/− mice, cartilage tumors developed (Figure S5 in Supplementary Material). The cartilage tumors in these mice can be detected at approximately the same age as the Shp-2fl/fl-CD4-Cre mice. Because SHP-2fl/fl-CD4-Cre-RAG−/− mice do not have T cells, we concluded that the phenotype observed is T cell independent. Altogether, these data demonstrate that Ptpn11 gene deletion in a non-T cell subset leads to cartilage tumors.
To mark cells that have deleted the Ptpn11 gene, we crossed the SHP-2fl/fl-CD4-Cre mice to RosaYFP reporter mice (39). In the resultant SHP-2fl/fl-CD4-Cre-RosaYFP mice, Cre activity simultaneously deletes the ptpn11 gene locus and removes a loxP-flanked stop cassette to drive constitutive expression of a YFP gene. This fate mapping strategy allowed us to identify SHP-2-deficient cells even if they no longer express CD4. Using these mice, we found that a low number of CD45+CD19−CD3−TCRβ− cells are YFP positive in spleens (Figure 5A), lymph nodes (data not shown), and liver (data not shown) of SHP-2fl/fl-CD4-Cre-RosaYFP mice. The proportion of these cells are low and appear to derive from different lineages. For instance, in the spleen a subset of the CD45+CD3− CD19+ cells (not shown) and a subset of the CD45+CD3− NK1.1+ cells express YFP (Figure 5A). In addition, within the YFP+ population, a CD3−CD19−NKp46−NK1.1−CD127+ subset can be identified, which can be subdivided into CD4+ and CD4− cells (Figure 5A). These cells are reminiscent of LTi-like cells (37, 38); however, only a few of these cells express CCR6 (Figure 5A).
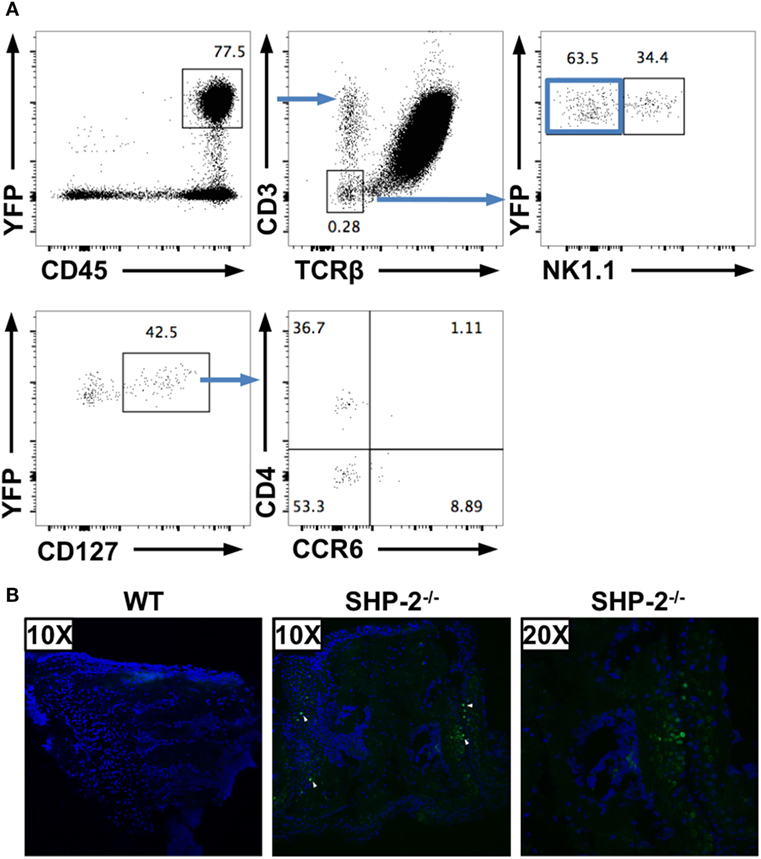
Figure 5. Several non-T cell subsets have an history of CD4 expression. (A) CD19−YFP+ splenocytes were gated for non-T cells (CD3−TCRβ−) and separated in NK1.1− and NK1.1+ cells. CD127+ cells from NK1.1− cells were subdivided based on CD4 and CCR6 expression. (B) 30 µm serial sections of frozen wrist tissues were prepared from SHP-2±CD4-Cre-RosaYFP and SHP-2−/−CD4-CreRosaYFP mice and imaged on a Nikon C1SI confocal microscope. Nuclei indicated by DAPI staining (blue) and YFP detected in GFP channel (green, white arrows).
