Abstract
Phagocytosis is a fundamental process of cells to capture and ingest foreign particles. Small unicellular organisms such as free-living amoeba use this process to acquire food. In pluricellular organisms, phagocytosis is a universal phenomenon that all cells are able to perform (including epithelial, endothelial, fibroblasts, etc.), but some specialized cells (such as neutrophils and macrophages) perform this very efficiently and were therefore named professional phagocytes by Rabinovitch. Cells use phagocytosis to capture and clear all particles larger than 0.5 µm, including pathogenic microorganisms and cellular debris. Phagocytosis involves a series of steps from recognition of the target particle, ingestion of it in a phagosome (phagocytic vacuole), maturation of this phagosome into a phagolysosome, to the final destruction of the ingested particle in the robust antimicrobial environment of the phagolysosome. For the most part, phagocytosis is an efficient process that eliminates invading pathogens and helps maintaining homeostasis. However, several pathogens have also evolved different strategies to prevent phagocytosis from proceeding in a normal way. These pathogens have a clear advantage to perpetuate the infection and continue their replication. Here, we present an overview of the phagocytic process with emphasis on the antimicrobial elements professional phagocytes use. We also summarize the current knowledge on the microbial strategies different pathogens use to prevent phagocytosis either at the level of ingestion, phagosome formation, and maturation, and even complete escape from phagosomes.
Introduction
Phagocytosis, in pluricellular organisms, is a complex process for the ingestion and elimination of pathogens. It is also important for elimination of apoptotic cells, and for maintaining tissue homeostasis (1, 2). All cells may, to some extent, perform phagocytosis; however, in mammals, phagocytosis is the hallmark of specialized cells including monocytes, macrophages, dendritic cells, osteoclasts, eosinophils, and polymorphonuclear neutrophils—these cells are collectively referred to as professional phagocytes (3). Professional phagocytes eliminate microorganisms and present them to cells of the adaptive immune system. Phagocytosis can be divided into several main steps: (i) microbial recognition, (ii) phagosome formation, and (iii) phagolysosome maturation.
Phagocytosis initiates with the recognition and ingestion of microbial pathogens larger than 0.5 µm into a plasma membrane-derived vesicle, known as phagosome. This recognition is achieved through several receptors that recognize precise molecular patterns associated with microorganisms. These receptors then trigger signaling cascades that induce phagocytosis. Receptors on phagocytes can be divided into non-opsonic or opsonic receptors. Non-opsonic receptors can directly identify pathogen-associated molecular patterns (PAMPs) on the surface of the microorganisms. Opsonic receptors bind to host-produced molecules called opsonins. These molecules bind to microorganisms and mark them for ingestion. Opsonins include antibodies, complement, fibronectin, mannose-binding lectin, and milk fat globulin (lactadherin) (4).
After receptor engagement, the plasma membrane covers the microorganism to be ingested and closes at the distal end, forming a vacuole where the microorganism is internalized (Figure 1). This vacuole, the early phagosome, then fuses with endocytic vesicles and at the same time secretory vesicles are separated from it, transforming the early phagosome into a late phagosome. This dynamic process consists of sequential fusion and fission events between the new phagosome and endosomes, and it is known as “the kiss-and-run” model (5). Later, the intermediary phagosome turns into a microbicidal vacuole, the phagolysosome, by fusing with lysosomes and changing its membrane and interior characteristics through a process named phagolysosome maturation (6). The results of this process are remodeling of the membrane, progressive acidification of the phagosome, and creation of an oxidative and degradative milieu (7, 8) (Figure 2).
Figure 1

Initiation of phagocytosis. After receptor engagement (A) (2), the plasma membrane covers the microorganism to be ingested (B) (9, 10) and closes at the distal end (C) (11, 12), forming a vacuole where the microorganism is internalized (D).
Figure 2

Phagosome maturation. The new phagosome quickly develops the characteristics of early endosomes, through a series of fusion and fission events with sorting and recycling endosomes (5, 6). The early phagosome is marked by the presence of the small GTPase Rab5 (13, 14), early endosome antigen 1 (EEA1) (15), and the class III PI-3K human vacuolar protein-sorting 34 (hvPS34) (16). The early phagosome also becomes a little acidic (pH 6.1–6.5) by the action of V-ATPase accumulating on its membrane (17, 18). The late phagosome is marked by the presence of Rab7 (19–21) and lysosomal-associated membrane proteins (LAMPs) (22, 23). Proteins that will be recycled are separated in sorting (recycling) vesicles, while proteins intended for degradation are eliminated in intraluminal vesicles (ILVs), directed into the lumen of the phagosome (24). The lumen of the late phagosome gets more acidic (pH 5.5–6.0), due to the action of more V-ATPase molecules on the membrane. Phagolysosomes are formed when late phagosomes fuse with lysosomes. Phagolysosomes are acidic (pH 5–5.5) and contain many degradative enzymes, including various cathepsins, proteases, lysozymes, and lipases. Other microbicidal component of the phagosome is the NADPH oxidase that generates reactive oxygen species (25).
The phagocytic process is usually very efficient and concludes with the destruction of the microorganism ingested. Nevertheless, several pathogens possess various anti-phagocytic strategies, which allow them to survive and escape phagocytes. These strategies can be directed to any step of the phagocytic process. However, most microorganisms interfere with phagosome maturation since the phagolysosome is the most destructive organelle. It is the purpose of this review to highlight the multiple anti-microbial effectors of professional phagocytes and to describe how various microbial pathogens hinder phagocytosis to continue the course of their infection.
Initiation of Phagocytosis
Microbial Recognition
The first step in phagocytosis is the detection of a microorganism by phagocytes. Microbial pathogens are recognized directly by receptors that bind PAMPs or indirectly by receptors that bind opsonins. Receptors that directly bind PAMPs are known as pattern-recognition receptors and among these receptors, we find lectin-like recognition molecules, such as CD169 and CD33; C-type lectins, such as Dectin-2, Monocyte-INducible C-type LEctin (Mincle), or DNGR-1; scavenger receptors (26), such as scavenger receptor A, which detects lipopolysaccharide (LPS) on some Gram-negative bacteria (27), and on Neisseria meningitidis (27); and Dectin-1, which is a receptor for fungal beta-glucan (28, 29). Mannose receptors bind to mannan (30), and CD14 binds to LPS-binding protein (31). Toll-like receptors (TLRs), although recognize microorganisms, do not function as phagocytic receptors (32), however, they can cooperate with other non-opsonic receptors to stimulate phagocytosis (33).
Opsonins are soluble molecules that bind to microorganisms, marking them for phagocytosis. Antibody [immunoglobulin (Ig)] molecules and complement components are important opsonins that induce efficient phagocytosis (2). The most studied opsonic phagocytic receptors are the Fc receptors (FcRs), and the complement receptors (CRs), respectively (34). FcγRs bind to the constant (Fc) portion of IgG (35–38), while FcαRs bind IgA antibodies (39). CRs, such as CR3, bind to iC3b deposited on the microorganism after complement activation (40). Crosslinking of FcγR on the surface of cells activates efficient phagocytosis and other effector functions. These effector functions, such as activation of the oxidative burst, cell degranulation, antibody-dependent cell-mediated cytotoxicity, and activation of genes for production of cytokines and chemokines, are aimed toward the destruction of pathogens and the induction of an inflammatory response that is beneficial during infections (37, 41, 42). CRs, such as the integrin αMβ2 (also known as CD11b/CD18, CR3, or Mac-1), bind the complement component iC3b deposited on pathogens to promote phagocytosis (34, 43).
Phagosome Formation
After phagocyte receptors engage a microorganism, signaling events are triggered to initiate phagocytosis. Important changes in membrane remodeling and the actin cytoskeleton take place leading to the formation of pseudopods that cover the microorganism. Lipids associate and dissociate from the membrane of phagosomes in an orderly fashion (44), and the small GTPases Rho, Rac, and cell division cycle 42 (Cdc42), which are important regulators of the actin cytoskeleton, get activated and recruited to the forming phagosome (9, 10). At the point of contact, a depression of the membrane (the phagocytic cup) is formed. Then, F-actin polymerization allows formation of pseudopods that surround the target microorganism and within few minutes, the membrane protrusions fuse at the distal end (11, 12, 45) to seal the new phagosome (Figure 1).
Phagosome Maturation
The new phagosome rapidly changes its membrane composition and its contents, to become a microbicidal vacuole, the phagolysosome. This process is known as phagosome maturation. The mechanism for transferring endocytosed material from endosomes to lysosomes is complex and not completely described. Four hypotheses have been proposed to explain the process of phagolysosome formation [reviewed in Ref. (46, 47)]. These include a maturation model where the endosome turns into a lysosome (48), a vesicular transport model where vesicles carry cargo from the endosome to the lysosome (49), a kiss-and-run model where endosomes and lysosomes engage in repeated transient fusions (50) and a direct fusion model where endosomes and lysosomes completely fuse giving rise to a hybrid compartment from which lysosomes reform (51, 52). Experimental evidence suggests that both the kiss-and-run and the complete fusion models contribute to the mechanism for delivery of endocytosed particles to the lysosome (53). The coordinated activities of endosomal sorting complex required for transport, homotypic fusion and vacuole protein sorting, and soluble N-ethylmaleimide-sensitive factor-attachment protein receptor protein complexes on the different vesicle membranes are required for efficient delivery of endocytosed macromolecules to lysosomes [reviewed in Ref. (54, 55)]. Phagosome maturation can be divided into three main stages, namely the early phagosome, the late phagosome, and the phagolysosome, as described later.
Early Phagosome
The new phagosome quickly develops the characteristics of early endosomes, through a series of fusion and fission events with endosomes (5, 6). The early phagosome is marked by the presence of the small GTPase Rab5 (13, 14). This membrane GTPase regulates the fusion events between the phagosome and early endosomes by recruiting early endosome antigen 1 (EEA1) (15). Rab5 also recruits the class III PI-3K human vacuolar protein-sorting 34, which in turn, generates phosphatidylinositol 3-phosphate (16). This lipid then promotes recruitment of other proteins involved in phagosome maturation, including Rab7, a marker of late endosomes (19, 20). The early phagosome also becomes a little acidic (pH 6.1–6.5) by the action of V-ATPase accumulating on its membrane and also by transient fusions with more acidic vesicles (56). This V-ATPase translocates protons (H+) into the lumen of the phagosome using cytosolic ATP as an energy source (17, 18) (Figure 2).
Late Phagosome
As maturation continues, Rab5 is lost, and Rab7 appears on the membrane. Rab7 mediates the fusion of the phagosome with late endosomes (21). At the same time, proteins that will be recycled are separated in sorting (recycling) vesicles, while proteins intended for degradation are eliminated in intraluminal vesicles, directed into the lumen of the phagosome (24). Furthermore, the lumen of the late phagosome gets more acidic (pH 5.5–6.0), due to the action of more V-ATPase molecules on the membrane (17) (Figure 2). Rab7 also recruits other proteins to the membrane. One such protein is Rab-interacting lysosomal protein (RILP), which brings the phagosome in contact with microtubules (57, 58), and lysosomes (57, 58). In addition, lysosomal-associated membrane proteins (LAMPs) and luminal proteases (cathepsins and hydrolases) are incorporated from fusion with late endosomes (7, 59) (Figure 2). LAMPs are a family of heavily glycosylated proteins that accumulate on the lysosomal membrane. They all contain a conserved intracytoplasmic tyrosine-based lysosome-targeting motif YXXφ (where φ represents a bulky hydrophobic residue) that directs the trafficking of the molecule through an endosome/lysosome pathway (60). LAMPs are fundamental in regulating membrane fusion events (61) and are required for fusion of lysosomes with phagosomes (22, 23).
Phagolysosome
The late phagosomes gradually fuse with lysosomes, to become phagolysosomes, the definitive microbicidal organelles (Figure 2) (47, 53, 62). Phagolysosomes possess many sophisticated mechanisms directed to eliminate and degrade microorganisms. They are acidic (pH 5–5.5) thanks to the large number of V-ATPase molecules on their membrane (18) and contain many degradative enzymes, including various cathepsins, proteases, lysozymes, and lipases (17). Other microbicidal components of the phagosome are scavenger molecules, such as lactoferrin that sequesters the iron required by some bacteria (63), and the NADPH oxidase that generates superoxide (25), and other reactive oxygen species (ROS) (64, 65) (Figure 2). Although the low pH is clearly microbicidal, it is important to note that phagosome acidification is highly regulated. The lysosomal pH may cycle between acidic and neutral conditions, allowing for the optimal activity of the different hydrolases (66). Within the hybrid degradative vesicle (phagolysosome) (46), most of these enzymes are active at pH 5–5.5; while in primary lysosomes that function as storage vesicles, the lower pH of 4.5 induces enzyme aggregation and inactivation (66).
Antimicrobial Effectors
The phagolysosome is the definitive antimicrobial organelle. The arsenal at its disposal is large and complex. The major destructive strategies will be presented next.
pH
The most distinctive characteristic of phagolysosomes is their low pH. During the maturation process, the membrane of the phagosome accumulates more and more active molecules of V-ATPase. The molecular complex of this ATPase translocates protons (H+) into the lumen of the phagosome using cytosolic ATP as an energy source (17, 18) (Figure 3). The acidic content of the phagosome creates an adverse environment for most microorganisms. The low pH disrupts the normal metabolism of many bacteria and fungi and prevents the use of several essential microbial nutrients (67). In addition, the low pH is required for the activation of many hydrolytic enzymes, which will degrade the ingested pathogen. The V-ATPase also favors the generation superoxide (25) by neutralizing the negative charges generated by the NADPH oxidase and by combining with H+ to generate other ROS (Figure 3) (as discussed later).
Figure 3
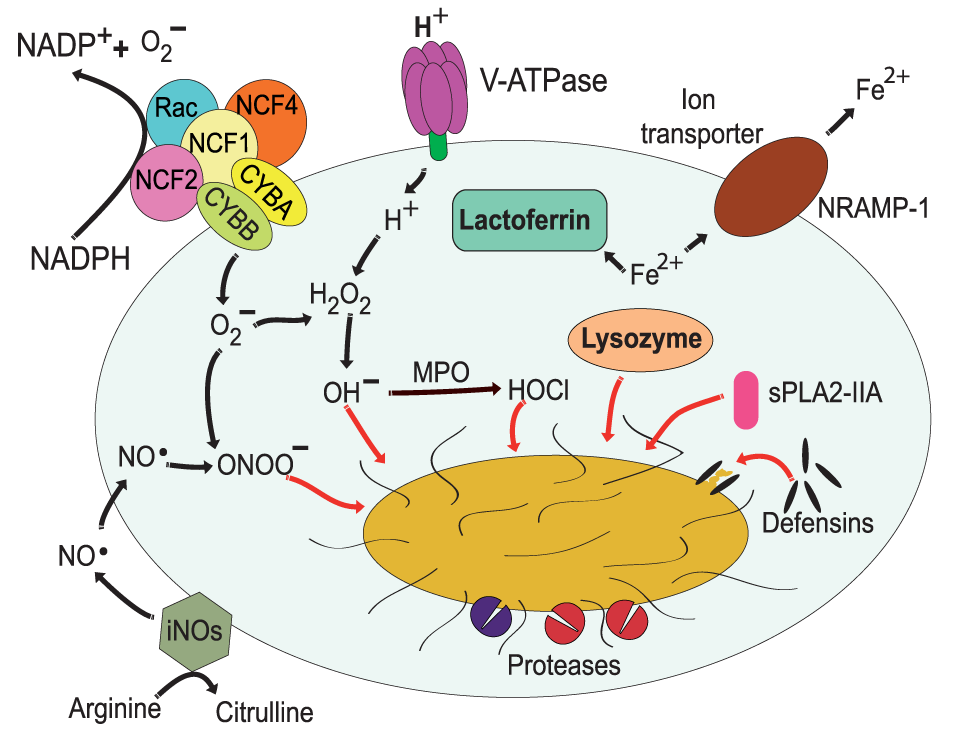
Antimicrobial effectors inside the phagolysosome. The most distinctive characteristic of phagolysosomes is their low pH. The V-ATPase translocates protons (H+) into the lumen of the phagosome (17, 18). The NADPH oxidase is an enzymatic complex formed by two transmembrane proteins, such as CYBB and CYBA, and three cytosolic components: NCF-4, NCF-1, and NCF-2 (68, 69). Rac is also required for efficient activation of the enzyme complex (70, 71). Myeloperoxidase (MPO) can transform H2O2 into hypochlorous acid (65). Nitric oxide radicals (NO⋅) are produced by the inducible nitric oxide synthase 2 (iNOS) (72), and NO⋅ reacts with to form peroxynitrite (ONOO−) (73, 74). Lactoferrin captures Fe2+ that is essential for bacterial growth (75), and the transporter natural resistance-associated macrophage protein 1 (NRAMP-1) takes Fe2+ out of the phagosome (76). Defensins are antimicrobial peptides that form multimeric ion-permeable channels on bacteria (77, 78). Cathepsins are lysosomal proteases (79, 80). Lysozyme (81, 82) degrades peptidoglycan, a primary building block of the cell wall of bacteria, and the type IIA secreted phospholipase A2 (sPLA2-IIA) (83) degrades anionic phospholipids such as phosphatidylglycerol, the main phospholipid component of bacterial membranes.
Reactive Oxygen and Nitrogen Species
In addition to an acid environment, the phagolysosome can concentrate ROS to destroy microorganisms. Production of ROS is achieved by the NADPH oxidase (NOX2) on the membrane of phagosomes (68, 69, 84). The relevance of this antimicrobial mechanism is evident in patients with chronic granulomatous disease, who have mutations that result in malfunction of the NADPH oxidase. These patients suffer from severe recurrent infections that can cause death in many cases (85, 86). The NADPH oxidase is an enzymatic complex formed by two transmembrane proteins, CYBB (cytochrome b-245 heavy chain/gp91-phox) and CYBA (cytochrome b-245 light chain/p22-phox), that upon activation bring together three cytosolic components: NCF-4 (neutrophil cytosol factor 4/p40-phox), NCF-1 (neutrophil cytosol factor 1/p47-phox), and NCF-2 (neutrophil cytosol factor 2/p67-phox) (69, 87) (Figure 3). In addition, Rac1 and Rac2 are also required for efficient activation of the enzyme complex (70, 71). The oxidase transfers electrons from cytosolic NADPH to molecular oxygen to generate inside the phagosome (25). In the acid conditions of the phagosome, quickly dismutate into H2O2, which can then produce hydroxyl radicals (OH−) by a Fenton reaction (88) catalyzed by iron released from proteins in the phagosome (89). Also, myeloperoxidase can transform H2O2 into hypochlorous acid and chloramines (65) (Figure 3). In addition, can also react with nitric oxide (NO) to form peroxynitrite (ONOO−), both of which are highly reactive agents. The various forms of ROS are together efficient antimicrobial agents because they can damage proteins, lipids, and DNA of microorganisms in the phagosome (89).
