 Greta Forlani
Greta Forlani Roberto S. Accolla
Roberto S. Accolla- Laboratories of General Pathology and Immunology “Giovanna Tosi”, Department of Medicine and Surgery, University of Insubria, Varese, Italy
Coevolution of the three basic mechanisms of immunity, intrinsic, innate and adaptive, is a constant feature of the host defense against pathogens. Within this frame, a peculiar role is played by restriction factors (RFs), elements of intrinsic immunity that interfere with viral life cycle. Often considered as molecules whose specific functions are distinct and unrelated among themselves recent results indicate instead, at least for some of them, a concerted action against the pathogen. Here we review recent findings on the antiviral activity of tripartite motif 22 (TRIM22) and class II transactivator (CIITA), first discovered as human immunodeficiency virus 1 RFs, but endowed with general antiviral activity. TRIM22 and CIITA provide the first example of cellular proteins acting together to potentiate their intrinsic immunity.
Restriction Factors (RFs): Key Elements of Intrinsic Immunity Against Viruses
In recent years, the role of viral RFs as potent effectors of intrinsic antiviral immunity has become more clear (1–5). Most studies have focused on the mechanisms of host-mediated restriction of human immunodeficiency virus 1 (HIV-1), in which RFs exert intrinsic antiviral activity by targeting different steps of the HIV-1 life cycle, from capsid uncoating to viral budding (4, 6–11). Although RFs can be expressed constitutively in the host cell, most of them are potently upregulated by molecules of innate immunity such as type I and type II interferons (IFN), reinforcing the concept that they are crucial players of the immune defense against retroviruses (12). The most extensively studied RFs are Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G (APOBEC3G), Tripartite motif 5-alpha (TRIM5α), tetherin (also known as BST-2, CD317, or HM1.24), and Sterile alpha motif domain and histidine aspartic domain containing protein1 (SAMHD1) (Figure 1).
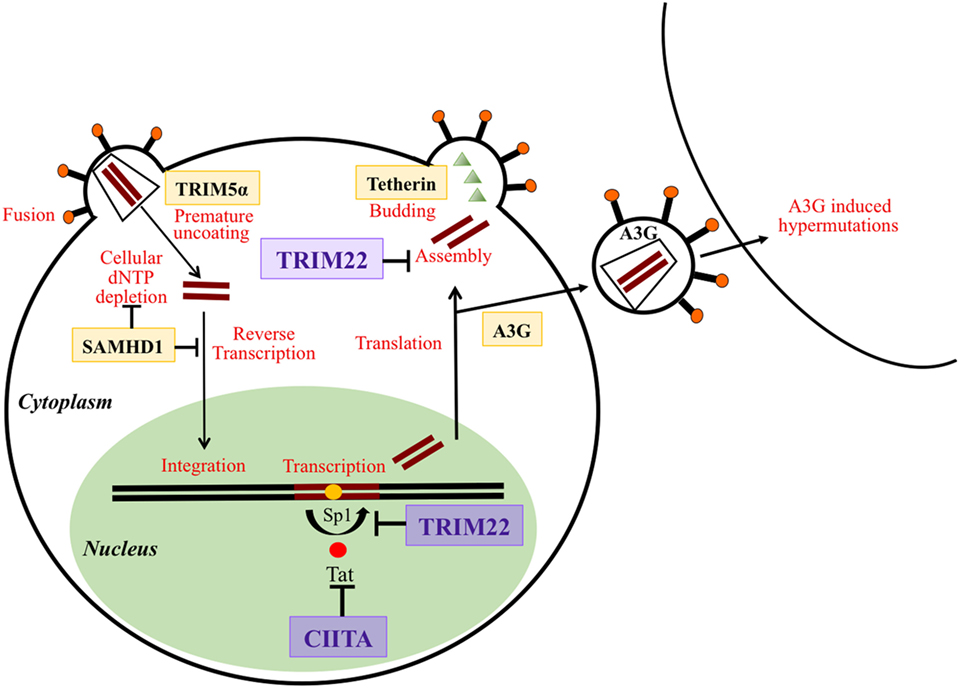
Figure 1. Schematic overview of the action of different restriction factors (RFs) at various stages of the human immunodeficiency virus life cycle. RFs are in bold and framed. Their targeted functions on the virus life cycle are indicated in red. Tripartite motif 22 (TRIM22) and class II transactivator (CIITA), the two RFs described in this review are magnified and written in violet. TRIM5α acts at early phases of virus infection by disturbing and inducing a premature uncoating. SAMHD1 inhibits reverse transcription of the viral RNA by either targeting the dNTP pool or reverse transcription products. CIITA acts by inhibiting the Tat-mediated activation of transcription and particularly the elongation of primary transcripts. TRIM22 acts both at the level of transcription of viral DNA by blocking the action of Sp1 and at the level of assembly of new viral particles. Tetherin blocks the budding of viral particles at the cell surface. APOBEC3G (A3G) incorporates into newly formed viral particles and after infection of a new cell A3G induces hypermutations during the process of reverse transcription thus inhibiting the viral replication cycle.
