 Katrin Habir
Katrin Habir Shahin Aeinehband
Shahin Aeinehband Fredrik Wermeling
Fredrik Wermeling Stephen Malin
Stephen Malin- Department of Medicine, Center for Molecular Medicine, Karolinska University Hospital, Karolinska Institutet, Stockholm, Sweden
The initiation, commitment, and terminal differentiation of the B cell lineage is stringently controlled by the coordinated action of various transcription factors. Among these, Arid3a has previously been implicated in regulating early B lymphopoiesis, humoral immune responses to phosphocholine, and furthermore to promote the B1 over the B2 cell lineage. We have now interrogated the function of Arid3a in the adult mouse using conditional mutagenesis. We demonstrate that loss of Arid3a does not affect early B cell development or lineage commitment but rather loss of this transcription factor results in a broad expansion of bone marrow B lymphopoiesis in a manner that reflects its developmental expression pattern. Furthermore, loss of Arid3a resulted in expanded splenic B cell numbers with the exception of the B1 lineage that was maintained at normal numbers. However, B1a lymphoyctes were reduced in the peritoneal cavity. In addition, antibody responses to phosphocholine were attenuated in the absence of Arid3a. Hence, functional Arid3a is required in mature B cells for specific immune responses and for generating normal numbers of B cells in a subset dependent manner.
Introduction
B lymphocytes form the humoral arm of adaptive immunity through their unique ability to construct and secrete antibodies. The B cell lineage can be broadly separated into the B1 and B2 lineages through the combination of surface markers, ontogeny, physiological location, and specificity for antigen (1). The microRNA Let-7 and RNA-binding protein LIN28b have a critical role in specifying this B1 versus B2 lymphocyte lineage (2).
Arid3a was identified through its ability to bind and transcriptionally regulate the immunoglobulin heavy chain gene (3, 4) and has been recently claimed to be a critical downstream target of the Let-7/LIN28b axis (5). There have been conflicting reports on the function of Arid3a within the B cell lineage. Germline deletion of Arid3a leads to multiple defects in both hematopoietic progenitors and bone marrow resident B cells, including B1 cells (6). Additional approaches using knockdown or overexpression of Arid3a followed by transplantation into lymphopenic hosts has led to the hypothesis that Arid3a is required for specifying the B1 cell fate over the B2 cell fate (5). Conversely, lymphocyte-specific deletion of Arid3a through RAG-deficient blastocyst complementation experiments did not reveal any significant differences in mature B cell development (6). Similarly, transgenic overexpression of a dominant-negative ARID3A protein had no effect on bone marrow B cell development or on the B1 versus B2 lineage in the peritoneum (7).
To circumvent the embryonic lethality of Arid3a germline deleted mice, we have created a conditional loss-of-function allele and combined this with B cell-specific Cre-mediated deletion. Comprehensive analysis of the B cell compartment in these mice revealed expansion of the bone marrow pro-B, pre-B, immature and recirculating stages of B cell development upon Arid3a deletion, as well as defects in humoral immunity. In notable contrast, adult B1 cells are reduced, but only in the peritoneal cavity, following the loss of Arid3a.
Results
Conditional Loss of Arid3a and B Cell Development
The expression pattern of the transcription factor Arid3a is believed to be largely restricted to the B cell lineage within the hematopoietic system (8). We first determined the pattern of Arid3a expression by examining the ImmGen database (9) and could confirm that Arid3a is restricted to B cells with the exception of prominent expression also within granulocytes. Surprisingly, Arid3a was not strongly expressed in hematopoietic stem cells or progenitors, but instead showed upregulation within B cell progenitors resident in the bone marrow (Figure 1A). Specifically, Arid3a expression was upregulated through successive maturation steps from the pro-B to immature B cell stages. Interestingly, B1 cells did show only moderately increased expression of Arid3a.
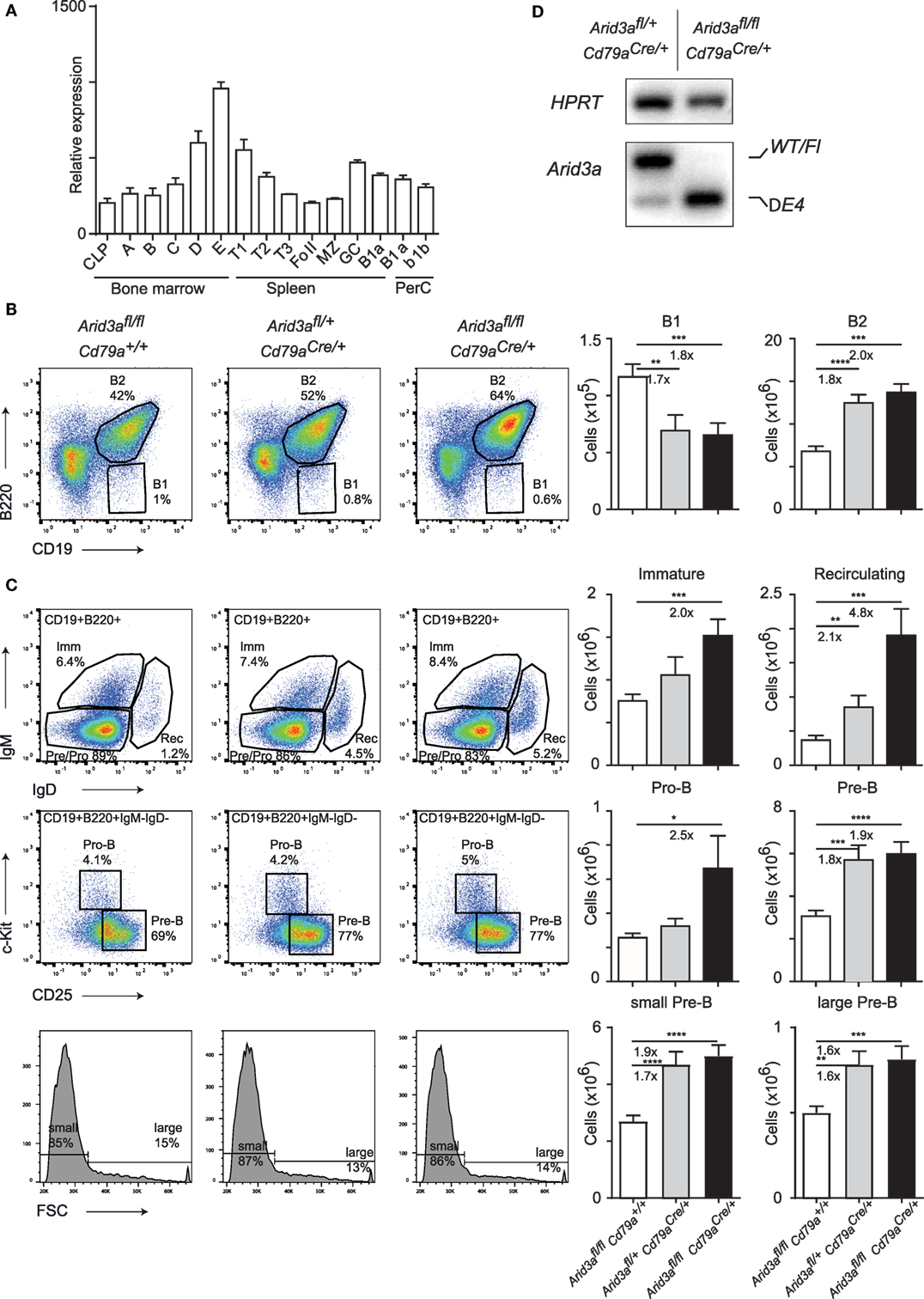
