Abstract
The gut microbiota is essential for the development and regulation of the immune system and the metabolism of the host. Germ-free animals have altered immunity with increased susceptibility to immunologic diseases and show metabolic alterations. Here, we focus on two of the major immune-mediated microbiota-influenced components that signal far beyond their local environment. First, the activation or suppression of the toll-like receptors (TLRs) by microbial signals can dictate the tone of the immune response, and they are implicated in regulation of the energy homeostasis. Second, we discuss the intestinal mucosal surface is an immunologic component that protects the host from pathogenic invasion, is tightly regulated with regard to its permeability and can influence the systemic energy balance. The short chain fatty acids are a group of molecules that can both modulate the intestinal barrier and escape the gut to influence systemic health. As modulators of the immune response, the microbiota-derived signals influence functions of distant organs and can change susceptibility to metabolic diseases.
Microbiota Shapes the Immune System and the Host Metabolism
The human microbiota comprises an enormous amount and variety of microorganisms. Among archea, eukarya, and viruses, bacteria are the most abundant inhabitants of the human host. The human gastrointestinal tract is one of the world’s most densely packed microbe habitats (1, 2). By coevolution with humans, a symbiotic relation evolved with the host providing a living environment and nutrients in exchange for support in nutrient degradation and protection from pathogenic microorganisms. The development of 16S rRNA sequencing allowed estimates of the abundance of microbiota in greater detail, revealing the gram-negative Bacteroidetes and gram-positive Firmicutes as the most abundant phyla in the human gut. Changes in the Firmicutes to Bacteroidetes ratio, as inducible among others by high-fat diet (3), can impact metabolism (4–7), and lead to symptoms of type 1 and 2 diabetes, colitis, and obesity (8). Interestingly, transplantation of microbiota from obese human or murine donors to germ-free (GF) mice is sufficient to induce insulin resistance and increased adiposity compared to the lean microbiota transplanted controls (9–12). The Firmicutes and the Bacteroidetes, together with the other inhabitants of the gut, can also modulate immune function (13). The importance of the microbiota for the formation of a fully functional immune system first became evident by studying the GF animals, which are bred and housed in an environment devoid of microorganisms. The immune tissues or local immune cell subsets that are in direct contact with microbiota, as in the gut, are different in GF animals. The gut of the GF mice shows fewer and smaller Payer’s patches (14), an altered mucus layer (15), not fully developed gut-associated lymphoid tissues, and no formation of isolated lymphoid follicles, which help inducing local immune responses (16). Counts of the immune cells residing within the gut are decreased, including the intraepithelial CD8+ T cells, the lamina propria CD4+ T cells, and their subsets type 17 T helper cells (Th17) and regulatory T cells (Tregs), as well as immunoglobulin G counts (17, 18). The absence of gut microbiota leads to functional alterations in immune and intestinal epithelial cells, which express less microbe sensing toll-like receptors (TLR) (19) and major histocompatibility complex II molecules for antigen presentation (20).
Certain aspects of an underdeveloped, less responsive gut immune system seem to have beneficial effects in obesity. GF, or microbiota depleted mice using antibiotics show improved glucose and insulin tolerance, accompanied by reduced adiposity (9), increased browning of their white fat depots (21), and are protected from diet-induced obesity (DIO) (22–24). The fat browning promotes energy dissipation in the form of heat, enabled by the uncoupling of oxidative phosphorylation from ATP biosynthesis (25). In part, the metabolic effects and the browning phenotype are mediated by the innate immune system and the increased M1 to M2 macrophage polarization that potentiates the fat browning either after cold (26, 27), or after microbiota depletion (21). The GF metabolic phenotype is partially reversible once mice are transplanted with microbiota from donor mice. These observations from GF animals, or from the mice transplanted with microbiota, illustrate that the microbiota is essential in the progression of metabolic imbalances and in the development of a functional immune system. This review will summarize how microbial molecules and changes in the microbiota composition are sensed by the immune system and how microbial products and consequently the immune response can spread into the circulation to cause systemic and metabolic consequences, starting in the gut and moving toward the periphery. We highlight TLR signaling, gut barrier modulation, and short chain fatty acid (SCFA)-mediated interactions as important mechanisms in this process.
TLRs Sense Microbial Products in the Gut and Periphery to Influence Systemic Immunity and Metabolism
One of the immune system’s tasks is to decide whether a microbe is a harmless commensal or an invading and potentially pathogenic one. As the first line of defense, the innate immune system will recognize both kinds of microbes as they include certain non-self patterns. This is particularly important for ensuring correct intestinal homeostasis (28). One class of molecules that recognizes such patterns is the pattern recognition receptor family that includes the TLRs. There are 10 TLRs in humans and 12 in mice found on the cell or endosomal membranes of different cell types including macrophages, dendritic cells (DCs), and non-immune cells such as epithelial cells, hepatocytes, or adipocytes. Activation of such receptors initiates downstream signaling cascades that often result in induction of cytokine expression. These cytokines can further influence other immune cells and accordingly dictate the tone of an immune response.
Toll-like receptor 2 signals as a heterodimer with TLR1 or TLR6 and recognizes a wide variety of signals on fungi and bacteria. One ligand of TLR2 is polysaccharide A (PSA) from Bacteroides fragilis that induces anti-inflammatory responses, activates plasmacytoid DCs, interleukin 10 (IL-10) production of CD4+ T cells, promotes clonal expansion and induction of Treg cells, and suppresses Th17 production in the gut (Figure 1) (29–31). These aspects potentially contribute to the amelioration of inflammation in animal models such as experimental autoimmune encephalomyelitis and colitis after PSA administration (29, 32, 33) or after colonization of GF mice with Bacteroidetes (34). Anti-inflammatory responses upon TLR2 activation were suggested as a way to recognize commensals as non-pathogenic. This suggests that specific microbial-derived mechanisms actively promote immunologic tolerance to symbiotic bacteria. In contrast, TLR2 signaling can also result in proinflammatory responses, as for instance upon detection of Lactobacillus plantarum teichoic acid d-alanylation (35). Although the highest encounter opportunity between microbes and immune cells is the gut, microbial products are also detected by TLRs at peripheral sites, influencing systemic and metabolic effects. Tlr2 expression is increased in visceral adipose tissue in mice fed a high fat diet (HFD) compared to a normal chow diet, resulting in tumor necrosis factor alpha (TNF-α) expression, thereby supporting a low grade inflammation in tissues (Table 1) that is typical of obesity (36). Tlr2 inhibition or ablation results in improved insulin sensitivity and decreased inflammation and adiposity in a DIO mouse model (37–39). These findings support the role of TLR2 as a proinflammatory mediator of metabolic symptoms.