To evade host restriction, HIV-1 has developed some countermeasures by using viral accessory proteins or by inducing specific mutations on protein interfaces (13). For example, the human APOBEC3 G/F enzymes are cytidine deaminases packaged into the virus particles during assembly. They inhibit reverse transcription by deaminating viral cDNA cytosines to uracils, thus introducing G-to-A hypermutations in the viral genome (14–17). The viral infectivity factor of HIV-1 counteracts the APOBEC3 activity, by mediating its proteasomal degradation. Similar to APOBEC3 enzymes SAMHD1 also inhibits retroviral transcription, by depleting the intracellular pool of deoxynucleoside triphosphates available during early reverse transcription (17). SAMHD1 activity is prevented in HIV-2 and related SIVs by the viral proteins Vpx and Vpr (18, 19). Different from the host factors mentioned above, TRIM5α acts as a species-specific antiviral factor. Indeed rhesus monkey TRIM5α (rhTRIM5α), but not human TRIM5α, potently limits HIV-1 infection in Old World monkeys by targeting the viral capsid, thus preventing the uncoating of the viral pre-integration complex (20). Tetherin targets the post-integration stages of HIV-1 replication and prevents the release of nascent viral particles by anchoring virions on the cell surface of HIV-1 infected cells. Viral proteins Vpu and Env overcome tetherin restriction by sequestering tetherin in cellular compartments away from sites of viral budding or by targeting it for degradation into lysosomes (21).
Often considered as independent elements in the action against virus infection, recent studies unveil, instead, that RFs may act in concert against the pathogen. This review will discuss the example of cooperativeness in HIV-1 restriction of two recently described RFs, the tripartite motif 22 (TRIM22) and the MHC class II transactivator (CIITA). Importantly, this functional synergy is mirrored by their cellular policy of getting together in common subcellular compartments where other crucial factors controlling HIV-1 replication convene. Thus, TRIM22 and CIITA represent a clear example of concerted action whose final goal is favored by compartmentalization of multiple players involved in fighting against HIV-1.
Mechanisms of TRIM22 and CIITA Viral Restriction
Tripartite motif 22 was first identified as an IFN-inducible protein that restricts HIV-1 transcription (Figure 1) (22). The first clinical evidence highlighting the antiviral function of TRIM22 in vivo was reported by Singh et al. demonstrating that in peripheral blood mononuclear cells (PBMCs) of infected patients, the expression of TRIM22 was significantly increased and correlated with lower viral loads and higher CD4+ T cell counts (23, 24). At present, several reports demonstrated that TRIM22 acts as RF against a broad spectrum of viruses. Besides inhibiting HIV-1 transcription, TRIM22 also inhibits Influenza A virus (25), Hepatitis B and C viruses (26, 27), and encephalomyocarditis virus (28), by using different mechanisms. TRIM22 belongs to TRIM family of proteins exerting various functions, including cellular proliferation, apoptosis, oncogenesis, and antiviral activity (29). TRIM proteins are characterized by the RBCC motif, consisting in a Really Interesting New Gene (RING) domain, one or two B-boxes followed by a coiled-coil (CC) region. The C terminal part is specific for each TRIM (6). The RING domain is involved in protein–protein interactions and is associated with E3 ubiquitin ligase activity (30). The CC region is crucial for the formation of protein complexes and promotes homo- and hetero-oligomerization that may cause dislocation in distinct subcellular compartments (31, 32). TRIM22, as other TRIM proteins, contains a C-terminal B30.2/SPRY domain, whose function has not been fully clarified. Some studies have shown that it is also critical for TRIM22 nuclear localization and formation of nuclear bodies (33). Furthermore, the B30.2/SPRY was shown to dictate the different subcellular distributions and thus specific functions of TRIM family members (34, 35). Indeed, the B30.2/SPRY domain is essential for TRIM22-mediated activation of nuclear factor kappa B (NF-κB) (26, 36) and possibly for the recently described TRIM22-mediated monocyte apoptosis (37). The intimate reasons of the distinct subcellular distribution of TRIM22 found in various studies, however, remain controversial. Indeed, several factors may influence the subcellular localization of the protein, such as the assessment of endogenous versus exogenous expression, the cell line analyzed, the cell cycle, being epitope-tagged or untagged, and the method of fixation used in the analysis (32). We and others have reported that TRIM22 localizes in the nucleus as punctate bodies (26, 38, 39). In some cells, these TRIM22 nuclear bodies partially overlap with Cajal bodies (34, 35) or centrosome (39). Other investigators have shown that TRIM22 may also localize in the cytoplasm with a diffuse and/or a speckled pattern (33–35). Here, TRIM22 localized in vimentin-containing structures (39).