Importantly, we have been able to detect few YFP-positive cells in frozen wrist sections of aged Shp-2fl/fl-CD4-Cre-RosaYFP mice by confocal microscopy (Figure 5B). No YFP-positive cells were found in frozen sections from the control heterozygote mice (Figure 5B), indicating that localization of the cells of interest are concomitant to the wrist tumors. These data confirm that several non-T cell subsets express CD4 or had expressed CD4 during development. In the spleen, we have shown that SHP-2 is efficiently deleted from YFP-positive cells (Figure 1), indicating that one of these subsets could be responsible for the phenotype observed.
Discussion
Src homology region 2 domain-containing phosphatase-2 is known to affect the quality of immune responses through its association with NK cell inhibitory receptors where the phosphatase is thought to dephosphorylate key players of cellular activation (9, 40, 41). However, inhibitory receptor-independent inhibition of cytolytic activity and IFN-γ secretion has also been reported in NK cells (42). Because iNKT cells and subsets of conventional T cells express inhibitory receptors, we hypothesized that SHP-2 deletion on T cells would have significant effects. Here, we report that conditional deletion of SHP-2 under the CD4 and Lck-distal promoters do not markedly affect conventional T cells and iNKT cells. This phenotype is relatively surprising given the well-documented necessity for SHP-2 during gastrulation, hematopoiesis, and embryonic development (3), as well as its reported association with the TCR immunoreceptor tyrosine-based activation motifs (43, 44). In both these conditional KO mice, thymic and peripheral T cell development, as well as effector CD8+ T cell differentiation and function, are comparable. iNKT cell development and function are also unaffected by a lack of SHP-2. Our results appear to contradict a previous report that used a different variant of SHP-2 conditional KO mice, where SHP-2 was deleted under the Lck promoter (8). In contrast to the authors’ findings, we did not detect any block in thymic T cell development. We also did not find reduced cellularity in the spleen and thymus or a decrease in TCR driven proliferation and cytokine production, as previously reported (8). Although we cannot explain the differences with the previous published study, another laboratory has found that T cell development is not affected by SHP-2 absence (Greta Guarda, personal communication).
The level of tyrosine phosphorylation and immune cell activation is determined by the opposing actions of cytoplasmic protein tyrosine kinases, phosphotyrosine phosphatases, and lipid phosphatases. Tyrosine phosphorylation is a reversible process, whereby phosphatases direct dephosphorylation of the tyrosyl residue. The SH2 domain-containing phosphotyrosine phosphatases SHP-2, and its related cellular tyrosine phosphatase SHP-1, have been shown to play essential roles in immune cell signaling (9, 45). Because our SHP-2−/− mice do not display an apparent T cell phenotype, it is possible that SHP-2 and SHP-1 play redundant functions in regulating immune signaling. SHP-1 is well expressed in hematopoietic cells and has been shown to limit the threshold of T cell activation and other immunological processes (3, 46, 47). We initially wanted to cross SHP-1 conditional knockout mice to our SHP-2−/−. Unfortunately, due to the fact that the Ptpn6 gene is located on chromosome 6, 4.2 megabases from the NK locus, there is no NK1.1 expression and the Ly49 repertoire is predominantly from 129 origin (data not shown) in the available mice (48), precluding the generation of double-deficient mice. Nevertheless, our preliminary analysis of SHP-1fl/fl-CD4-Cre conditional mice suggests that there is no gross defect in T cell and iNKT cell development, thus reinforcing a possible redundancy between SHP-1 and SHP-2 in T cell development.
The development of cartilage tumors in aged SHP-2−/− mice was unexpected. Using a variety of genetic and cellular approaches, our findings indicate that these tumors result from loss of SHP-2 in a cell subset(s) expressing CD4 during their development. Although the precise identification of these cells is beyond the scope of this manuscript, our data using RAG−/− and Lck-Cre transgenic mice rule out T cells as responsible for the phenotype. Using reporter mice for CD4 expression, we identified several non-T cell subsets that express YFP, a surrogate marker for prior CD4-Cre activity. Interestingly, low number of YFP+ cells are found in a variety or organs, including wrist sections. As mentioned above, LTi-like cells can express CD4 (37) and CD4 RNA expression has been reported in chondrocytes (49). Therefore, CD4+ LTi-like cells and CD4+ chondrocytes are among the potential candidates responsible for the observed skeleton abnormalities.