Figure 1
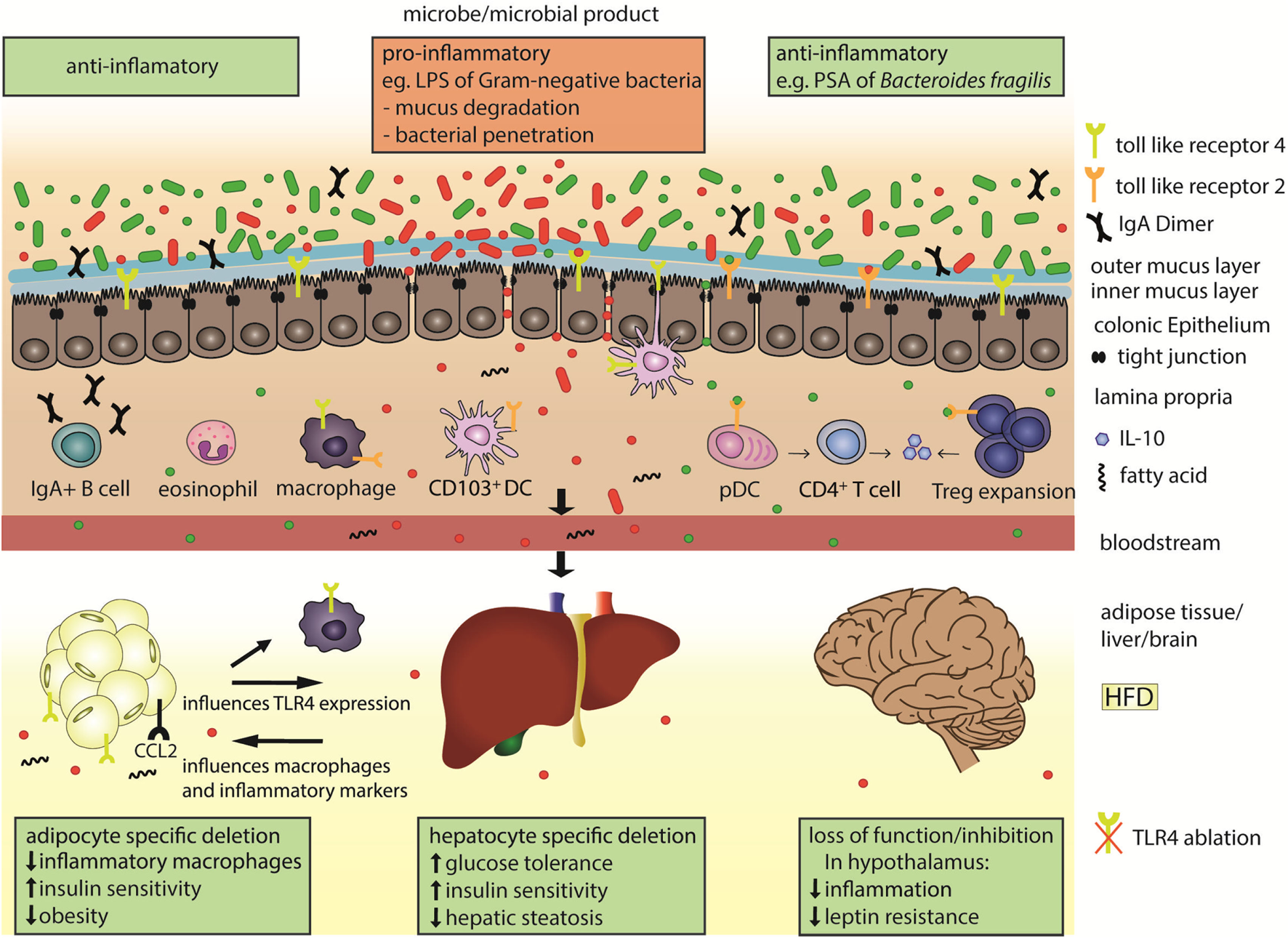
Microbial products are sensed by toll-like receptors (TLRs) in the gut and in the periphery to influence systemic immunity and metabolism. The colonic epithelial barrier is composed of an outer mucus layer that is habitat for microbes and an inner, impenetrable layer, in addition to tight-junction connected epithelial cells. Under normal physiologic conditions and when sufficient anti-inflammatory microbial products (green) are present in the gut, the gut lining is well protected, with plasma B cell secreted Immunoglobulin A (IgA), regulatory T cells (Tregs), and eosinophils. The polysaccharide A (PSA) of Bacteroides fragilis is a TLR2 ligand that promotes secretion of the anti-inflammatory cytokine interleukin 10 (IL-10) from the plasmacytoid dendritic cells (pDCs)-activated CD4+ T cells, or from the expanded Tregs cells. Under microbial dysbiosis, proinflammatory microbes (red) predominate in the gut and can be sensed, e.g., by TLR4 on CD103+ dendritic cells (DCs) or macrophages. Such microbes can degrade and invade the second colonic mucus layer and escape the gut when the epithelial cell lining and tight junctions are disrupted. Subsequently the inflammatory microbial products, like the LPS of gram-negative bacteria, can reach distant organs through circulation. LPS and fatty acids are elevated under high fat diet (HFD) and activate TLR4 signaling. Organ-specific effects after HFD include upregulation of TLR4 and macrophage attractant CCL2 in the adipose tissue, which leads to inflammation and supports obesity.
Table 1
| Target | Effect | Reference | |
|---|---|---|---|
| TLRs | |||
| TLR2 | Insulin resistance, inflammation | (37–39) | |
|
|
|||
| TLR4 | Adipose tissue | Inflammation, macrophage accumulation | (40–42) |
| Hypothalamus | Inflammation, leptin resistance | (43) | |
|
|
|||
| TLR5 | Liver | Protection from liver disease | (44) |
|
|
|||
| SCFAs | |||
|
|
|||
| SCFAs | Intestine | Gut-derived hormone secretion, gluconeogenesis | (45–47) |
| Intestine | DIO, insulin resistance | (48, 49) | |
| Lymphoid tissues | Gut barrier, Tregs | (50) | |
| Autoreactive T cells (T1D) | |||
|
|
|||
| Acetate | Adipose tissue | Fat accumulation, insulin signaling, adipose tissue browning, lipid storage | (51–53) |
| Liver | Hepatic function | (52) | |
| Various | Energy expenditure, insulin secretion, ghrelin secretion, hyperphagia | (51, 53) | |
|
|
|||
| Butyrate | Intestine | M2 macrophage polarization, colonic mucus secretion | (54, 55) |
|
|
|||
| Proprionate | Intestine | Gut hormones | (49) |
| Brain | Food intake | ||
TLRs and SCFA bridge microbiota with host metabolism as initiators of immunologic processes.
TLR and SCFA signaling occurs in response to microbiota changes in the intestine and can reach distant organs, influencing the inflammatory and metabolic state to initiate systemic effects.
DIO, diet-induced obesity; HFD, high-fat diet; TLR, toll-like receptor; Tregs, regulatory T cells; T1D, type 1 diabetes.