The considerations expressed above are crucial for a precise delineation of additional mechanisms of inhibition of HIV-1 replication mediated by TRIM22. Indeed, Barr et al. reported that cytoplasmic TRIM22 ectopically expressed in human epithelial HeLa or in osteosarcoma HOS-CD4-CXCR4 cell lines blocked the release of HIV-1 particles by targeting Gag and thus inhibiting HIV particle assembly (Figure 1). This inhibitory function depended on its RING domain (40). Other reports demonstrated that TRIM22 overexpressed in COS-1, human macrophages or in 293 T cells impaired basal as well as phorbol-12-myristate13-acetate (PMA)-ionomycin induced HIV-1 long terminal repeat (LTR)-mediated transcription when present in the nucleus (7, 22, 41). TRIM22 suppressed transcription from HIV-1 LTR independent of its E3 ubiquitin ligase activity and did not inhibit neither Tat nor NF-kB-activated HIV-1 transcription (7). Recently, this effect has been attributed to the capacity of TRIM22 to affect the binding of Specific protein 1 (Sp1) to the HIV-1 LTR promoter region (42) (Figure 1). Although most of the studies demonstrating the antiviral function of TRIM22 have been conducted by using ectopically expressed proteins, some reports indicated that physiologic levels of TRIM22 could indeed interfere with HIV-1 replication (7, 24). In particular, Kajaste-Rudnitski et al. identified TRIM22 as the HIV-1 RF expressed in a subset of U937 promonocytic cell clones poorly permissive to HIV-1 replication, and not expressed in the isogenic HIV-1 permissive U937 cell clones (7, 43). In this regard, they found that the depletion of TRIM22 in non-permissive U937 clones increased viral LTR transcription to levels closer to those observed in the permissive cells, thus suggesting that TRIM22 contributed to HIV-1 refractory phenotype of poorly permissive cells (7). Consistently, exogenous expression of TRIM22 in permissive clones reduced HIV-1 LTR transcription.
The MHC class II transcriptional activator, also designed CIITA, was originally discovered as a master regulator of major histocompatibility complex (MHC) class II gene expression (44–46). Both constitutive and IFNγ-inducible expression of MHC-II are under the control of CIITA (47). By regulating the expression of all MHC class II genes, CIITA controls antigen presentation to CD4+ T helper (TH) cells, thus playing a critical role in the triggering of the adaptive immune response against a wide variety of antigens including pathogens (48, 49) and tumors (50). CIITA is a large protein characterized by distinct functional domains critical for its transactivating function: the N-terminal transcription activation domain (AD); the proline/serine/threonine-rich region (P/S/T); the GTP-binding domain (GBD), and the C-terminal leucine-rich repeats (LRR) important for the subcellular localization of the protein (51–53).
Besides its role in antigen presentation, via the regulation of MHC-II genes expression, it was demonstrated that CIITA restricts HIV-1 infection in human T cells by acting at the level of viral LTR transcription (Figure 1). CIITA binds to CyclinT1 of the positive transcription elongation factor b (P-TEFb) (54) and competes with the viral transactivator Tat for the binding to CyclinT1 (55). Subsequent studies have shown that CIITA acts as a general RF against retroviruses (49). Indeed, CIITA inhibits human T lymphotropic virus 2 (HTLV-2) replication, by targeting the viral transactivator Tax-2 (56). This inhibition occurs by preventing the association of Tax-2 to the common binding element nuclear factor Y (NF-Y), used by the virus to promote viral transcription (57, 58). Importantly, CIITA blocks the replication of the HTLV-1, the first discovered human oncogenic retrovirus (59), responsible for a severe form of T cell leukemia-lymphoma of the adult (ATL). Here, CIITA exerts a double function: it competes with the viral transactivator Tax-1 for binding with key cellular factors required for Tax-1 transactivating function on the viral LTR, and it binds directly to the viral transactivator, greatly limiting its intracellular migration (49, 60). Thus, CIITA restricts HTLV-1 viral replication by physically and functionally excluding Tax-1 transactivator from its crucial action in recruiting cellular transcription factors on virus LTR promoter to initiate viral replication. Tax-1 is crucial not only for the regulation of viral replication but also for its key action on cellular transformation predisposing to ATL. In this regard, it has been recently demonstrated that CIITA affects not only Tax-1 transactivating capacity but also Tax-1-mediated NF-kB activation, the crucial molecular event in initiation of leukemogenesis (61). Here, CIITA exerts its inhibitory function mainly by retaining Tax-1-NF-kB complex in the cytoplasm, thus preventing the translocation of NF-kB to the nucleus and the consequent activation of NF-kB responsive genes. In this regard, CIITA may counteract the oncogenic potential of HTLV-1 (52, 53).
The Importance of Subcellular Compartmentalization to Counteract Viral Replication
As mentioned above, the first clue that CIITA may act as an RF was made when it was found that CIITA inhibited HIV-1 replication in T cells (55). Relevant to HIV infection, subsequent recent studies have shown that CIITA, like TRIM22, was expressed in HIV-1 poorly permissive U937 myeloid cell clones, and absent in the permissive U937 myeloid parental cells (52, 53). Importantly, transfection of CIITA in HIV-1 permissive U937 clones resulted in the inhibition of Tat-dependent HIV-1 replication independent of TRIM22 (52, 53). Thus, CIITA is an HIV-1 RF for both lymphoid and myeloid cells.
