 Alexander N. R. Weber1*
Alexander N. R. Weber1* Zsofia Bittner1
Zsofia Bittner1 Xiao Liu1
Xiao Liu1 Truong-Minh Dang1
Truong-Minh Dang1 Markus Philipp Radsak2
Markus Philipp Radsak2 Cornelia Brunner3
Cornelia Brunner3
- 1Department of Immunology, Interfaculty Institute for Cell Biology, University of Tübingen, Tübingen, Germany
- 2Department of Internal Medicine III, University Medical Center of the Johannes Gutenberg-University Mainz, Mainz, Germany
- 3Department of Otorhinolaryngology, Ulm University Medical Center, Ulm, Germany
Bruton’s tyrosine kinase (BTK) was initially discovered as a critical mediator of B cell receptor signaling in the development and functioning of adaptive immunity. Growing evidence also suggests multiple roles for BTK in mononuclear cells of the innate immune system, especially in dendritic cells and macrophages. For example, BTK has been shown to function in Toll-like receptor-mediated recognition of infectious agents, cellular maturation and recruitment processes, and Fc receptor signaling. Most recently, BTK was additionally identified as a direct regulator of a key innate inflammatory machinery, the NLRP3 inflammasome. BTK has thus attracted interest not only for gaining a more thorough basic understanding of the human innate immune system but also as a target to therapeutically modulate innate immunity. We here review the latest developments on the role of BTK in mononuclear innate immune cells in mouse versus man, with specific emphasis on the sensing of infectious agents and the induction of inflammation. Therapeutic implications for modulating innate immunity and critical open questions are also discussed.
Introduction
Since the first description of X-linked agammaglobulinemia (XLA, OMIM entry 300300) (1) and the identification of Bruton’s tyrosine kinase (BTK) as its genetic cause (2), BTK has been widely characterized as a critical mediator of B cell receptor (BCR) signaling and thus adaptive immunity (3). In the murine Btk-mutated (R28C) X-linked immunodeficiency (Xid) mutant strain CBA/N (4) B cell numbers and functionality are reduced but detectable [e.g., unaffected B-1b cell levels (5)]. In contrast, in humans BTK’s pivotal role is highlighted by the fact that a wide spectrum of BTK loss-of-function mutations [reviewed by Ref. (6) and documented in the ‘BTKbase’ database] lead to an almost complete absence of peripheral B cells and antibodies in XLA. BTK catalytic activity typically drives the activation of at least three key signaling pathways, phospholipase C, phosphatidalyinositol-3-kinase/Akt and NF-κB, giving B cells a very strong survival signal upon BCR engagement. Totaling a molecular weight of approximately 77 kDa, BTK also contains an N-terminal Pleckstrin homology domain that binds membrane phosphatidylinositol (3,4,5)-trisphosphate (PIP3), and Tec homology, Src homology (SH) 3, and SH2 domains involved in protein-protein interactions. Y223 and Y551 represent two critical tyrosine phosphorylation sites in the SH3 and kinase domain (7). Y551 is phosphorylated by the kinases Syk or Lyn during BCR signaling and promotes the catalytic activity of BTK and subsequent Y223 autophosphorylation. The strong dependence of malignant B cells on BTK activity for survival (3), made BTK a key target for the development of small molecule inhibitors (8) in B cell malignancies. Nevertheless, BTK is being increasingly studied for its role in myeloid and other innate immune cells (Figure 1). Here, we summarize the emerging multi-faceted roles of this versatile and therapeutically tractable kinase in innate immunity.