In addition to ROS, phagocytes, such as macrophages, can also generate nitrogen-based radicals or reactive nitrogen species (RNS) that contribute to microbial destruction. Nitric oxide radicals (NO⋅) are produced by the inducible nitric oxide synthase 2 (iNOS or NOS2) (72). This enzyme is not present in the resting phagocyte and is only produced in response to proinflammatory agonists (90). NOS2 catalyzes the conversion of l-arginine and oxygen into l-citrulline and NO⋅ (Figure 3). Contrary to , NO⋅ is produced on the cytoplasmic side of phagosomes, but it can diffuse across membranes and accumulate inside the phagosome (91). As mentioned earlier, once inside the phagosome, NO⋅ can react with to generate the highly toxic peroxynitrite (ONOO−) that can alter proteins and DNA of ingested microorganisms (73, 74). Also, NO⋅ can directly impair bacterial enzymes and affect microbial growth (92).
Nutrient Capture
Not only the low pH and the oxidative conditions are used to harm the ingested pathogen but also a series of enzymes and peptides are delivered into the phagosome to limit its growth. Microbial growth can be limited by reducing the amount of essential nutrients inside the phagosome. Nutrients are eliminated from the phagosome by special capture molecules delivered into the phagosome or by transporters present on the phagosome membrane. Perhaps, lactoferrin is the best characterized capture molecule that prevents growth of some bacteria (75). Lactoferrin is a non-heme Fe2+-binding glycoprotein present in the specific granules of neutrophils (93), and it can be delivered into the phagolysosome. In there, lactofferin captures Fe2+ that is essential for bacterial growth (75, 94) (Figure 3). Other metals, such as Mn2+, are also important for microbial growth. Thus, during maturation, phagosomes become gradually depleted of Fe2+ and Mn2+ by the action the transporter natural resistance-associated macrophage protein 1 (NRAMP-1; also known as SlC11A1) (76) (Figure 3). NRAMP-1 is a membrane protein present on phagolysosomes that transports divalent cations, such as Fe2+, Zn2+, and Mn2+ out of the phagolysosome. This transporter requires H+ ions to function, thus NRAMP-1 is more efficient in the acid environment of the phagolysosome (76). Removal of these metal ions prevents microbial growth. However, some microorganisms present a mechanism to counteract the phagocyte function and retain these nutrients (see next section). Microorganisms secrete siderophores, specialized molecules that capture Fe2+ and target it for pathogen use (95). The phagocyte in turn presents yet another strategy to control microbial growth. The phagocyte protein lipocalin binds selectively catechol type siderophores expressed by some bacteria, such as Escherichia coli and Mycobacterium tuberculosis. Consequently, the Fe2+-loaded siderophore is still sequestered from the bacteria (96, 97).
Microorganism Destruction
As described earlier, the phagolysosome interior is an inhospitable environment for most microorganisms. Enzymes and peptides delivered to the phagolysosome have potent antimicrobial effects by destroying the different components of the microbial cell.
Antimicrobial peptides are small (<10 kDa), cationic, and amphipathic polypeptides, often broadly classified based on structure (82, 98). In phagocytes, the main types include defensins (disulfide-stabilized peptides) (77, 78) and cathelicidins (α-helical or extended peptides) (99). Defensins are subdivided into α and β groups and are stored in primary granules of neutrophils. These peptides disrupt the integrity of pathogens by attaching to negatively charged molecules on their membrane. Defensins form multimeric ion-permeable channels that cause membrane permeabilization on both Gram-positive and Gram-negative bacteria (100) (Figure 3). Cathelicidins are stored in secondary granules of neutrophils as inactive precursors. They are then converted into active antimicrobial peptides in the lumen of the phagosome by elastase (99), a primary granule enzyme (93). Active cathelicidins permeabilize the cell wall and inner membrane of Gram-positive bacteria (100). In particular, the cathelicidin LL-37 (hCAP) has drawn particular attention because of its multiple functions. Not only LL-37 works as a broad-spectrum antibiotic but also it has potent chemotactic and immunomodulatory properties (101). Neutrophil-produced LL-37 can be internalized by macrophages (102) and can induce phagocytosis of IgG-opsonized Gram-negative and Gram-positive bacteria by these phagocytes (103). LL-37 also promotes leukotriene B4 and thromboxane A2 generation by human monocyte-derived macrophages (104), thus regulating the inflammation response during infections. Recently, it was also found that macrophages could also produce LL-37. Mice deficient in the Cramp (cathelicidin-related antimicrobial peptide) gene, the murine functional homolog of human LL-37, had increased susceptibility to M. tuberculosis; and macrophages from these mice were unable to control M. tuberculosis growth in an in vitro infection model (105).
Among the many degradative enzymes, the cathepsins are perhaps the most extensively characterized group of lysosomal proteases. These are cysteine proteases that play a direct role in microbial killing by inducing the disruption of bacterial membranes (Figure 3). For example, cathepsins L and K were found to be involved in phagocytosis and non-oxidative killing of Staphylococcus aureus (80), while cathepsin D controlled Listeria monocytogenes intracellular growth (79), probably by degrading the pore-forming toxin listeriolysin O of L. monocytogenes and thus preventing bacterial escape from the phagosome (see next section).
The phagolysosome also contains many lysosomal hydrolases, which help destroy ingested pathogens (106). An important enzyme of this group is lysozyme, an antibacterial protein that can be expressed and secreted by several cell types (81, 82). Lysozyme degrades peptidoglycan, a primary building block of the cell wall of bacteria (Figure 3). By breaking the bonds between N-acetylmuramic acid and N-acetyl-d-glucosamine, lysozyme disrupts the peptidoglycan integrity (107), and then other enzymes can complete the lysis of bacterial cells. One such enzyme is the type IIA secreted phospholipase A2 (sPLA2-IIA) (Figure 3), which exhibits potent antimicrobial activity against Gram-positive and Gram-negative bacteria (83). This remarkable property is due to the unique preference of sPLA2-IIA for anionic phospholipids such as phosphatidylglycerol, the main phospholipid component of bacterial membranes. The importance of this mechanism is highlighted by the fact that transgenic mice overexpressing human sPLA2-IIA are resistant to infection by S. aureus, E. coli, and Bacillus anthracis, the etiological agent of anthrax (83). Thus, antimicrobial peptides and degradative enzymes work together in the lumen of the phagolysosome to completely degrade most phagocytized microorganisms (Figure 3).
Microbial Control of Phagocytosis
The discussion presented earlier clearly shows that phagocytosis is an efficient process (1, 2, 4, 108, 109) that culminates with the generation of the phagolysosome and its very harsh environment for most microorganisms (6). Therefore, it is not surprising that many successful pathogens have evolved multiple strategies to prevent and/or inhibit phagocytosis (110, 111). These strategies include prevention of phagocytosis, interference of phagosome maturation, resistance to phagolysosome contents, and even physical escape from the phagosome. Our knowledge comes mainly from studies of important extracellular and intracellular pathogens, such as S. aureus (112–114), M. tuberculosis (115–117), and L. monocytogenes (118, 119). However, many other microbial pathogens also have tactics for interfering with phagocytosis. The mechanisms for controlling phagocytosis employed by these model pathogens, as we understand them today, will be described next. In addition, information available on microbial control of phagocytosis by some other pathogens will also be presented.
Prevention of Phagocytosis
The best way to escape from the destructive power of phagocytosis would be just to prevent ingestion by phagocytes from happening. Some pathogens try just to do that by producing substances that extracellularly intoxicate phagocytes. S. aureus can secrete various membrane damaging toxins that will cause cell lysis and death. These toxins include the leukocidins (120) (named this way because they can kill leukocytes) and α-hemolysin (121) (Figure 4). Although there are different leukocidins, they all are dimer proteins (e.g., LukAB, LukED, HlgAB, HlgCB, and LukSF-PV) that induce membrane permeability by the formation of octameric β-barrel pores on the cell membrane (120, 122, 123). Interestingly, leukocidins do not attack any membrane indiscriminately. They must bind first to specific membrane receptors, and therefore, only cells with these receptors are targeted for intoxication (124, 125). For example, LukE binds to the chemokine receptor CCR5 on macrophages, marking these cells for lysis by the active leukocidin LukED (124, 126). Similarly, LukA binds only to the CD11b subunit of the CR Mac-1, which is expressed on both macrophages and neutrophils (125) (Figure 4). The α-hemolysin is another toxin from S. aureus that also forms pores on the membrane of macrophages. It uses phagocyte protein ADAM10 (a disintegrin and metalloproteinase domain-containing protein 10) as a receptor (127, 128), and then it assembles into a β-barrel pore of seven identical monomers across the cell membrane (129, 130) (Figure 4).
Figure 4
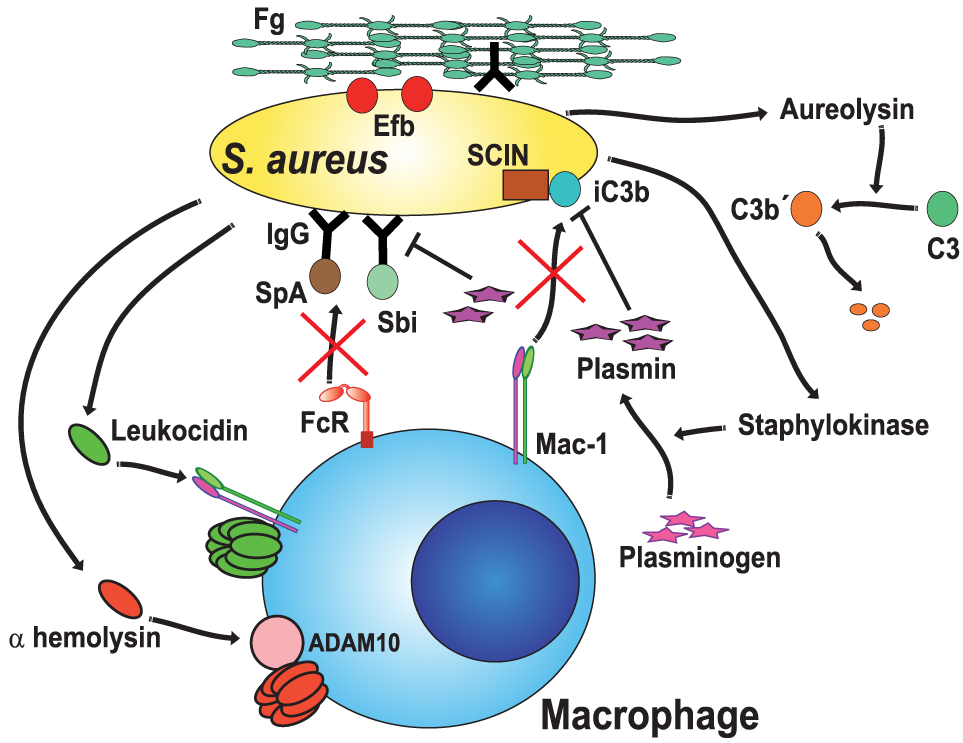
Staphylococcus aureus blocks opsonic phagocytosis. S. aureus secrete toxins, leukocidins (120, 125) and α-hemolysin (121), which induce membrane permeability by forming pores on the cell membrane. To be fully active, leukocidin A binds to the complement receptor Mac-1 (125), while α-hemolysin binds to protein ADAM10 (a disintegrin and metalloproteinase domain-containing protein 10) (127, 128). Staphylokinase converts host plasminogen to the active serine protease plasmin, which in turn degrades IgG or iC3b on the bacteria (127, 129). Protein A (SpA) (131) and staphylococcal binder of IgG (Sbi) protein specifically bind to the Fc region of IgG (132–134), blocking Fc receptor (FcR) engagement and activation. Aureolysin functions as a C3 convertase, leaving non-functional C3b′ fragments (135). Also, the staphylococcal complement inhibitor (SCIN) prevents complement activation on the bacteria (136). Finally, the extracellular fibrinogen binding protein (Efb) binds the serum protein fibrinogen (Fg), creating a proteinaceous shield that covers surface-bound opsonins (137, 138).
Phagocytosis is most efficient when microorganisms have been opsonized by antibodies or complement. Microorganisms have also evolved mechanisms to prevent opsonization. The first strategy displayed by S. aureus to block opsonization is simply to degrade opsonins. Some staphylococcal proteases seem capable of directly attacking opsonins. However, a more efficient instrument for this function is the protein staphylokinase, a bacterial plasminogen activator that converts host plasminogen to the active serine protease plasmin. Activated plasmin can then degrade IgG or C3b on the bacterial surface (139) (Figure 4). Another mechanism is to capture the opsonin, so that it does not bind to the bacteria. S. aureus Protein A is a well-known protein expressed on the bacterial cell wall. Protein A specifically binds to the Fc region of IgG, preventing the antibody from engaging FcγRs. In consequence, phagocytosis is effectively blocked (131) (Figure 4). In addition, Protein A can obstruct complement activation by the classical pathway, since the Fc portion of IgG is no longer accessible to the complement component C1q. This will result in less deposition of C3b on the bacteria (140). In addition to Protein A, most S. aureus strains express the Staphylococcal binder of IgG (Sbi) protein, which also binds to the Fc portion of IgG (132–134) (Figure 4). Inhibition of complement activation is an important strategy also used by Staphylococcus. The secreted metalloprotease aureolysin functions as an effective C3 convertase. Aureolysin cuts C3 in the α-chain at a different cleavage site from the C3 convertase, leaving C3a′ and C3b′ fragments (135). Unfortunately, the aureolysin-generated C3b′ fragment is rapidly degraded and not deposited on the bacteria (Figure 4). In addition, the staphylococcal complement inhibitor, a 10-kDa protein, can inhibit complement activation and efficiently prevent phagocytosis and killing of staphylococci (136) (Figure 4). As if all this were not enough, S. aureus can also hide the C3b deposited on its surface. The bacteria secrete the extracellular fibrinogen binding protein (Efb), which binds the serum protein fibrinogen (137). In this way, the bacterium creates a proteinaceous shield that covers the surface bound opsonin and prevents phagocytosis (137, 138) (Figure 4). This impressive array of anti-phagocytic effectors has been described for individual molecules. However, there is not enough information on when and how bacteria decide to use each one of them. The external elements that regulate the expression of each factor are not known. Novel techniques, such as expression profiling, should bring new light into these topics, as discussed later.
Another way to prevent ingestion by phagocytes from happening is to inactivate the cell machinery that creates the phagosome around the microorganism. Some pathogens have developed strategies to prevent actin polymerization and thus avoiding phagocytosis (141). The role of the actin cytoskeleton is fundamental for constructing a phagocytic cup and then extending membrane protrusions around the target particle. The small GTPase Rho family (10) controls formation of F-actin fibers required for phagocytosis. The GTPases Rho, Rac1, and Cdc42 act as molecular switches alternating between an active (GTP-bound) state and an inactive (GDP-bound) state (142, 143). For activation, they need to release GDP and replace it with GTP. This action is catalyzed by guanine nucleotide exchange factors (GEFs). Later, GTP is hydrolyzed to GDP returning the GTPase to its inactive state. This last step is enhanced through interactions with GTPase-activating proteins (GAPs). During phagocytosis, these GTPases are activated and recruited to the forming phagosome, where they activate nucleation-promoting factors such as Wiskott–Aldrich Syndrome protein (WASp) (144). WASp, in turn activates the actin-related protein 2/3 (Arp2/3) complex for actin polymerization (145, 146). As the new actin fibers grow, the plasma membrane is forced out, extending the membrane as pseudopodia around the particle to be ingested. Due to their central role in controlling actin dynamics, these small GTPases are the chosen target of some bacterial toxins. These toxins can alter the activity of the GTPases through covalent modifications or regulation of the nucleotide state. For example, the bacterium Clostridium difficile, which causes pseudomembranous colitis and is responsible for many cases of nosocomial antibiotic-associated diarrhea, produces two glycosylating exotoxins. Toxin A and toxin B modify Rho by glycosylation and inactivate its function. Rho inactivation causes disorganization of actin reducing phagocyte cell migration and phagocytosis (147). Similarly, the bacterium Photorhabdus asymbiotica, an emerging pathogen in humans, produces a toxin (PaTox) that tyrosine glycosylates Rho causing its inactivation. PaTox actions result in actin disassembly and inhibition of phagocytosis (148).
Another group of bacterial toxins regulate the nucleotide state and thus the function of the GTPases by functioning as GAPs or GEFs. For example, the enteropathogenic bacteria Yersinia spp. have type III secretion systems that inject Rho GAP toxins into cells. One such toxin (virulence factor) is YopO, which prevents Rac activation and in consequence prevents phagocytosis (149). Similarly, the Gram-negative bacteria Pseudomonas aeruginosa, an opportunistic pathogen that causes life-threatening infections in cystic fibrosis patients, burn victims, and immunosuppressed individuals, produces the type III virulence factor ExoS that is injected into cells. ExoS is a Rho GAP for Rho, Rac, and Cdc42 that causes the reorganization of the actin cytoskeleton by inhibition of Rac and Cdc42, and actin stress fiber formation by inhibition of Rho (150). An additional example recently described of pathogens disrupting Rho GTPase function comes from the opportunistic bacteria Burkholderia cenocepacia that has a propensity to infect cystic fibrosis patients. B. cenocepacia was shown to disrupt Rac and Cdc42 activation through perturbation of GEF function. Inactive Rac and Cdc42 led to inhibition of phagocyte function (151).
Besides bacteria, several fungal pathogens also display mechanisms for evading phagocytosis. Candida albicans, a commensal ascomycete, is part of the normal microbiota associated with mucosal tissues. It causes opportunistic infections, known as thrush, on superficial mucosas, and systemic infections, named candidiasis. C. albicans is normally phagocytized by macrophages, but it can decrease being recognized by phagocytes with a thick cell wall. The cell wall antigen, β-glucan is hidden among manno-proteins, thus reducing phagocytosis (152). In addition, C. albicans can limit phagocyte chemotaxis during transition from the yeast to the hyphal forms (153). Another fungus, Aspergillus fumigatus, also can mask antigenic proteins and carbohydrates to avoid recognition by phagocytes. RodA hydrophobin is a hydrophobic protein expressed on the surface of A. fumigatus conidia. This hydrophobin efficiently prevents recognition and phagocytosis (154). Similarly, the yeast basidiomycete Cryptococcus neoformans can also avoid recognition by macrophages. The basidiospores of C. neoformans produce a polysaccharide coat (capsule) that forms a thick barrier from phagocytes (155). This capsule can also be shed to prevent macrophage detection and phagocytosis (155). In addition, C. neoformans secretes antiphagocytic protein 1, a protein that binds to CR Mac-1 and inhibits phagocytosis (156).