Toll-like receptor 4 mainly recognizes the Gram-negative bacterial cell wall component lipopolysaccharide (LPS). LPS levels in blood are increased in obesity or after high caloric diet (HCD) feeding (56) and are associated with increased Firmicutes to Bacteroidetes ratios. TLR4 signals through the MyD88-dependent pathway, or by TIR-domain-containing adapter-inducing interferon-β regulation of type 1 interferons via interferon regulatory factor 3 (IRF3). Tlr4 expression is increased in adipose tissues, peripheral blood or muscle of obese or type 2 diabetes patients (57, 58) and in adipose tissues of obese db/db mice (59) and correlates with insulin resistance. LPS induces adipose tissue inflammation through TLR4 (Figure 1), and increases Ccl2 expression on adipocytes. This chemokine contributes to a microbiota-induced macrophage accumulation and WAT inflammation in lard-fed mice (40). Loss of function mutation in Tlr4, or TLR4 deletion reduces macrophage infiltration into the adipose tissue, promotes their anti-inflammatory M2 polarization (41), decreases tissue and circulating inflammatory marker levels, and diminishes inflammation in the streptozotocin-induced mouse model for type 1 diabetes (42). Irf3 knockdown improves insulin sensitivity, mediates an anti-inflammatory phenotype, and also increases white fat browning (60). Intestinal epithelial cell-specific MyD88 deletion decreases fat mass accumulation, body weight gain and glucose intolerance in diet-induced obese mice (61). Apart from LPS, fatty acids can also increase TLR4 signaling in several cell types including adipocytes and macrophages (62) and promote visceral obesity and insulin resistance (63). Interestingly, adipocyte-specific Tlr4 KO mice demonstrate two distinct effects of TLR4 on the adipose tissue. Specifically, these mice show increased whole body and muscle insulin resistance after HFD feeding, but also improved insulin sensitivity after an acute lipid challenge during a hyperinsulinemic euglycemic clamp (64). Adipocyte-specific Tlr4 deletion also modifies Tlr4 expression in other tissues, as it decreases Tlr4 expression in peritoneal macrophages and liver. Alteration of TLR levels in the liver has important systemic metabolic effects. Hepatocyte-specific Tlr4 deletion improves glucose tolerance, insulin sensitivity, and hepatic steatosis in HFD fed obese mice (65). In addition to the local signaling in the gut or the peripheral tissues, the TLR-mediated effects also reach the brain. TLR4 inhibition decreases inflammation and leptin resistance in the hypothalamus (43). Deletion of the TLR downstream signaling molecule MyD88 in the central nervous system prevents obesity and leptin resistance (66). Furthermore, in older or HCD-fed mice, Tlr4 expression was increased in the pro-opiomelanocortin neurons from the arcuate nucleus of the hypothalamus, a central metabolism regulating brain area (67).
Toll-like receptor 5 recognizes bacterial flagellin, a component of the bacterial locomotion system. This signaling pathway causes an anti-inflammatory response by inducing interleukin 1 receptor antagonist secretion and diminishing IL-1β and inflammasome activity (68). Tlr5 KO mice were first reported to be prone to develop metabolic syndrome, including insulin resistance and increased adiposity, which correlated with changes in their microbiota composition (69). This phenotype was also confirmed in an epithelial cell-specific Tlr5 KO (70) and was associated with low-grade inflammation. Interestingly, these initial findings could not be reproduced in the same Tlr5 KO mouse line by a different research group (71, 72), nor in a new Tlr5 KO model (73). While a clear explanation for this discrepancy is missing, a possible reason could lay in the differences in the microbiota composition between different mouse facilities, and/or may depend on whether initially pro- or anti-inflammatory molecules are predominant in the local gut environment. Specific Tlr5 deletion in hepatocytes confers predisposition to diet-induced liver pathology. This is accompanied by elevated expression of proinflammatory cytokines and dependent on the Nod-like-receptor C4 inflammasome and rescued by microbiota depletion (44). Similarly, hepatocyte-specific deletion of MyD88 leads to inflammation, glucose intolerance, and hepatic insulin resistance, accompanied by alterations in the microbiota composition (74). These observations suggest that hepatocyte TLR5 plays a role in protecting the liver and in preventing diet-induced liver disease.
Dysregulation of Gut Mucosal Surfaces Can Contribute to Systemic Microbial Effects
Changes in the gut microbiota can modulate systemic microbe-derived metabolite levels by affecting their biosynthesis, or by changing the intestinal permeability and gut barrier. The mucosal surfaces are another first line defense component of the innate immune system, at which most of the microbe–host interactions take place. In the gastrointestinal tract, the mucosal surface consists of an epithelial cell layer, in which the mucus-secreting goblet cells are responsible for forming a second protective barrier, the mucus layer. Due to its physiologic function in the gut for food absorption, the mucosal surface is naturally a thin and permeable layer. The protection against microbial invaders under natural physiologic conditions is ensured by several mechanisms, including tight junctions between the epithelial cells, which are constantly renewed from stem cells allowing immediate repair in case of cell loss or damage. Mucus is composed of charged glycoproteins called mucins and its density and stickiness traps microbes and their products, preventing contact to the epithelial cells. Within the large intestinal mucus, there are two layers, a loose outer layer that is a natural habitat for microbes and a dense inner layer, impermeable for bacteria (15).
Dysregulation of the mucus structure by changes in its thickness or penetrability can lead to inflammation. This is evident in the mucin 2 (Muc2) KO mice where microbiota is in direct contact with the epithelial cells (75, 76). TLRs were shown to mediate MUC2 secretion (77) and were implied in epithelial homeostasis (28, 78). MyD88 deletion in intestinal epithelial cells leads to defects in mucosal barrier functions with reduced Muc2 mRNA expression and increases epithelial cell penetration by bacteria (79, 80). GF mice have easily penetrable mucus in the large intestine, whereas conventionally raised mice show a thick, impenetrable mucus layer (15, 81). The level of penetration and degradation depends largely on the gut microbiota composition (82). Although most bacteria metabolize non-digested food polysaccharides (83, 84), many bacteria can use mucin glycans as an energy source (85–87). This substantial role of the microbiota also indicates that an imbalance in the bacterial species composition can influence the protective capacity of the mucus barrier. This kind of dysbiosis can result from a variety of different factors, including a decreased bacterial diversity, lack of secretory antibodies that line the mucus layer, and lack of Tregs (88) or eosinophils (89, 90). Once the mucus barrier is invaded and microbes are in closer contact to epithelial cells, mucus production and stem cell number is increased in the intact epithelium, and this is triggered by MyD88 signaling (91). This illustrates that the epithelium counteracts the potential threat of microbes invading the mucus layer. However, similar to the mucus layer, the epithelial cell lining can also become permeable. Damage to the epithelial layer can originate from altered tight junction composition or epithelial cell stress (92). Autophagy, a process in which a phagosome is formed in the cytoplasm to engulf various contents, is also important for maintaining an intact epithelial barrier and in the regulation of mucus secretion (93, 94). Disruption of autophagy induced by coding polymorphism (Thr300Ala) in the autophagy-related 16-like 1 leads to decreased antibacterial autophagy, induces epithelial cell stress, and allows systemic penetration of bacteria and inflammation (95).