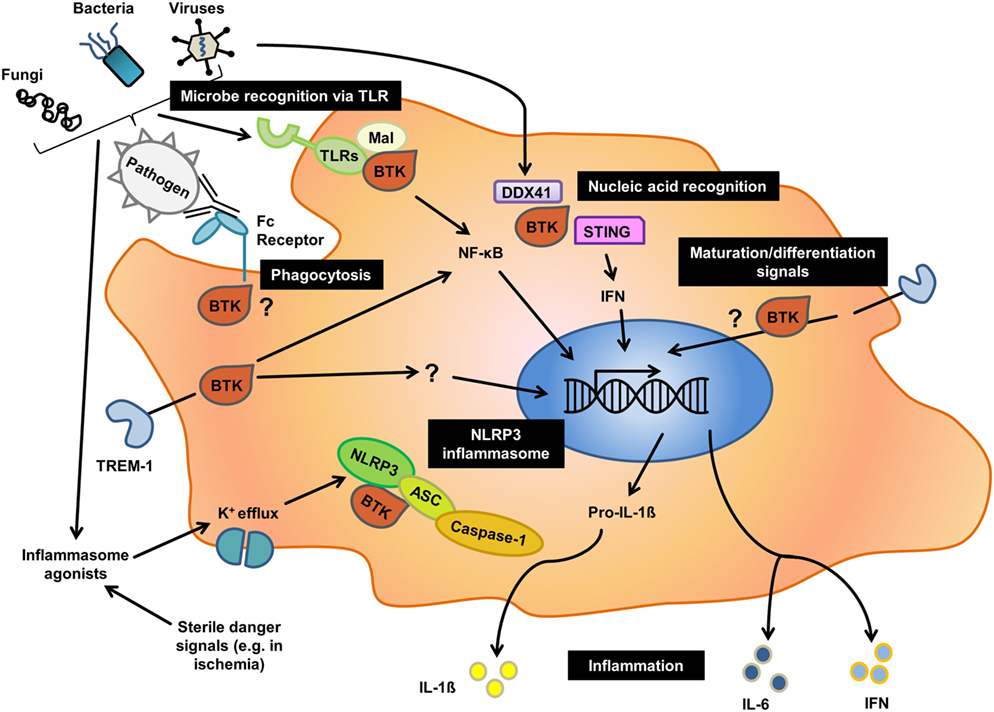
Figure 1. Overview of the different roles of Bruton’s tyrosine kinase (BTK) in innate immunity. Black boxes indicate major cellular processes for which an involvement of BTK has been reported in human or mice, or both. As outlined in the text, for processes such as phagocytosis there is contradictory evidence illustrating that the nature of function-modifying mutations, cellular context and species may have a profound effect on the role of BTK in a given process.
BTK in Infection and Danger Recognition by Cell Surface Receptors in Innate Immune Cells
Although innate immune contributions for BTK in in vivo infection models with Btk gene knockout or Xid mice have to be interpreted with care (see below), a role for BTK/Btk in the sensing of multiple microbes has been reported: Sensing and antimicrobial responses to Listeria monocytogenes (9), Staphylococcus aureus (10), dengue virus (11), and Aspergillus fumigatus (12) were shown to depend on BTK. This effect may in part be due to BTK’s involvement in the sensing of microbes via multiple Toll-like receptors (TLRs)—TLR2 (13, 14), TLR3 (11), TLR4 (14, 15), TLR7/8 (14, 16, 17), and TLR9 (9, 17, 18) on human and mouse macrophages and dendritic cells (DC). However, some TLR studies, especially those involving XLA patients, have been contradictory with regard to specific TLRs requiring BTK (19). Potentially, the functional requirements for BTK function during B cell development are higher, leading to an XLA phenotype in a broader range of mutations and thus patients; conversely, it seems that for TLR signaling only certain BTK mutations may cause a significant impairment of signaling. Within the vast spectrum of BTK mutations reported in XLA patients the functional impact can oftentimes not adequately be predicted. On a postreceptor level, BTK is thought to interface with canonical TLR pathways at the level of the TLR/MyD88 bridging adaptor Mal/TIRAP, one suggested direct BTK substrate (15, 20, 21) apart from TLR3 (11). TLR-dependent BTK-activation promotes NF-κB and interferon-regulatory factor-dependent transcription of inflammatory cytokines and interferons (IFNs) (15, 17). BTK was also linked with the cytosolic nucleic acid sensor DDX41 (11) and promoted its cooperation with the important IFN response regulator STING. BTK also operates downstream of the myeloid receptor TREM-1 for cytokine production (22, 23). On a more global immunoregulatory level, downregulation of innate immune-related genes and an upregulation of oxidative phosphorylation and apoptosis-related genes was observed in XLA patients (24). In contrast to these proimmune innate functions of BTK, the kinase was also shown to negatively regulate TLR-induced cytokine release from primary human innate immune cells (25). Moreover, in other DC studies, hepatocyte growth factor (HGF) as well as T cell Ig and mucin protein-3 (TIM-3)-induced BTK function blocked NF-κB