Interference with Phagosome Maturation
Once a microorganism is ingested, it will be exposed to the very harsh environment of the phagolysosome. Thus, many pathogens present strategies directed to avoid the formation of this final antimicrobial organelle. Phagosome maturation can be blocked at different points and there are examples of pathogens blocking acidification, reducing activation of the NADPH oxidase, and preventing phagosome to lysosome fusion. Perhaps the most studied example of inhibition of phagosome maturation occurs in M. tuberculosis. The first report was published more than 40 years ago (157), and since then several mycobacterial factors interfering with the process have been found, such as mannose-capped lipoarabinomannan (ManLAM), phosphatidyl-myo-inositol-mannosides (PIMs) (115, 158, 159), and trehalose-6,6′-dimycolate (TDM) (160).
As mentioned earlier, one of the earlier features of phagosome maturation is the rapid and gradual acidification of the phagosome. The number of V-ATPase molecules increases on the phagosome membrane as the maturation process takes place. The low pH directly affects many pathogens (67), and it is also required for the activation of many hydrolytic enzymes. In the case of M. tuberculosis, acidification is inhibited by preventing the accumulation of V-ATPase on the phagosome membrane (161) (Figure 5). Although the complete mechanism is unknown, the M. tuberculosis secreted protein tyrosine phosphatase (PtpA) plays an important role. PtpA binds to subunit H of the macrophage vacuolar V-ATPase (162), and then it dephosphorylates human vacuolar protein sorting 33B (VPS33B) (163), leading to subsequent exclusion of the V-ATPase from the phagosome (Figure 5).
Figure 5
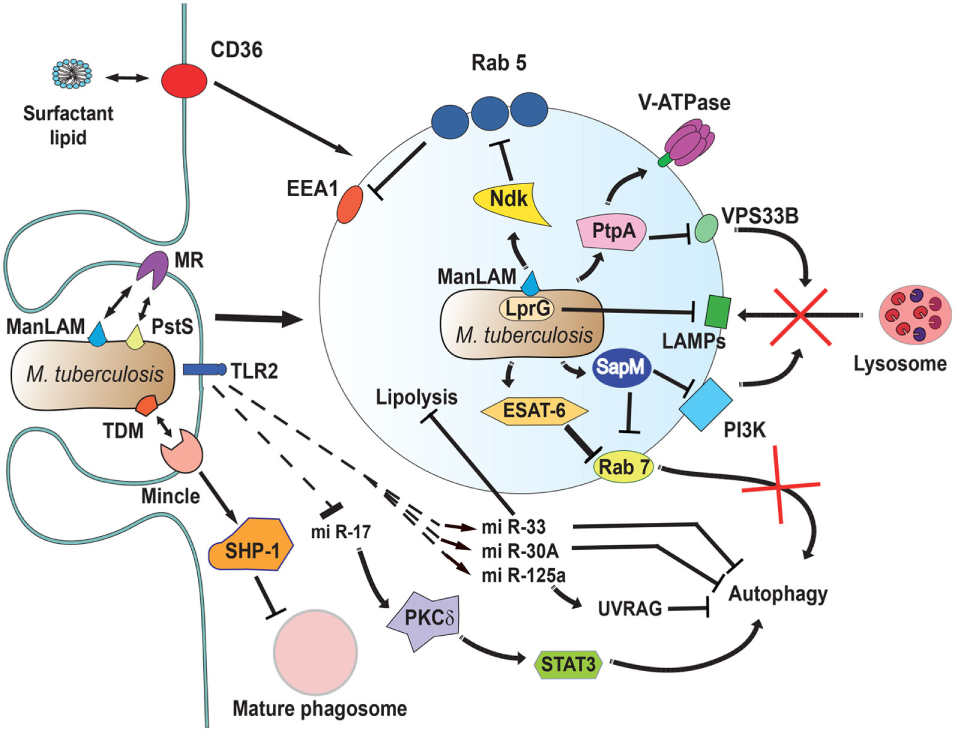
Mycobacterium tuberculosis interferes with phagosome maturation. M. tuberculosis inhibits acidification by preventing the accumulation of V-ATPase on the phagosome membrane (161), in part through the action of protein tyrosine phosphatase (PtpA) (162). PtpA also dephosphorylates human vacuolar protein sorting 33B (VPS33B) leading to the inhibition of phagosome-lysosome fusion (163). The nucleoside diphosphate kinase (Ndk) is a GAP for Rab5, and by inactivating this GTPase (164), it prevents recruitment of early endosome antigen 1 (EEA1) to the membrane (165). The lipoprotein LprG increases the surface-expression of mannose-capped lipoarabinomannan (ManLAM) (166) and can directly bind to lysosomal-associated membrane proteins (LAMPs) to modulate the traffic machinery of the cell (167, 168). Also, ManLAM (169) and the adhesin PstS-1 (170) bind the mannose receptor, which is involved in the lysosome fusion machinery by an unknown mechanism (171). The mycobacterial glycolipid TDM binds the receptor Monocyte-INducible C-type LEctin (Mincle) (172), activating the SH2-domain-containing inositol polyphosphate 5′ phosphatase (SHP-1) to interfere with phagosome maturation (160). The virulence factor early secretory antigenic target-6 (ESAT-6) inhibits recruitment of Rab7 to the phagosome membrane, preventing autophagy-mediated degradation (173). Also, the secretory acid phosphatase (SapM) direct binds to Rab7 (174) and prevents autophagosome-lysosome fusion (174). In addition, SapM can block the effects of phosphotidylinositol 3-kinase (PI3K) present on phagosomes (158). Upon infection, mycobacteria induce upregulation of several microRNAs (miRNAs) (175–177) and downregulation of others (178) to block autophagy. miR-125a targets UV radiation resistance-associated gene (UVRAG) (176) to block autophagy, while miR-17 activates a protein kinase Cδ (PKCδ)/signal transducer and activator of transcription 3 (STAT3) pathway to regulate autophagy (178). The miR-33 also inhibits fatty acid oxidation to support bacterial replication by a mechanism not yet described (177). How M. tuberculosis alters cell signaling to control miRNAs is not known, but the initial signal might come from TLR2 (176, 179). Finally, the scavenger receptor CD36 participates in surfactant lipid uptake by alveolar macrophages, and M. tuberculosis exploits this function for growth (180).
The Gram-positive bacteria Streptococcus pyogenes blocks the V-ATPase activity through expression of surface proteins regulated by the virulence factor Mga (a transcription factor) (181). Similarly, Rhodococcus equi, Gram-positive bacteria that cause severe pneumonia in horses, and the dimorphic fungus Histoplasma capsulatum are also able to maintain a non-acidic phagosome by excluding the V-ATPase (182, 183) (Figure 6). Other pathogens can avoid acidification of phagosomes, including Yersenia pestis, the Gram-negative bacteria causing bubonic plague (184), and C. albicans (185), by mechanisms not completely described.
Figure 6

Inhibition of phagosome maturation. (a) Several pathogens, such as Mycobacterium tuberculosis (161), Histoplasma capsulatum (182), and Rhodococcus equi (183) inhibit acidification by preventing the accumulation of V-ATPase on the phagosome membrane. M. tuberculosis also blocks early endosome antigen 1 (EEA1) on the membrane (165), while Neisseria gonorrhoeae express a porin that induces large amounts of Rab5 (186) and also proteases that digest lysosomal-associated membrane proteins (LAMPs) (187). Another bacteria, Streptococcus pyogenes, express the virulence factor M1, which regulates vesicle trafficking (188). Each of these actions effectively will block lysosome fusion to the phagosome. (b) Other pathogens, such as Legionella pneumophila (189, 190) and Brucella melitensis (191), induce the rapid association of the phagosome with the endoplasmic reticulum (ER). (c) The bacteria Coxiella burnetti (192, 193), and the parasite Leishmania reside inside a phagolysosome-like vesicle known as parasitophorous vacuole (PV) that concentrates Rab5 on the membrane. Leishmania promastigotes also insert lipophosphoglycan (LPG) into the phagosome membrane (194). These actions, in consequence, prevent lysosome fusion (195).
Phagosome maturation is also inhibited by interfering with the proper accumulation of molecules responsible for vesicle fusion, thus keeping the new phagosome with characteristics of an early phagosome. M. tuberculosis blocks phagosome maturation at a stage between the expression of Rab5 and Rab7, by preventing the delivery of the molecule EEA1 to the membrane (165) (Figure 5). This effect is mediated in part by the action of nucleoside diphosphate kinase (Ndk), which exhibits GAP activity toward Rab5 and Rab7. Ndk inactivates both Rab5 and Rab7 thus preventing recruitment of their respective effectors EEA1 and RILP and in consequence inhibits phagosome maturation and fusion with lysosomes (164) (Figure 5). This blockage also involves ManLAM (171), and it seems to require binding of ManLAM to the mannose receptor (169). Recently, the adhesin PstS-1, a 38-kDa mannosylated glycolipoprotein, was also found to bind the mannose receptor (170) (Figure 5). The connection between the mannose receptor and the lysosome fusion machinery is obscure. Because, capping of the ManLAM with mannose receptor was necessary during phagocytosis to maintain the blockade (169), it seems that the initial engagement of the mannose receptor directs, in an unclear manner, M. tuberculosis to a selective initial phagosomal niche, where other molecules can be excluded. Also, Mincle was recently identified as a receptor for the mycobacterial glycolipid TDM (172). Recruitment of Mincle by TDM coupled to IgG-opsonized beads during FcγR-mediated phagocytosis interfered with phagosome maturation (160). This inhibition involved the SH2-domain-containing inositol polyphosphate 5′ phosphatase (SHP-1) and the FcγRIIb (160), strongly suggesting an inhibitory downstream signaling of Mincle during phagosome formation (Figure 5). Without EEA1, delivery of the V-ATPase or enzymes such as cathepsin D does not take place (196). Therefore, the M. tuberculosis-containing phagosome is kept with a pathogen-friendly environment (Figure 5). Other microorganisms can also arrest phagosome maturation at early stages. For example, the Gram-negative bacteria Neisseria gonorrhoeae express a porin that induces phagosomes to keep larger amounts of Rab5 and low levels of Rab7 (186) (Figure 6). In addition, this bacterium also secretes proteases that digest LAMPs (187) (Figure 6). As mentioned earlier, LAMPs are fundamental for fusion of lysosomes to phagosomes (22), thus its degradation prevents formation of a mature phagolysosome (187). Similarly, the Gram-negative bacteria Legionella pneumophila intercepts vesicular traffic from endoplasmic reticulum (ER) (189) to create an organelle that allows the bacteria to have access to cysteine for survival (190) (Figure 6). This bacterium is the cause of Legionnaires’ disease, a severe form of pneumonia. When the bacteria are phagocytized, the phagosome is rapidly associated with mitochondria and the rough ER, thus getting decorated with ribosomes (197). This effect seems to be mediated by DotA, a bacterial product that is part of the type IV secretion system (T4SS) transporter. T4SS exports various bacterial effector proteins, including RalF, a GEF for the phagocyte ADP-ribosylation factor (ARF1) (198). Active ARF1 promotes vesicle traffic between the ER and the Golgi (199). Therefore, the ER-like phagosome does not get acidic and it does not fuse with lysosomes. Another example of phagosomes fusing with the ER is found in the Gram-negative bacteria Brucella melitensis (Figure 6). This bacterium is the etiological agent of brucellosis, a zoonotic infection that can cause muscle pain, fever, weight loss, and fatigue in people, but can also induce abortion and infertility in animals. In the macrophage cell line J774, B. melitensis alters vesicle trafficking (200) to create a modified phagosome known as a Brucella-containing vacuole (BCV) that fuses with the ER (191) (Figure 6). The mechanism for creating a BCV is not completely known, but it involves several virulence factors such as VirB, an element of the bacterial type III secretion system (191), and cyclic β-1,2-glucan, a cell wall component (201).
Since the phagolysosome is the most harmful organelle for microorganisms, many pathogens have mechanisms to prevent fusion of lysosomes with the phagosome. The best-known example is again M. tuberculosis that avoids lysosome fusion by maintaining an early phagosome (115) (Figure 5). The mechanism for this effect is multifactorial and complex. We only have a partial understanding of it with the identification of some key virulent factors involved. One such virulent factor is the lipoprotein LprG, which binds to lipoglycans, such as lipoarabinomannan (LAM), increasing the surface expression of LAM (166). A M. tuberculosis null mutant for LprG (Mtb ΔlprG) had lower levels of surface-exposed LAM and impaired phagosome–lysosome fusion (167). How LprG prevents phagosome–lysosome fusion is only partially known. It is possible that its effect is indirect via Ndk, which inactivates both Rab5 and Rab7 (164), or is direct by binding to LAMP-3 and modulating the traffic machinery in the host cell (168) (Figure 5). One more virulent factor is PtpA which, as mentioned earlier, dephosphorylates VPS33B, a regulator of membrane fusion events and leads to inhibition of phagosome–lysosome fusion (163) (Figure 5). Another way M. tuberculosis prevents phagosome–lysosome fusion involves inhibition of Rab7 recruitment to prevent autophagy-mediated degradation. The maturation of mycobacteria-containing autophagosomes into autolysosomes requires recruitment of Rab7, but this is blocked by the virulence factor early secretory antigenic target-6 (ESAT-6) (173) (Figure 5). Again, the molecular events for this blockage are not known. However, for another virulence factor of M. tuberculosis, the secretory acid phosphatase (SapM) the inhibition of autophagosome-lysosome fusion (202) is achieved via direct binding to Rab7 (174). Molecularly, Rab7 is blocked by SapM through its cytoplasmic domain preventing its involvement in autophagosome–lysosome fusion (174) (Figure 5). In addition, SapM is known to dephosphorylate phosphotidylinositol 3-phosphate present on phagosomes (158). This phospholipid is also required for membrane fusion events, thus SapM also prevents lysosome fusion in this manner (Figure 5).
Mycobacterium tuberculosis has also evolved other ways to prevent autophagy from happening. One recently described way is the activation or inhibition of cell host microRNAs (miRNAs). Upon infection, macrophages increased several miRNAs and inhibited pathways involved in autophagy. These miRNAs include miR-30A (175), miR-33 (177), and miR-125a (176) (Figure 5). At the same time, another miRNA, miR-17, is downregulated with the same result, blockage of autophagy (178). The signaling pathways affected by these miRNAs are only beginning to be described. For example, miR-125a targets UV radiation resistance-associated gene (UVRAG) (176) to block autophagy, while miR-17 activates a PKCδ/STAT3 pathway to regulate autophagy (178). Thus, inhibition of miR-17 leads also to reduce autophagy (Figure 5). How M. tuberculosis usurps cell host signaling pathways to alter expression of these miRNAs is not known. It seems, however, that the initial signal for this comes from TLRs (176) (Figure 5).
Similarly, S. pyogenes can also prevent lysosome fusion by expressing the virulence factor M1, which regulates vesicle trafficking (188) (Figure 6). M1 can also inhibit activation of the nuclear factor κB and in consequence reduce the macrophage inflammatory response (188). The Gram-negative bacteria Coxiella burnetti, the causative agent of Q fever, resides inside a large phagolysosome-like vesicle known as parasitophorous vacuole (192). This modified phagosome concentrates Rab5 on the membrane and avoids lysosome fusion (193) (Figure 6). The fungi A. fumigatus (203) and the parasitic protozoa Leishmania (204) seem also able to avoid being killed by macrophages by preventing fusion between phagosomes and lysosomes. In the case of A. fumigatus, the molecule dihydroxynaphthalene–melanin on the surface of the pathogen has been reported as responsible for altering vesicle fusion events (205). For Leishmania, the promastigote is efficiently internalized by receptor-mediated phagocytosis (204). Complement and mannose receptors participate in macrophage ingestion (195). Once internalized, promastigotes insert lipophosphoglycan (LPG) into the phagosome membrane. LPG inhibits depolymetization of F-actin (194), and in consequence prevents lysosome fusion (195) (Figure 6). This allows enough time for the promastigote to transform into the other life-cycle form, the amastigote, which can then replicate inside the phagosome.
Resistance to Phagolysosome Contents
In addition to preventing phagolysosome formation, pathogens also possess various mechanisms to resist the microbial components found in the phagolysosome lumen. A prominent example is S. aureus that can resist the lytic effect of lysozyme on the cell wall peptidoglycan. These bacteria express the enzyme O-acetyltransferase A (OatA), which causes O-acetylation of the peptidoglycan. This modification makes the peptidoglycan resistant to the muramidase activity of lysozyme (206, 207) (Figure 7). S. aureus also can block the action of antimicrobial peptides. First, the enzyme staphylokinase directly binds α-defensins, blocking almost completely their bactericidal effect (208) (Figure 7). Second, bacteria alter the composition of its membrane. Phosphatidylglycerol is modified with l-lysine, causing a reduction in the negative charge of the membrane (209). In addition, the cell wall is also modified by incorporation of teichoic acids and lipoteichoic acids (210), making it more positively charged. These modifications reduce interaction of α-defensins with the bacterial surface. Third, the metalloprotease aureolysin can degrade LL-37, an antimicrobial peptide with potent activity against staphylococci (211) (Figure 7).
Figure 7

Resistance of Staphylococcus aureus to phagolysosome contents. The bacteria S. aureus modifies the composition of its cell wall to resist the action of lysozyme (206, 207) and alters the composition of its membrane, with l-lysine and lipoteichoic acids, to reduce the negative charge of the membrane (209, 210); thus resisting antimicrobial peptides, such as the cathelicidin LL-37. Also, it secretes staphylokinase and aureolysin to block α-defensins and LL-37, respectively (208, 211). In addition, S. aureus has the golden pigment staphyloxanthin (Sx), which works as an antioxidant (212), two super oxide dismutases (Sod) (213), and a catalase (214, 215) that together protect against reactive oxygen species. In addition, flavohemoglobin functions as an NO⋅ scavenger (216, 217). The bacterial urease catalyzes the hydrolysis of urea to form ammonia, resulting in pH neutralization (218). Finally, S. aureus produces siderophores (SA) (219, 220) that trap enough Fe2+ to allow bacterial survival.
Also, several pathogens express urease, an enzyme that catalyzes the hydrolysis of urea to form ammonia, resulting in the pH neutralization of the phagosome (Figure 7). Important examples of microorganisms using this strategy to survive in the phagosome are S. aureus (218), Helicobacter pylori, bacteria known for causing gastric and duodenal ulcers (221), C. neoformans (222), and Coccidioides posadasii (223).