Gut permeability is increased with HFD, in obesity and in diabetes (96). HFD-induced nonalcoholic fatty liver disease (NAFLD) at thermoneutral housing conditions (30°C) is associated with an altered microbiome, increased intestinal permeability and induction of proinflammatory responses that are active in this disease in humans. Depletion of hematopoietic Tlr4 or gram-negative microbiota leads to altered immune responsiveness and protects from NAFLD at thermoneutrality (97). With dietary fiber deficiency, a subset of mucin-degrading bacteria increases, which express mucin-degrading carbohydrate-active enzymes (CAZymes) that enable access to host-secreted mucus glycoproteins as a nutrient (98), resulting in the degradation of the colonic mucus barrier. This increases LPS levels systemically. Glucagon like peptide (GLP)-1 and 2 levels, which have beneficial metabolic effects (99) and could affect gastric barrier function (100), can be regulated by LPS. In addition, obese mice have impaired IL-22 induction from innate lymphoid and CD4+ T cells under immune challenge, and IL-22 reverses the HFD-induced epithelial cell stress (92). IL-22 depletion results in defects in mucosal defense and metabolic symptoms, many of which can also be reversed by exogenous IL-22 (101).
A group of molecules that can both modulate the intestinal barrier and escape the gut to influence systemic health are the SCFAs. SCFAs acetate, butyrate, and propionate are produced by bacterial anaerobic metabolism of indigestible dietary components, including fiber. They signal via G-protein-coupled-receptors such as GPR41, GPR43, and GPR109a (102, 103) and are important regulators of gut homeostasis and epithelial barrier maintenance. Acetate enhances protection against infection (104), while butyrate is an energy source for colonocytes (105–107) and can regulate stem cell proliferation and anti-inflammatory macrophage polarization (54, 108) with its histone deacetylase inhibiting function. Butyrate also increases colonic mucus secretion, both directly by promoting Muc2 and glycoltransferase expression (55, 109), and indirectly by promoting autophagy (93, 94, 110, 111). Further protection against inflammation through SCFAs is accomplished by their regulation of Tregs (112). The elevated circulating SCFA levels in mice fed a high-fiber diet result in allergy protection by impaired Th2 differentiation and increased phagocytosis by DCs, while low-fiber diet promotes allergic inflammation (113). In type 1 diabetes, the SCFA amount correlates with improvement of symptoms via limitation of autoreactive T cells, induction of Tregs, and enhanced gut barrier (50). The beneficial SCFA effects (52, 114) can act through local induction of gut-derived hormone secretion, including GLP-1 and peptide YY (45–47), and through the circulation (115). SCFA-mediated beneficial metabolic effects on health can be mediated by induction of intestinal gluconeogenesis (48). Specifically, supplementation with acetate in drinking water or through nanoparticles, butyrate gavage alone or combined with propionate, or adding propionate, butyrate, and acetate to the diet, improve host physiology and glucose metabolism (49, 51, 52, 116) (Table 1), which in the case of propionate seems to be mediated by vagus-nerve stimulation by the peripheral nervous system (48). Conversely, the microbiota-mediated increase in acetate turnover that occurs during exposure to a HFD diet might mediate a feedback loop between the gut microbiota and the parasympathetic nervous system, promoting hyperphagia owing to increased ghrelin secretion, and increased energy storage (53). The site of stimulation seem to be important for the outcome of SCFA-mediated effects (117, 118) and points to the need for further exploration of the general role of SCFAs in regulating obesity.
Conclusion
The encounter of microbial molecules by TLRs initially provokes a local immune response in the gut. Such local reactions can spread by escape of the microbial products from the gut, which can then reach distant organs. In addition to immune cells, the adipose tissue, liver, and brain are important TLR-expressing targets and are all potent metabolic regulators with systemic effects. The escape of microbial factors from the gut can result from perturbations in the gut microbiota and its interaction with immune system components that weakens the mucosal and epithelial lining of the gut. When the integrity of the mucus layer and intestinal epithelium is impaired, harmless commensals can become a threat, crossing the epithelium to invade the bloodstream, causing systemic infection or inflammation, which favors development of immune-mediated and metabolic diseases. Consequently, peripheral anti- or proinflammatory responses take place and modulate host metabolism, emphasizing to which substantial extent microbiota can influence systemic health.
In addition to the above discussed mechanisms, bile acids, which can be modified by microbiota or result from microbiota changes, could modulate host metabolism (119, 120) and also have immunomodulatory effects (121). Further gut microbiota derived metabolites, including tryptophan, phenylalanine, tyrosine, and polyamines, can regulate the immune system and the host metabolism with potential effects on health. For instance, the molecular signature of such interactions is described for the regulation of mucosal integrity and inflammatory signaling (122–124) by bacterial indoles binding to the pregnane X receptor (125) via pathways involving TLR4 (126) and NFkB. Indoles (127–132) and dietary ligands such as flavonoids (133) can be sensed by aryl hydrocarbon receptor nuclear translocator 2, which modulates epithelial barrier integrity (134–139) and provokes immune changes (140–144). Further immunologic components that can bridge microbiota with host metabolism include the nucleotide-binding oligomerization domain-like receptors (NOD-like receptors) and innate lymphoid cells that have been reviewed elsewhere (145). While linking host metabolism with microbiota alterations is a challenging task, the integrative metabolomics–metagenomics approaches are a promising way to provide a better understanding of these interactions. For example, using such approaches Pedersen and colleagues identified Prevotella copri and Bacteroides vulgatus as the main species driving the association between biosynthesis of branched-chain amino acids and insulin resistance, where P. copri increased their circulating levels, and induced insulin resistance and glucose intolerance (146).
Reprogramming of microbiota in patients can be an accessible and promising anti-obesity treatment. Thus, understanding the complex and reciprocal interplay between the microbiota and the immune system, and how this relationship can modulate metabolic parameters could result in advances toward the treatment of metabolic diseases.
Statements
Author contributions
The authors reviewed literature and conceived the manuscript. MS wrote the manuscript and prepared the figure. DM provided insightful comments and corrections. MT wrote paragraphs, corrected the manuscript, and provided guidance.
Funding
This work was supported by the CONFIRM grant of the Hôpitaux Universitaires de Genève (HUG) Foundation (no. RC2-09) and the Swiss Multiple Sclerosis Society Grant to DM and MT; the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement no. 336607 (ERC-2013-StG-336607) to MT; Swiss National Science Foundation (SNSF) grant to DM (310030_173010); and SNSF Professorship grants (PP00P3_144886 and PP00P3_172906) to MT.