Figure 1. Conditional loss of Arid3a and B cell development. (A) Relative expression of Arid3a in B cell subsets in the bone marrow (common lymphoid progenitor and fractions A to E), spleen (transitional and mature stages), and peritoneal cavity using data from the ImmGen database. (B,C) Flow cytometry of bone marrow B cell development in Arid3afl/fl, Arid3afl/+ Cd79aCre/+, and Arid3afl/fl Cd79aCre/+ mice. Flow cytometry plots are shown on the left. Absolute cell numbers for B1 (CD19+B220lo), B2 (CD19+B220+), immature B (CD19+B220+IgM+IgD−), recirculating B (CD19+B220+IgM−IgD+), pro-B (CD19+B220+IgM−IgD− c-Kit+CD25−), and pre-B (CD19+B220+IgM−IgD−c-Kit−CD25+) cell populations are shown on the right. Error bars represent SEM and n = 17–24 for each group. p-Values were determined by Student’s t-test and fold changes are indicated. (D) Deletion of the conditional Arid3a allele determined by RT-polymerase chain reaction in sorted pro-B cells from the bone marrow of Arid3afl/+ Cd79aCre/+ and Arid3afl/fl Cd79aCre/+ aged 4–6 weeks of age. The full length wild-type and floxed allele (WT/Fl) and exon 4 deleted allele (ΔE4) are indicated. HPRT was used as a loading control.
We next wished to interrogate the in vivo function of Arid3a by genetic loss-of-function analysis. We generated a floxed allele of Arid3a by first removing the Lacz/Neo cassette of Arid3atm1a(KOMP)Wtsi by FLPE-mediated recombination to create an allele that contains Loxp sites flanking exon 4 (Figure S1A in Supplementary Material). This encodes for amino acids 237–261 that are crucial for Arid3a DNA binding (4), and additionally loss of exon 4 will result in an out of frame allele and a nonsense protein. We then crossed this floxed allele with the B cell-specific Cre line Cd79aCre (Mb1Cre), which initiates deletion at the transition from the common lymphoid progenitor to the pro-B cell stage of development (10). We therefore created littermate cohorts of Arid3afl/fl (herein referred to as control mice), Arid3afl/+Cd79aCre/+ (herein referred to as heterozygous mice), and Arid3afl/flCd79aCre/+ (herein referred to as homozygous mice) and analyzed the B cell compartment in the bone marrow of these mice at 4–6 weeks of age. We found that loss of Arid3a influenced the relative abundance of the B1 (defined B220loCD19+) populations in the bone marrow, with approximate 1.7-fold decreases in B1 cells in heterozygous versus control mice (p = 0.0047) and 1.8-fold decreases in homozygous versus control mice (p = 0.0006). In contrast, the B2 population (defined B220+CD19+) was expanded 1.7-fold in heterozygous versus control (p < 0.0001) and 2-fold increased in the homozygous versus control cohorts (p = 0.0006) (Figure 1B). We next determined which stage of B cell development was responsible for this increase in B2 cells. The first committed pro-B stage of B cell development (defined B220+CD19+IgM−IgD−c-Kit+CD25−) was increased 2.5-fold in homozygous mice upon loss of Arid3a (p = 0.038). Later stages of B cell development were also expanded. The pre-B population (defined B220+CD19+IgM−IgD−c-Kit−CD25+) including the large pre-B and small and pre-B subpopulations increased approximately 1.9-fold in both heterozygous and homozygous mice (control versus heterozygous p = 0.0001: control versus homozygous p < 0.0001). Immature-B (defined B220+CD19+IgM+IgD−) were increased 2-fold in the homozygous versus control mice (p = 0.038), whereas recirculating B cells (defined B220+CD19+IgM−IgD+) demonstrated dosage-dependent increases in cell number upon loss of Arid3a ranging from 2.1-fold (control versus heterozygous p = 0.0043) to 4.8-fold (control versus homozygous p = 0.0002) (Figure 1C; Figure S1B in Supplementary Material), resulting in an increase in bone marrow cellularity (Figure S1C in Supplementary Material).
Cd79aCre is well established as an efficient deleter of floxed alleles in B cells. We further confirmed this by sorting pro-B cells by fluorescence-activated cell sorting and assessing deletion of Arid3a at the mRNA level. Cells from heterozygous mice contained both wild-type and exon-4 deleted Arid3a, whereas cells sorted from homozygous mice only contained the deleted allele (Figure 1D). We further sequenced this deleted allele and confirmed that an out of frame mRNA is produced upon loss of exon 4 resulting in a protein with multiple premature stop codons (Figure S1D in Supplementary Material). In summary, loss of Arid3a leads to reductions in bone marrow B1 cell numbers and conversely a more noticeable expansion of all stages of B2 cell development.