The oxidative environment of the phagolysosome is also very damaging to most microorganisms. Yet, some pathogens have evolved ways to fight back the effects of ROS and RNS. For example, S. aureus has the golden pigment staphyloxanthin, which works as an antioxidant and prevents damage from peroxide (212) (Figure 7). Also, the protein SOK (surface factor promoting resistance to oxidative killing), that is expressed on the bacteria surface, was recently described as a virulence factor that blocks the effects of ROS (224). In addition, S. aureus express the enzymes super oxide dismutases, sodA and sodM, which convert into H2O2 (213), and the enzyme catalase (KatA), which breaks down H2O2 into oxygen and water (214, 215) (Figure 7). A phagocytized bacterium has also to prevent the effects of iNOS-derived RNS. S. aureus can detect NO⋅ by the two component system SsrAB (225), which regulates the expression of the gene hmp coding for a flavohemoglobin that functions as an NO⋅ scavenger (216, 217) (Figure 7).
Similarly, M. tuberculosis can resist in various ways the microbicidal components within the phagolysosome. A novel glycosylated and surface-localized lipoprotein, Lprl can inhibit the lytic activity of lysozyme (226) (Figure 8). Also, at least two proteins have been found to prevent the formation of ROS by inhibiting the NADPH oxidase. The type I NADH dehydrogenase (NDH-1) blocks ROS production to inhibit tumor necrosis factor alpha (TNF-α)-mediated host cell apoptosis (227) (Figure 8), while the enhanced intracellular survival (eis) gene product (Eis) abrogates production of both ROS and proinflammatory cytokines leading to arrest in apoptosis. These effects seem to depend on the N-acetyltransferase domain of the Eis protein (228) (Figure 8). In both cases, apoptosis is inhibited, but the mechanisms are different. In the case of NDH-1, apoptosis is dependent on caspase-3 and caspase-8 (227), while for Eis, apoptosis seems to be caspase independent (228). M. tuberculosis can also block RNS by interfering with EBP50, a scaffolding protein that controls the recruitment of iNOS at the membrane of phagosomes in macrophages. Interestingly, overexpression of EBP50 by a recombinant lentivirus had no effect on the iNOS recruitment to M. tuberculosis-containing phagosomes, but significantly increased the generation of NO⋅ and the level of apoptosis in macrophages (229). The EBP50-induced apoptosis was NO⋅-dependent and mediated by Bax and caspase-3 (229) (Figure 8). The mechanism for iNOS inhibition is not completely elucidated, but it seems to involve both having less iNOS on the membrane and blocking its enzymatic activity. The way M. tuberculosis prevents EBP50 functions remains a mystery.
Figure 8
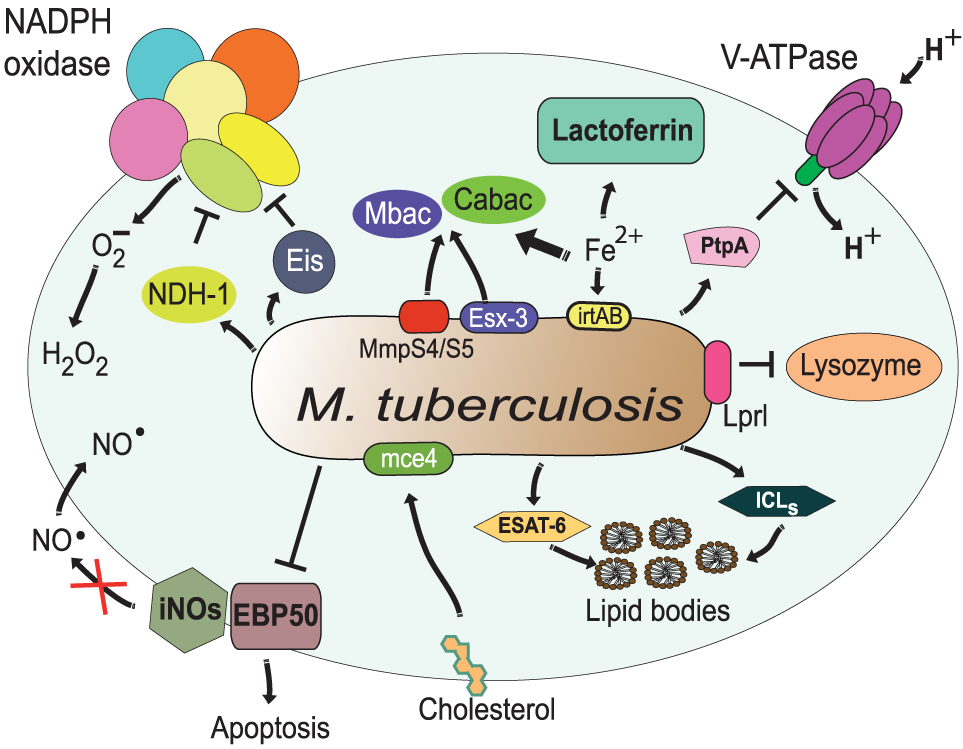
Resistance of Mycobacterium tuberculosis to phagolysosome contents. M. tuberculosis inhibits acidification by preventing the accumulation of V-ATPase on the phagosome membrane (161), in part through the action of protein tyrosine phosphatase (PtpA) (162). The bacterial lipoprotein, Lprl, can inhibit the lytic activity of lysozyme (226). The secretion system Esx-3 (230, 231) and the MmpS4/S5 transporters (232) are required for biosynthesis and secretion of the siderophores mycobactins (Mbac) and carboxymycobactins (Cabac), which seize Fe2+ from host proteins, such as lactoferrin (233). Then, the transporter system irtAB takes Fe2+ from Fe2+-carboxymycobactin into the bacterium (234, 235). The type I NADH dehydrogenase (NDH-1) (227) and the Eis protein (228) inhibit the NADPH oxidase, preventing formation of ROS. Also, M. tuberculosis prevents the generation of NO⋅ and apoptosis by interfering with EBP50, a scaffolding protein that controls the recruitment of iNOS at the membrane of phagosomes (229). In addition, M. tuberculosis alters the phagosome to divert host lipids for its own benefit through mce4, a cholesterol import system (236), and through accumulation of lipid bodies via the early secretory antigenic target-6 (ESAT-6) (237). The enzymes isocitrate lyases (ICLs) allow bacteria survival on even (acetate) and odd (propionate) chain fatty acids in lipid bodies (238).
Other pathogens are also known to display similar mechanisms against ROS and RNS. Streptococcus agalactiae (Group B Streptococcus) is an important cause of pneumonia and meningitis in neonatal humans (239). S. agalactiae expresses a superoxide dismutase (SodA), an orange carotenoid pigment, and glutathione. The latter two compounds functions as ROS scavengers (240, 241). H. pylori can also express a superoxide dismutase (SodB) (242), a catalase (KatA) (243), and the arginase RocF, which transforms the iNOS substrate arginine into urea (244, 245). Similarly, the yeast C. albicans expresses a copper and zinc containing superoxide dismutase (Sod1) (246), and a catalase (Cta1p) (247, 248), while H. capsulatum also secretes two catalases, CatB and CatP (249). The fungus C. neoformans produces a superoxide dismutase (250) and covers itself in a thick polysaccharide and melanin capsule that absorbs ROS (251). Also, the dimorphic fungus Blastomyces dermatitidis seems to be able to inhibit the enzyme iNOS to prevent the production of RNS (252). In all these pathogens, the expression of these enzymes and virulent factors effectively reduces the levels of ROS and RNS within the phagosome. Yet, very little is known about the mechanisms that induce expression of these virulent factors in each pathogen and the molecular details by which they inhibit NADPH oxidase and iNOS enzymes.
Resistance to Nutrient Capture
The phagolysosome is a place where microbial nutrients are eliminated to arrest pathogen growth. As mentioned earlier, divalent cations, such as Fe2+, Zn2+, and Mn2+, are actively transported out of the phagolysosome (76). In response to this, several microorganisms have evolved mechanisms to retain these important nutrients. One strategy to acquire Fe2+ relies on the production of siderophores, which are low-molecular weight Fe2+-binding molecules of extremely high affinity, that remove Fe2+ from host proteins, such as hemoglobin, and transferrin (233). S. aureus produces two citrate-based siderophores, staphyloferrin A (SA) and staphyloferrin B (SB) (219, 220) (Figure 7). Together, SA and SB can trap enough Fe2+ to allow bacterial survival. These siderophores are very efficient because they avoid detection by the phagocyte siderophore-binding protein lipocalin (96, 97). In addition, S. aureus is also able to acquire Mn2+ through the action of Mn2+ transporters encoded by the bacterial gene loci mntABC and mntH (253). In M. tuberculosis, two groups of siderophores, mycobactins and carboxymycobactins, exist to overcome Fe2+ deficiency. The type VII secretion system Esx-3 contributes to siderophore production and release from these bacteria (230, 231) (Figure 8). Recently, another siderophore export system was identified in M. tuberculosis. The MmpS4 and MmpS5 transporters are required for biosynthesis and secretion of siderophores (Figure 8). Because a M. tuberculosis mutant lacking the mmpS4 and mmpS5 genes did not grow under low Fe2+ conditions and experienced Fe2+ starvation even under high-Fe2+ conditions, it seems that these transporters are the primary source of siderophores in mycobacteria (232). The importance of siderophore synthesis for Fe2+ acquisition is clear, but Fe2+ must find a way back into the bacteria. In M. tuberculosis an ABC transporter system, irtAB (product of the genes irtA and irtB), has been described for efficient utilization of Fe2+ from Fe2+ carboxymycobactin (Figure 8). Inactivation of the irtAB system decreases the ability of M. tuberculosis to survive Fe2+-deficient conditions (234, 235). Similarly, other microorganisms such as A. fumigatus (254) and H. capsulatum (255) can produce siderophores for Fe2+ capture.
Intracellular bacteria have also evolved various means to take nutrients from the host cell. Lipids are important building blocks for bacterial membrane formation and an energy source (256). Upon infection, M. tuberculosis alters the phagosome to divert host lipids for its own benefit. A virulent factor was identified within the gene cluster, mce4, because it was specifically required for bacterial survival during prolonged infection. It was found that mce4 encodes a cholesterol import system that enables these bacteria to derive both carbon and energy from this lipid in host membranes (236) (Figure 8). Also, mycobacteria-infected macrophages acquire a “foamy” phenotype characterized by the accumulation of lipid bodies, which serve as source of nutrients. This foamy phenotype is caused by bacterial manipulation of host cellular metabolism to divert the glycolytic pathway toward ketone body synthesis (237). This deregulation results in feedback activation of the anti-lipolytic G protein-coupled receptor GPR109A, causing changes in lipid homeostasis and accumulation of lipid bodies in the cell. ESAT-6, a secreted M. tuberculosis virulence factor, mediates the enforcement of this feedback loop via an unknown mechanism (237) (Figure 8). Another strategy used by M. tuberculosis to exploit host lipids involves the bacterial enzymes isocitrate lyases (ICLs). These ICLs are catalytically bifunctional isocitrate and methylisocitrate lyases that allow bacteria survival on even (acetate) and odd (propionate) chain fatty acids (238) (Figure 8). Moreover, the miR-33 induced by M. tuberculosis also inhibited fatty acid oxidation to support bacterial growth by a mechanism not yet described (177) (Figure 5). In addition, M. tuberculosis has yet another strategy to acquire lipids even from outside the cell in the lung environment. Alveolar macrophages are not only responsible for phagocytosis of these bacteria but also for catabolizing lung surfactant, a lipid–protein complex that lines the alveolar spaces. Recently, it was found that the scavenger receptor CD36 is redistributed to the macrophage cell membrane following exposure to surfactant lipids and participated in surfactant lipid uptake by these cells (180) (Figure 5). These macrophages also supported better bacterial growth in a CD36-dependent manner (180). Thus, it seems that CD36 mediates surfactant lipid uptake by human macrophages and that M. tuberculosis exploits this function for growth.
Physical Escape from the Phagosome
In addition to resisting all the microbial effectors within a phagolysosome, several pathogens such as C. neoformans, L. monocytogenes, or M. tuberculosis can also completely escape from it. By getting out of the phagosome, these microorganisms can in the cytoplasm travel to other cell sites and finally leave the host cell.
As mentioned earlier, the fungus C. neoformans is well equipped to replicate inside the phagosome. In addition, it can subsequently escape the cell by a non-lytic tactic known as vomocytosis (257, 258). Vomocytosis allows for the pathogen escape leaving the phagocytic cell alive (259). Although the molecular details of vomocytosis are not completely described, the process involves an exocytic fusion of the phagosome with the plasma membrane, thus releasing the fungus (259) (Figure 9). Vomocytosis also involves microtubules, but apparently not actin polymerization. Nevertheless, the formation of dynamic actin cages (“actin flashes”) around the phagosome is observed in many cases. These actin structures actually prevent vomocytosis. Yet, fungus strains with high rates of vomocytosis induce more actin flashes, suggesting that these flashes are a reaction from the cell to contain the phagosome. Still, at the end, the fungal phagosome is fused with the cell membrane and the pathogen is liberated (259). Also, the secreted phospholipase B1 (PLB1) is required for vomocytosis (260). It is thought that PLB1 helps permeabilizing the fungal phagosome to neutralize its lumen and to allow nutrients to come in (111, 261). Although vomocytosis is a unique escape function known only for cryptococci, a similar process has recently been described for C. albicans (262) and Candida krusei (263).
Figure 9

Escape from the phagosome. Several pathogens escape from the phagosome to persist in the less harsh environment of the cytoplasm. (a) The intracellular pathogen Listeria monocytogenes uses its virulent factor listeriolysin O (LLO) (264) and phospholipases (PLC and PLD) (265) to escape the phagosome. Once in the cytosol, the bacterium is propelled by the formation of actin “comet tails” that push it across the cell, allowing it to transfer between cells (266–268). (b) Staphylococcus aureus can escape from neutrophil phagosomes (269) by producing phenol soluble modulins (PSM), which are peptides with lytic activity toward many mammalian cells (270). (c) The fungus Cryptococcus neoformans escapes the cell by vomocytosis (257, 258). Here, the phagosome fuses with the cell membrane with assistance of the secreted phospholipase B1 (PLB1) (260), leaving the pathogen free and the phagocytic cell alive.
Another intracellular pathogen capable of escaping from the phagosome and then from the infected cell is L. monocytogenes (268). This bacterium uses its virulent factor listeriolysin O (LLO) to escape the phagosome (264) (Figure 9). LLO is a pore-forming toxin that permeabilizes the phagosome membrane. It is a potent toxin capable of also degrading the cell membrane, thus its expression and activity are strictly regulated. LLO expression is limited to the intraphagosomal phase of the bacteria, where it is induced by the low pH and high Ca2+ conditions of the phagosome (264). Also, LLO activation requires cooperation of host factor such as GILT (γ-inducible lysosomal thiol reductase) (271). In addition, several phospholipases are activated to completely degrade the phagosomal membrane and allow the bacterial escape (265). Once in the cytosol, the bacterium is propelled by the formation of actin tails that push it across the cell. This process is known as “actin rocketing” and it is initiated by the Listeria surface protein ActA (266, 267) (Figure 9). The actin fibers pushing the bacteria are called “comet tails” and propel the bacteria with enough force, allowing it to transfer between cells (268). In the same way, the Gram-negative bacteria Shigella flexneri can disrupt the phagosome membrane and escape into the cytosol (272), where it induces “comet tails” similar to Listeria. The bacterial protein IscA induces activation of N-WASp to initiate actin polymerization by the complex Arp2/3 (273). The actin “comet tails” then propel the bacteria across the cytosol and into neighboring cells. Bacteria from the genus Rickettsia are obligate intracellular pathogens that can also escape phagosomes. Rickettsia uses a secreted phospholipase A2 to disturb the phagosome membrane (274). Once in the cytosol, Rickettsia produce actin tails that allow them direct cell to cell transfer. The bacterial protein RickA is able to activate the Arp2/3 complex to initiate actin polymerization (275). Another microorganism that seems capable of phagosome escaping from neutrophils but not macrophages is S. aureus (269). These bacteria produce phenol soluble modulins (PSMs), which are peptides with lytic activity toward many mammalian cells (270). In particular, the α-PSM was found to induce a strong destruction of neutrophils after phagocytosis, allowing the escape of the phagocytized bacteria (276) (Figure 9).
Other bacteria, such as M. tuberculosis (277) and Mycobacterium marinum (278), can also escape phagosomes. After escaping the phagosome into the cytosol, M. marinum is able to move around by actin-mediated propulsion (279). The M. marinum actin tail formation involves activation of WASp proteins (280) and requires a functional region of difference 1 (RD1) loci (281). This RD1 locus encodes for a secretion system called the ESAT-6 system-1 (ESX-1) or type VII secretion system, which can induce pore formation on host-cell membranes (282). Thus, it was thought that all mycobacteria could escape from phagosomes using the pore-forming activity of ESX-1. However, this has to be formally proven experimentally. M. tuberculosis could be found in increasing numbers in the cytosol of dendritic cells and macrophages when infection was allowed to proceed beyond 2 days in culture (283), and the presence of cytosolic bacteria was also shown to occur in vivo (284). Therefore, there is no doubt about the capacity of mycobacteria to escape into the cytosol but the significance of this phenomenon is still a matter of debate. A simple idea is that bacteria need to leave the phagosome to replicate and then leave the cell. However, bacilli escape the phagosome at later times of infection and this is followed by cell lysis and release of bacilli (278). In consequence, escaping from the vacuole is not a requirement for either survival or growth of M. tuberculosis (285). Instead, it was proposed that the escape from the vacuole represents a transient state that could be critical to the rapid expansion of the bacterial population (285). If this is the case, then escaping from the phagosome is just an important step in the pathology that accompanies progression of tuberculosis infection to active disease. How, mycobacteria kill the cell to allow its release is not clear. Yet, recently, it was reported that the M. tuberculosis protein Rv3903c (channel protein with necrosis-inducing toxin, CpnT) is required for survival and cytotoxicity of M. tuberculosis in macrophages (286). CpnT consists of an N-terminal channel domain that is used for uptake of nutrients across the outer membrane and a secreted toxic C-terminal domain. This secreted portion is also named tuberculosis-necrotizing toxin (287). It can, in the cytosol of mycobacteria-infected macrophages, hydrolyze the essential coenzyme NAD(+) and induce cell necrosis. However, the mechanism for this cell lysis remains to be elucidated. Clearly, CpnT has a dual function in M. tuberculosis. It is used for uptake of nutrients within the phagosome and for induction of host cell lysis in the cytosol. The regulation of CpnT functions becomes then a topic of important research for controlling M. tuberculosis infections. Another M. tuberculosis virulence factor has also been found to participate in phagosome escape. The unique cell wall lipid phthiocerol dimycocerosates greatly augmented the bacteria escape from its intracellular vacuole (288), by a process not well understood. The mechanism for phagosome lysis is clearly complex as indicated by the fact that host molecules are also recruited by the bacteria to aid in its escape. Activation of host cytosolic phospholipase A2 rapidly led to phagosome lysis for bacteria moving into the cytoplasm of the host cell (116).