A phenotype reminiscent of the tumors observed in the current study has been recently described in humans (6, 50). Using linkage analysis and parallel sequencing of bar-coded DNAs from several families, mutations in Ptpn11 were identified as a cause of metachondromatosis, a rare tumor syndrome leading to cartilage tumors (6). Notably, although patients with metachondromatosis have one mutant copy and one normal copy of Ptpn11 in all cells, the normal copy is lost in cartilage cells that form tumors (6), reinforcing the critical tumor suppressor role of SHP-2 in cartilages. In mice, it has been found that SHP-2 regulates the proliferation of Ctsk chondroid progenitors, which are responsible for cartilage growth (36). In another study, deletion of SHP-2 in mesenchymal stem cells, which are progenitors of both osteoblasts and chondrocytes, was shown to cause multiple skeleton abnormalities (51). However, in these two studies, the phenotype is more severe and occurs within 8 weeks of birth, while our SHP-2−/− mice do not develop tumors before 4 months of age, suggesting that SHP-2 deletion occurs at a later stage of chondrocyte differentiation. In mice, using a mosaic postnatal inactivation of Ptpn11, it has been shown that SHP-2-deficient cells affect SHP-2+ chondrocytes, via a paracrine mechanism. This leads to a delay in terminal differentiation of growth plate chondrocytes (52). Additionally, the authors demonstrated that SHP-2-deficient fibroblast precursors can inappropriately differentiate into chondrocyte-like cells (52). We have been able to detect chondrocyte-like cells (Figure 4) and YFP+ cells (Figure 5) in the cartilage tumors. We therefore hypothesize that the SHP-2-deficient cell subset may either inappropriately differentiate into chondrocyte-like cells or cause neighboring SHP-2-sufficient cells to contribute to exostoses and enchondromas via paracrine signals. For instance, Bowen and colleagues showed that mosaic inactivation of Ptpn11 in growth plates affect Ptpn11 deficient and WT chondrocytes (52). Therefore, it is possible that SHP-2-deficient cells directly or indirectly activate mesenchymal stem cells to undergo chondrogenesis. Interestingly, a similar phenotype was recently reported when Sos1/2 or Erk1/2 were deleted from CD4+ cells (49, 53). Sos proteins are Ras-guanine exchange factors, which can be recruited downstream of SHP-2, and promote the activation of ERK. It is therefore likely that the SHP-2/SOS/ERK pathway is critically involved in the regulation of a subset of CD4+ cells, leading to cartilage tumors.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals as defined by the National Institutes of Health (PHS Assurance #A3284-01). Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Brown University. All animals were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with trained husbandry, technical, and veterinary personnel.
Author Contributions
SM, CJ, ER, and CF conceived, performed, and analyzed the experiments. AS conceived, performed, analyzed the experiments, and wrote the article. WY provided reagents. QC conceived and analyzed the experiments. LB conceived, analyzed the experiments, and wrote the article. The authors have no conflict of interest to declare.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health Research Grants AI46709 and AI122217 (to LB) and GM104937 (to QC). We thank Kevin Carlson for cell sorting.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01326/full#supplementary-material.