Expanded Peripheral B2 Cell Populations in Arid3a-Deficient Mice
The expansion of B cell numbers in the bone marrow could result in further abnormalities in peripheral B cell numbers. We analyzed control, heterozygous, and homozygous mice at 10–12 weeks of age and quantified the absolute cell numbers of various B cell subsets in the spleen. We observed a twofold expansion (p = 0.0001) in B2 cell numbers in homozygous versus control mice, but no difference in B1 cell numbers. This increase in B2 cells was dosage dependent as loss of one Arid3a allele also resulted in increases in absolute numbers of B cells (p = 0.0032) (Figure 2A; Figure S2A in Supplementary Material), with subsequent increases in spleen cellularity and CD3+ cell numbers (Figures S2B,C in Supplementary Material). We next wished to determine which subset was responsible for the increase in B2 cell number. Transitional 1 B cells (B220+CD19+IgM+CD23−) were increased in both heterozygous (p = 0.0024) and homozygous (p = 0.0027) versus control mice. Transitional 2 B cells (B220+CD19+IgM+CD23+) were increased up to 2.4-fold (p = 0.0036) in homozygous mice versus controls (Figure 2B). An alternative gating strategy incorporating CD93 also revealed increases in immature splenic populations (Figure S2D in Supplementary Material). Similarly, the number of follicular B cells (defined B220+CD21+CD21loCD23+) was also increased in cell numbers and again appeared to be dosage sensitive to loss of Arid3a (control versus heterozygous p = 0.0022: control versus homozygous p < 0.0001). Marginal zone B cells (defined B220+CD21+CD21hiCD23lo) were also increased in homozygous versus control mice (p = 0.0095) (Figure 2C). Interestingly, surface IgM levels were decreased on follicular and marginal zone B cells in homozygous mice (Figure 2D). In summary, peripheral B cell numbers are expanded upon loss of Arid3a in similar fashion to B cell progenitors in the bone marrow.
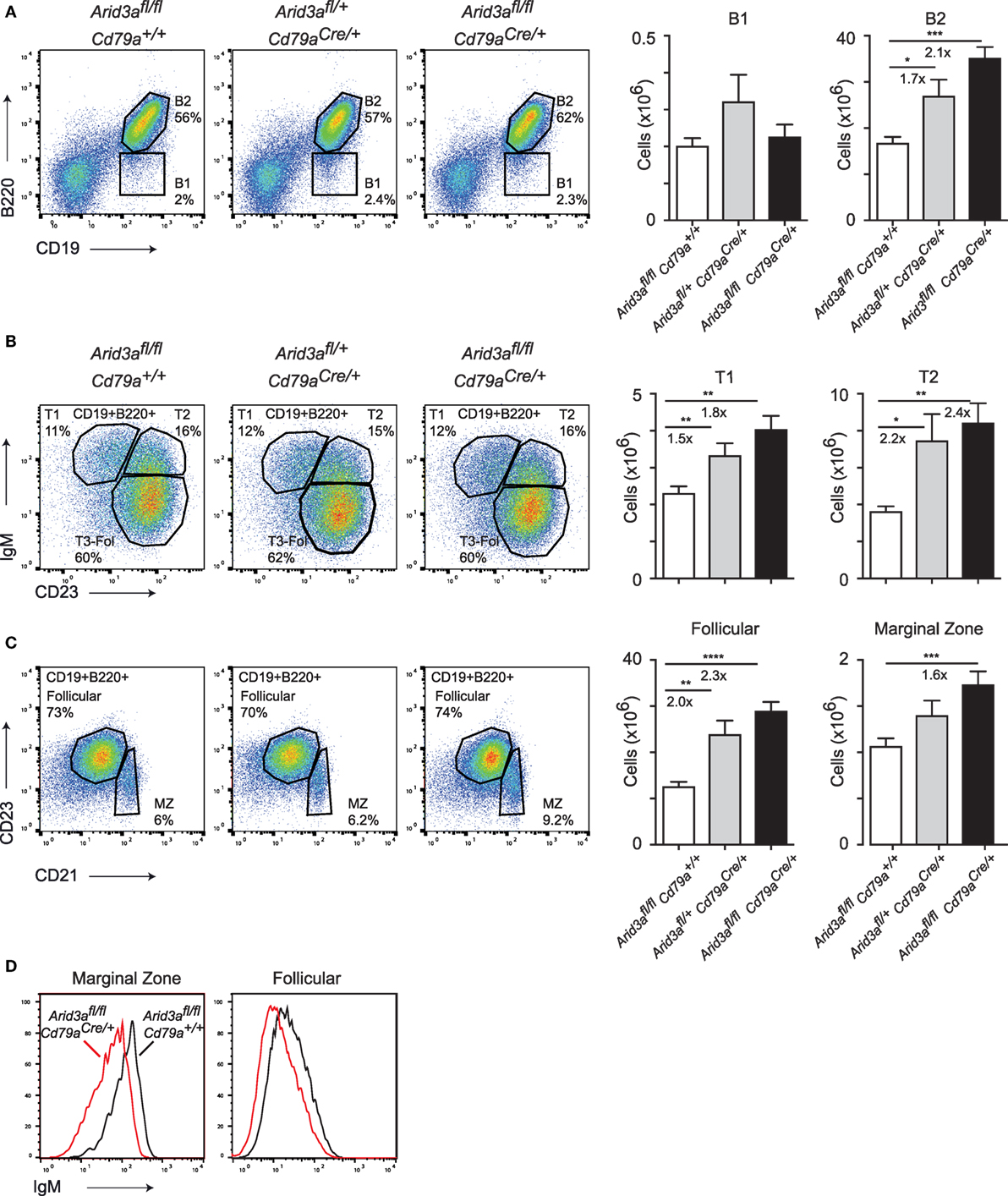
Figure 2. The in vivo function of Arid3a in splenic B cell development. (A) Flow cytometry of splenic B1 cells (CD19+B220lo) and B2 cells (CD19+B220+) in the left panel with quantification shown on the right for the indicated genotypes from mice aged 10–12 weeks. (B) Analysis of Transitional 1 (CD19+B220+IgM+CD23−) and Transitional 2 (CD19+B220+IgM+CD23+) B cells. (C) Follicular B (CD19+B220+CD21loCD23+) and marginal zone B (CD19+B220+CD21hi CD23lo) cell populations in the spleen. Error bars represent SEM and n = 9–32 for each group. p-Values were determined by Student’s t-test and fold changes are indicated. (D) Expression of IgM on marginal zone and follicular B cells.
The In Vivo Role of Arid3a in the B1 versus B2 Cell Development
Arid3a has recently been positioned as a key regulator of the B1 versus B2 lineage (5, 11). The reduction of B1 cells in the bone marrow indicated that this phenotype is also present upon conditional loss of Arid3a in the adult mouse. We therefore analyzed a key reservoir of B1 cells in the mouse, namely the peritoneal cavity. We observed a strong decrease in the relative abundance of B1a (CD19+B220loCD5a+) cells in homozygous mice, being 5.2-fold (p = 0.0306) reduced relative to controls. B2 cells and B1b (CD19+B220loCD5a−) cells were not significantly reduced in numbers (Figure 3A). These B1a cells were further confirmed to have a cell surface phenotype of IgMhi, IgDlo, and CD11b+ (Figure 3B). An alternative gating strategy to define B1 cells (CD19+CD11b+) produced similar results (Figures S3A,B in Supplementary Material). Finally, we confirmed that peritoneal lymphocytes were deleted for Arid3a by sorting B1 and B2 cells and determining Arid3a deletion. Similar to the results from bone marrow pro-B cells, homozygous mice displayed efficient deletion of exon 4 of Arid3a (Figure 3C). We could therefore confirm that Arid3a is required for normal peritoneal B1a cell development.