Novel Therapeutic Opportunities
The study of the many mechanisms used by microbial pathogens to control phagocytosis provides opportunity for detecting novel potential targets of clinical intervention. Promising therapeutic approaches will be designed based on our new understanding of the tactics pathogens use to interfere with phagocytosis. For example, studies with miRNA in mycobacteria infections identified TLR2 as a potential target to prevent the blockage of phagosome maturation (179) (Figure 5). Recently, it was also found by gene expression profiling of human macrophages treated with glucocorticoids and/or IFN-γ that glucocorticoids, in contrast to IFN-γ, failed to trigger expression and phagolysosome recruitment of V-ATPase (289). This explained the increased risk for mycobacterial infections associated with the use of glucocorticoids. Moreover, this group also found that giving imatinib, a tyrosine kinase inhibitor, to glucocorticoid-treated macrophages induced lysosome acidification and antimicrobial activity without reversing the anti-inflammatory effects of glucocorticoids (289). Thus, an improved therapy would be to administer glucocorticoids together with drugs that induce phagosome acidification. In another recent report, a phagosome maturation assay using confocal microscopy in THP-1-derived macrophages infected with an attenuated M. tuberculosis strain was used to test the effects of Saxifragifolin D, a traditional Chinese medicine (290). Saxifragifolin D (a pentacyclic triterpenoid compound first isolated from the rockjasmine Androsace umbellata) reduced the inhibition of phagosome maturation. Using assays of this type, new potential drugs can be tested for future therapies.
Another potential therapeutic approach would be to modulate macrophage function to improve their antimicrobial potential against bacterial infections. The feasibility of such an approach has been suggested in a recent report of macrophage phagocytosis of L. monocytogenes (291). In this study, the engagement of receptor T cell immunoglobulin mucin-3 (Tim-3) on macrophages inhibited phagocytosis of L. monocytogenes by blocking nuclear erythroid 2-related factor 2 (Nrf2) signaling. In contrast, inhibition of Tim-3 augmented phagocytosis (291). Thus, modulating the Tim-3 pathway to alter macrophage function is a promising tool for treating infectious diseases, such as Listeria infections.
Phagocytosis of opsonized particles is, in general, more efficient and more efficacious in eliminating microorganisms. The idea to generate opsonizing antibodies for controlling infections is another promising area of opportunity for novel therapeutics. The value of this approach has been suggested in studies where opsonizing antibodies improve elimination of bacteria. In a study with five apparently healthy Indian donors having high titers of serum antibodies against M. tuberculosis cell membrane antigens, it was found that phagocytosis and killing of bacilli by the donor macrophages was significantly enhanced following their opsonization with antibody-rich, heat-inactivated autologous sera (292). Another study showed that antibodies directed at the R domain of S. aureus secreted coagulase could trigger phagocytosis and killing of staphylococci (293). This coagulase activates host prothrombin and generates fibrin fibers that cover the bacteria and prevent phagocytosis. These antibodies directed the fibrin-covered bacteria to phagocytes and also protected mice against lethal bloodstream infections caused by methicillin-resistant S. aureus isolates (293). Yet, another study, showed that a monoclonal antibody (mAb) directed at the Protein A could protect neonatal mice against S. aureus sepsis and create protective immunity against subsequent staphylococcal infection (294). A humanized version of this mAb was developed, and it is proposed as a potential new therapy for S. aureus-induced sepsis and meningitis in very-low-birth-weight infants (294). These reports encourage the development of novel vaccines that favor the formation of opsonizing antibodies against bacterial antigens to activate phagocyte innate immunity.
Future Directions
Phagocytosis is a fundamental biological process (109) that in multicellular organisms is required for proper homeostasis and for fighting infections (1, 2). Therefore, it is not surprising that many microbial pathogens have mechanisms to counteract phagocytosis. As we have discussed here, for some model pathogens, namely S. aureus (295), M. tuberculosis (117), and L. monocytogenes (119), particular virulence factors that affect phagocytosis have been identified and to some extent the way they work is described. For many other microbial pathogens, their tactics for interfering with phagocytosis are only beginning to be defined. Despite the tremendous amount of published studies on microbial phagocytosis or knowledge on microbial control of this biological process is still incipient and fragmented. We know that some pathogens block phagocytosis at one step or another, but no information is available on how this blockage is accomplished. Some molecules have been identified but their mechanisms of action are not yet described. Future research will serve to fill these gaps and will provide clues on how to improve antimicrobial therapeutics.
An important element for future research is the implementation of novel techniques. Great advances have been achieved by application of proteomics analysis to phagosomes formed under different infection conditions (296). Earlier studies on M. tuberculosis phagosomes with high-resolution two-dimensional gel electrophoresis and mass spectrometry revealed unique bacterial proteins associated with the intracellular stage of the bacteria (297). The effect of a particular protein of the phagocytic machinery identified by proteomics can then also be tested by RNA-mediated interference (298). By comparing the protein profile of phagosomes formed with virulent and avirulent variants of a pathogen, relevant molecules for pathogenesis can be identified. For example, comparing phagosomes containing highly virulent L. pneumophila to phagosomes with avirulent L. hackeliae revealed a lack of Rho GDP-dissociation inhibitor (RhoGDI) in L. pneumophila replicative phagosomes (299). Similarly, comparing macrophage phagosomes formed after triggering different receptors, it was found that phagosome outcome was regulated by the individual receptors triggered for phagocytosis (300). This is in agreement with recent findings that indicate particular FcRs promote particular cell responses on neutrophil phagocytes (42). Thus, phagocytosis is clearly modified according to the receptor involved. We have a good understanding on how opsonic phagocytic receptors signal, but very little is known about the signaling pathways activated by other phagocytic receptors. This is an area of research that needs much further exploration in the future.
Other techniques that have been instrumental for our present understanding of phagocytosis are fluorescence microscopy coupled to particular probes to measure phagosome pH (301), to describe phospholipid dynamics during phagosome formation (302), and to quantify antibody-dependent phagocytosis (303). Together with these, the use of confocal microscopy coupled to fluorescence resonance energy transfer-based assays has been helpful to investigate the mechanisms of L. monocytogenes for phagosome escaping (304). Equally important, the use of novel microbial readouts of bacterial fitness have been developed to probe the host cell environments that promote or control bacterial growth (305). In particular, M. tuberculosis strains that express GFP under certain environmental signals relevant to the infection status of the macrophage, permitted identify infected phagocytes and demonstrated that bacteria in immune-activated phagocytes presented higher drug tolerance than bacteria in resting phagocytes (306). These assays will be very useful in future studies on phagocytosis of other microbial pathogens. To implement these assays, the proper fluorescent probes will need to be developed.
During phagocytosis, both the phagocyte and the microorganism adapt to fight and overcome each other. These changes, important to the final outcome of an infection, can be studied by modern techniques such as transcriptional analysis via RNA sequencing (RNA-seq). Changes in pathogen phenotype under various conditions are revealed when the total transcriptome is analyzed. For example, it is known that cigarette smoke predisposes exposed individuals to respiratory infections by enhancing the virulence of pathogenic bacteria. A recent study on the effect of cigarette smoke on S. aureus gene expression using RNA-seq revealed that these bacteria increased twofold the expression of protein A with the consequent reduction in phagocytosis (307). A similar comparative transcriptome study with RNA-seq of Brucella melitensis grown in normal-medium culture and in acid-medium (pH 4.4) culture revealed that 113 genes were differentially expressed. Among these genes, a two-component response regulator gene in the transcriptional regulation pathway was identified as important for acid resistance and virulence of Brucella (308). Also, an analysis of RNA-seq data from in vivo and in vitro cultures of Cryptococcus gattii identified highly expressed genes and pathways of amino acid metabolism that would enable these bacteria to survive and proliferate in vivo (309). Hence, particular genes expressed under particular conditions can be identified as potential therapeutic targets for controlling infections. Likewise, changes in cell phenotype can be analyzed by RNA-seq. For example, increased susceptibility to bacterial pneumonia is found after influenza infections. A recent RNA-seq analysis of alveolar macrophages revealed that the virus infection caused a reduction in the phagocytic receptor MARCO. This effect could be reversed after IFNγ treatment of monocyte-derived macrophages and THP-1 macrophages. Moreover, treatment with sulforaphane or SC79, activators of Nrf2 and Akt, respectively, caused increased MARCO expression and MARCO-dependent phagocytosis (310). Therefore, a promising strategy for controlling postinfluenza bacterial pneumonia would be to increase MARCO expression by targeting Nrf2 and Akt signaling in alveolar macrophages. Another example of RNA-seq analysis of macrophages in two different conditions, namely infection with virulent or avirulent strains of M. tuberculosis, revealed extensive remodeling of alternative splicing in macrophage transcriptome (311). This led to considerable increase in truncated/non-translatable variants of several genes with a decline in the corresponding protein levels. The product of one such gene, RAB8B that is required for phagosome maturation, was reduced due to elevated levels of truncated RAB8B variants in cells with virulent mycobacteria (311). Alternative splicing is a new mechanism that M. tuberculosis uses to control macrophages. The molecular details of this mechanism are not known and will certainly become an area of interesting research in the near future.
We have described phagocytosis as a general model based mainly on macrophages. However, there are important differences among diverse types of phagocytes and even between phenotypes of the same phagocyte. As indicated earlier, environmental cues can alter the functioning of a phagocyte, and no much is known about the mechanisms involved in these cell changes. Hence, this is an area of great interest, as shown by some recent studies. Metabolic conditions can alter macrophage function (312), and in the case of diabetes mellitus it was found that phagocytosis was reduced (313). This disease is also associated with increased tuberculosis risk and severity. Recently, it was also reported that alveolar murine macrophages from diabetic mice have a reduced expression of MARCO (314). The lack of this receptor could be the reason for inefficient phagocytosis in diabetic cells. Future research should determine whether other phagocytic receptors are also altered in diabetic macrophages. Nothing is known about the metabolic mechanisms that control phagocyte receptor expression.
The role of other phagocytes besides macrophages in controlling some intracellular bacterial infections is just beginning to be appreciated. For example, neutrophils also participate in controlling M. tuberculosis by autophagy (315) and are mobilized from the bone marrow to perform phagocytosis and secrete antimicrobial factors against L. monocytogenes (119). In addition, other cells such as dendritic cells can also perform phagocytosis by mechanisms that are different from those of macrophages (316). The particular role of these various phagocytic cells in different infection settings will also become an area of fruitful research in the future.
Macrophages not only perform phagocytosis of microbial pathogens but also ingest dead and dying host cells. The process of engulfing apoptotic cells is called efferocytosis, and it has an important role in the resolution of inflammation (317). Although efferocytosis of M. tuberculosis-infected cells leads to pathogen destruction, efferocytosis of Leishmania-infected neutrophils may promote infection (318). Understanding how macrophages, neutrophils, and dendritic cells process pathogens within a dying cell is another area for future research. Discoveries in this field should lead to novel therapeutics that simultaneously suppress inflammation and promote pathogen clearance.
Conclusion
Elimination of pathogens by macrophages and neutrophils is an essential function of our innate defenses. These phagocytic leukocytes clear microorganisms from tissues via phagocytosis. Once inside the phagocyte, the microorganism is destroyed by a series of degrading mechanisms inside the phagosome. Despite this, many pathogens have evolved means to prevent phagocytosis or to resist its effects inside the phagocytic cells. Thus, these pathogens remain a considerable health threat. We have presented the main mechanisms phagocytes have for eliminating microbes and then we discussed the strategies used by some pathogens to interfere with each step of the phagocytic process. Our list of pathogens is not complete, since there are many microorganisms capable of resisting phagocytosis in ways, we do not completely recognize. Technical advances have allowed us to make significant advances toward understanding the molecular details of the interaction between some pathogens and phagocytes, but important questions remain. Future research in this area will certainly bring us interesting surprises that will help us conceive novel therapeutic approaches that could render pathogens more susceptible to phagocyte attack.
Statements
Author contributions
CR and EU-Q both equally conceived the issues, which formed the content of the manuscript, prepared the figures, and wrote the manuscript.
Funding
Research in the authors’ laboratory was supported by grant 254434 from Consejo Nacional de Ciencia y Tecnología (CONACyT), Mexico.
Conflict of interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
LimJJGrinsteinSRothZ. Diversity and versatility of phagocytosis: roles in innate immunity, tissue remodeling, and homeostasis. Front Cell Infect Microbiol (2017) 7:191.10.3389/fcimb.2017.00191
2
RosalesCUribe-QuerolE. Phagocytosis: a fundamental process in immunity. Biomed Res Int (2017) 2017:9042851.10.1155/2017/9042851
3
RabinovitchM. Professional and non-professional phagocytes: an introduction. Trends Cell Biol (1995) 5:85–7.10.1016/S0962-8924(00)88955-2
4
FlannaganRSJaumouilléVGrinsteinS. The cell biology of phagocytosis. Annu Rev Pathol (2012) 7:61–98.10.1146/annurev-pathol-011811-132445
5
DesjardinsM. Biogenesis of phagolysosomes: the ‘kiss and run’ hypothesis. Trends Cell Biol (1995) 5:183–6.10.1016/0962-8924(95)80001-W
6
LevinRGrinsteinSCantonJ. The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev (2016) 273:156–79.10.1111/imr.12439
7
FairnGGrinsteinS. How nascent phagosomes mature to become phagolysosomes. Trends Immunol (2012) 33:397–405.10.1016/j.it.2012.03.003
8
CantonJKhezriRGlogauerMGrinsteinS. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell (2014) 25:3330–41.10.1091/mbc.E14-05-0967
9
HoppeADSwansonJA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell (2004) 15:3509–19.10.1091/mbc.E03-11-0847
10
MaoYFinnemannSC. Regulation of phagocytosis by Rho GTPases. Small GTPases (2015) 6:89–99.10.4161/21541248.2014.989785
11
ArakiNHataeTFurukawaASwansonJA. Phosphoinositide-3-kinase-independent contractile activities associated with Fcγ-receptor-mediated phagocytosis and macropinocytosis in macrophages. J Cell Sci (2003) 116:247–57.10.1242/jcs.00235
12
DartAETollisSBrightMDFrankelGEndresRG. The motor protein myosin 1G functions in FcγR-mediated phagocytosis. J Cell Sci (2012) 125:6020–9.10.1242/jcs.109561
13
KitanoMNakayaMNakamuraTNagataSMatsudaM. Imaging of Rab5 activity identifies essential regulators for phagosome maturation. Nature (2008) 453:241–5.10.1038/nature06857
14
GutierrezMG. Functional role(s) of phagosomal Rab GTPases. Small GTPases (2013) 4:148–58.10.4161/sgtp.25604
15
ChristoforidisSMcBrideHMBurgoyneRDZerialM. The Rab5 effector EEA1 is a core component of endosome docking. Nature (1999) 397:621–5.10.1038/17618
16
VieiraOVBotelhoRJRamehLBrachmannSMMatsuoTDavidsonHWet alDistinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol (2001) 155:19–25.10.1083/jcb.200107069
17
KinchenJMRavichandranKS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol (2008) 9:781–95.10.1038/nrm2515
18
MarshanskyVFutaiM. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol (2008) 20:415–26.10.1016/j.ceb.2008.03.015
19
ArakiNJohnsonMTSwansonJA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol (1996) 135:1249–60.10.1083/jcb.135.5.1249
20
VieiraOVBucciCHarrisonRETrimbleWSLanzettiLGruenbergJet alModulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol (2003) 23:2501–14.10.1128/MCB.23.7.2501-2514.2003
21
RinkJGhigoEKalaidzidisYZerialM. Rab conversion as a mechanism of progression from early to late endosomes. Cell (2005) 122:735–49.10.1016/j.cell.2005.06.043
22
HuynhKKEskelinenELScottCCMalevanetsASaftigPGrinsteinS. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J (2007) 26:313–24.10.1038/sj.emboj.7601511
23
BeertsenWWillenborgMEvertsVZirogianniAPodschunRSchröderBet alImpaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J Immunol (2008) 180:475–82.10.4049/jimmunol.180.1.475
24
HansonPIRothRLinYHeuserJE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol (2008) 180:389–402.10.1083/jcb.200707031
25
BabiorBM. NADPH oxidase. Curr Opin Immunol (2004) 16:42–7.10.1016/j.coi.2003.12.001
26
CantonJNeculaiDGrinsteinS. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol (2013) 13:621–34.10.1038/nri3515
27
PeiserLGoughPJKodamaTGordonS. Macrophage class A scavenger receptor-mediated phagocytosis of Escherichia coli: role of cell heterogeneity, microbial strain, and culture conditions in vitro. Infect Immun (2000) 68:1953–63.10.1128/IAI.68.4.1953-1963.2000
28
HerreJMarshallASCaronEEdwardsADWilliamsDLSchweighofferEet alDectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood (2004) 104:4038–45.10.1182/blood-2004-03-1140
29
DambuzaIMBrownGD. C-type lectins in immunity: recent developments. Curr Opin Immunol (2015) 32:21–7.10.1016/j.coi.2014.12.002
30
EzekowitzRASastryKBaillyPWarnerA. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J Exp Med (1990) 172:1785–94.10.1084/jem.172.6.1785
31
SchiffDEKlineLSoldauKLeeJDPuginJTobiasPSet alPhagocytosis of Gram-negative bacteria by a unique CD14-dependent mechanism. J Leukoc Biol (1997) 62:786–94.