References
1. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J (1997) 16(9):2352–64. doi:10.1093/emboj/16.9.2352
2. Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Mol Cell Biol (1997) 17(9):5499–507. doi:10.1128/MCB.17.9.5499
3. Lai L, Zhao C, Zhang E, Feng GS. The Shp-2 tyrosine phosphatase. Protein Phosphatases (2004) 5:275–99. doi:10.1007/978-3-540-40035-6_14
4. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet (2001) 29(4):465–8. doi:10.1038/ng772
5. Jiang C, Hu F, Tai Y, Du J, Mao B, Yuan Z, et al. The tumor suppressor role of Src homology phosphotyrosine phosphatase 2 in hepatocellular carcinoma. J Cancer Res Clin Oncol (2012) 138(4):637–46. doi:10.1007/s00432-011-1143-5
6. Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafe L, Superti-Furga A, et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet (2011) 7(4):e1002050. doi:10.1371/journal.pgen.1002050
7. Qu CK, Nguyen S, Chen J, Feng GS. Requirement of Shp-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood (2001) 97(4):911–4. doi:10.1182/blood.V97.4.911
8. Nguyen TV, Ke Y, Zhang EE, Feng G-S. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol (2006) 177(9):5990–6. doi:10.4049/jimmunol.177.9.5990
9. Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev (2009) 228(1):342–59. doi:10.1111/j.1600-065X.2008.00760.x
10. Liu X, Qu C-K. Protein tyrosine phosphatase SHP-2 (PTPN11) in hematopoiesis and leukemogenesis. J Signal Transduct (2011) 2011:195239. doi:10.1155/2011/195239
11. Ketroussi F, Giuliani M, Bahri R, Azzarone B, Charpentier B, Durrbach A. Lymphocyte cell-cycle inhibition by HLA-G is mediated by phosphatase SHP-2 and acts on the mTOR pathway. PLoS One (2011) 6(8):e22776. doi:10.1371/journal.pone.0022776
12. Neel BG, Gu H, Pao L. The “shp”ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci (2003) 28(6):284–93. doi:10.1016/S0968-0004(03)00091-4
13. Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell (1998) 92(4):441–50. doi:10.1016/S0092-8674(00)80938-1
14. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9(5):495–502. doi:10.1038/ni1581
15. McMahon CW, Raulet DH. Expression and function of NK cell receptors in CD8+ T cells. Curr Opin Immunol (2001) 13(4):465–70. doi:10.1016/S0952-7915(00)00242-9
16. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
17. Godfrey DI, Berzins SP. Control points in NKT-cell development. Nat Rev Immunol (2007) 7(7):505–18. doi:10.1038/nri2116
18. Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell (2006) 10(3):317–27. doi:10.1016/j.devcel.2006.01.002
19. Reilly EC, Thompson EA, Aspeslagh S, Wands JR, Elewaut D, Brossay L. Activated iNKT cells promote memory CD8+ T cell differentiation during viral infection. PLoS One (2012) 7(5):e37991. doi:10.1371/journal.pone.0037991
20. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278(5343):1626–9. doi:10.1126/science.278.5343.1626
21. Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol (2006) 177(1):450–8. doi:10.4049/jimmunol.177.1.450
22. Stankovic S, Gugasyan R, Kyparissoudis K, Grumont R, Banerjee A, Tsichlis P, et al. Distinct roles in NKT cell maturation and function for the different transcription factors in the classical NF-kappaB pathway. Immunol Cell Biol (2011) 89(2):294–303. doi:10.1038/icb.2010.93
23. Sawada S, Scarborough JD, Killeen N, Littman DR. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell (1994) 77(6):917–29. doi:10.1016/0092-8674(94)90140-6
24. Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity (2001) 15(5):763–74. doi:10.1016/S1074-7613(01)00227-8
25. Anfossi N, Robbins SH, Ugolini S, Georgel P, Hoebe K, Bouneaud C, et al. Expansion and function of CD8+ T cells expressing Ly49 inhibitory receptors specific for MHC class I molecules. J Immunol (2004) 173(6):3773–82. doi:10.4049/jimmunol.173.12.7223
26. Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol (2012) 188(7):2957–65. doi:10.4049/jimmunol.1100038
27. Rahim MM, Tu MM, Mahmoud AB, Wight A, Abou-Samra E, Lima PD, et al. Ly49 receptors: innate and adaptive immune paradigms. Front Immunol (2014) 5:145. doi:10.3389/fimmu.2014.00145
28. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an a-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med (1998) 188(8):1521–8. doi:10.1084/jem.188.8.1521
29. Chiu YH, Jayawardena J, Weiss A, Lee D, Park SH, Dautry-Varsat A, et al. Distinct subsets of CD1d-restricted T cells recognize self-antigens loaded in different cellular compartments. J Exp Med (1999) 189(1):103–10. doi:10.1084/jem.189.1.103