It was apparent, however, that neither CIITA nor TRIM22 alone could restore completely the level of HIV-1 inhibition of replication observed in poorly permissive cells (7, 52, 53), suggesting that the simultaneous expression of these two RF could be required for a more effective HIV-1 restriction (52, 53). To better delineate the biological basis of the possible cooperativeness between CIITA and TRIM22, further experiments were designed to assess possible interaction and subcellular localization of the two RFs. It was found that TRIM22 interacted with CIITA and recruited it in newly defined compartments that we designated as TRIM22 nuclear bodies (Figure 2) (38). Interestingly TRIM19, another member of TRIM family, also known as promyleocitic leukemia protein (PML), and reported to inhibit the replication of various viruses, including HIV-1 (62–66), co-localized to a significant number of TRIM22 bodies. Relevant to this point, previous studies showed that PML and CIITA homed to the same nuclear bodies when cells were treated with IFN-γ (67). Thus, it was important to assess whether in cells co-expressing TRIM22 and CIITA, the latter was recruited by TRIM22 in nuclear bodies containing also endogenous PML. Indeed, this was the case (38). Of further relevance was the fact that TRIM22 nuclear bodies hosted also CyclinT1 of the P-TEFb complex, a key component of the trascription machinery used by HIV-1 to promote viral gene expression. CyclinT1 was previously shown not only to be bound by CIITA to inhibit Tat-mediated HIV-1 transcription (55), but also to localize in PML bodies where aggregation with PML negatively affected Tat-induced LTR transcription (68). Interestingly, silenced but transcriptionally competent HIV-1 proviruses were shown to reside in close proximity to PML NBs and this association inhibited HIV-1 gene expression (69).
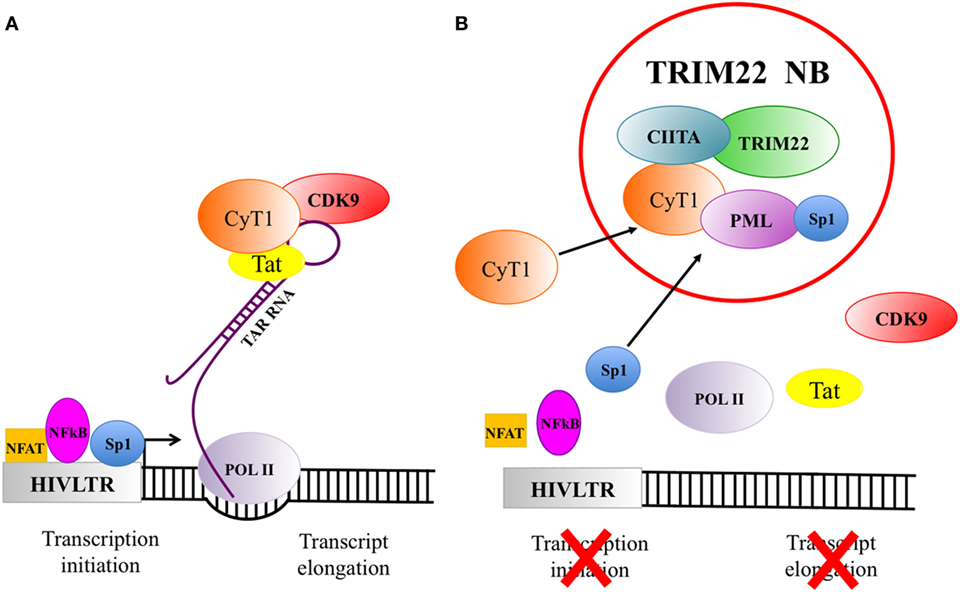
Figure 2. The recruitment of factors in tripartite motif 22 (TRIM22) nuclear bodies strongly inhibits viral gene expression. (A) Human immunodeficiency virus 1 (HIV-1) gene transcription and expression are essential steps in the viral life cycle. HIV-1 transcription is under the control of several factors among which NFAT, NFkB, and Sp1 (symbolized in the figure as a square, oval and circle, respectively) are particularly important. They bind to specific sequences of the 5′LTR promoter/enhancer region to activate viral genome transcription. The cellular transcription factor Sp1 is one of the most potent inducer of HIV-1 gene expression and it is crucial to initiate the basal transcription of viral RNA. The viral transactivator protein Tat plays a central role in sustaining a high level of HIV-1 replication. When Tat is present and binds to the bulge of the Trans-Activation Response (TAR) RNA element, the cellular transcription elongation factor P-TEFb, composed by the regulatory subunit CyclinT1 (CyT1) and the kinase subunit CDK9, will be recruited. Upon phosphorylation of RNA polymerase II (POL II), TAR can be elongated to the full length viral RNA. (B) TRIM22 nuclear bodies, containing class II transactivator (CIITA) and PML, recruit also CyT1 and Sp1, thus impairing both HIV gene transcription initiation and transcription elongation.