32
KawaiTAkiraS. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50.10.1016/j.immuni.2011.05.006
33
IwasakiAMedzhitovR. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16:343–53.10.1038/ni.3123
34
RosalesC. Fc receptor and integrin signaling in phagocytes. Signal Transduct (2007) 7:386–401.10.1002/sita.200700141
35
NimmerjahnFRavetchJ. Antibodies, Fc receptors and cancer. Curr Opin Immunol (2007) 19:239–45.10.1016/j.coi.2007.01.005
36
PowellMSHogarthPM. Fc receptors. Adv Exp Med Biol (2008) 640:22–34.10.1007/978-0-387-09789-3_3
37
RosalesCUribe-QuerolE. Antibody-Fc receptor interactions in antimicrobial functions. Curr Immunol Rev (2013) 9:44–55.10.2174/1573395511309010006
38
RosalesCUribe-QuerolE. Fc receptors: cell activators of antibody functions. Adv Biosci Biotechnol (2013) 4:21–33.10.4236/abb.2013.44A004
39
BakemaJEGanzevlesSHFluitsmaDMSchilhamMWBeelenRHValeriusTet alTargeting FcαRI on polymorphonuclear cells induces tumor cell killing through autophagy. J Immunol (2011) 187:726–32.10.4049/jimmunol.1002581
40
van Lookeren CampagneMWiesmannCBrownEJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol (2007) 9:2095–102.10.1111/j.1462-5822.2007.00981.x
41
NimmerjahnFRavetchJV. FcγRs in health and disease. Curr Top Microbiol Immunol (2011) 350:105–25.10.1007/82_2010_86
42
RosalesC. Fcγ receptor heterogeneity in leukocyte functional responses. Front Immunol (2017) 8:280.10.3389/fimmu.2017.00280
43
TohyamaYYamamuraH. Complement-mediated phagocytosis – the role of Syk. IUBMB Life (2006) 58:304–8.10.1080/15216540600746377
44
FlannaganRGrinsteinS. The application of fluorescent probes for the analysis of lipid dynamics during phagocytosis. Methods Mol Biol (2010) 591:121–34.10.1007/978-1-60761-404-3_7
45
SwansonJAJohnsonMTBeningoKPostPMoosekerMArakiN. A contractile activity that closes phagosomes in macrophages. J Cell Sci (1999) 112:307–16.
46
LuzioJPPryorPRBrightNA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol (2007) 8:622–32.10.1038/nrm2217
47
PryorPRLuzioJP. Delivery of endocytosed membrane proteins to the lysosome. Biochim Biophys Acta (2009) 1793:615–24.10.1016/j.bbamcr.2008.12.022
48
MullinsCBonifacinoJS. The molecular machinery for lysosome biogenesis. Bioessays (2001) 23:333–43.10.1002/bies.1048
49
VidaTGerhardtB. A cell-free assay allows reconstitution of Vps33p-dependent transport to the yeast vacuole/lysosome. J Cell Biol (1999) 146:85–98.10.1083/jcb.146.1.85
50
StorrieBDesjardinsM. The biogenesis of lysosomes: is it a kiss and run, continuous fusion and fission process?Bioessays (1996) 18:895–903.10.1002/bies.950181108
51
BrightNAReavesBJMullockBMLuzioJP. Dense core lysosomes can fuse with late endosomes and are re-formed from the resultant hybrid organelles. J Cell Sci (1997) 110:2027–40.
52
MullockBMBrightNAFearonCWGraySRLuzioJP. Fusion of lysosomes with late endosomes produces a hybrid organelle of intermediate density and is NSF dependent. J Cell Biol (1998) 140:591–601.10.1083/jcb.140.3.591
53
BrightNAGratianMJLuzioJP. Endocytic delivery to lysosomes mediated by concurrent fusion and kissing events in living cells. Curr Biol (2005) 15:360–5.10.1016/j.cub.2005.01.049
54
LuzioJPGraySRBrightNA. Endosome-lysosome fusion. Biochem Soc Trans (2010) 38:1413–6.10.1042/BST0381413
55
LuzioJPHackmannYDieckmannNMGriffithsGM. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb Perspect Biol (2014) 6:a016840.10.1101/cshperspect.a016840
56
SteinbergBEHuynhKKGrinsteinS. Phagosomal acidification: measurement, manipulation and functional consequences. Biochem Soc Trans (2007) 35:1083–7.10.1042/BST0351083
57
JordensIFernandez-BorjaMMarsmanMDusseljeeSJanssenLCalafatJet alThe Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol (2001) 11:1680–5.10.1016/S0960-9822(01)00531-0
58
HarrisonREBucciCVieiraOVSchroerTAGrinsteinS. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol (2003) 23:6494–506.10.1128/MCB.23.18.6494-6506.2003
59
CantonJ. Phagosome maturation in polarized macrophages. J Leukoc Biol (2014) 96:729–38.10.1189/jlb.1MR0114-021R
60
GuarnieriFGArterburnLMPennoMBChaYAugustJT. The motif Tyr-X-X-hydrophobic residue mediates lysosomal membrane targeting of lysosome-associated membrane protein 1. J Biol Chem (1993) 268:1941–6.
61
SchwakeMSchröderBSaftigP. Lysosomal membrane proteins and their central role in physiology. Traffic (2013) 14:739–48.10.1111/tra.12056
62
LuzioJPParkinsonMDGraySRBrightNA. The delivery of endocytosed cargo to lysosomes. Biochem Soc Trans (2009) 37:1019–21.10.1042/BST0371019
63
MassonPLHeremansJFSchonneE. Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J Exp Med (1969) 130:643–58.10.1084/jem.130.3.643
64
MinakamiRSumimotoaH. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int J Hematol (2006) 84:193–8.10.1532/IJH97.06133
65
NauseefWM. Myeloperoxidase in human neutrophil host defence. Cell Microbiol (2014) 16:1146–55.10.1111/cmi.12312
66
PillayCSElliottEDennisonC. Endolysosomal proteolysis and its regulation. Biochem J (2002) 363:417–29.10.1042/bj3630417
67
JabadoNJankowskiADougaparsadSPicardVGrinsteinSGrosP. Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J Exp Med (2000) 192:1237–48.10.1084/jem.192.9.1237
68
PandayASahooMKOsorioDBatraS. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol (2015) 12:5–23.10.1038/cmi.2014.89
69
El-BennaJHurtado-NedelecMMarzaioliVMarieJCGougerot-PocidaloMADangPM. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev (2016) 273:180–93.10.1111/imr.12447
70
AboAPickEHallATottyNTeahanCGSegalAW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature (1991) 353:668–70.10.1038/353668a0
71
KnausUGHeyworthPGEvansTCurnutteJTBokochGM. Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science (1991) 254:1512–5.10.1126/science.1660188
72
BogdanCRöllinghoffMDiefenbachA. The role of nitric oxide in innate immunity. Immunol Rev (2000) 173:17–26.10.1034/j.1600-065X.2000.917307.x
73
RichardsonARSolivenKCCastorMEBarnesPDLibbySJFangFC. The base excision repair system of Salmonella enterica serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog (2009) 5:e1000451.10.1371/journal.ppat.1000451
74
RichardsonARPayneECYoungerNKarlinseyJEThomasVCBeckerLAet alMultiple targets of nitric oxide in the tricarboxylic acid cycle of Salmonella enterica serovar typhimurium. Cell Host Microbe (2011) 10:33–43.10.1016/j.chom.2011.06.004
75
WardPPUribe-LunaSConneelyOM. Lactoferrin and host defense. Biochem Cell Biol (2002) 80:95–102.10.1139/o01-214
76
CellierMFCourvillePCampionC. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect (2007) 9:1662–70.10.1016/j.micinf.2007.09.006
77
ZhaoLLuW. Defensins in innate immunity. Curr Opin Hematol (2014) 21:37–42.10.1097/MOH.0000000000000005
78
Suarez-CarmonaMHubertPDelvennePHerfsM. Defensins: “simple” antimicrobial peptides or broad-spectrum molecules?Cytokine Growth Factor Rev (2015) 26:361–70.10.1016/j.cytogfr.2014.12.005
79
del Cerro-VadilloEMadrazo-TocaFCarrasco-MarínEFernandez-PrietoLBeckCLeyva-CobiánFet alCutting edge: a novel nonoxidative phagosomal mechanism exerted by cathepsin-D controls Listeria monocytogenes intracellular growth. J Immunol (2006) 176:1321–5.10.4049/jimmunol.176.3.1321
80
MüllerSFaulhaberASieberCPfeiferDHochbergTGanszMet alThe endolysosomal cysteine cathepsins L and K are involved in macrophage-mediated clearance of Staphylococcus aureus and the concomitant cytokine induction. FASEB J (2014) 28:162–75.10.1096/fj.13-232272
81
MiyauchiJSasadairaHWatanabeKWatanabeY. Ultrastructural immunocytochemical localization of lysozyme in human monocytes and macrophages. Cell Tissue Res (1985) 242:269–77.10.1007/BF00214539
82
GanzT. Antimicrobial polypeptides. J Leukoc Biol (2004) 75:34–8.10.1189/jlb.0403150
83
WuYRaymondBGoossensPLNjamkepoEGuisoNPayaMet alType-IIA secreted phospholipase A2 is an endogenous antibiotic-like protein of the host. Biochimie (2010) 92:583–7.10.1016/j.biochi.2010.01.024
84
BehePSegalAW. The function of the NADPH oxidase of phagocytes, and its relationship to other NOXs. Biochem Soc Trans (2007) 35:1100–3.10.1042/BST0351100
85
GoldblattD. Recent advances in chronic granulomatous disease. J Infect (2014) 69:S32–5.10.1016/j.jinf.2014.07.013
86
RoosD. Chronic granulomatous disease. Br Med Bull (2016) 118:50–63.10.1093/bmb/ldw009
87
El-BennaJDangPMGougerot-PocidaloMAMarieJCBraut-BoucherF. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med (2009) 41:217–25.10.3858/emm.2009.41.4.058
88
WinterbournCC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett (1995) 82-83:969–74.10.1016/0378-4274(95)03532-X
89
LamGYHuangJBrumellJH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol (2010) 32:415–30.10.1007/s00281-010-0221-0
90
FangFC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol (2004) 2:820–32.10.1038/nrmicro1004
91
WebbJLHarveyMWHoldenDWEvansTJ. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect Immun (2001) 69:6391–400.10.1128/IAI.69.10.6391-6400.2001
92
GrosserMRWeissAShawLNRichardsonAR. Regulatory requirements for Staphylococcus aureus nitric oxide resistance. J Bacteriol (2016) 198:2043–55.10.1128/JB.00229-16
93
HägerMCowlandJBBorregaardN. Neutrophil granules in health and disease. J Intern Med (2010) 268:25–34.10.1111/j.1365-2796.2010.02237.x
94
WardPPPazEConneelyOM. Multifunctional roles of lactoferrin: a critical overview. Cell Mol Life Sci (2005) 62:2540–8.10.1007/s00018-005-5369-8
95
LuoMFadeevEAGrovesJT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol (2005) 1:149–53.10.1038/nchembio717
96
GoetzDHHolmesMABorregaardNBluhmMERaymondKNStrongRK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell (2002) 10:1033–43.10.1016/S1097-2765(02)00708-6
97
FloTHSmithKDSatoSRodriguezDJHolmesMAStrongRKet alLipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature (2004) 432:917–21.10.1038/nature03104
98
YountNYYeamanMR. Peptide antimicrobials: cell wall as a bacterial target. Ann N Y Acad Sci (2013) 1277:127–38.10.1111/nyas.12005
99
KościuczukEMLisowskiPJarczakJStrzałkowskaNJóźwikAHorbańczukJet alCathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep (2012) 39:10957–70.10.1007/s11033-012-1997-x
100
LehrerRILuW. α-Defensins in human innate immunity. Immunol Rev (2012) 245:84–112.10.1111/j.1600-065X.2011.01082.x
101
FabisiakAMurawskaNFichnaJ. LL-37: cathelicidin-related antimicrobial peptide with pleiotropic activity. Pharmacol Rep (2016) 68:802–8.10.1016/j.pharep.2016.03.015
102
TangXBasavarajappaDHaeggströmJZWanM. P2X7 receptor regulates internalization of antimicrobial peptide LL-37 by human macrophages that promotes intracellular pathogen clearance. J Immunol (2015) 195:1191–201.10.4049/jimmunol.1402845
103
WanMvan der DoesAMTangXLindbomLAgerberthBHaeggströmJZ. Antimicrobial peptide LL-37 promotes bacterial phagocytosis by human macrophages. J Leukoc Biol (2014) 95:971–81.10.1189/jlb.0513304
104
WanMSoehnleinOTangXvan der DoesAMSmedlerEUhlénPet alCathelicidin LL-37 induces time-resolved release of LTB4 and TXA2 by human macrophages and triggers eicosanoid generation in vivo. FASEB J (2014) 28:3456–67.10.1096/fj.14-251306
105
GuptaSWingleeKGalloRBishaiWR. Bacterial subversion of cAMP signalling inhibits cathelicidin expression, which is required for innate resistance to Mycobacterium tuberculosis. J Pathol (2017) 242:52–61.10.1002/path.4878
106
SchröderBAWrocklageCHasilikASaftigP. The proteome of lysosomes. Proteomics (2010) 10:4053–76.10.1002/pmic.201000196
107
GanzTLehrerRI. Antimicrobial peptides of leukocytes. Curr Opin Hematol (1997) 4:53–8.10.1097/00062752-199704010-00009
108
BotelhoRJGrinsteinS. Phagocytosis. Curr Biol (2011) 21:R533–8.10.1016/j.cub.2011.05.053
109
GordonS. Phagocytosis: an immunobiologic process. Immunity (2016) 44:463–75.10.1016/j.immuni.2016.02.026
110
FlannaganRSCosíoGGrinsteinS. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol (2009) 7:355–66.10.1038/nrmicro2128
111
SmithLMMayRC. Mechanisms of microbial escape from phagocyte killing. Biochem Soc Trans (2013) 41:475–90.10.1042/BST20130014
112
SpaanANSurewaardBGNijlandRvan StrijpJA. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol (2013) 67:629–50.10.1146/annurev-micro-092412-155746
113
FlannaganRSHeitBHeinrichsDE. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens (2015) 4:826–68.10.3390/pathogens4040826
114
LacomaACanoVMorantaDRegueiroVDomínguez-VillanuevaDLaabeiMet alInvestigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line. Virulence (2017) 1–15.10.1080/21505594.2017.1361089
115
VergneIGilleronMNigouJ. Manipulation of the endocytic pathway and phagocyte functions by Mycobacterium tuberculosis lipoarabinomannan. Front Cell Infect Microbiol (2015) 4:187.10.3389/fcimb.2014.00187
116
JamwalSVMehrotraPSinghASiddiquiZBasuARaoKV. Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci Rep (2016) 6:23089.10.1038/srep23089
117
AwuhJAFloTH. Molecular basis of mycobacterial survival in macrophages. Cell Mol Life Sci (2017) 74:1625–48.10.1007/s00018-016-2422-8
118
ArnettEVadiaSNackermanCCOghumuSSatoskarARMcLeishKRet alThe pore-forming toxin listeriolysin O is degraded by neutrophil metalloproteinase-8 and fails to mediate Listeria monocytogenes intracellular survival in neutrophils. J Immunol (2014) 192:234–44.10.4049/jimmunol.1301302
119
WitterAROkunnuBMBergRE. The essential role of neutrophils during infection with the intracellular bacterial pathogen Listeria monocytogenes. J Immunol (2016) 197:1557–65.10.4049/jimmunol.1600599
120
AlonzoFIIITorresVJ. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev (2014) 78:199–230.10.1128/MMBR.00055-13
121
BerubeBJBubeck WardenburgJ. Staphylococcus aureus α-toxin: nearly a century of intrigue. Toxins (Basel) (2013) 5:1140–66.10.3390/toxins5061140
122
YamashitaKKawaiYTanakaYHiranoNKanekoJTomitaNet alCrystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc Natl Acad Sci U S A (2011) 108:17314–9.10.1073/pnas.1110402108
123
BadarauARouhaHMalafaSLoganDTHåkanssonMStulikLet alStructure-function analysis of heterodimer formation, oligomerization, and receptor binding of the Staphylococcus aureus bi-component toxin LukGH. J Biol Chem (2015) 290:142–56.10.1074/jbc.M114.598110
124
AlonzoFIIIKozhayaLRawlingsSAReyes-RoblesTDuMontALMyszkaDGet alCCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature (2013) 493:51–5.10.1038/nature11724
125
DuMontALYoongPDayCJAlonzoFIIIMcDonaldWHJenningsMPet alStaphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A (2013) 110:10794–9.10.1073/pnas.1305121110
126
Reyes-RoblesTAlonzoFIIIKozhayaLLacyDBUnutmazDTorresVJ. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe (2013) 14:453–9.10.1016/j.chom.2013.09.005
127
WilkeGABubeck WardenburgJ. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A (2010) 107:13473–8.10.1073/pnas.1001815107
128
BeckerREBerubeBJSampedroGRDeDentACBubeck WardenburgJ. Tissue-specific patterning of host innate immune responses by Staphylococcus aureus α-toxin. J Innate Immun (2014) 6:619–31.10.1159/000360006
129
SongLHobaughMRShustakCCheleySBayleyHGouauxJE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science (1996) 274:1859–66.10.1126/science.274.5294.1859
130
SugawaraTYamashitaDKatoKPengZUedaJKanekoJet alStructural basis for pore-forming mechanism of staphylococcal α-hemolysin. Toxicon (2015) 108:226–31.10.1016/j.toxicon.2015.09.033
131
SulicaAMedesanCLakyMOnicăDSjöquistJGhetieV. Effect of protein A of Staphylococcus aureus on the binding of monomeric and polymeric IgG to Fc receptor-bearing cells. Immunology (1979) 38:173–9.
132
ZhangLJacobssonKVasiJLindbergMFrykbergL. A second IgG-binding protein in Staphylococcus aureus. Microbiology (1998) 144:985–91.10.1099/00221287-144-4-985
133
SmithEJVisaiLKerriganSWSpezialePFosterTJ. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect Immun (2011) 79:3801–9.10.1128/IAI.05075-11
134
SmithEJCorriganRMvan der SluisTGründlingASpezialePGeogheganJAet alThe immune evasion protein Sbi of Staphylococcus aureus occurs both extracellularly and anchored to the cell envelope by binding lipoteichoic acid. Mol Microbiol (2012) 83:789–804.10.1111/j.1365-2958.2011.07966.x
135
LaarmanAJRuykenMMaloneCLvan StrijpJAHorswillARRooijakkersSH. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J Immunol (2011) 186:6445–53.10.4049/jimmunol.1002948
136
RooijakkersSHWuJRuykenMvan DomselaarRPlankenKLTzekouAet alStructural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol (2009) 10:721–7.10.1038/ni.1756
137
KoYPKuipersAFreitagCMJongeriusIMedinaEvan RooijenWJet alPhagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog (2013) 9:e1003816.10.1371/journal.ppat.1003816
138
KuipersAStapelsDAWeerwindLTKoYPRuykenMLeeJCet alThe Staphylococcus aureus polysaccharide capsule and Efb-dependent fibrinogen shield act in concert to protect against phagocytosis. Microbiology (2016) 162:1185–94.10.1099/mic.0.000293
139
RooijakkersSHvan WamelWJRuykenMvan KesselKPvan StrijpJA. Anti-opsonic properties of staphylokinase. Microbes Infect (2005) 7:476–84.10.1016/j.micinf.2004.12.014
140
LakyMSjöquistJMoraruIGheţieV. Mutual inhibition of the binding of C1q and protein A to rabbit IgG immune complexes. Mol Immunol (1985) 22:1297–302.10.1016/0161-5890(85)90049-5
141
AnesE. Acting on actin during bacterial infection, cytoskeleton – structure, dynamics, function and disease. In: Jimenez-LopezJC, editor. Biochemistry, Genetics and Molecular Biology. Rijeka, Croacia: InTech d.o.o. (2017). p. 257–78.