activity (26, 27). In phagocytosis BTK was found essential for the clearance of infectious agents by mouse macrophages (12, 28); for humans, both data supporting a requirement for BTK in phagocytosis (24, 29, 30) as well as data arguing for a redundant role of BTK in this process (19, 31) have been reported based on studies of cells from XLA patients. Off-target effects in studies involving BTK inhibitors and the aforementioned unpredictability of naturally occurring BTK mutations or gene alterations1 are likely to contribute to these controversial findings. The breadth of this multifaceted body of evidence certainly highlights the complexity of BTK function and regulation. Specific mutation site, receptor pathway, cell type and species are thus important factors, rendering the more systematic exploration of BTK’s role in innate immunity a formidable challenge.
BTK in the Maturation, Recruitment and Function of Innate Immune Cells
Given its role in B cell development, a role for BTK in the development of myeloid cells, which depends on many cues provided by cell surface receptors (32), is not surprising. Interestingly, in mice GM-CSF receptor α-chain expression was required for macrophage maturation and survival. In mice, Btk deficiency also correlated with reduced monocyte/macrophage numbers (33) but favored granulopoiesis (34, 35). However, these granulocytes were immature, had inefficient granule function and impaired recruitment of neutrophils to sites of sterile inflammation. Similarly, in humans BTK seems to be implicated in the maturation of neutrophils, since in XLA patients, who are frequently neutropenic, neutrophils were arrested at the myelocyte/promyelocyte stage (36–38). Conversely, Marron et al. (19) and Cavaliere et al. (31) suggested that BTK is dispensable for human neutrophil function; Honda et al. (39) even found an increased TLR or tumor necrosis factor receptor-induced ROS production of XLA neutrophils, albeit at higher levels of neutrophil apoptosis. Although DC numbers in Btk-deficient animals were unaffected, these DC had defects in maturation and DC-mediated antigen presentation (40). In human DC, the aforementioned HGF- and TIM-3-induced BTK-mediated NF-κB inhibition impaired DC activation as well as maturation leading to impaired CpG-induced anti-tumor responses (26, 27). In tumor infiltrating macrophages BTK was found to exert immune-inhibitory and tumor-promoting effects (41, 42). In contrast, inhibition of Btk activity promoted DC maturation and CD4+ T cell activating functions (43, 44). Together, these data suggest BTK may serve as an important target for immunomodulatory-based anticancer therapy. The unexpected description of (so far) cancer-specific alternative isoforms, p65 and p80, in breast (45), brain (46), prostate (47), gastric (48), and colon cancer (49) as well as reports for a role of BTK in NK cells (50), and platelets (51) also deserve mention and warrant further research.