Taken together, these recent observations strongly point to the possibility that CIITA and PML cooperate in the inhibition of Tat-mediated HIV-1 LTR transactivation by competing with Tat for the binding to CyclinT1. Moreover, TRIM22 inhibits basal HIV-1 LTR transcription by affecting the binding of Sp1 to the viral promoter (42), and PML interacts with and sequesters Sp1 in PML nuclear bodies, suppressing the transcriptional function of Sp1 to its target sequence (70). Thus it is also possible that PML, by associating with Sp1, contributes to TRIM22-mediated inhibition of basal HIV-1 transcription as well (Figure 2). Within this frame, TRIM22 nuclear bodies may then be seen as the first example of an intracellular hub where several RFs may convene and act in concert to inhibit HIV-1 viral replication. This may be relevant for the mechanism of latency and persistence as the regulation of HIV-1 transcription might correlate not only with the localization of proviral DNA (69) but also with the recruitment-dependent synergy of key transcription factors with inhibitory function on viral transcription.
Concluding Remarks and Future Perspective
Although several studies have focused at clarifying the antiviral mechanisms of host RFs, much research is still required particularly on those aspects related to the possible cooperative action of RF during viral infection. In this review, we summarized the recent acquisition on the function of two newly defined RFs, CIITA and TRIM22, against retroviruses, particularly HIV-1, highlighting peculiar aspects that strongly suggest the existence of a concerted action against HIV-1.
First, the generation of specific TRIM22 nuclear bodies and the accumulation of TRIM22 and CIITA in these compartments.
Second, the recruitment of CIITA and CyclinT1, a key component in the HIV-1 Tat-mediated elongation of HIV-1 primary transcripts, in the same TRIM22 nuclear bodies, an event that may ensure a potent inhibition of HIV-1 transcription by acting at basal (through TRIM22) and Tat-promoted (through CIITA) LTR transcription.
Third, the co-localization in TRIM22 nuclear bodies of PML, previously shown to affect Tat-dependent LTR transcription and to bind Sp1, can synergize with the inhibitory action of both CIITA and TRIM22.
The above findings now open up the field to novel investigations and additional interesting new questions related, for example, to the intimate molecular mechanisms that drive distinct RFs to migrate to the same endocellular compartments and to retain them in the endocellular compartment. Additionally, are these mechanisms of co-localization of molecules with potential synergic action against the virus an active response of the infected cells to the insult or, paradoxically, are used by the virus to tentatively counteract or even neutralize its intracellular opponents? In the latter case may RFs be used by the virus to favor a state of latency by reducing its capacity to replicate? Is the co-localization of several RFs in the same compartments the mirror of a recently acquired evolutionary action to better fight virus infection, or a mechanism to serve other diverse functions in cell homeostasis? CIITA is one of the most important regulators of adaptive immunity through its activating function on the expression of MHC class II genes and thus on antigen presentation to CD4+ TH cells. Is its function as RF against retroviruses a recent diversification of its duties or an old function preceeding its role on adaptive immunity?
Future research on the mechanisms of subcellular localization and redistribution as well as functional cooperativity between distinct RFs will certainly enlarge our knowledge on the complexity of host cell–pathogen interaction and provide new ideas and strategies to better counteract viral infectivity and spreading.
Author Contributions
GF and RA participated in the conception and design of the review; revised the manuscript; read, critiqued, and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the members of the Laboratory of General Pathology and Immunology “Giovanna Tosi,” in particular Dr. Alessandra Tedeschi, Dr. Marco Baratella, and Dr. Farah Bou Nasser Eddine for their support in conceiving and writing this review. A particular thank to Drs. Elisa Vicenzi and Guido Poli and their research team, Institute San Raffaele, Milan, for their collaboration in the research work related to TRIM22.