Acknowledgments
We wish to thank our colleagues from the Faculty of Medicine, University of Geneva for discussions. We apologize that due to space limitations, we could not cite all relevant literature.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Ley RE Peterson DA Gordon JI . Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell (2006) 124:837–48.10.1016/j.cell.2006.02.017
2
Whitman WB Coleman DC Wiebe WJ . Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A (1998) 95:6578–83.10.1073/pnas.95.12.6578
3
Ley RE Turnbaugh PJ Klein S Gordon JI . Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3.10.1038/4441022a
4
Hwang I Park YJ Kim YR Kim YN Ka S Lee HY et al Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J (2015) 29:2397–411.10.1096/fj.14-265983
5
Rosenbaum M Knight R Leibel RL . The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol Metab (2015) 26:493–501.10.1016/j.tem.2015.07.002
6
Verdam FJ Fuentes S de Jonge C Zoetendal EG Erbil R Greve JW et al Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) (2013) 21:E607–15.10.1002/oby.20466
7
Vrieze A Van Nood E Holleman F Salojarvi J Kootte RS Bartelsman JF et al Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology (2012) 143:913–6.e7.10.1053/j.gastro.2012.06.031
8
Clemente JC Ursell LK Parfrey LW Knight R . The impact of the gut microbiota on human health: an integrative view. Cell (2012) 148:1258–70.10.1016/j.cell.2012.01.035
9
Backhed F Ding H Wang T Hooper LV Koh GY Nagy A et al The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A (2004) 101:15718–23.10.1073/pnas.0407076101
10
Ley RE Backhed F Turnbaugh P Lozupone CA Knight RD Gordon JI . Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A (2005) 102:11070–5.10.1073/pnas.0504978102
11
Ridaura VK Faith JJ Rey FE Cheng J Duncan AE Kau AL et al Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341:1241214.10.1126/science.1241214
12
Turnbaugh PJ Ley RE Mahowald MA Magrini V Mardis ER Gordon JI . An obesity-associated gut microbiome with increased capacity for energy harvest. Nature (2006) 444:1027–31.10.1038/nature05414
13
van den Elsen LW Poyntz HC Weyrich LS Young W Forbes-Blom EE . Embracing the gut microbiota: the new frontier for inflammatory and infectious diseases. Clin Transl Immunology (2017) 6:e125.10.1038/cti.2016.91
14
Pollard M Sharon N . Responses of the Peyer’s patches in germ-free mice to antigenic stimulation. Infect Immun (1970) 2:96–100.
15
Johansson ME Phillipson M Petersson J Velcich A Holm L Hansson GC . The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A (2008) 105:15064–9.10.1073/pnas.0803124105
16
Bouskra D Brezillon C Berard M Werts C Varona R Boneca IG et al Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature (2008) 456:507–10.10.1038/nature07450
17
Kamada N Seo SU Chen GY Nunez G . Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13:321–35.10.1038/nri3430
18
Niess JH Leithauser F Adler G Reimann J . Commensal gut flora drives the expansion of proinflammatory CD4 T cells in the colonic lamina propria under normal and inflammatory conditions. J Immunol (2008) 180:559–68.10.4049/jimmunol.180.1.559
19
Lundin A Bok CM Aronsson L Bjorkholm B Gustafsson JA Pott S et al Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol (2008) 10:1093–103.10.1111/j.1462-5822.2007.01108.x
20
Umesaki Y Okada Y Matsumoto S Imaoka A Setoyama H . Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol Immunol (1995) 39:555–62.10.1111/j.1348-0421.1995.tb02242.x
21
Suarez-Zamorano N Fabbiano S Chevalier C Stojanovic O Colin DJ Stevanovic A et al Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med (2015) 21:1497–501.10.1038/nm.3994
22
Backhed F Manchester JK Semenkovich CF Gordon JI . Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A (2007) 104:979–84.10.1073/pnas.0605374104
23
Ding S Chi MM Scull BP Rigby R Schwerbrock NM Magness S et al High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One (2010) 5:e12191.10.1371/journal.pone.0012191
24
Rabot S Membrez M Bruneau A Gerard P Harach T Moser M et al Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J (2010) 24:4948–59.10.1096/fj.10-164921
25
Cannon B Nedergaard J . Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84:277–359.10.1152/physrev.00015.2003
26
Nguyen KD Qiu Y Cui X Goh YP Mwangi J David T et al Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480:104–8.10.1038/nature10653
27
Qiu Y Nguyen KD Odegaard JI Cui X Tian X Locksley RM et al Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell (2014) 157:1292–308.10.1016/j.cell.2014.03.066
28
Rakoff-Nahoum S Paglino J Eslami-Varzaneh F Edberg S Medzhitov R . Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118:229–41.10.1016/j.cell.2004.07.002
29
Dasgupta S Erturk-Hasdemir D Ochoa-Reparaz J Reinecker HC Kasper DL . Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe (2014) 15:413–23.10.1016/j.chom.2014.03.006
30
Johnson JL Jones MB Cobb BA . Polysaccharide A from the capsule of Bacteroides fragilis induces clonal CD4+ T cell expansion. J Biol Chem (2015) 290:5007–14.10.1074/jbc.M114.621771
31
Round JL Lee SM Li J Tran G Jabri B Chatila TA et al The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science (2011) 332:974–7.10.1126/science.1206095
32
Ochoa-Reparaz J Mielcarz DW Wang Y Begum-Haque S Dasgupta S Kasper DL et al A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol (2010) 3:487–95.10.1038/mi.2010.29
33
Wang Y Telesford KM Ochoa-Reparaz J Haque-Begum S Christy M Kasper EJ et al An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun (2014) 5:4432.10.1038/ncomms5432
34
Mazmanian SK Round JL Kasper DL . A microbial symbiosis factor prevents intestinal inflammatory disease. Nature (2008) 453:620–5.10.1038/nature07008
35
Smelt MJ de Haan BJ Bron PA van Swam I Meijerink M Wells JM et al The impact of Lactobacillus plantarum WCFS1 teichoic acid D-alanylation on the generation of effector and regulatory T-cells in healthy mice. PLoS One (2013) 8:e63099.10.1371/journal.pone.0063099
36
Murakami K Bujo H Unoki H Saito Y . High fat intake induces a population of adipocytes to co-express TLR2 and TNFalpha in mice with insulin resistance. Biochem Biophys Res Commun (2007) 354:727–34.10.1016/j.bbrc.2007.01.039
37
Caricilli AM Nascimento PH Pauli JR Tsukumo DM Velloso LA Carvalheira JB et al Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol (2008) 199:399–406.10.1677/JOE-08-0354