BTK and the NLRP3 Inflammasome
The NLRP3 inflammasome, a multiprotein complex involving NLRP3, the adaptor ASC and the proteolytic enzyme, caspase-1, has recently emerged as a key molecular machinery for the processing and thus activation of bioactive IL-1β (52, 53) and a major pathophysiological regulator in infection, myocardial infarction, stroke, Alzheimer’s and diabetes (53). Reports by us (10) and others (54) recently identified BTK as a direct regulator in NLRP3 inflammasome activation (Figure 2): Ito et al. demonstrated that BTK was critically required for NLRP3 inflammasome-dependent IL-1β release from murine macrophages. BTK physically interacted with NLRP3 and its adaptor ASC, resulting in the induction of ASC oligomerization and caspase-1 activation in a kinase activity-dependent manner in vitro. In both studies, BTK was rapidly phosphorylated upon NLRP3 activation. We additionally observed that inflammasome activity was impaired in PBMC from XLA patients, suggesting that a genetic inflammasome deficiency may contribute to the immunocompromised XLA phenotype. Pharmacological BTK inhibitors in vivo affected S. aureus clearance in mice and IL-1β release in cancer patients, which was associated with a reduced ability of isolated PBMC to secrete IL-1β. Excessive IL-1β release in PBMC from Muckle-Wells Syndrome MWS (OMIM entry 191900) patients could also be blocked by BTK inhibitors (10). In a brain ischemia/reperfusion in vivo model Btk was activated in infiltrating macrophages/neutrophils, and Btk inhibition protected against brain injury (54). In combination, these results warrant the exploration of BTK inhibition as a strategy to target the NLRP3 inflammasome therapeutically. Mechanistically, the emerging role of NRF2, a protein shown separately to interact with both BTK (55) and NLRP3 (56), will also be interesting to study further. Likewise, the observed link with caspase-11 (33) may indicate an additional role for BTK in the non-canonical NLRP3 inflammasome that depends on caspase-11 in mice and caspase-4/-5 in humans for intracellular LPS sensing (57)—a notion intriguing for further study.
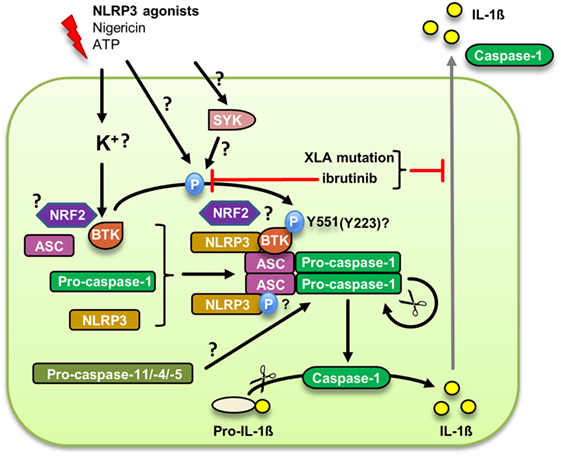
Figure 2. Bruton’s tyrosine kinase (BTK) regulation of the canonical NLRP3 inflammasome. Upon an upstream signal potentially linked to membrane integrity or K+ efflux, BTK is phosphorylated at Y551, presumably by Syk, and subsequently is activated. The supposed phosphorylation of ASC promotes inflammasome assembly and caspase-1 autoproteolytic activation leading to the cleavage and secretion of mature IL-1β. Whether BTK also plays a role in the alternative NLRP3 inflammasome dependent on caspase-11 remains to be investigated.
Therapeutic Opportunities in Innate Immunity
Undoubtedly, the existence of and first clinical data for an FDA-approved BTK inhibitor, ibrutinib (also known as PCI-32765), in oncology (8) make preclinical and translational research into BTK’s innate functions highly interesting, for example in arthritis (30), thromboinflammation (51), or in ischemic stroke, as aforementioned (52, 54). Compared to other strategies proposed to target the pathologically relevant NLRP3 inflammasome/IL-1 axis—for example, the inhibitor MCC950, whose target is however unknown (58), or IL-1 blockade which only neutralizes the inflammatory potential of certain inflammasome-dependent mediators—targeting NLRP3 via BTK is highly intriguing since BTK is a well-known (if incompletely understood) molecular target with inhibitors approved or in clinical trials. In cancer immunotherapies, first results on BTK inhibition modulating DC and subsequent CD4+ T cell activation (43) or upregulation of the inhibitory receptor TIM-3 on DCs are also noteworthy (59). On the other hand, targeting BTK with ibrutinib causes significant immunosuppression associated with an increased risk of infections (60) indicating that BTK dependent innate immunity is severely impaired (23). In addition, leukostasis as well as bleeding complications have been reported indicating that BTK inhibition by ibrutinib also affects leukocyte adhesion and platelet functions in a clinically relevant way (51, 61). Increased rates of atrial fibrillation (62) as a non-immune adverse event in patients receiving ibrutinib advises caution when exploring the novel opportunities of BTK blockade in various disease entities. Potentially, transient use of inhibitors, e.g., only during phases of acute adverse inflammation (e.g., shortly after ischemic brain or heart injury), may nevertheless offer advantageous therapeutic windows in non-chronic diseases. Nonetheless, much further work will be required to safely harness the potential of BTK for treating additional innate immune-related disorders.