Funding
This work was supported by University of Insubria intramural grant FAR 2014-2016 to RA and GF; by The Institutional Grant 2017, University of Insubria, to RA; by European Community FP7 project HepaVAC (Grant Number 602893) to RA (www.hepavac.eu). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
1. Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol (2015) 6:546–53. doi:10.1038/ni.3156
2. Porter SS, Stepp WH, Stamos JD, McBride AA. Host cell restriction factors that limit transcription and replication of human papillomavirus. Virus Res (2017) 231:10–20. doi:10.1016/j.virusres.2016.11.014
3. Landolfo S, De Andrea M, Dell’Oste V, Gugliesi F. Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape. World J Virol (2016) 5:87–96. doi:10.5501/wjv.v5.i3.87
4. Bieniasz PD. Intrinsic immunity: a front-line defence against viral attack. Nat Immunol (2004) 5(11):1109–15. doi:10.1038/ni1125
5. Wolf D, Goff SP. Host restriction factors blocking retroviral replication. Annu Rev Genet (2008) 42:143–63. doi:10.1146/annurev.genet.42.110807.091704
6. Nisole S, Stoye JP, Saib A. Trim family proteins: retroviral restriction and antiviral defense. Nat Rev Microbiol (2005) 3:799–808. doi:10.1038/nrmicro1248
7. Kajaste-Rudnitski A, Marelli SS, Pultrone C, Pertel T, Uchil PD, Mechti N, et al. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappa B-responsive long terminal repeat elements. J Virol (2011) 85:5183–96. doi:10.1128/JVI.02302-10
8. Neil SJ, Zang T, Beniaz PD. Theterin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature (2007) 452:425–30. doi:10.1038/nature06553
9. Strebel K, Luban J, Jeang KT. Human cellular restriction factors that target HIV-1 replication. BMC Med (2009) 7:48. doi:10.1186/1741-7015-7-48
10. Berger G, Durand S, Fargier G, Nguyen XN, Cordeil S, Bouaziz S, et al. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog (2011) 7:e1002221. doi:10.1371/journal.ppat.1002221
11. Bianco-Melo D, Venkatesh S, Bieniasz PD. Intrinsic cellular defence against human immunodeficency viruses. Immunity (2012) 37(3):399–411. doi:10.1016/j.immuni.2012.08.013
12. Harris RS, Hultquist JF, Evans DT. The restriction factor of human immunodeficiency virus. J Biol Chem (2012) 287(49):40875–83. doi:10.1074/jbc.R112.416925
13. Jia X, Zhao Q, Xiong Y. HIV suppression by host restriction factors and immune evasion. Curr Opin Struct Biol (2015) 31:106–14. doi:10.1016/j.sbi.2015.04.004
14. Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, et al. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol (2004) 11:435–42. doi:10.1038/nsmb758
15. Cuevas JM, Geller R, Garijo R, Lopez-Aldeguer J, Sanjuan R. Extremely high mutation rate of HIV-1 in vivo. PLoS Biol (2015) 13:e1002251. doi:10.1371/journal.pbio.1002251
16. Izumi T, Shirakawa K, Takaori-Kondo A. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini Rev Med Chem (2008) 8(3):231–8. doi:10.2174/138955708783744047
17. Laguette N, Benkirane N. How Samhd1 changes our view of viral restriction. Trends Immunol (2012) 33(1):26–33. doi:10.1016/j.it.2011.11.002
18. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature (2011) 474:658–61. doi:10.1038/nature10195
19. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature (2011) 474:654–7. doi:10.1038/nature10117
20. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature (2004) 427(6977):848–53. doi:10.1038/nature02343
21. Hotter D, Sauter D, Kirchhoff F. Emerging role of the host restriction factor tethering in viral immune sensing. J Mol Biol (2013) 425:4956–64. doi:10.1016/j.jmb.2013.09.029
22. Tissot C, Mechti N. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J Biol Chem (1995) 270:14891–8. doi:10.1074/jbc.270.25.14891
23. Singh R, Gaiha G, Werner L, McKim K, Mlisana K, Luban J, et al. Association of TRIM22 with the type 1 interferon response and viral control during primary HIV-1 infection. J Virol (2011) 85(1):208–16. doi:10.1128/JVI.01810-10
24. Singh R, Patel V, Mureithi MW, Naranbhai V, Ramsuran D, Tulsi S, et al. TRIM5α and TRIM22 are differentially regulated according to HIV-1 infection phase and compartment. J Virol (2014) 88(8):4291–303. doi:10.1128/JVI.03603-13
25. Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, et al. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J Virol (2013) 87:4523–33. doi:10.1128/JVI.02548-12
26. Gao B, Duan Z, Xu W, Xiong S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology (2009) 50:424–33. doi:10.1002/hep.23011
27. Yang C, Zhao X, Sun D, Yang L, Chong C, Pan Y, et al. Interferon alpha (IFNalpha)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5A. Cell Mol Immunol (2015) 13(1):94–102. doi:10.1038/cmi.2014.131
28. Eldin P, Papon L, Oteiza A, Brocchi E, Lawson TG, Mechti N. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J Gen Virol (2009) 90:536–45. doi:10.1099/vir.0.006288-0