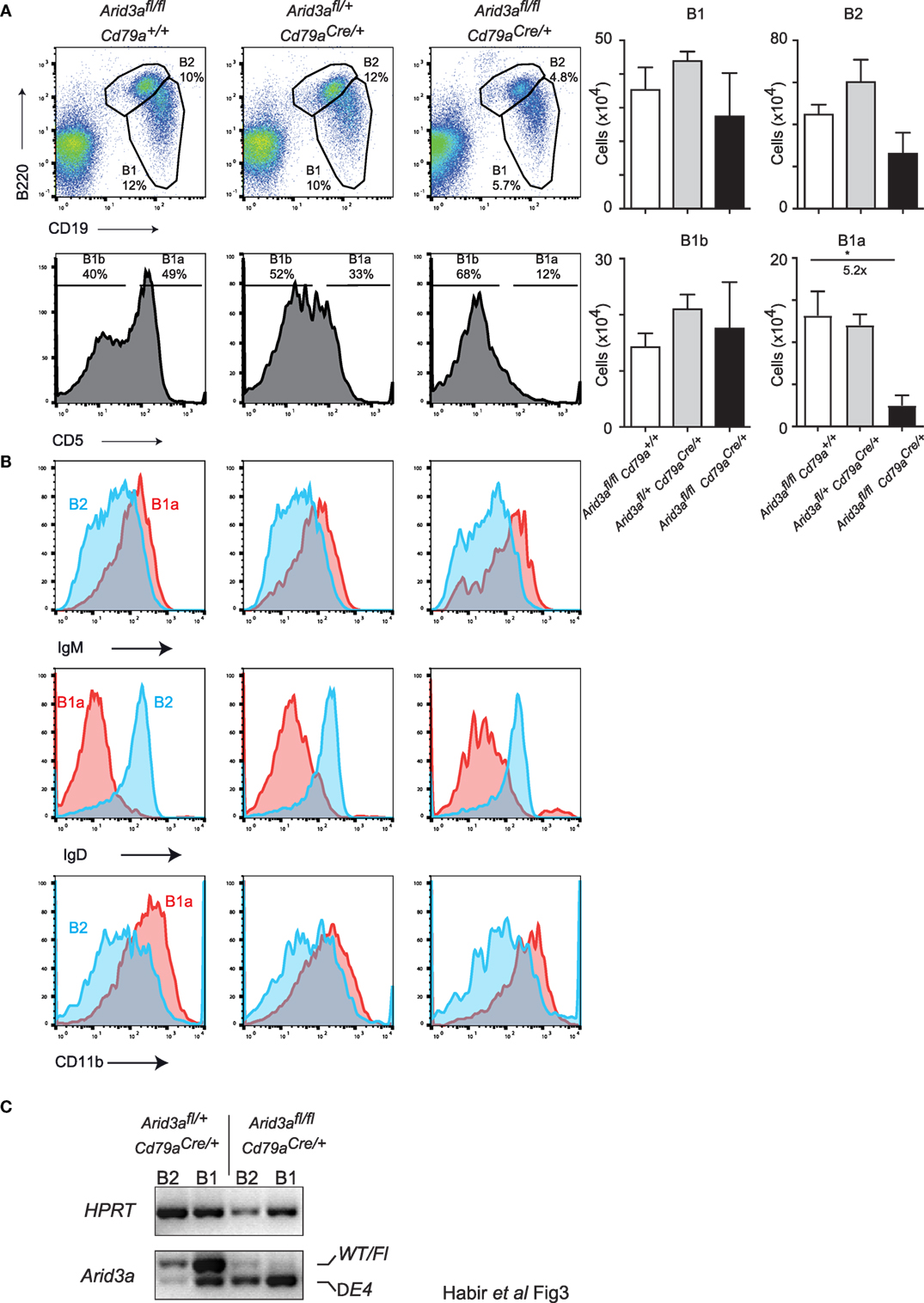
Figure 3. Arid3a is required for normal peritoneal cavity B1a cell development. (A) Flow cytometry of B1 cells (CD19+B220lo) and B2 cells (CD19+B220+) taken from peritoneal lavages of mice aged 10–12 weeks old. B1 cells were further gated for CD5 to determine B1a (CD19+B220loCD5+) and B1b (CD19+B220loCD5−) populations shown in the bottom panel. Error bars represent SEM and n = 3–8 for each group. p-Values were determined by Student’s t-test and fold changes are indicated. (B) Cell surface phenotype of B1a (CD19+B220loCD5+) and B2 (CD19+B220+) cells. (C) Deletion of the conditional Arid3a allele was shown by RT-polymerase chain reaction in B1 and B2 cells sorted from peritoneal cavity in Arid3afl/+ Cd79aCre/+ and Arid3afl/fl Cd79aCre/+ mice, 10–12 weeks of age. The full length wild-type and floxed allele (WT/Fl) and exon 4 deleted allele (ΔE4) are indicated. HPRT was used as a loading control.
The Function of Arid3a in Humoral Immunity
The humoral immune response of mouse B cells has been reported to be influenced by the transcription factor Arid3a. We measured total plasma immunoglobulin levels in naïve mice and found significant decreases in IgM (p = 0.0003), IgA (p = 0.039), and IgG (p = 0.0001) in homozygous mice compared to controls (Figure 4A), similar to that reported in mice expressing dominant-negative Arid3a (7) and the very rare survivor mice from the germline deleted Arid3a strain (6). Within the IgG fraction, levels of IgG1, IgG2b, and IgG3 were all significantly reduced (Figure 4B). We did not observe any significant differences in heterozygous mice relative to control mice.