142
HallA. Rho family GTPases. Biochem Soc Trans (2012) 40:1378–82.10.1042/BST20120103
143
BiroMMunozMAWeningerW. Targeting Rho-GTPases in immune cell migration and inflammation. Br J Pharmacol (2014) 171:5491–506.10.1111/bph.12658
144
HiggsHNPollardTD. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich Syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J Cell Biol (2000) 150:1311–20.10.1083/jcb.150.6.1311
145
MarchandJBKaiserDAPollardTDHiggsHN. Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat Cell Biol (2001) 3:76–82.10.1038/35050590
146
PollardTDCooperJA. Actin, a central player in cell shape and movement. Science (2009) 326:1208–12.10.1126/science.1175862
147
JankTGiesemannTAktoriesK. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology (2007) 17:15R–22R.10.1093/glycob/cwm004
148
JankTBogdanovićXWirthCHaafESpoernerMBöhmerKEet alA bacterial toxin catalyzing tyrosine glycosylation of Rho and deamidation of Gq and Gi proteins. Nat Struct Mol Biol (2013) 20:1273–80.10.1038/nsmb.2688
149
GrovesERittingerKAmstutzMBerrySHoldenDWCornelisGRet alSequestering of Rac by the Yersinia effector YopO blocks Fcγ receptor-mediated phagocytosis. J Biol Chem (2010) 285:4087–98.10.1074/jbc.M109.071035
150
KrallRSunJPedersonKJBarbieriJT. In vivo Rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS. Infect Immun (2002) 70:360–7.10.1128/IAI.70.1.360-367.2002
151
FlannaganRS. Burkholderia cenocepacia infection: disruption of phagocyte immune functions through Rho GTPase inactivation. Cell Adh Migr (2012) 6:297–301.10.4161/cam.20487
152
WheelerRTKombeDAgarwalaSDFinkGR. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog (2008) 4:e1000227.10.1371/journal.ppat.1000227
153
BrothersKMGratacapRLBarkerSENewmanZRNorumAWheelerRT. NADPH oxidase-driven phagocyte recruitment controls Candida albicans filamentous growth and prevents mortality. PLoS Pathog (2013) 9:e1003634.10.1371/journal.ppat.1003634
154
AimaniandaVBayryJBozzaSKniemeyerOPerruccioKElluruSRet alSurface hydrophobin prevents immune recognition of airborne fungal spores. Nature (2009) 460:1117–21.10.1038/nature08264
155
BoseIReeseAJOryJJJanbonGDoeringTL. A yeast under cover: the capsule of Cryptococcus neoformans. Eukaryot Cell (2003) 2:655–63.10.1128/EC.2.4.655-663.2003
156
StanoPWilliamsVVillaniMCymbalyukESQureshiAHuangYet alApp1: an antiphagocytic protein that binds to complement receptors 3 and 2. J Immunol (2009) 182:84–91.10.4049/jimmunol.182.1.84
157
ArmstrongJAHartPD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med (1971) 134:713–40.10.1084/jem.134.3.713
158
VergneIChuaJLeeHHLucasMBelisleJDereticV. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2005) 102:4033–8.10.1073/pnas.0409716102
159
RussellDG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev (2011) 240:252–68.10.1111/j.1600-065X.2010.00984.x
160
PatinECGeffkenACWillcocksSLeschczykCHaasANimmerjahnFet alTrehalose dimycolate interferes with FcγR-mediated phagosome maturation through Mincle, SHP-1 and FcγRIIB signalling. PLoS One (2017) 12:e0174973.10.1371/journal.pone.0174973
161
Sturgill-KoszyckiSSchlesingerPHChakrabortyPHaddixPLCollinsHLFokAKet alLack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science (1994) 263:678–81.10.1126/science.8303277
162
WongDBachHSunJHmamaZAv-GayY. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A (2011) 108:19371–6.10.1073/pnas.1109201108
163
BachHPapavinasasundaramKGWongDHmamaZAv-GayY. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe (2008) 3:316–22.10.1016/j.chom.2008.03.008
164
SunJWangXLauALiaoTYBucciCHmamaZ. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS One (2010) 5:e8769.10.1371/journal.pone.0008769
165
FrattiRABackerJMGruenbergJCorveraSDereticV. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol (2001) 154:631–44.10.1083/jcb.200106049
166
GaurRLRenKBlumenthalABhamidiSGonzález-NiloFDJacksonMet alLprG-mediated surface expression of lipoarabinomannan is essential for virulence of Mycobacterium tuberculosis. PLoS Pathog (2014) 10:e1004376.10.1371/journal.ppat.1004376
167
ShuklaSRichardsonETAthmanJJShiLWearschPAMcDonaldDet alMycobacterium tuberculosis lipoprotein LprG binds lipoarabinomannan and determines its cell envelope localization to control phagolysosomal fusion. PLoS Pathog (2014) 10:e1004471.10.1371/journal.ppat.1004471
168
VázquezCLBiancoMVBlancoFCForrelladMAGutierrezMGBigiF. Mycobacterium bovis requires P27 (LprG) to arrest phagosome maturation and replicate within bovine macrophages. Infect Immun (2017) 85:e00720–16.10.1128/IAI.00720-16
169
KangPBAzadAKTorrellesJBKaufmanTMBeharkaATibesarEet alThe human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med (2005) 202:987–99.10.1084/jem.20051239
170
EsparzaMPalomaresBGarcíaTEspinosaPZentenoEMancillaR. PstS-1, the 38-kDa Mycobacterium tuberculosis glycoprotein, is an adhesin, which binds the macrophage mannose receptor and promotes phagocytosis. Scand J Immunol (2015) 81:46–55.10.1111/sji.12249
171
WelinAWinbergMEAbdallaHSärndahlERasmussonBStendahlOet alIncorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun (2008) 76:2882–7.10.1128/IAI.01549-07
172
IshikawaEIshikawaTMoritaYSToyonagaKYamadaHTakeuchiOet alDirect recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med (2016) 206:2879–88.10.1084/jem.20091750
173
ChandraPGhanwatSMattaSKYadavSSMehtaMSiddiquiZet alMycobacterium tuberculosis inhibits Rab7 recruitment to selectively modulate autophagy flux in macrophages. Sci Rep (2015) 5:16320.10.1038/srep16320
174
HuDWuJWangWMuMZhaoRXuXet alAutophagy regulation revealed by SapM-induced block of autophagosome-lysosome fusion via binding Rab7. Biochem Biophys Res Commun (2015) 461:401–7.10.1016/j.bbrc.2015.04.051
175
ChenZWangTLiuZZhangGWangJFengSet alInhibition of autophagy by miR-30A induced by Mycobacteria tuberculosis as a possible mechanism of immune escape in human macrophages. Jpn J Infect Dis (2015) 68:420–4.10.7883/yoken.JJID.2014.466
176
KimJKYukJMKimSYKimTSJinHSYangCSet alMicroRNA-125a inhibits autophagy activation and antimicrobial responses during mycobacterial infection. J Immunol (2015) 194:5355–65.10.4049/jimmunol.1402557
177
OuimetMKosterSSakowskiERamkhelawonBvan SolingenCOldebekenSet alMycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol (2016) 17:677–86.10.1038/ni.3434
178
KumarRSahuSKKumarMJanaKGuptaPGuptaUDet alMicroRNA 17-5p regulates autophagy in Mycobacterium tuberculosis-infected macrophages by targeting Mcl-1 and STAT3. Cell Microbiol (2016) 18:679–91.10.1111/cmi.12540
179
GuXGaoYMuDGFuEQ. miR-23a-5p modulates mycobacterial survival and autophagy during Mycobacterium tuberculosis infection through TLR2/MyD88/NF-κB pathway. Exp Cell Res (2017) 354:71–7.10.1016/j.yexcr.2017.03.039
180
DoddCEPyleCJGlowinskiRRajaramMVSchlesingerLS. CD36-mediated uptake of surfactant lipids by human macrophages promotes intracellular growth of Mycobacterium tuberculosis. J Immunol (2016) 197:4727–35.10.4049/jimmunol.1600856
181
NordenfeltPGrinsteinSBjorckLTapperH. V-ATPase-mediated phagosomal acidification is impaired by Streptococcus pyogenes through Mga-regulated surface proteins. Microbes Infect (2012) 14:1319–29.10.1016/j.micinf.2012.08.005
182
StrasserJENewmanSLCiraoloGMMorrisREHowellMLDeanGE. Regulation of the macrophage vacuolar ATPase and phagosome-lysosome fusion by Histoplasma capsulatum. J Immunol (1999) 162:6148–54.
183
ToyookaKTakaiSKirikaeT. Rhodococcus equi can survive a phagolysosomal environment in macrophages by suppressing acidification of the phagolysosome. J Med Microbiol (2005) 54:1007–15.10.1099/jmm.0.46086-0
184
PujolCKleinKARomanovGAPalmerLECirotaCZhaoZet alYersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect Immun (2009) 77:2251–61.10.1128/IAI.00068-09
185
Fernández-ArenasEBleckCKNombelaCGilCGriffithsGDiez-OrejasR. Candida albicans actively modulates intracellular membrane trafficking in mouse macrophage phagosomes. Cell Microbiol (2009) 11:560–89.10.1111/j.1462-5822.2008.01274.x
186
MoslehIMHuberLASteinleinPPasqualiCGuntherDMeyerTF. Neisseria gonorrhoeae porin modulates phagosome maturation. J Biol Chem (1998) 273:35332–8.10.1074/jbc.273.52.35332
187
BinkerMGCosen-BinkerLITerebiznikMRMalloGVMcCawSEEskelinenELet alArrested maturation of Neisseria-containing phagosomes in the absence of the lysosome-associated membrane proteins, LAMP-1 and LAMP-2. Cell Microbiol (2007) 9:2153–66.10.1111/j.1462-5822.2007.00946.x
188
HertzenEJohanssonLWallinRSchmidtHKrollMRehnAPet alM1 protein-dependent intracellular trafficking promotes persistence and replication of Streptococcus pyogenes in macrophages. J Innate Immun (2010) 2:534–45.10.1159/000317635
189
KaganJCRoyCR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol (2002) 4:945–54.10.1038/ncb883
190
EwannFHoffmanPS. Cysteine metabolism in Legionella pneumophila: characterization of an L-cystine-utilizing mutant. Appl Environ Microbiol (2006) 72:3993–4000.10.1128/AEM.00684-06
191
CelliJde ChastellierCFranchiniDMPizarro-CerdaJMorenoEGorvelJP. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med (2003) 198:545–56.10.1084/jem.20030088
192
GhigoEColomboMIHeinzenRA. The Coxiella burnetii parasitophorous vacuole. Adv Exp Med Biol (2012) 984:141–69.10.1007/978-94-007-4315-1_8
193
GrahamJGMacDonaldLJHussainSKSharmaUMKurtenRCVothDE. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol (2013) 15:1012–25.10.1111/cmi.12096
194
HolmATejleKMagnussonKEDescoteauxARasmussonB. Leishmania donovani lipophosphoglycan causes periphagosomal actin accumulation: correlation with impaired translocation of PKCα and defective phagosome maturation. Cell Microbiol (2001) 3:439–47.10.1046/j.1462-5822.2001.00127.x
195
RodriguezNEGaur DixitUAllenLAWilsonME. Stage-specific pathways of Leishmania infantum chagasi entry and phagosome maturation in macrophages. PLoS One (2011) 6:e19000.10.1371/journal.pone.0019000
196
FrattiRAChuaJDereticV. Induction of p38 mitogen-activated protein kinase reduces early endosome autoantigen 1 (EEA1) recruitment to phagosomal membranes. J Biol Chem (2003) 278:46961–7.10.1074/jbc.M305225200
197
HorwitzMA. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med (1983) 158:1319–31.10.1084/jem.158.4.1319
198
CoersJMonahanCRoyCR. Modulation of phagosome biogenesis by Legionella pneumophila creates an organelle permissive for intracellular growth. Nat Cell Biol (1999) 1:451–3.10.1038/15687
199
NagaiHKaganJCZhuXKahnRARoyCR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science (2002) 295:679–82.10.1126/science.1067025
200
ArenasGNGrilliDJSamartinoLEMagadanJMayorgaLS. Brucella alters endocytic pathway in J774 macrophages. Virulence (2010) 1:376–85.10.4161/viru.1.5.12861
201
Arellano-ReynosoBLapaqueNSalcedoSBrionesGCiocchiniAEUgaldeRet alCyclic β-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nat Immunol (2005) 6:618–25.10.1038/ni1202
202
PuriRVReddyPVTyagiAK. Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues. PLoS One (2013) 8:e70514.10.1371/journal.pone.0070514
203
MortonCOBouzaniMLoefflerJRogersTR. Direct interaction studies between Aspergillus fumigatus and human immune cells; what have we learned about pathogenicity and host immunity?Front Microbiol (2012) 3:413.10.3389/fmicb.2012.00413
204
SacksDSherA. Evasion of innate immunity by parasitic protozoa. Nat Immunol (2002) 3:1041–7.10.1038/ni1102-1041
205
ThywissenAHeinekampTDahseHMSchmaler-RipckeJNietzscheSZipfelPFet alConidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis pathway. Front Microbiol (2011) 2:96.10.3389/fmicb.2011.00096
206
BeraAHerbertSJakobAVollmerWGotzF. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol (2005) 55:778–87.10.1111/j.1365-2958.2004.04446.x
207
HerbertSBeraANerzCKrausDPeschelAGoerkeCet alMolecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog (2007) 3:e102.10.1371/journal.ppat.0030102
208
JinTBokarewaMFosterTMitchellJHigginsJTarkowskiA. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol (2004) 172:1169–76.10.4049/jimmunol.172.2.1169
209
PeschelAJackRWOttoMCollinsLVStaubitzPNicholsonGet alStaphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L-lysine. J Exp Med (2001) 193:1067–76.10.1084/jem.193.9.1067
210
WeidenmaierCPeschelA. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol (2008) 6:276–87.10.1038/nrmicro1861
211
Sieprawska-LupaMMydelPKrawczykKWojcikKPukloMLupaBet alDegradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother (2004) 48:4673–9.10.1128/AAC.48.12.4673-4679.2004
212
LiuGYEssexABuchananJTDattaVHoffmanHMBastianJFet alStaphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med (2005) 202:209–15.10.1084/jem.20050846
213
BallalAMannaAC. Regulation of superoxide dismutase (sod) genes by SarA in Staphylococcus aureus. J Bacteriol (2009) 191:3301–10.10.1128/JB.01496-08
214
MandellGL. Catalase, superoxide dismutase, and virulence of Staphylococcus aureus. In vitro and in vivo studies with emphasis on staphylococcal-leukocyte interaction. J Clin Invest (1975) 55:561–6.10.1172/JCI107963
215
CosgroveKCouttsGJonssonIMTarkowskiAKokai-KunJFMondJJet alCatalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol (2007) 189:1025–35.10.1128/JB.01524-06
216
GonçalvesVLNobreLSVicenteJBTeixeiraMSaraivaLM. Flavohemoglobin requires microaerophilic conditions for nitrosative protection of Staphylococcus aureus. FEBS Lett (2006) 580:1817–21.10.1016/j.febslet.2006.02.039
217
RichardsonARDunmanPMFangFC. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol (2006) 61:927–39.10.1111/j.1365-2958.2006.05290.x
218
BoreELangsrudSLangsrudORodeTMHolckA. Acid-shock responses in Staphylococcus aureus investigated by global gene expression analysis. Microbiology (2007) 153:2289–303.10.1099/mic.0.2007/005942-0
219
BeasleyFCVinesEDGriggJCZhengQLiuSLajoieGAet alCharacterization of staphyloferrin A biosynthetic and transport mutants in Staphylococcus aureus. Mol Microbiol (2009) 72:947–63.10.1111/j.1365-2958.2009.06698.x
220
CheungJBeasleyFCLiuSLajoieGAHeinrichsDE. Molecular characterization of staphyloferrin B biosynthesis in Staphylococcus aureus. Mol Microbiol (2009) 74:594–608.10.1111/j.1365-2958.2009.06880.x
221
SchwartzJTAllenLA. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J Leukoc Biol (2006) 79:1214–25.10.1189/jlb.0106030
222
JohnstonSAMayRC. Cryptococcus interactions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen. Cell Microbiol (2013) 15:403–11.10.1111/cmi.12067
223
Mirbod-DonovanFSchallerRHungCYXueJReichardUColeGT. Urease produced by Coccidioides posadasii contributes to the virulence of this respiratory pathogen. Infect Immun (2006) 74:504–15.10.1128/IAI.74.1.504-515.2006
224
MalachowaNKohlerPLSchlievertPMChuangONDunnyGMKobayashiSDet alCharacterization of a Staphylococcus aureus surface virulence factor that promotes resistance to oxidative killing and infectious endocarditis. Infect Immun (2011) 79:342–52.10.1128/IAI.00736-10
225
KinkelTLRouxCMDunmanPMFangFC. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio (2013) 4:e00696–13.10.1128/mBio.00696-13
226
SethiDMahajanSSinghCLamaAHadeMDGuptaPet alLipoprotein LprI of Mycobacterium tuberculosis acts as a lysozyme inhibitor. J Biol Chem (2016) 291:2938–53.10.1074/jbc.M115.662593
227
MillerJLVelmuruganKCowanMJBrikenV. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog (2010) 6:e1000864.10.1371/journal.ppat.1000864
228
ShinDMJeonBYLeeHMJinHSYukJMSongCHet alMycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog (2010) 6:e1001230.10.1371/journal.ppat.1001230
229
GuoYDengYHuangZLuoQPengYChenJet alEBP50 induces apoptosis in macrophages by upregulating nitric oxide production to eliminate intracellular Mycobacterium tuberculosis. Sci Rep (2016) 6:18961.10.1038/srep18961
230
SerafiniABoldrinFPalùGManganelliR. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol (2009) 191:6340–4.10.1128/JB.00756-09
231
SiegristMSSteigedalMAhmadRMehraADragsetMSSchusterBMet alMycobacterial Esx-3 requires multiple components for iron acquisition. MBio (2014) 5:e01073–14.10.1128/mBio.01073-14
232
WellsRMJonesCMXiZSpeerADanilchankaODoornbosKSet alDiscovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog (2013) 9:e1003120.10.1371/journal.ppat.1003120
233
BeasleyFCMaroldaCLCheungJBuacSHeinrichsDE. Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by staphyloferrin A, staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect Immun (2011) 79:2345–55.10.1128/IAI.00117-11