38
Ehses JA Meier DT Wueest S Rytka J Boller S Wielinga PY et al Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia (2010) 53:1795–806.10.1007/s00125-010-1747-3
39
Himes RW Smith CW . Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J (2010) 24:731–9.10.1096/fj.09-141929
40
Caesar R Tremaroli V Kovatcheva-Datchary P Cani PD Backhed F . Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab (2015) 22:658–68.10.1016/j.cmet.2015.07.026
41
Tanti JF Ceppo F Jager J Berthou F . Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front Endocrinol (2013) 3:181.10.3389/fendo.2012.00181
42
Devaraj S Tobias P Jialal I . Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine (2011) 55:441–5.10.1016/j.cyto.2011.03.023
43
Milanski M Degasperi G Coope A Morari J Denis R Cintra DE et al Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci (2009) 29:359–70.10.1523/JNEUROSCI.2760-08.2009
44
Etienne-Mesmin L Vijay-Kumar M Gewirtz AT Chassaing B . Hepatocyte toll-like receptor 5 promotes bacterial clearance and protects mice against high-fat diet-induced liver disease. Cell Mol Gastroenterol Hepatol (2016) 2:584–604.10.1016/j.jcmgh.2016.04.007
45
Brooks L Viardot A Tsakmaki A Stolarczyk E Howard JK Cani PD et al Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol Metab (2017) 6:48–60.10.1016/j.molmet.2016.10.011
46
Psichas A Sleeth ML Murphy KG Brooks L Bewick GA Hanyaloglu AC et al The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int J Obes (Lond) (2015) 39:424–9.10.1038/ijo.2014.153
47
Tolhurst G Heffron H Lam YS Parker HE Habib AM Diakogiannaki E et al Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes (2012) 61:364–71.10.2337/db11-1019
48
De Vadder F Kovatcheva-Datchary P Goncalves D Vinera J Zitoun C Duchampt A et al Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell (2014) 156:84–96.10.1016/j.cell.2013.12.016
49
Lin HV Frassetto A Kowalik EJ Jr Nawrocki AR Lu MM Kosinski JR et al Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS One (2012) 7:e35240.10.1371/journal.pone.0035240
50
Marino E Richards JL McLeod KH Stanley D Yap YA Knight J et al Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol (2017) 18:552–62.10.1038/ni.3713
51
Kimura I Ozawa K Inoue D Imamura T Kimura K Maeda T et al The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun (2013) 4:1829.10.1038/ncomms2852
52
Sahuri-Arisoylu M Brody LP Parkinson JR Parkes H Navaratnam N Miller AD et al Reprogramming of hepatic fat accumulation and ’browning’ of adipose tissue by the short-chain fatty acid acetate. Int J Obes (Lond) (2016) 40:955–63.10.1038/ijo.2016.23
53
Perry RJ Peng L Barry NA Cline GW Zhang D Cardone RL et al Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature (2016) 534:213–7.10.1038/nature18309
54
Ji J Shu D Zheng M Wang J Luo C Wang Y et al Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci Rep (2016) 6:24838.10.1038/srep24838
55
Willemsen LE Koetsier MA van Deventer SJ van Tol EA . Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E(1) and E(2) production by intestinal myofibroblasts. Gut (2003) 52:1442–7.10.1136/gut.52.10.1442
56
Cani PD Neyrinck AM Fava F Knauf C Burcelin RG Tuohy KM et al Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia (2007) 50:2374–83.10.1007/s00125-007-0791-0
57
Ahmad R Al-Mass A Atizado V Al-Hubail A Al-Ghimlas F Al-Arouj M et al Elevated expression of the toll like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. J Inflamm (Lond) (2012) 9:48.10.1186/1476-9255-9-48
58
Reyna SM Ghosh S Tantiwong P Meka CS Eagan P Jenkinson CP et al Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes (2008) 57:2595–602.10.2337/db08-0038
59
Song MJ Kim KH Yoon JM Kim JB . Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun (2006) 346:739–45.10.1016/j.bbrc.2006.05.170
60
Kumari M Wang X Lantier L Lyubetskaya A Eguchi J Kang S et al IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest (2016) 126:2839–54.10.1172/JCI86080
61
Everard A Geurts L Caesar R Van Hul M Matamoros S Duparc T et al Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun (2014) 5:5648.10.1038/ncomms6648
62
Shi H Kokoeva MV Inouye K Tzameli I Yin H Flier JS . TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116:3015–25.10.1172/JCI28898
63
Boden G . Obesity and free fatty acids. Endocrinol Metab Clin North Am (2008) 37:635–46, viii–ix.10.1016/j.ecl.2008.06.007
64
Tao C Holland WL Wang QA Shao M Jia L Sun K et al Short-term vs. long-term effects of adipocyte toll-like receptor 4 activation on insulin resistance in male mice. Endocrinology (2017) 158(5):1260–70.10.1210/en.2017-00024
65
Jia L Vianna CR Fukuda M Berglund ED Liu C Tao C et al Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun (2014) 5:3878.10.1038/ncomms4878
66
Kleinridders A Schenten D Konner AC Belgardt BF Mauer J Okamura T et al MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab (2009) 10:249–59.10.1016/j.cmet.2009.08.013
67
Shechter R London A Kuperman Y Ronen A Rolls A Chen A et al Hypothalamic neuronal toll-like receptor 2 protects against age-induced obesity. Sci Rep (2013) 3:1254.10.1038/srep01254
68
Carvalho FA Aitken JD Gewirtz AT Vijay-Kumar M . TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal Immunol (2011) 4:102–11.10.1038/mi.2010.57
69
Vijay-Kumar M Aitken JD Carvalho FA Cullender TC Mwangi S Srinivasan S et al Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science (2010) 328:228–31.10.1126/science.1179721
70
Chassaing B Ley RE Gewirtz AT . Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology (2014) 147:1363–77.e17.10.1053/j.gastro.2014.08.033
71
Ubeda C Lipuma L Gobourne A Viale A Leiner I Equinda M et al Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med (2012) 209:1445–56.10.1084/jem.20120504
72
Letran SE Lee SJ Atif SM Flores-Langarica A Uematsu S Akira S et al TLR5-deficient mice lack basal inflammatory and metabolic defects but exhibit impaired CD4 T cell responses to a flagellated pathogen. J Immunol (2011) 186:5406–12.10.4049/jimmunol.1003576
73
Zhang W Hartmann R Tun HM Elson CO Khafipour E Garvey WT . Deletion of the Toll-like receptor 5 gene per se does not determine the gut microbiome profile that induces metabolic syndrome: environment trumps genotype. PLoS One (2016) 11:e0150943.10.1371/journal.pone.0150943
74