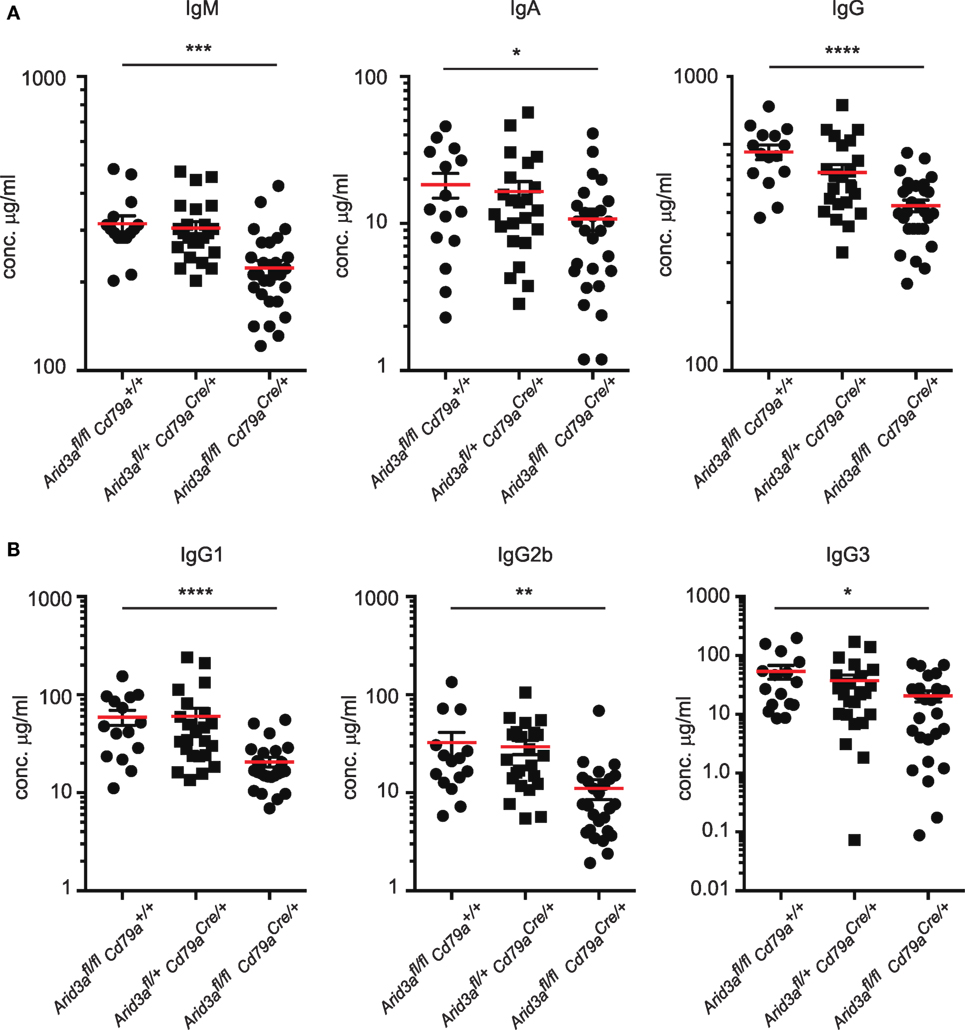
Figure 4. A requirement for Arid3a in humoral immunity. (A) Plasma was collected from naïve mice of the indicated genotypes aged 10–12 weeks old and analyzed for total IgM, IgA, and IgG levels with Enzyme-Linked ImmunoSorbent Assay. (B) IgG1, IgG2b, and IgG3 antibody titers. n = 15–26 for each group and p-values were determined by Student’s t-test.
A consistent feature of mice with perturbed Arid3a function is reduced antibody titers against the hapten phosphocholine, which in C57/Bl6 mice is partially dependent on V(D)J rearrangements that have incorporated VHS107.1.42 (12, 13). We assayed titers of IgM against phosphocholine and observed significant decreases in naïve homozygous mice versus that of controls (p = 0.0006) (Figure 5A). We next immunized mice with the antigen phosphocholine linked to Keyhole limpet hemocyanin (PC-KLH) emulsified in Alum to study germinal centre formation and specific T cell-dependent immune responses. We observed a reduction in anti-phosphocholine IgM responses 14 days after immunization (p = 0.0056) (Figure 5B). We next tested if this reduced anti-PC response was due to an inability to form germinal centers. Activated B cells (CD19+B220+IgDloCD95+GL7+) could be readily detected in the spleen following immunization in all genotypes and were also present in normal numbers in the Peyer’s patches and mesenteric lymph nodes regardless of the Arid3a genotype (Figure 5C).
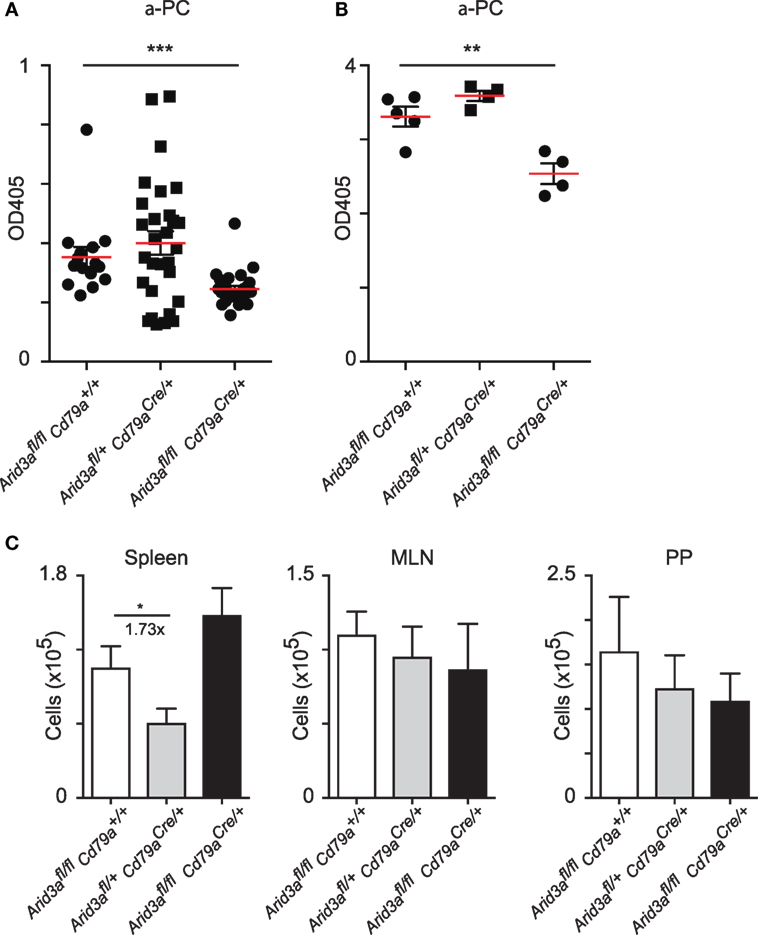
Figure 5. Normal antibody production against phosphocholine requires Arid3a. (A) Enzyme-Linked ImmunoSorbent Assay measurements of anti-phosphocholine IgM in naïve mice from the indicated genotypes n = 15–28 for each group. (B) Anti-phosphocholine IgM levels 14 days after PC-KLH immunization n = 4–5 for each group. (C) Absolute number of cells with a germinal center phenotype (CD19+B220+IgD−GL7+CD95+) in spleen, Peyer’s patches, and mesenteric lymph nodes after immunization with PC-KLH/Alum. n = 4–6 for each group. p-Values determined by Student’s t-test.