Open Questions and Outlook
Although much progress on deciphering the molecular function of BTK in various innate cell types has been made, specific BTK interactors and substrates in the different aforementioned processes have to be studied more systematically as highlighted by the many apparent controversies. Additionally, whether BTK functions as a bona fide kinase or more as a scaffold protein requires clarification, e.g., in the NLRP3 inflammasome process. In cell lines, well-characterized loss and gain of function mutants of BTK may be useful tools (22). Conditional and/or inducible gain- or loss-of-function mouse alleles, which surprisingly have not been described, will be essential for innate immunologists to meaningfully study BTK further in vivo and to exclude confounding effects from impaired B cell function, e.g., in in vivo infection studies. Furthermore, conditional alleles would help flesh out cell-specific and hematopoietic roles of BTK more precisely. The resulting in vivo mouse models should complement urgently needed additional studies on human BTK that may help to solve some of the apparent discrepancies between human and murine studies and decipher some of the profound complexity surrounding BTK. Such vital research could be done within ongoing studies in the cancer field or of ex vivo studies on biomaterial from healthy volunteers or XLA patients. Concomitant and standardized kinase and expression level assays conducted on XLA samples may help to gauge the penetrance and severity of naturally occurring variants better and, by incorporating these results, may allow drawing more generally valid conclusions from these patient studies.
In conclusion, BTK has emerged as a key node in many immunological signaling networks in innate immunity, some of which have profound therapeutic potential. Future efforts in both academia and industry may help to explore and subsequently harness the potential of this intriguing yet highly complex kinase for innate immunity. This may offer therapeutic opportunities comparable or potentially exceeding those already envisaged for oncology.
Author Contributions
All authors collected and analyzed data, AW coordinated the study and drafted the manuscript, and all authors contributed toward and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the German Research Foundation (DFG)-funded CRC 685 “Immunotherapy” and CRC/TR 156 “The skin as a sensor and effector organ orchestrating local and systemic immune responses,” the Else-Kröner-Fresenius Stiftung, the University of Tübingen, and the University Hospital Tübingen (Fortüne Grant 2310-0-0 to XL and AW).
Footnote
- ^Gross deletion within the BTK genomic locus could affect not only expression and function of BTK itself but also that of adjacent genes like TIMM8A, the genetic cause of the Mohr-Tranebjærg syndrome (MTS, OMIM entry 304700), a neurodegenerative disorder leading to sensorineural deafness. Additionally, beside isolated BTK-deficiency (XLA OMIM entry 300300), patients were reported with growth hormone deficiency (GHD) associated with mutations within the BTK gene (XLH-GHD, OMIM entry 307200). The reason for GHD in XLA remains obscure.