29. Ozato K, Shin DM, Chang TH, Morse HC III. Trim family proteins and their emerging roles in innate immunity. Nat Rev Immunol (2008) 8(11):849–60. doi:10.1038/nri2413
30. Joazeiro CA, Weissman AM. RING finger proteins: mediators of ubiquitin ligase activity. Cell (2000) 102:549–52. doi:10.1016/S0092-8674(00)00077-5
31. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J (2001) 20:2140–51. doi:10.1093/emboj/20.9.2140
32. Hattlmann CJ, Kelly JN, Barr SD. TRIM22: a diverse and dynamic antiviral protein. Mol Biol Int (2012) 2012:153415. doi:10.1155/2012/153415
33. Herr AM, Dressel R, Walter L. Different subcellular localisations of TRIM22 suggest species-specific function. Immunogenetics (2009) 61:271–80. doi:10.1007/s00251-009-0357-z
34. Sivaramakrishnan G, Sun Y, Rajmohan R, Lin VCL. B30.2/SPRY domain in tripartite motif-containing 22 is essential for the formation of distinct nuclear bodies. FEBS Lett (2009) 583(12):2093–9. doi:10.1016/j.febslet.2009.05.036
35. Sivaramakrishnan G, Sun Y, Tan SK, Lin VCL. Dynamic localization of tripartite motif-containing 22 in nuclear and nucleolar bodies. Exp Cell Res (2009) 315(8):1521–32. doi:10.1016/j.yexcr.2009.01.028
36. Yu S, Gao B, Duan Z, Xu W, Xiong S. Identification of tripartite motif-containing 22 (TRIM22) as a novel NF-kB activator. Biochem Biophys Res Commun (2011) 410:247–51. doi:10.1016/j.bbrc.2011.05.124
37. Chen C, Zhao D, Fang S, Chen Q, Cheng B, Fang X, et al. TRIM22-mediated apoptosis is associated with Bak oligomerization in monocytes. Sci Rep (2017) 7:39961. doi:10.1038/srep39961
38. Forlani G, Tosi G, Turrini F, Poli G, Vicenzi E, Accolla RS. Tripartite motif-containing protein 22 interacts with class II transactivator and orchestrates its recruitment in nuclear bodies containing TRIM19/PML and cyclin T1. Front Immunol (2017) 8:564. doi:10.3389/fimmu.2017.00564
39. Petersson J, Lonnbro P, Herr AM, Morgelin M, Gullberg U, Drott K. The human IFN-inducible p53 target geneTRIM22 colocalized with the centrosome independently of cell cycle phase. Exp Cell Res (2010) 316(4):568–79. doi:10.1016/j.yexcr.2009.12.007
40. Barr SD, Smiley JR, Bushman FD. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog (2008) 4:e1000007. doi:10.1371/journal.ppat.1000007
41. Bouazzaoui A, Kreutz M, Eisert V, Dinauer N, Heinzelmann A, Hallenberger S, et al. Stimulated trans-acting factor of 50 kDa (Staf50) inhibits HIV-1 replication in human monocyte-derived macrophages. Virology (2006) 356:79–94. doi:10.1016/j.virol.2006.07.025
42. Turrini F, Marelli S, Kajaste-Rudnitski A, Lusic M, Van Lint C, Das AT, et al. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology (2015) 12(1):104. doi:10.1186/s12977-015-0230-0
43. Franzoso G, Biswas P, Poli G, Carlson LM, Brown KD, Tomita-Yamaguchi M, et al. A family of serine proteases expressed exclusively in myelo-monocytic cells specifically processes the nuclear factor-kappa B subunit p65 in vitro and may impair human immunodeficiency virus replication in these cells. J Exp Med (1994) 180:1445–56. doi:10.1084/jem.180.4.1445
44. Accolla RS, Jotterand-Bellomo M, Scarpellino L, Maffei A, Carra G, Guardiola J. Air-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. J Exp Med (1986) 164:369–74. doi:10.1084/jem.164.1.369
45. Guardiola J, Scarpellino L, Carra G, Accolla RS. Stable integration of mouse DNA into Ia-negative human B cells causes re-expression of the human Ia-positive phenotype. Proc Natl Acad Sci U S A (1986) 83:7415–8. doi:10.1073/pnas.83.19.7415
46. Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell (1993) 75:135–46. doi:10.1016/S0092-8674(05)80090-X
47. Reith W, LeibundGut-landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol (2005) 5:793–806. doi:10.1038/nri1708
48. Accolla RS, De Lerma Barbaro A, Mazza S, Casoli C, De Maria A, Tosi G. The MHC class II transactivator: prey and hunter in infectious diseases. Trends Immunol (2001) 22:560–3. doi:10.1016/S1471-4906(01)02003-8
49. Forlani G, Abdallah R, Accolla RS, Tosi G. The MHC-II transactivator CIITA, a restriction factor against oncogenic HTLV-1 and HTLV-2 retroviruses: similarities and differences in the inhibition of Tax-1 and Tax-2 viral transactivators. Front Microbiol (2013) 4:234. doi:10.3389/fmicb.2013.00234
50. Bou Nasser Eddine F, Forlani G, Lombardo L, Tedeschi A, Tosi G, Accolla RS. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4+ TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. Oncoimmunology (2016) 6(1):e1261777. doi:10.3389/fimmu.2017.00564
51. Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell (2002) 109:S21–33. doi:10.1016/S0092-8674(02)00696-7