Duparc T Plovier H Marrachelli VG Van Hul M Essaghir A Stahlman M et al Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut (2017) 66:620–32.10.1136/gutjnl-2015-310904
75
Heazlewood CK Cook MC Eri R Price GR Tauro SB Taupin D et al Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med (2008) 5:e54.10.1371/journal.pmed.0050054
76
van der Sluis S Dolan CV Neale MC Boomsma DI Posthuma D . Detecting genotype-environment interaction in monozygotic twin data: comparing the Jinks and Fulker test and a new test based on marginal maximum likelihood estimation. Twin Res Hum Genet (2006) 9:377–92.10.1375/183242706777591218
77
Birchenough GM Nyström EE Johansson ME Hansson GC . A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science (2016) 352(6293):1535–42.10.1126/science.aaf7419
78
Vijay-Kumar M Sanders CJ Taylor RT Kumar A Aitken JD Sitaraman SV et al Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest (2007) 117:3909–21.10.1172/JCI33084
79
Bhinder G Stahl M Sham HP Crowley SM Morampudi V Dalwadi U et al Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun (2014) 82:3753–63.10.1128/IAI.02045-14
80
Frantz AL Rogier EW Weber CR Shen L Cohen DA Fenton LA et al Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol (2012) 5:501–12.10.1038/mi.2012.23
81
Johansson ME Jakobsson HE Holmen-Larsson J Schutte A Ermund A Rodriguez-Pineiro AM et al Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe (2015) 18:582–92.10.1016/j.chom.2015.10.007
82
Jakobsson HE Rodriguez-Pineiro AM Schutte A Ermund A Boysen P Bemark M et al The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep (2015) 16:164–77.10.15252/embr.201439263
83
Ley RE Hamady M Lozupone C Turnbaugh PJ Ramey RR Bircher JS et al Evolution of mammals and their gut microbes. Science (2008) 320:1647–51.10.1126/science.1155725
84
Sonnenburg JL Xu J Leip DD Chen CH Westover BP Weatherford J et al Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science (2005) 307:1955–9.10.1126/science.1109051
85
Li H Limenitakis JP Fuhrer T Geuking MB Lawson MA Wyss M et al The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun (2015) 6:8292.10.1038/ncomms9292
86
Marcobal A Southwick AM Earle KA Sonnenburg JL . A refined palate: bacterial consumption of host glycans in the gut. Glycobiology (2013) 23:1038–46.10.1093/glycob/cwt040
87
Pudlo NA Urs K Kumar SS German JB Mills DA Martens EC . Symbiotic human gut bacteria with variable metabolic priorities for host mucosal glycans. MBio (2015) 6:e1282–315.10.1128/mBio.01282-15
88
He B Hoang TK Wang T Ferris M Taylor CM Tian X et al Resetting microbiota by Lactobacillus reuteri inhibits T reg deficiency-induced autoimmunity via adenosine A2A receptors. J Exp Med (2017) 214:107–23.10.1084/jem.20160961
89
Chu VT Beller A Rausch S Strandmark J Zanker M Arbach O et al Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity (2014) 40:582–93.10.1016/j.immuni.2014.02.014
90
Johnson AM Costanzo A Gareau MG Armando AM Quehenberger O Jameson JM et al High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS One (2015) 10:e0122195.10.1371/journal.pone.0122195
91
Skoczek DA Walczysko P Horn N Parris A Clare S Williams MR et al Luminal microbes promote monocyte-stem cell interactions across a healthy colonic epithelium. J Immunol (2014) 193:439–51.10.4049/jimmunol.1301497
92
Gulhane M Murray L Lourie R Tong H Sheng YH Wang R et al High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep (2016) 6:28990.10.1038/srep28990
93
Tsuboi K Nishitani M Takakura A Imai Y Komatsu M Kawashima H . Autophagy protects against colitis by the maintenance of normal gut microflora and secretion of mucus. J Biol Chem (2015) 290:20511–26.10.1074/jbc.M114.632257
94
Wlodarska M Thaiss CA Nowarski R Henao-Mejia J Zhang JP Brown EM et al NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell (2014) 156:1045–59.10.1016/j.cell.2014.01.026
95
Lassen KG Kuballa P Conway KL Patel KK Becker CE Peloquin JM et al Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A (2014) 111:7741–6.10.1073/pnas.1407001111
96
Cani PD Bibiloni R Knauf C Waget A Neyrinck AM Delzenne NM et al Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes (2008) 57:1470–81.10.2337/db07-1403
97
Giles DA Moreno-Fernandez ME Stankiewicz TE Graspeuntner S Cappelletti M Wu D et al Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat Med (2017) 23:829–38.10.1038/nm1017-1241c
98
Desai MS Seekatz AM Koropatkin NM Kamada N Hickey CA Wolter M et al A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell (2016) 167:1339–53.e21.10.1016/j.cell.2016.10.043
99
Janssen P Rotondo A Mule F Tack J . Review article: a comparison of glucagon-like peptides 1 and 2. Aliment Pharmacol Ther (2013) 37:18–36.10.1111/apt.12092
100
Cani PD Possemiers S Van de Wiele T Guiot Y Everard A Rottier O et al Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut (2009) 58:1091–103.10.1136/gut.2008.165886
101
Wang X Ota N Manzanillo P Kates L Zavala-Solorio J Eidenschenk C et al Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature (2014) 514:237–41.10.1038/nature13564
102
Brown AJ Goldsworthy SM Barnes AA Eilert MM Tcheang L Daniels D et al The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem (2003) 278:11312–9.10.1074/jbc.M211609200
103
Koh A De Vadder F Kovatcheva-Datchary P Backhed F . From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell (2016) 165:1332–45.10.1016/j.cell.2016.05.041
104
Fukuda S Toh H Hase K Oshima K Nakanishi Y Yoshimura K et al Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature (2011) 469:543–7.10.1038/nature09646
105
Donohoe DR Garge N Zhang X Sun W O’Connell TM Bunger MK et al The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab (2011) 13:517–26.10.1016/j.cmet.2011.02.018
106
Gaudier E Jarry A Blottiere HM de Coppet P Buisine MP Aubert JP et al Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am J Physiol Gastrointest Liver Physiol (2004) 287:G1168–74.10.1152/ajpgi.00219.2004
107
Roediger WE . Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology (1982) 83:424–9.
108
Park JS Lee EJ Lee JC Kim WK Kim HS . Anti-inflammatory effects of short chain fatty acids in IFN-gamma-stimulated RAW 264.7 murine macrophage cells: involvement of NF-kappaB and ERK signaling pathways. Int Immunopharmacol (2007) 7:70–7.10.1016/j.intimp.2006.08.015
109
Paulson JC Colley KJ . Glycosyltransferases. Structure, localization, and control of cell type-specific glycosylation. J Biol Chem (1989) 264:17615–8.