234
RodriguezGMSmithI. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol (2006) 188:424–30.10.1128/JB.188.2.424-430.2006
235
RyndakMBWangSSmithIRodriguezGM. The Mycobacterium tuberculosis high-affinity iron importer, IrtA, contains an FAD-binding domain. J Bacteriol (2010) 192:861–9.10.1128/JB.00223-09
236
PandeyAKSassettiCM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A (2008) 105:4376–80.10.1073/pnas.0711159105
237
SinghVJamwalSJainRVermaPGokhaleRRaoKV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe (2012) 12:669–81.10.1016/j.chom.2012.09.012
238
EohHRheeKY. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids. Proc Natl Acad Sci U S A (2014) 111:4976–81.10.1073/pnas.1400390111
239
VornhagenJAdams WaldorfKMRajagopalL. Perinatal group B streptococcal infections: virulence factors, immunity, and prevention strategies. Trends Microbiol (2017).10.1016/j.tim.2017.05.013
240
MaiseyHCDoranKSNizetV. Recent advances in understanding the molecular basis of group B Streptococcus virulence. Expert Rev Mol Med (2008) 10:e27.10.1017/S1462399408000811
241
RajagopalL. Understanding the regulation of group B streptococcal virulence factors. Future Microbiol (2009) 4:201–21.10.2217/17460913.4.2.201
242
BorlaceGNKeepSJProdoehlMJJonesHFButlerRNBrooksDA. A role for altered phagosome maturation in the long-term persistence of Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol (2012) 303:G169–79.10.1152/ajpgi.00320.2011
243
BasuMCzinnSJBlanchardTG. Absence of catalase reduces long-term survival of Helicobacter pylori in macrophage phagosomes. Helicobacter (2004) 9:211–6.10.1111/j.1083-4389.2004.00226.x
244
GobertAPMcGeeDJAkhtarMMendzGLNewtonJCChengYet alHelicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A (2001) 98:13844–9.10.1073/pnas.241443798
245
GobertAPWilsonKT. The immune battle against Helicobacter pylori infection: NO offense. Trends Microbiol (2016) 24:366–76.10.1016/j.tim.2016.02.005
246
HwangCSRhieGEOhJHHuhWKYimHSKangSO. Copper- and zinc-containing superoxide dismutase (Cu/ZnSOD) is required for the protection of Candida albicans against oxidative stresses and the expression of its full virulence. Microbiology (2002) 148:3705–13.10.1099/00221287-148-11-3705
247
EnjalbertBMacCallumDMOddsFCBrownAJ. Niche-specific activation of the oxidative stress response by the pathogenic fungus Candida albicans. Infect Immun (2007) 75:2143–51.10.1128/IAI.01680-06
248
MayerFLWilsonDHubeB. Candida albicans pathogenicity mechanisms. Virulence (2013) 4:119–28.10.4161/viru.22913
249
HolbrookEDSmolnyckiKAYouseffBHRappleyeCA. Redundant catalases detoxify phagocyte reactive oxygen and facilitate Histoplasma capsulatum pathogenesis. Infect Immun (2013) 81:2334–46.10.1128/IAI.00173-13
250
BrownSMCampbellLTLodgeJK. Cryptococcus neoformans, a fungus under stress. Curr Opin Microbiol (2007) 10:320–5.10.1016/j.mib.2007.05.014
251
ZaragozaOChrismanCJCastelliMVFrasesSCuenca-EstrellaMRodriguez-TudelaJLet alCapsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell Microbiol (2008) 10:2043–57.10.1111/j.1462-5822.2008.01186.x
252
RoccoNMCarmenJCKleinBS. Blastomyces dermatitidis yeast cells inhibit nitric oxide production by alveolar macrophage inducible nitric oxide synthase. Infect Immun (2011) 79:2385–95.10.1128/IAI.01249-10
253
Kehl-FieTEZhangYMooreJLFarrandAJHoodMIRathiSet alMntABC and MntH contribute to systemic Staphylococcus aureus infection by competing with calprotectin for nutrient manganese. Infect Immun (2013) 81:3395–405.10.1128/IAI.00420-13
254
SchrettlMHaasH. Iron homeostasis – Achilles’ heel of Aspergillus fumigatus?Curr Opin Microbiol (2011) 14:400–5.10.1016/j.mib.2011.06.002
255
InglisDOVoorhiesMHocking MurrayDRSilA. Comparative transcriptomics of infectious spores from the fungal pathogen Histoplasma capsulatum reveals a core set of transcripts that specify infectious and pathogenic states. Eukaryot Cell (2013) 12:828–52.10.1128/EC.00069-13
256
VrommanFSubtilA. Exploitation of host lipids by bacteria. Curr Opin Microbiol (2014) 17:38–45.10.1016/j.mib.2013.11.003
257
AlvarezMCasadevallA. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr Biol (2006) 16:2161–5.10.1016/j.cub.2006.09.061
258
NicolaAMRobertsonEJAlbuquerquePDerengowski LdaSCasadevallA. Nonlytic exocytosis of Cryptococcus neoformans from macrophages occurs in vivo and is influenced by phagosomal pH. MBio (2011) 2:e00167–11.10.1128/mBio.00167-11
259
JohnstonSAMayRC. The human fungal pathogen Cryptococcus neoformans escapes macrophages by a phagosome emptying mechanism that is inhibited by Arp2/3 complex-mediated actin polymerisation. PLoS Pathog (2010) 6:e1001041.10.1371/journal.ppat.1001041
260
ChayakulkeereeMJohnstonSAOeiJBLevSWilliamsonPRWilsonCFet alSEC14 is a specific requirement for secretion of phospholipase B1 and pathogenicity of Cryptococcus neoformans. Mol Microbiol (2011) 80:1088–101.10.1111/j.1365-2958.2011.07632.x
261
SmithLMDixonEFMayRC. The fungal pathogen Cryptococcus neoformans manipulates macrophage phagosome maturation. Cell Microbiol (2015) 17:702–13.10.1111/cmi.12394
262
BainJMLewisLEOkaiBQuinnJGowNAErwigLP. Non-lytic expulsion/exocytosis of Candida albicans from macrophages. Fungal Genet Biol (2012) 49:677–8.10.1016/j.fgb.2012.01.008
263
García-RodasRGonzález-CamachoFRodríguez-TudelaJLCuenca-EstrellaMZaragozaO. The interaction between Candida krusei and murine macrophages results in multiple outcomes, including intracellular survival and escape from killing. Infect Immun (2011) 79:2136–44.10.1128/IAI.00044-11
264
SchnupfPPortnoyDA. Listeriolysin O: a phagosome-specific lysin. Microbes Infect (2007) 9:1176–87.10.1016/j.micinf.2007.05.005
265
GoldfineHWadsworthSJJohnstonNC. Activation of host phospholipases C and D in macrophages after infection with Listeria monocytogenes. Infect Immun (2000) 68:5735–41.10.1128/IAI.68.10.5735-5741.2000
266
KocksCGouinETabouretMBerchePOhayonHCossartP. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell (1992) 68:521–31.10.1016/0092-8674(92)90188-I
267
PistorSGröbeLSechiASDomannEGerstelBMacheskyLMet alMutations of arginine residues within the 146-KKRRK-150 motif of the ActA protein of Listeria monocytogenes abolish intracellular motility by interfering with the recruitment of the Arp2/3 complex. J Cell Sci (2000) 113:3277–87.
268
PortnoyDAAuerbuchVGlomskiIJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol (2002) 158:409–14.10.1083/jcb.200205009
269
WangRBraughtonKRKretschmerDBachTHQueckSYLiMet alIdentification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med (2007) 13:1510–4.10.1038/nm1656
270
PeschelAOttoM. Phenol-soluble modulins and staphylococcal infection. Nat Rev Microbiol (2013) 11:667–73.10.1038/nrmicro3110
271
SinghRJamiesonACresswellP. GILT is a critical host factor for Listeria monocytogenes infection. Nature (2008) 455:1244–7.10.1038/nature07344
272
MarteynBGaziASansonettiP. Shigella: a model of virulence regulation in vivo. Gut Microbes (2012) 3:104–20.10.4161/gmic.19325
273
EgileCLoiselTPLaurentVLiRPantaloniDSansonettiPJet alActivation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol (1999) 146:1319–32.10.1083/jcb.146.6.1319
274
WalkerDHFengHMPopovVL. Rickettsial phospholipase A2 as a pathogenic mechanism in a model of cell injury by typhus and spotted fever group rickettsiae. Am J Trop Med Hyg (2001) 65:936–42.10.4269/ajtmh.2001.65.936
275
GouinEEgileCDehouxPVilliersVAdamsJGertlerFet alThe RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature (2004) 427:457–61.10.1038/nature02318
276
SurewaardBGde HaasCJVervoortFRigbyKMDeLeoFROttoMet alStaphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol (2013) 15:1427–37.10.1111/cmi.12130
277
MyrvikQNLeakeESWrightMJ. Disruption of phagosomal membranes of normal alveolar macrophages by the H37Rv strain of Mycobacterium tuberculosis. A correlate of virulence. Am Rev Respir Dis (1984) 129:322–8.
278
SimeoneRBobardALippmannJBitterWMajlessiLBroschRet alPhagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog (2012) 8:e1002507.10.1371/journal.ppat.1002507
279
StammLMMorisakiJHGaoLYJengRLMcDonaldKLRothRet alMycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med (2003) 198:1361–8.10.1084/jem.20031072
280
StammLMPakMAMorisakiJHSnapperSBRottnerKLommelSet alRole of the WASP family proteins for Mycobacterium marinum actin tail formation. Proc Natl Acad Sci U S A (2005) 102:14837–42.10.1073/pnas.0504663102
281
GaoLYGuoSMcLaughlinBMorisakiHEngelJNBrownEJ. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol (2004) 53:1677–93.10.1111/j.1365-2958.2004.04261.x
282
SmithJManoranjanJPanMBohsaliAXuJLiuJet alEvidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun (2008) 76:5478–87.10.1128/IAI.00614-08
283
van der WelNHavaDHoubenDFluitsmaDvan ZonMPiersonJet alM. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell (2007) 129:1287–98.10.1016/j.cell.2007.05.059
284
SimeoneRSayesFSongOGröschelMIBrodinPBroschRet alCytosolic access of Mycobacterium tuberculosis: critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PLoS Pathog (2015) 6:e1004650.10.1371/journal.ppat.1004650
285
RussellDG. The ins and outs of the Mycobacterium tuberculosis-containing vacuole. Cell Microbiol (2016) 18:1065–9.10.1111/cmi.12623
286
DanilchankaOSunJPavlenokMMaueröderCSpeerASiroyAet alAn outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc Natl Acad Sci U S A (2014) 111:6750–5.10.1073/pnas.1400136111
287
SunJSiroyALokareddyRKSpeerADoornbosKSCingolaniGet alThe tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat Struct Mol Biol (2015) 22:672–8.10.1038/nsmb.3064
288
QuigleyJHughittVKVelikovskyCAMariuzzaRAEl-SayedNMBrikenV. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. MBio (2017) 8:e00148–17.10.1128/mBio.00148-17
289
SteigerJStephanAInkelesMSRealegenoSBrunsHKröllPet alImatinib triggers phagolysosome acidification and antimicrobial activity against Mycobacterium bovis bacille Calmette-Guérin in glucocorticoid-treated human macrophages. J Immunol (2016) 197:222–32.10.4049/jimmunol.1502407
290
ZhouJXuRDuXZZhouXDLiQ. Saxifragifolin D attenuates phagosome maturation arrest in Mycobacterium tuberculosis-infected macrophages via an AMPK and VPS34-dependent pathway. AMB Express (2017) 7:11.10.1186/s13568-016-0317-6
291
WangZSunDChenGLiGDouSWangRet alTim-3 inhibits macrophage control of Listeria monocytogenes by inhibiting Nrf2. Sci Rep (2017) 7:42095.10.1038/srep42095
292
KumarSKSinghPSinhaS. Naturally produced opsonizing antibodies restrict the survival of Mycobacterium tuberculosis in human macrophages by augmenting phagosome maturation. Open Biol (2015) 5:150171.10.1098/rsob.150171
293
ThomerLEmoloCThammavongsaVKimHKMcAdowMEYuWet alAntibodies against a secreted product of Staphylococcus aureus trigger phagocytic killing. J Exp Med (2016) 213:293–301.10.1084/jem.20150074
294
ThammavongsaVRauchSKimHKMissiakasDMSchneewindO. Protein A-neutralizing monoclonal antibody protects neonatal mice against Staphylococcus aureus. Vaccine (2015) 33:523–6.10.1016/j.vaccine.2014.11.051
295
McGuinnessWAKobayashiSDDeLeoFR. Evasion of neutrophil killing by Staphylococcus aureus. Pathogens (2016) 5:E32.10.3390/pathogens5010032
296
HerwegJAHansmeierNOttoAGeffkenACSubbarayalPPrustyBKet alPurification and proteomics of pathogen-modified vacuoles and membranes. Front Cell Infect Microbiol (2015) 5:48.10.3389/fcimb.2015.00048
297
MattowJSiejakFHagensKBecherDAlbrechtDKrahAet alProteins unique to intraphagosomally grown Mycobacterium tuberculosis. Proteomics (2006) 6:2485–94.10.1002/pmic.200500547
298
StuartLMBoulaisJCharriereGMHennessyEJBrunetSJutrasIet alA systems biology analysis of the Drosophila phagosome. Nature (2007) 445:95–101.10.1038/nature05380
299
ShevchukOBatzillaCHägeleSKuschHEngelmannSHeckerMet alProteomic analysis of Legionella-containing phagosomes isolated from Dictyostelium. Int J Med Microbiol (2009) 299:489–508.10.1016/j.ijmm.2009.03.006
300
DillBDGierlinskiMHärtlovaAArandillaAGGuoMClarkeRGet alQuantitative proteome analysis of temporally resolved phagosomes following uptake via key phagocytic receptors. Mol Cell Proteomics (2015) 14:1334–49.10.1074/mcp.M114.044594
301
CantonJGrinsteinS. Measuring phagosomal pH by fluorescence microscopy. Methods Mol Biol (2017) 1519:185–99.10.1007/978-1-4939-6581-6_12
302
SteinbergBMagalhaesMGrinsteinS. Expression of genetically encoded fluorescent probes to monitor phospholipid dynamics in live neutrophils. Methods Mol Biol (2014) 1124:269–77.10.1007/978-1-62703-845-4_17
303
LuSMGrinsteinSFairnGD. Quantitative live-cell fluorescence microscopy during phagocytosis. Methods Mol Biol (2017) 1519:79–91.10.1007/978-1-4939-6581-6_6
304
QueredaJJSachseMBalestrinoDGrenierTFredlundJDanckaertAet alAssessing vacuolar escape of Listeria monocytogenes. Methods Mol Biol (2017) 1535:173–95.10.1007/978-1-4939-6673-8_11
305
TanSYatesRMRussellDG. Mycobacterium tuberculosis: readouts of bacterial fitness and the environment within the phagosome. Methods Mol Biol (2017) 1519:333–47.10.1007/978-1-4939-6581-6_23
306
LiuYTanSHuangLAbramovitchRBRohdeKHZimmermanMDet alImmune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J Exp Med (2016) 213:213.10.1084/jem.20151248
307
KulkarniRCaskeyJSinghSKPaudelSBaralPSchexnayderMet alCigarette smoke extract-exposed methicillin-resistant Staphylococcus aureus regulates leukocyte function for pulmonary persistence. Am J Respir Cell Mol Biol (2016) 55:586–601.10.1165/rcmb.2015-0397OC
308
LiuQLiuXYanFHeYWeiJZhangYet alComparative transcriptome analysis of Brucella melitensis in an acidic environment: identification of the two-component response regulator involved in the acid resistance and virulence of Brucella. Microb Pathog (2016) 91:92–8.10.1016/j.micpath.2015.11.007
309
FerrarezePAGStreitRSASantosPRDSantosFMDAlmeidaRMCSchrankAet alTranscriptional analysis allows genome reannotation and reveals that Cryptococcus gattii VGII undergoes nutrient restriction during infection. Microorganisms (2017) 5:E49.10.3390/microorganisms5030049
310
WuMGibbonsJGDeLoidGMBedugnisASThimmulappaRKBiswalSet alImmunomodulators targeting MARCO expression improve resistance to postinfluenza bacterial pneumonia. Am J Physiol Lung Cell Mol Physiol (2017) 313:L138–53.10.1152/ajplung.00075.2017
311
KalamHFontanaMFKumarD. Alternate splicing of transcripts shape macrophage response to Mycobacterium tuberculosis infection. PLoS Pathog (2017) 13:e1006236.10.1371/journal.ppat.1006236
312
O’NeillLAPearceEJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213:15–23.10.1084/jem.20151570
313
LecubeAPachónGPetrizJHernándezCSimóR. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS One (2011) 6:e23366.10.1371/journal.pone.0023366
314
MartinezNKetheesanNWestKVallerskogTKornfeldH. Impaired recognition of Mycobacterium tuberculosis by alveolar macrophages from diabetic mice. J Infect Dis (2016) 214:1629–37.10.1093/infdis/jiw436
315
KimmeyJMHuynhJPWeissLAParkSKambalADebnathJet alUnique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature (2015) 528:565–9.10.1038/nature16451
316
RengarajanMHayerATheriotJA. Endothelial cells use a formin-dependent phagocytosis-like process to internalize the bacterium Listeria monocytogenes. PLoS Pathog (2016) 12:e1005603.10.1371/journal.ppat.1005603
317
Greenlee-WackerMC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev (2016) 273:357–70.10.1111/imr.12453
318
MartinCJPetersKNBeharSM. Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol (2014) 17:17–23.10.1016/j.mib.2013.10.007
Summary
Keywords
macrophage, neutrophil, bacteria, infection, inflammation, phagosome maturation, phagolysosome
Citation
Uribe-Querol E and Rosales C (2017) Control of Phagocytosis by Microbial Pathogens. Front. Immunol. 8:1368. doi: 10.3389/fimmu.2017.01368
Received
13 August 2017
Accepted
05 October 2017
Published
24 October 2017
Volume
8 - 2017
Edited by
Elsa Anes, Universidade de Lisboa, Portugal
Reviewed by
Anthony George Tsolaki, Brunel University London, United Kingdom; Carmen Judith Serrano, Instituto Mexicano del Seguro Social, Mexico
Updates
Copyright
© 2017 Uribe-Querol and Rosales.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Rosales, carosal@unam.mx
Specialty section: This article was submitted to Microbial Immunology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.