52. Forlani G, Abdallah R, Accolla RS, Tosi G. The major histocompatibility complex class II transactivator CIITA inhibits the persistent activation of NF-kB by the human T cell lymphotropic virus type 1 tax-1 oncoprotein. J Virol (2016) 90(7):3708–21. doi:10.1128/JVI.03000-15
53. Forlani G, Turrini F, Ghezzi S, Tedeschi A, Poli G, Accolla RS, et al. The MHC-II transactivator CIITA inhibits Tat function and HIV-1 replication in human myeloid cells. J Trasl Med (2016) 14:94. doi:10.1186/s12967-016-0853-5
54. Kanazawa S, Okamoto T, Peterlin BM. Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity (2000) 12:61–70. doi:10.1016/S1074-7613(00)80159-4
55. Accolla RS, Mazza S, De Lerma Barbaro A, De Maria A, Tosi G. The HLA class II transcriptional activator blocks the function of HIV-1 Tat and inhibits viral replication. Eur J Immunol (2002) 32:278391. doi:10.1002/1521-4141(2002010)32:10<2783::AID-IMMU2783>3.0.CO;2-E
56. Casoli C, De Lerma Barbaro A, Pilotti E, Bertazzoni U, Tosi G, Accolla RS. The MHC class II transcriptional activator (CIITA) inhibits HTLV-2 viral replication by blocking the function of the viral transactivator Tax-2. Blood (2004) 103:995–1001. doi:10.1182/blood-2003-07-2503
57. Tosi G, Pilotti E, Mortara L, De Lerma Barbaro A, Casoli C, Accolla RS. Inhibition of human T cell leukemia virus type 2 replication by the suppressive action of class II transactivator and nuclear factor Y. Proc Natl Acad Sci U S A (2006) 103:12861–6. doi:10.1073/pnas.0601589103
58. Orlandi C, Forlani G, Tosi G, Accolla RS. Molecular and cellular correlates of the CIITA-mediated inhibition of HTLV-2 tax-2 transactivator function resulting in loss of viral replication. J Transl Med (2011) 9:106. doi:10.1186/1479-5876-9-106
59. Poiesz B, Ruscetti F, Gazdar A, Bunn P, Minna J, Gallo R. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A (1980) 77:7415–9. doi:10.1073/pnas.77.12.7415
60. Tosi G, Forlani G, Andresen V, Turci M, Bertazzoni U, Franchini G, et al. Major histocompatibility complex class II transactivator CIITA is a viral restriction factor that targets human T-cell lymphotropic virus type 1 Tax-1 function and inhibits viral replication. J Virol (2011) 85(20):10719–29. doi:10.1128/JVI.00813-11
61. Matsuoka M, Jeang KT. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene (2011) 30:1379–89. doi:10.1038/onc.2010.537
62. Dutrieux J, Maarifi G, Portilho DM, Arhel NJ, Chelbi-Alix MK, Nisole S. PML/TRIM19-dependent inhibition of retroviral reverse transcription by Daxx. PLoS Pathog (2015) 11(11):e1005280. doi:10.1371/journal.ppat.1005280
63. Masroori N, Merindol N, Berthoux L. The interferon-induced antiviral protein PML (TRIM19) promotes the restriction and transcriptional silencing of lentiviruses in a context-specific, isoform-specific fashion. Retrovirology (2016) 13:19. doi:10.1186/s12977-016-253-1
64. Turelli P, Doucas V, Craig E, Mangeat B, Klages N, Evans R, et al. Cytoplasmic recruitment of INI1 and PML on incoming HIV preintegration complexes: interference with early steps of viral replication. Mol Cell (2001) 7:1245–54. doi:10.1016/S1097-2765(01)00255-6
65. Bonilla WV, Pinschewer DD, Klenerman P, Rousson V, Gaboli M, Pandolfi PP, et al. Effects of promyelocytic leukemia protein on virus-host balance. J Virol (2002) 76:3810–8. doi:10.1128/JVI.76.8.3810-3818.2002
66. Chee AV, Lopez P, Pandolfi PP, Roizman B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol (2003) 77:7101–5. doi:10.1128/JVI.77.12.7101-7105.2003
67. Ulbricht T, Alzrigat M, Horch A, Reuter N, von Mikecz A, Steimle V, et al. PML promotes MHC class II gene expression by stabilizing the class II trasactivator. J Cell Biol (2012) 199:49–63. doi:10.1083/jcb.201112015
68. Marcello A, Ferrari A, Pellegrini V, Pegoraro G, Lusic M, Beltram F, et al. Recruitment of human cyclin T1 to nuclear bodies through direct interaction with the PML protein. EMBO J (2003) 22:2156–66. doi:10.1093/emboj/cdg205
69. Lusic M, Marini B, Ali H, Lucic B, Luzzati R, Giacca M. Proximity to PML nuclear bodies regulates HIV-1 latency in CD4+ T cells. Cell Host Microbe (2013) 13:665–77. doi:10.1016/j.chom.2013.05.006
Keywords: restriction factors, tripartite motif 22, class II transactivator, promyleocitic leukemia protein, CyclinT1, nuclear bodies, human immunodeficiency virus 1
Citation: Forlani G and Accolla RS (2017) Tripartite Motif 22 and Class II Transactivator Restriction Factors: Unveiling Their Concerted Action against Retroviruses. Front. Immunol. 8:1362. doi: 10.3389/fimmu.2017.01362
Received: 19 July 2017; Accepted: 04 October 2017;
Published: 18 October 2017
Edited by:
Annapurna Vyakarnam, King’s College London, United KingdomReviewed by:
Marina Lusic, Universität Heidelberg, GermanyJianzhong Zhu, Yangzhou University, China
Copyright: © 2017 Forlani and Accolla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto S. Accolla, cm9iZXJ0by5hY2NvbGxhQHVuaW5zdWJyaWEuaXQ=