Discussion
The regulation of the B1 versus the B2 cell fate is thought to be controlled either through antigen receptor specificity within the immature and transitional B cell stages, as ontogenically separate lineages, or a combination of these two models (14). Several transcription factors have been shown to effect the relative abundance of B1 versus B2 cells in secondary lymphoid organs, including Ebf1 (15), classical NF-kappaB signaling (16), and most recently Bhlhe41 (17). Arid3a has also been proposed to critically regulate the B1 versus B2 cell fate (11). Using conditional mutagenesis in the adult mouse, we found no evidence that expression of Arid3a is required for the production of B1 lymphocytes at the expense of B2 cells in the spleen, in agreement with studies that used RAG-deficient blastocyst complementation with Arid3a germline deleted ES cells (6) and overexpression of dominant-negative form of Arid3a (7). However, we did observe a loss of B1a cells in the peritoneal cavity, indicating that Arid3a has a very specific function in the generation of peritoneal B1a lymphocytes and is possibly responsible for the reduction in the IgM titers we observed. One possibility is that Arid3a may regulate migration of B1a cells to the peritoneal cavity.
Loss of Arid3a resulted in the expansion of all the B cell developmental stages in the bone marrow. This stage specific affect mirrors the expression pattern of Arid3a. Previous analysis of the less than l% of germline deleted Arid3a mice that survived to adulthood had indicated a possible role for this transcription factor in early B cell development (6); however, this germline-deficient strain has strong reductions in the common lymphoid progenitor compartment. Mature B cells were generated normally from Arid3a−/− when using RAG-deficient blastocyst complementation, indicating that bone marrow B cell development had proceeded normally (6). Hence, the loss of B cell progenitors in Arid3a−/− is probably due to the profound developmental defects in hematopoietic progenitors that occur in this strain. Similar results were obtained by conditional deletion of the related Arid3b in the mouse, which also resulted in a reduction of common lymphoid progenitors (18). Interestingly, follicular B cells were also expanded in this strain. It is possible that Arid3a and Arid3b have redundant functions during B lymphopoiesis, and combined deletion of these two factors will be required to confirm this.
Although we could demonstrate that the mRNA from our deleted allele should result in a missense protein, we were not able to measure ARID3A protein levels in our experiments through western blot experiments despite using multiple commercially available antibodies. We cannot therefore formally rule out that a truncated protein is produced through evasion of nonsense-mediated decay. However, we note that loss of exon 4 in the highly related Arid3b deleted locus resulted in loss of this protein (18).
Heterozygous or homozygous loss of Arid3a resulted in a complex set of phenotypes. Within bone marrow B cell development, heterozygous animals had similar increases in pre-B cells as homozygous mice, whereas for pro-B, immature and recirculating cells, loss of both alleles produced a more prominent expansion. This dosage pattern was conserved in spleen populations. This expansion of B cell progenitors we observed may have implications for B cell acute lymphoblastic leukemia (B-ALL). Knockdown of ARID3A in 70Z/3 and Ba/F3 B cell progenitor cell lines resulted in increased proliferation, and ARID3A was downregulated in a subset of B-All patients (19). Our results would be consistent with Arid3a suppressing proliferation in B cell progenitors. Alternatively, Arid3a may also have a function in cell survival. Regardless, expansion of certain B-ALL clones may be sensitive to the dosage of ARID3A, as normal B lymphopoiesis is expanded upon loss of one or two Arid3a alleles.
Loss of Arid3a results in specific defects in humoral responses, especially a partial defect in IgM responses against phosphocholine. The defects observed in immature bone marrow B cell development in Arid3a-deficient B cells may partially be the consequence of a positive feedback loop, as mice that cannot secrete IgM have increased immature B cells (20). Hence, in Arid3a-deleted B cells, there is diminished production of phosphocholine binding natural IgM. This is probably resulting from reduced VHS107.1.42 expression, which is regulated by ARID3A (6). In contrast to bone marrow progenitors, however, the reduction in peritoneal B1a cells and plasma immunoglobulin levels was only observed in homozygous mice.
Although the defects we observed upon loss of Arid3a may appear subtle, increased ARID3A expression in human B cells correlates with the severity of systemic lupus erythematosus (21), which may be indicative of a more general function of this transcription factor in autoimmunity. The reduction in “natural” IgM against phosphocholine could make the Arid3afl/flCd79aCre/+ strain useful in disease models where these natural antibodies have been implicated, for example, in SLE models and atherosclerosis.
Materials and Methods
Mice
The Arid3atm1a(KOMP)Wtsi allele was imported from the KOMP resource and in vitro fertilized with sperm from the B6.Cg-Tg(ACTFLPe)9205Dym/J strain to remove the Frt flanked selection and reporter cassette to create Arid3afl/+ mice. Mice were then further crossed with the Cd79aCre/+ strain to create control and homozygous mice. All experiments used mice at 4–6 or 10–12 weeks of age and were approved by the Stockholm Regional Board for Animal Ethics, permit numbers N46/14, N164/14, and N111/11. Mice were regularly controlled by the Swedish Veterinary Authorities.
Genotyping with Polymerase Chain Reaction (PCR)
Polymerase chain reaction was performed after DNA extraction from ear biopsies of mice. The Arid3afl/fl and Arid3afl/+ DNA fragments were amplified with the primers: 5′-CTCCTTCCTCTTGTCTCCTGTGTGG-3′ (ttR) and 5′-TGCCTGTTGGAAGGATGATCTGG-3′ (F); and with the primers 5′-GAGATGGCGCAACGCAATTAATG-3′ (loxF) and 5′-GATAGCTCAGGAGCTTCCTCACACC-3′ (R). The Cd79aCre/+ fragment was amplified with the following primers: 5′-CCTTGCGAGGTCAGGGAGCC-3′ (WT reverse), 5′-GTCCTGGCATCTGTCAGAG-3′ (Cre reverse) and 5′-GGCTCTGACCCATCTGTCTC-3′ (common forward). Primers ttR and F amplified a 354-bp wild-type fragment and a 470-bp postFlp fragment. Primers loxF and R amplified a 253-bp floxed fragment. Primers WT reverse and common forward gave a 477-bp wild-type fragment, and primers Cre reverse and common forward gave a 500-bp Cd79aCre/specific product. PCR products were subsequently separated by electrophoresis through agarose gels and visualized by GelRed nucleic acid gel stain (Biotium).