110
Tang Y Chen Y Jiang H Nie D . Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ (2011) 18:602–18.10.1038/cdd.2010.117
111
Zhang J Yi M Zha L Chen S Li Z Li C et al Sodium butyrate induces endoplasmic reticulum stress and autophagy in colorectal cells: implications for apoptosis. PLoS One (2016) 11:e0147218.10.1371/journal.pone.0147218
112
Smith PM Howitt MR Panikov N Michaud M Gallini CA Bohlooly YM et al The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73.10.1126/science.1241165
113
Trompette A Gollwitzer ES Yadava K Sichelstiel AK Sprenger N Ngom-Bru C et al Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med (2014) 20:159–66.10.1038/nm.3444
114
Canfora EE Jocken JW Blaak EE . Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol (2015) 11:577–91.10.1038/nrendo.2015.128
115
Bloemen JG Venema K van de Poll MC Olde Damink SW Buurman WA Dejong CH . Short chain fatty acids exchange across the gut and liver in humans measured at surgery. Clin Nutr (2009) 28:657–61.10.1016/j.clnu.2009.05.011
116
den Besten G Bleeker A Gerding A van Eunen K Havinga R van Dijk TH et al Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARgamma-dependent switch from lipogenesis to fat oxidation. Diabetes (2015) 64:2398–408.10.2337/db14-1213
117
Trajkovski M Wollheim CB . Physiology: microbial signals to the brain control weight. Nature (2016) 534:185–7.10.1038/534185a
118
van der Beek CM Canfora EE Lenaerts K Troost FJ Damink SW Holst JJ et al Distal, not proximal, colonic acetate infusions promote fat oxidation and improve metabolic markers in overweight/obese men. Clin Sci (Lond) (2016) 130:2073–82.10.1042/CS20160263
119
Wahlstrom A Sayin SI Marschall HU Backhed F . Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab (2016) 24:41–50.10.1016/j.cmet.2016.05.005
120
Zietak M Kovatcheva-Datchary P Markiewicz LH Stahlman M Kozak LP Backhed F . Altered microbiota contributes to reduced diet-induced obesity upon cold exposure. Cell Metab (2016) 23:1216–23.10.1016/j.cmet.2016.05.001
121
Sipka S Bruckner G . The immunomodulatory role of bile acids. Int Arch Allergy Immunol (2014) 165:1–8.10.1159/000366100
122
Mencarelli A Renga B Palladino G Claudio D Ricci P Distrutti E et al Inhibition of NF-kappaB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur J Pharmacol (2011) 668:317–24.10.1016/j.ejphar.2011.06.058
123
Shah YM Ma X Morimura K Kim I Gonzalez FJ . Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol (2007) 292:G1114–22.10.1152/ajpgi.00528.2006
124
Zhou C Tabb MM Nelson EL Grun F Verma S Sadatrafiei A et al Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest (2006) 116:2280–9.10.1172/JCI26283
125
Ranhotra HS Flannigan KL Brave M Mukherjee S Lukin DJ Hirota SA et al Xenobiotic receptor-mediated regulation of intestinal barrier function and innate immunity. Nucl Receptor Res (2016) 3:101199.10.11131/2016/101199
126
Venkatesh M Mukherjee S Wang H Li H Sun K Benechet AP et al Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity (2014) 41:296–310.10.1016/j.immuni.2014.06.014
127
DiNatale BC Murray IA Schroeder JC Flaveny CA Lahoti TS Laurenzana EM et al Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci (2010) 115:89–97.10.1093/toxsci/kfq024
128
Jin UH Lee SO Sridharan G Lee K Davidson LA Jayaraman A et al Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol Pharmacol (2014) 85:777–88.10.1124/mol.113.091165
129
Miller CA III . Expression of the human aryl hydrocarbon receptor complex in yeast. Activation of transcription by indole compounds. J Biol Chem (1997) 272:32824–9.10.1074/jbc.272.52.32824
130
Oberg M Bergander L Hakansson H Rannug U Rannug A . Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci (2005) 85:935–43.10.1093/toxsci/kfi154
131
Opitz CA Litzenburger UM Sahm F Ott M Tritschler I Trump S et al An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197–203.10.1038/nature10491
132
Schroeder JC Dinatale BC Murray IA Flaveny CA Liu Q Laurenzana EM et al The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry (2010) 49:393–400.10.1021/bi901786x
133
Zhang S Qin C Safe SH . Flavonoids as aryl hydrocarbon receptor agonists/antagonists: effects of structure and cell context. Environ Health Perspect (2003) 111:1877–82.10.1289/ehp.6322
134
Arsenescu R Arsenescu V Zhong J Nasser M Melinte R Dingle RW et al Role of the xenobiotic receptor in inflammatory bowel disease. Inflamm Bowel Dis (2011) 17:1149–62.10.1002/ibd.21463
135
Fukumoto S Toshimitsu T Matsuoka S Maruyama A Oh-Oka K Takamura T et al Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol Cell Biol (2014) 92:460–5.10.1038/icb.2014.2
136
Li Y Innocentin S Withers DR Roberts NA Gallagher AR Grigorieva EF et al Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell (2011) 147:629–40.10.1016/j.cell.2011.09.025
137
Qiu J Heller JJ Guo X Chen ZM Fish K Fu YX et al The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36:92–104.10.1016/j.immuni.2011.11.011
138
Takamura T Harama D Fukumoto S Nakamura Y Shimokawa N Ishimaru K et al Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol Cell Biol (2011) 89:817–22.10.1038/icb.2010.165
139
Takamura T Harama D Matsuoka S Shimokawa N Nakamura Y Okumura K et al Activation of the aryl hydrocarbon receptor pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunol Cell Biol (2010) 88:685–9.10.1038/icb.2010.35
140
Chng SH Kundu P Dominguez-Brauer C Teo WL Kawajiri K Fujii-Kuriyama Y et al Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci Rep (2016) 6:23820.10.1038/srep23820
141
Climaco-Arvizu S Dominguez-Acosta O Cabanas-Cortes MA Rodriguez-Sosa M Gonzalez FJ Vega L et al Aryl hydrocarbon receptor influences nitric oxide and arginine production and alters M1/M2 macrophage polarization. Life Sci (2016) 155:76–84.10.1016/j.lfs.2016.05.001
142
Rothhammer V Mascanfroni ID Bunse L Takenaka MC Kenison JE Mayo L et al Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22:586–97.10.1038/nm.4106
143
Vaidyanathan B Chaudhry A Yewdell WT Angeletti D Yen WF Wheatley AK et al The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J Exp Med (2017) 214:197–208.10.1084/jem.20160789
144
Yamada T Horimoto H Kameyama T Hayakawa S Yamato H Dazai M et al Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat Immunol (2016) 17:687–94.10.1038/ni.3422
145
Thaiss CA Zmora N Levy M Elinav E . The microbiome and innate immunity. Nature (2016) 535:65–74.10.1038/nature18847
146
Pedersen HK Gudmundsdottir V Nielsen HB Hyotylainen T Nielsen T Jensen BA et al Human gut microbes impact host serum metabolome and insulin sensitivity. Nature (2016) 535:376–81.10.1038/nature18646
Summary
Keywords
gut microbiota, immune system, toll-like receptors, short chain fatty acids, mucosal barrier, metabolism, dysbiosis
Citation
Spiljar M, Merkler D and Trajkovski M (2017) The Immune System Bridges the Gut Microbiota with Systemic Energy Homeostasis: Focus on TLRs, Mucosal Barrier, and SCFAs. Front. Immunol. 8:1353. doi: 10.3389/fimmu.2017.01353
Received
25 July 2017
Accepted
03 October 2017
Published
30 October 2017
Volume
8 - 2017
Edited by
Marina I. Arleevskaya, Kazan State Medical Academy, Russia
Reviewed by
Angela M. Zivkovic, University of California, Davis, United States; Francois-Pierre Martin, Nestle Institute of Health Sciences, Switzerland
Updates
Copyright
© 2017 Spiljar, Merkler and Trajkovski.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirko Trajkovski, mirko.trajkovski@unige.ch
Specialty section: This article was submitted to Microbial Immunology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.