30. Wei DG, Lee H, Park SH, Beaudoin L, Teyton L, Lehuen A, et al. Expansion and long-range differentiation of the NKT cell lineage in mice expressing CD1d exclusively on cortical thymocytes. J Exp Med (2005) 202(2):239–48. doi:10.1084/jem.20050413
31. Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity (2008) 29(4):650–9. doi:10.1016/j.immuni.2008.07.017
32. Sierro S, Rothkopf R, Klenerman P. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. Eur J Immunol (2005) 35(4):1113–23. doi:10.1002/eji.200425534
33. Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest (2011) 121(12):4758–74. doi:10.1172/JCI59492
34. Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal (2012) 5(252):ra87. doi:10.1126/scisignal.2003365
35. Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J Exp Med (2013) 210(7):1419–31. doi:10.1084/jem.20122239
36. Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature (2013) 499(7459):491–5. doi:10.1038/nature12396
37. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science (2010) 330(6004):665–9. doi:10.1126/science.1194597
38. Bar-Ephraim YE, Mebius RE. Innate lymphoid cells in secondary lymphoid organs. Immunol Rev (2016) 271(1):185–99. doi:10.1111/imr.12407
39. Srinivas S, Watanabe T, Lin C-S, William C, Tanabe Y, Jessell T, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol (2001) 1(1):4. doi:10.1186/1471-213X-1-4
40. Yusa S, Campbell KS. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J Immunol (2003) 170(9):4539–47. doi:10.4049/jimmunol.170.9.4539
41. Yusa S, Catina TL, Campbell KS. KIR2DL5 can inhibit human NK cell activation via recruitment of Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2). J Immunol (2004) 172(12):7385–92. doi:10.4049/jimmunol.172.12.7385
42. Purdy AK, Campbell KS. SHP-2 expression negatively regulates NK cell function. J Immunol (2009) 183(11):7234–43. doi:10.4049/jimmunol.0900088
43. Frearson JA, Yi T, Alexander DR. A tyrosine-phosphorylated 110-120-kDa protein associates with the C-terminal SH2 domain of phosphotyrosine phosphatase-1D in T cell receptor-stimulated T cells. Eur J Immunol (1996) 26(7):1539–43. doi:10.1002/eji.1830260720
44. Frearson JA, Alexander DR. The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T cells. J Exp Med (1998) 187(9):1417–26. doi:10.1084/jem.187.9.1417
45. Pao LI, Badour K, Siminovitch KA, Neel BG. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol (2007) 25:473–523. doi:10.1146/annurev.immunol.23.021704.115647
46. Lorenz U, Ravichandran KS, Burakoff SJ, Neel BG. Lack of SHPTP1 results in src-family kinase hyperactivation and thymocyte hyperresponsiveness. Proc Natl Acad Sci U S A (1996) 93(18):9624–9. doi:10.1073/pnas.93.18.9624
47. Zhang J, Somani AK, Yuen D, Yang Y, Love PE, Siminovitch KA. Involvement of the SHP-1 tyrosine phosphatase in regulation of T cell selection. J Immunol (1999) 163(6):3012–21.
48. Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity (2007) 27(1):35–48. doi:10.1016/j.immuni.2007.04.016
49. Wehenkel M, Corr M, Guy CS, Edwards BA, Castellaw AH, Calabrese C, et al. Extracellular signal-regulated kinase signaling in CD4-expressing cells inhibits osteochondromas. Front Immunol (2017) 8:482. doi:10.3389/fimmu.2017.00482
50. Bauler TJ, Kamiya N, Lapinski PE, Langewisch E, Mishina Y, Wilkinson JE, et al. Development of severe skeletal defects in induced SHP-2-deficient adult mice: a model of skeletal malformation in humans with SHP-2 mutations. Dis Model Mech (2011) 4(2):228–39. doi:10.1242/dmm.006130
51. Lapinski PE, Meyer MF, Feng GS, Kamiya N, King PD. Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis Model Mech (2013) 6(6):1448–58. doi:10.1242/dmm.012849
52. Bowen ME, Ayturk UM, Kurek KC, Yang W, Warman ML. SHP2 regulates chondrocyte terminal differentiation, growth plate architecture and skeletal cell fates. PLoS Genet (2014) 10(5):e1004364. doi:10.1371/journal.pgen.1004364
Keywords: Src homology region 2 domain-containing phosphatase-2, T cells, invariant natural killer T cells, cartilage tumor, chondrocytes
Citation: Miah SMS, Jayasuriya CT, Salter AI, Reilly EC, Fugere C, Yang W, Chen Q and Brossay L (2017) Ptpn11 Deletion in CD4+ Cells Does Not Affect T Cell Development and Functions but Causes Cartilage Tumors in a T Cell-Independent Manner. Front. Immunol. 8:1326. doi: 10.3389/fimmu.2017.01326
Received: 07 September 2017; Accepted: 29 September 2017;
Published: 16 October 2017
Edited by:
Bernard Malissen, Centre d’Immunologie de Marseille Luminy, FranceCopyright: © 2017 Miah, Jayasuriya, Salter, Reilly, Fugere, Yang, Chen and Brossay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Brossay, bGF1cmVudF9icm9zc2F5QGJyb3duLmVkdQ==
†These authors have contributed equally to this work.