Flow Cytometry
Bone marrow from Femur and Tibia of the two hind legs of 4–6 weeks old mice, and spleen and peritoneal cavity from 10 to 12 weeks old mice was isolated. Tissues were then fractioned to single-cell suspensions and filtered through 100-µm cell strainers (Sigma-Aldrich) with phosphate-buffered saline (PBS) containing 0.5% fetal bovine serum (FBS). Erythrocytes were thereafter lysed with Ammonium-Chloride-Potassium Lysing Buffer. Single-cell suspensions of bone marrow, spleen, and peritoneal cavity were pre-incubated at a density of 100 × 106 cells/ml with Fc Block (BD Biosciences), and cells were subsequently analyzed by a CyAn ADP (Beckman Coulter) flow cytometer with the following antibodies B220 (APC-Cy7 clone: RA3-6B2), anti-CD19 (Alexa Fluor 647 clone: eBio1D3), anti-CD25 (PE-Cy7 clone: PC61.5), anti-c-kit (PE clone: 2B8), anti-CD5 (PE clone: 53-7.3), anti-GL7 (APC clone: GL-7), anti-CD95 (PE-Cy7 clone: Jo2), anti-IgD (PerCP clone: 11-26c.2a), anti-IgM (Pacific Blue clone: Il/41), anti-CD23 (PE-Cy7 clone: B3B4), anti-CD21 (FITC clone: 7G6), and anti-CD11b (PerCPclone: M1/70). Immune cell populations were defined as follows: B-1 (CD19+B220lo) or (CD19+CD11b+), B-1a (CD19+B220loCD5+) or (CD19+CD11b+CD5+), B-1b (CD19+B220loCD5−) or (CD19+CD11b+CD5−), B-2 (CD19+B220+), pro-B (CD19+B220+IgM−IgD−c-Kit+CD25−), pre-B (CD19+B220+IgM−IgD−c-Kit−CD25+), immature-B cells (CD19+B220+IgM+IgD−) recirculating-B cells (CD19+B220+IgM−IgD+), Transitional-1 (CD19+B220+IgM+CD23−) or (CD19+B220+CD93+IgM+CD23−), Transitional-2 (CD19+B220+IgM+CD23+) or (CD19+B220+CD93+IgM+CD23+), marginal zone B cells (CD19+B220+CD21hiCD23lo), follicular B cells (CD19+B220+CD21intCD23hi) germinal center B cells (CD19+B220+IgD−GL7+CD95+). Absolute cell numbers were calculated by first counting live cells using a BioRad TC-20 cell counter based on trypan blue exclusion. Cells within a defined gated quadrant were divided by the cells contained in a large gate encompassing all live cells and excluding debris as determined by forward and side scatter properties from the flow cytometry. This ratio was then multiplied by the cell count to give the absolute cell number.
Cell Sorting and Deletion Analysis
Pro-B cells from bone marrow and B1 and B2 peritoneal cavity cells of Arid3afl/+Cd79aCre/+ and Arid3afl/flCd79aCre/+ mice were sorted into PBS containing 0.5% FBS before RNA extraction with the RNeasy Mini Kit (Qiagen). cDNA was synthesized with High capacity RNA-to-cDNA Kit (Applied Biosystems) followed by PCR with primers with the following Arid3a oligonucleotides: 5′-TGGACCTTTGAGGAGCAGTT-3′ (Primer 1) and 5′-GATGGAGGTAGGCAGGTTGA-3′ (Primer 2). A 255-bp PCR product was amplified from the floxed Arid3a allele and a 182-bp product from the deleted Arid3a allele. The Hypoxanthine guanine phosphoribosyl transferase (HPRT) gene was used as a control with primers GGGGGCTATAAGTTCTTTGC and TCCAACACTTCGAGAGGTCC generating a 312-bp PCR product. PCR products were then separated by electrophoresis through agarose gels and visualized by GelRed nucleic acid gel stain.
Immunization and Enzyme-Linked ImmunoSorbent Assay (ELISA)
For immunization with PC-KLH (Biosearch Technologies Inc.), 100 µg of PC-KLH was precipitated on Alum and subsequently injected intraperitoneally in mice. Spleens were analyzed by flow cytometry 14 days after immunization. Specific IgM responses against PC-KLH was determined by ELISA using plates coated with PC-BSA (Biosearch Technologies), alkaline phosphatase coupled goat anti-mouse IgM (Southern Biotechnology Associates), and alkaline phosphatase yellow Liquid Substrate (Sigma-Aldrich). Purified rat anti-mouse IgG (H + L) (Sigma-Aldrich) was used as capture antibody to measure total plasma IgM, IgA, and IgG levels. AP-labeled goat anti-mouse IgM, IgA and IgG, IgG1, IgG2b, and IgG3 (Sigma-Aldrich) was used for detection.
Statistical Analysis
Data were analyzed using GraphPad Prism. p-Values of <0.05 from Student’s t-tests were reported.
Ethics Statement
All experiments were approved by the Stockholm Regional Board for Animal Ethics, permit numbers N46/14, N164/14, and N111/11. Mice were regularly controlled by the Swedish Veterinary Authorities.
Author Contributions
KH performed most of the experiments. SA assisted with ELISA measurements. FW provided mouse lines and contributed to the planning of the experiments. SM devised the study, interpreted data and wrote the manuscript with contributions from all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Michael Reth and Meinrad Busslinger for providing the Cd79aCre/+ strain. This research was supported by the Swedish Medical Research Council (Vetenskapsrådet) to the Linné Center CERIC (Center of Excellence for Research on Inflammation and Cardiovascular disease), the EU FP7-Health-2013-innovation-1 programme Athero-B-Cell.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01387/full#supplementary-material.
Figure S1. Schematic of the targeted Arid3a locus and bone marrow flow cytometry. (A) The Frt flanked Neo/LacZ cassette was removed through FLPE-mediated excision to create an allele that contains LoxP sites flanking exon 4. Exon 4 partially encodes for the DNA-binding domain and loss of this exon results in an out of frame allele. (B) Expanded bone marrow flow cytometry gating strategy. (C) Absolute numbers of live bone marrow cells. Error bars represent SEM and n = 23–36 for each group. p-Values were determined by Student’s t-test and fold changes are indicated. (D) Sequence of the wild-type and deleted Arid3a locus across exons 3–5 with the translated sequence underneath. The nonsense protein predicted from the exon 4 deleted Arid3a was confirmed by sequencing of the mRNA.