References
2. Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature (1993) 361:226–33. doi:10.1038/361226a0
3. Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood (2012) 120:1175–84. doi:10.1182/blood-2012-02-362624
4. Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science (1993) 261:358–61. doi:10.1126/science.8332901
5. Riggs J, Howell K, Matechin B, Matlack R, Pennello A, Chiasson R. X-chromosome-linked immune-deficient mice have B-1b cells. Immunology (2003) 108:440–51. doi:10.1046/j.1365-2567.2003.01624.x
6. Valiaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat (2006) 27:1209–17. doi:10.1002/humu.20410
7. Wahl MI, Fluckiger AC, Kato RM, Park H, Witte ON, Rawlings DJ. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc Natl Acad Sci U S A (1997) 94:11526–33. doi:10.1073/pnas.94.21.11526
8. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med (2015) 21:922–6. doi:10.1038/nm.3884
9. Koprulu AD, Kastner R, Wienerroither S, Lassnig C, Putz EM, Majer O, et al. The tyrosine kinase Btk regulates the macrophage response to Listeria monocytogenes infection. PLoS One (2013) 8:e60476. doi:10.1371/journal.pone.0060476
10. Liu X, Pichulik T, Wolz OO, Dang TM, Stutz A, Dillen C, et al. Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J Allergy Clin Immunol (2017) 140:1054–67.e10. doi:10.1016/j.jaci.2017.01.017
11. Lee KG, Xu S, Kang ZH, Huo J, Huang M, Liu D, et al. Bruton’s tyrosine kinase phosphorylates toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci U S A (2012) 109:5791–6. doi:10.1073/pnas.1119238109
12. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med (2015). doi:10.15252/emmm.201404556
13. Horwood NJ, Page TH, Mcdaid JP, Palmer CD, Campbell J, Mahon T, et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol (2006) 176:3635–41. doi:10.4049/jimmunol.176.6.3635
14. Taneichi H, Kanegane H, Sira MM, Futatani T, Agematsu K, Sako M, et al. Toll-like receptor signaling is impaired in dendritic cells from patients with X-linked agammaglobulinemia. Clin Immunol (2008) 126:148–54. doi:10.1016/j.clim.2007.10.005
15. Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem (2003) 278:26258–64. doi:10.1074/jbc.M301484200
16. Sochorova K, Horvath R, Rozkova D, Litzman J, Bartunkova J, Sediva A, et al. Impaired toll-like receptor 8-mediated IL-6 and TNF-alpha production in antigen-presenting cells from patients with X-linked agammaglobulinemia. Blood (2007) 109:2553–6. doi:10.1182/blood-2006-07-037960
17. Li YF, Lee KG, Ou X, Lam KP. Bruton’s tyrosine kinase and protein kinase C micro are required for TLR7/9-induced IKKalpha and IRF-1 activation and interferon-beta production in conventional dendritic cells. PLoS One (2014) 9:e105420. doi:10.1371/journal.pone.0105420
18. Lougaris V, Baronio M, Vitali M, Tampella G, Cattalini M, Tassone L, et al. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation. J Allergy Clin Immunol (2014) 133:1644–50.e4. doi:10.1016/j.jaci.2013.12.1085
19. Marron TU, Rohr K, Martinez-Gallo M, Yu J, Cunningham-Rundles C. TLR signaling and effector functions are intact in XLA neutrophils. Clin Immunol (2010) 137:74–80. doi:10.1016/j.clim.2010.06.011
20. Gray P, Dunne A, Brikos C, Jefferies CA, Doyle SL, O’neill LA. MyD88 adapter-like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signal transduction. J Biol Chem (2006) 281:10489–95. doi:10.1074/jbc.M508892200
21. Semaan N, Alsaleh G, Gottenberg JE, Wachsmann D, Sibilia J. Etk/BMX, a Btk family tyrosine kinase, and Mal contribute to the cross-talk between MyD88 and FAK pathways. J Immunol (2008) 180:3485–91. doi:10.4049/jimmunol.180.5.3485
22. Ormsby T, Schlecker E, Ferdin J, Tessarz AS, Angelisova P, Koprulu AD, et al. Btk is a positive regulator in the TREM-1/DAP12 signaling pathway. Blood (2011) 118:936–45. doi:10.1182/blood-2010-11-317016
23. Stadler N, Hasibeder A, Aranda Lopez P, Teschner D, Desuki A, Kriege O, et al. The Bruton tyrosine kinase inhibitor ibrutinib abrogates triggering receptor on myeloid cells 1 mediated neutrophil activation. Haematologica (2017) 102:e191–4. doi:10.3324/haematol.2016.152017
24. Mirsafian H, Ripen AM, Leong WM, Chear CT, Bin Mohamad S, Merican AF. Transcriptome profiling of monocytes from XLA patients revealed the innate immune function dysregulation due to the BTK gene expression deficiency. Sci Rep (2017) 7:6836. doi:10.1038/s41598-017-06342-5
25. Marron TU, Martinez-Gallo M, Yu JE, Cunningham-Rundles C. Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines. J Allergy Clin Immunol (2012) 129:184–90.e1–4. doi:10.1016/j.jaci.2011.10.009
26. Singhal E, Kumar P, Sen P. A novel role for Bruton’s tyrosine kinase in hepatocyte growth factor-mediated immunoregulation of dendritic cells. J Biol Chem (2011) 286:32054–63. doi:10.1074/jbc.M111.271247
27. Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P. Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein-3 via Bruton’s tyrosine kinase and c-Src. J Immunol (2014) 193:3417–25. doi:10.4049/jimmunol.1400395
28. Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol (2008) 181:288–98. doi:10.4049/jimmunol.181.1.288
29. Amoras AL, Kanegane H, Miyawaki T, Vilela MM. Defective Fc-, CR1- and CR3-mediated monocyte phagocytosis and chemotaxis in common variable immunodeficiency and X-linked agammaglobulinemia patients. J Investig Allergol Clin Immunol (2003) 13:181–8.
30. Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol (2011) 7:41–50. doi:10.1038/nchembio.481
31. Cavaliere FM, Prezzo A, Bilotta C, Iacobini M, Quinti I. The lack of BTK does not impair monocytes and polymorphonuclear cells functions in X-linked agammaglobulinemia under treatment with intravenous immunoglobulin replacement. PLoS One (2017) 12:e0175961. doi:10.1371/journal.pone.0175961
32. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24:801–12. doi:10.1016/j.immuni.2006.04.008
33. Melcher M, Unger B, Schmidt U, Rajantie IA, Alitalo K, Ellmeier W. Essential roles for the Tec family kinases Tec and Btk in M-CSF receptor signaling pathways that regulate macrophage survival. J Immunol (2008) 180:8048–56. doi:10.4049/jimmunol.180.12.8048
34. Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, et al. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood (2011) 117:1329–39. doi:10.1182/blood-2010-04-281170
35. Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity (2016) 44:73–87. doi:10.1016/j.immuni.2015.11.011
36. Kozlowski C, Evans DI. Neutropenia associated with X-linked agammaglobulinaemia. J Clin Pathol (1991) 44:388–90. doi:10.1136/jcp.44.5.388
37. Farrar JE, Rohrer J, Conley ME. Neutropenia in X-linked agammaglobulinemia. Clin Immunol Immunopathol (1996) 81:271–6. doi:10.1006/clin.1996.0188
38. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) (2006) 85:193–202. doi:10.1097/01.md.0000229482.27398.ad
39. Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol (2012) 13:369–78. doi:10.1038/ni.2234
40. Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL-10 and Stat3. Proc Natl Acad Sci U S A (2006) 103:153–8. doi:10.1073/pnas.0509784103
41. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov (2016) 6:270–85. doi:10.1158/2159-8290.CD-15-0827
42. Ping LY, Ding N, Shi YF, Feng LX, Li J, Liu YL, et al. The Bruton’s tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget (2017) 8:39218–29. doi:10.18632/oncotarget.16836
43. Natarajan G, Oghumu S, Terrazas C, Varikuti S, Byrd JC, Satoskar AR. A Tec kinase BTK inhibitor ibrutinib promotes maturation and activation of dendritic cells. Oncoimmunology (2016) 5:e1151592. doi:10.1080/2162402X.2016.1151592
44. Natarajan G, Terrazas C, Oghumu S, Varikuti S, Dubovsky JA, Byrd JC, et al. Ibrutinib enhances IL-17 response by modulating the function of bone marrow derived dendritic cells. Oncoimmunology (2016) 5:e1057385. doi:10.1080/2162402X.2015.1057385
45. Eifert C, Wang X, Kokabee L, Kourtidis A, Jain R, Gerdes MJ, et al. A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosomes Cancer (2013) 52:961–75. doi:10.1002/gcc.22091
46. Wei L, Su YK, Lin CM, Chao TY, Huang SP, Huynh TT, et al. Preclinical investigation of ibrutinib, a Bruton’s kinase tyrosine (Btk) inhibitor, in suppressing glioma tumorigenesis and stem cell phenotypes. Oncotarget (2016) 7:69961–75. doi:10.18632/oncotarget.11572
47. Kokabee L, Wang X, Sevinsky CJ, Wang WL, Cheu L, Chittur SV, et al. Bruton’s tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol Ther (2015) 16:1604–15. doi:10.1080/15384047.2015.1078023
48. Wang JD, Chen XY, Ji KW, Tao F. Targeting Btk with ibrutinib inhibit gastric carcinoma cells growth. Am J Transl Res (2016) 8:3003–12.
49. Grassilli E, Pisano F, Cialdella A, Bonomo S, Missaglia C, Cerrito MG, et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene (2016) 35:4368–78. doi:10.1038/onc.2015.504
50. Bao Y, Zheng J, Han C, Jin J, Han H, Liu Y, et al. Tyrosine kinase Btk is required for NK cell activation. J Biol Chem (2012) 287:23769–78. doi:10.1074/jbc.M112.372425
51. Murthy P, Durco F, Miller-Ocuin JL, Takedai T, Shankar S, Liang X, et al. The NLRP3 inflammasome and Bruton’s tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem Biophys Res Commun (2017) 483:230–6. doi:10.1016/j.bbrc.2016.12.161
52. Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol (2015) 10:395–424. doi:10.1146/annurev-pathol-012414-040431
53. Dubois H, Wullaert A, Lamkanfi M. General strategies in inflammasome biology. Curr Top Microbiol Immunol (2016) 397:1–22. doi:10.1007/978-3-319-41171-2_1
54. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun (2015) 6:7360. doi:10.1038/ncomms8360
55. Vijayan V, Baumgart-Vogt E, Naidu S, Qian G, Immenschuh S. Bruton’s tyrosine kinase is required for TLR-dependent heme oxygenase-1 gene activation via Nrf2 in macrophages. J Immunol (2011) 187:817–27. doi:10.4049/jimmunol.1003631
56. Garstkiewicz M, Strittmatter GE, Grossi S, Sand J, Fenini G, Werner S, et al. Opposing effects of Nrf2 and Nrf2-activating compounds on the NLRP3 inflammasome independent of Nrf2-mediated gene expression. Eur J Immunol (2017) 47:806–17. doi:10.1002/eji.201646665
57. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol (2015) 45:2911–7. doi:10.1002/eji.201545523
58. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi:10.1038/nm.3806
59. Gujar R, Maurya N, Yadav V, Gupta M, Arora S, Khatri N, et al. c-Src suppresses dendritic cell antitumor activity via T cell Ig and mucin protein-3 receptor. J Immunol (2016) 197:1650–62. doi:10.4049/jimmunol.1600104
60. Williams AM, Baran AM, Meacham PJ, Feldman MM, Valencia HE, Newsom-Stewart C, et al. Analysis of the risk of infection in patients with chronic lymphocytic leukemia in the era of novel therapies. Leuk Lymphoma (2017) 11:1–8. doi:10.1080/10428194.2017.1347931
61. Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia (2015) 29:783–7. doi:10.1038/leu.2014.247
Keywords: Bruton’s tyrosine kinase, macrophage, dendritic cell, Toll-like receptor, NLRP3 inflammasome, ibrutinib, X-linked agammaglobulinemia
Citation: Weber AN, Bittner Z, Liu X, Dang T-M, Radsak MP and Brunner C (2017) Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front. Immunol. 8:1454. doi: 10.3389/fimmu.2017.01454
Received: 10 August 2017; Accepted: 18 October 2017;
Published: 08 November 2017
Edited by:
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Pradip Sen, Institute of Microbial Technology (CSIR), IndiaIsabella Quinti, Sapienza Università di Roma, Italy
Amir Feisal Merican Bin Aljunid Merican, University of Malaya, Malaysia
Copyright: © 2017 Weber, Bittner, Liu, Dang, Radsak and Brunner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander N. R. Weber, YWxleGFuZGVyLndlYmVyQHVuaS10dWViaW5nZW4uZGU=