Figure S2. Expanded flow cytometry analysis of the spleen. (A) An example of an expanded flow cytometry gating strategy for splenic B cells, indicating lymphocyte and large gates. (B) Absolute numbers of live splenic cells. Error bars represent SEM and n = 9–33 for each group. p-Values were determined by Student’s t-test and fold changes are indicated. (C) Absolute numbers of splenic CD3+ cells. n = 8–15 for each group. (D) Absolute cell numbers of transitional B cells gated as Immature (CD19+B220+CD93+), T1 (CD19+B220+CD93+IgM+CD23−), T2 (CD19+B220+CD93+IgM+CD23+), and T3 (CD19+B220+CD93+IgMloCD23+). n = 9–15 for each group.
Figure S3. Alternative gating analysis for peritoneal cavity B1 cells. (A) Flow cytometry of B1 cells (CD19+CD11b+) taken from peritoneal lavages of mice aged 10–12 weeks old, gated for CD5, to determine B1a (CD19+CD11b+CD5+) and B1b (CD19+B220loCD5−) populations. Error bars represent SEM and n = 3–8 for each group. p-Values were determined by Student’s t-test and fold changes are indicated. (B) Cell surface phenotype of B1a (CD19+CD11b+CD5+) and B2 cells (CD19+CD11b+).
References
1. Kantor A. A new nomenclature for B cells. Immunol Today (1991) 12:388. doi:10.1016/0167-5699(91)90135-G
2. Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science (2012) 335:1195. doi:10.1126/science.1216557
3. Das C, Eaton S, Calame K, Tucker PW. Novel protein-DNA interactions associated with increased immunoglobulin transcription in response to antigen plus interleukin-5. Mol Cell Biol (1991) 11:5197. doi:10.1128/MCB.11.10.5197
4. Herrscher RF, Kaplan MH, Lelsz DL, Das C, Scheuermann R, Tucker PW. The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: a B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev (1995) 9:3067. doi:10.1101/gad.9.24.3067
5. Zhou Y, Li YS, Bandi SR, Tang L, Shinton SA, Hayakawa K, et al. Lin28b promotes fetal B lymphopoiesis through the transcription factor Arid3a. J Exp Med (2015) 212:569–80. doi:10.1084/jem.20141510
6. Webb CF, Bryant J, Popowski M, Allred L, Kim D, Harriss J, et al. The ARID family transcription factor bright is required for both hematopoietic stem cell and B lineage development. Mol Cell Biol (2011) 31:1041–53. doi:10.1128/MCB.01448-10
7. Nixon JC, Ferrell S, Miner C, Oldham AL, Hochgeschwender U, Webb CF. Transgenic mice expressing dominant-negative bright exhibit defects in B1 B cells. J Immunol (2008) 181:6913. doi:10.4049/jimmunol.181.10.6913
8. Webb CF, Smith EA, Medina KL, Buchanan KL, Smithson G, Dou SS. Expression of bright at two distinct stages of B lymphocyte development. J Immunol (1998) 160:4747–54.
9. Heng TS, Painter MW; Immunological Genome Project Consortium. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
10. Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A (2006) 103:13789–94. doi:10.1073/pnas.0605944103
11. Li Y-S, Zhou Y, Tang L, Shinton SA, Hayakawa K, Hardy RR. A developmental switch between fetal and adult B lymphopoiesis. Ann N Y Acad Sci (2015) 1362:8–15. doi:10.1111/nyas.12769
12. Centa M, Gruber S, Nilsson D, Polyzos KA, Johansson DK, Hansson GK, et al. Atherosclerosis susceptibility in mice is independent of the V1 immunoglobulin heavy chain gene. Arterioscler Thromb Vasc Biol (2016) 36(1):25–36. doi:10.1161/ATVBAHA.115.305990
13. Mi QS, Zhou L, Schulze DH, Fischer RT, Lustig A, Rezanka LJ, et al. Highly reduced protection against Streptococcus pneumoniae after deletion of a single heavy chain gene in mouse. Proc Natl Acad Sci U S A (2000) 97:6031–6. doi:10.1073/pnas.110039497
14. Rajewsky K. The Herzenberg lecture: how to make a B-1 cell? Ann N Y Acad Sci (2015) 1362:6. doi:10.1111/nyas.12767
15. Vilagos B, Hoffmann M, Souabni A, Sun Q, Werner B, Medvedovic J, et al. Essential role of EBF1 in the generation and function of distinct mature B cell types. J Exp Med (2012) 209:775–92. doi:10.1084/jem.20112422
16. Ádori M, Hedestam G. NF-κB signaling in B-1 cell development. Ann N Y Acad Sci (2015) 1362:39. doi:10.1111/nyas.12800
17. Kreslavsky T, Vilagos B, Tagoh H, Poliakova DK, Schwickert TA, Wöhner M, et al. Essential role for the transcription factor Bhlhe41 in regulating the development, self-renewal and BCR repertoire of B-1a cells. Nat Immunol (2017) 18:442–55. doi:10.1038/ni.3694
18. Kurkewich JL, Klopfenstein N, Hallas WM, Wood C, Sattler RA, Das C, et al. Arid3b is critical for B lymphocyte development. PLoS One (2016) 11:e0161468. doi:10.1371/journal.pone.0161468
19. Puissegur MP, Eichner R, Quelen C, Coyaud E, Mari B, Lebrigand K, et al. B-cell regulator of immunoglobulin heavy-chain transcription (Bright)/ARID3a is a direct target of the oncomir microRNA-125b in progenitor B-cells. Leukemia (2012) 26:2224–32. doi:10.1038/leu.2012.95
20. Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol (1998) 160:4776–87.
Keywords: Arid3a, B cell development, spleen, bone marrow, antibody formation
Citation: Habir K, Aeinehband S, Wermeling F and Malin S (2017) A Role for the Transcription Factor Arid3a in Mouse B2 Lymphocyte Expansion and Peritoneal B1a Generation. Front. Immunol. 8:1387. doi: 10.3389/fimmu.2017.01387
Received: 12 June 2017; Accepted: 09 October 2017;
Published: 24 October 2017
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, United StatesCopyright: © 2017 Habir, Aeinehband, Wermeling and Malin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Malin, c3RlcGhlbi5tYWxpbkBraS5zZQ==