 Angelique Hölzemer
Angelique Hölzemer Wilfredo F. Garcia-Beltran
Wilfredo F. Garcia-Beltran Marcus Altfeld
Marcus Altfeld- 1First Department of Internal Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2German Center for Infection Research (DZIF), Partner site Hamburg-Lübeck-Borstel-Riems, Hamburg, Germany
- 3Harvard Medical School, Boston, MA, United States
- 4Institute for Immunology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Natural killer (NK) cells are effector lymphocytes of the innate immune system that are able to mount a multifaceted antiviral response within hours following infection. This is achieved through an array of cell surface receptors surveilling host cells for alterations in human leukocyte antigen class I (HLA-I) expression and other ligands as signs of viral infection, malignant transformation, and cellular stress. This interaction between HLA-I ligands and NK-cell receptor is not only important for recognition of diseased cells but also mediates tuning of NK-cell-effector functions. HIV-1 alters the expression of HLA-I ligands on infected cells, rendering them susceptible to NK cell-mediated killing. However, over the past years, various HIV-1 evasion strategies have been discovered to target NK-cell-receptor ligands and allow the virus to escape from NK cell-mediated immunity. While studies have been mainly focusing on the role of polymorphic HLA-A, -B, and -C molecules, less is known about how HIV-1 affects the more conserved, non-classical HLA-I molecules HLA-E, -G, and -F. In this review, we will focus on the recent progress in understanding the role of non-classical HLA-I ligands in NK cell-mediated recognition of HIV-1-infected cells.
Introduction
Untreated HIV-1 infection will lead to progressive, severe, and mostly fatal immune deficiency in the vast majority of individuals. Protective HIV-1 immunity is observed in a small subset of subjects whose immune system can naturally control HIV-1 infection and who are termed “elite controllers.” Despite intense research in this area over the past decades, the correlates leading to protective immunity are still insufficiently understood. Host genetics alone can only explain approximately 20% of the variable outcomes between individuals observed in the natural course of infection (1). Nonetheless, a consistently documented key genetic determinant of HIV-1 control is the presence of particular human leukocyte antigen (HLA) class I alleles. This strong association between classical HLA-I alleles and HIV-1 disease outcome has been identified in genome-wide association studies (1, 2) as well as in large cohorts studying the immunogenetics of HIV-1 disease (3, 4). The protective effects of certain HLA-I alleles have mostly been attributed to enhanced CD8+ T lymphocyte-mediated immunity (5–7). HLA-I presentation of HIV-1 epitopes derived from conserved sequences of HIV-1 to CD8+ T cells can pressure the virus to select for mutations in these epitopes, but viral escape can be associated with costs in viral fitness (8). Indeed, early CD8+ T-lymphocyte responses contribute to the initial drop in HIV-1 peak viremia and with this, first HIV-1 escape mutations arise (9). Other protective factors in HIV-1 infection include enhanced proliferation potential of T lymphocytes (10, 11), polyfunctional immune responses (12, 13), variations in host restriction factors (14), and variants in HIV-1 coreceptors, in particular, of CCR5 (15, 16).
Over the past years, the role of antiviral innate immune responses mediated by natural killer (NK) cells in HIV-1 infection has been increasingly appreciated (17, 18). In vitro, NK cells can inhibit HIV-1 replication in autologous CD4+ T cells as effectively as CD8+ T cells (19). Additionally, the strong protective effect of host HLA-I alleles on disease progression has been linked to receptor families recognizing HLA-I. These include killer-cell immunoglobulin-like receptors (KIRs), predominantly expressed on NK cells (20), and leukocyte immunoglobulin-like receptors (LILRs), expressed on professional antigen presenting cells such as dendritic cells (DCs), monocytes, macrophages, and B cells, but also on T cells and NK cells (21). Indeed, accumulating data from population studies have identified certain KIR, LILR, and HLA-I allele combinations associated with slower HIV-1 disease progression (22–24), which has helped decipher a further piece of complex host genetics in HIV-1 disease variability.
Natural killer cells comprise 5–15% of the circulating lymphocytes (25) and their role in controlling viral infections has been long established (26). Two major subsets exist: CD56brightCD16dim/neg and CD56dimCD16pos NK cells (25). These differ in their expression of key NK-cell receptors, response to soluble factors and cellular targets, capacity for cytotoxicity, and production of immunomodulatory cytokines (27). NK cells are a crucial first line of defense that detect infected cells before antigen sensitization has occurred (28, 29), and therefore, they precede adaptive immunity in the early phases of HIV-1 infection. Indeed, there is evidence that the early events following infection prior to the development of a specific immune response can determine the viral set point and influence the clinical course of infection (30). In acute HIV-1 infection, a rapid expansion occurs in predominantly cytotoxic CD56dim NK cells, prior to CD8+ T cell expansion (31). On the other hand, in chronic HIV-1 infection, a redistribution of NK cells toward less functional subsets can be observed (32–35) and the presence of persistent viremia appears to deteriorate NK-cell function (19, 34, 36). Overall, the full extent of receptor-ligand interactions between NK cells and HIV-1–infected target cells in HIV-1 infection leading to either NK-cell expansion/killing or exhaustion is highly complex and not yet fully understood.
Natural killer cells, as members of the innate immune system, express a plethora of germline-encoded receptors, and their effector function is determined by integration of inhibitory and activating NK-cell receptor signaling, whereby inhibitory signals tend to be dominant (27). Major NK-cell receptor families are (i) natural cytotoxicity receptors (i.e., NKp46, NKp44, and NKp30), which deliver mainly activating signals, (ii) the KIR family, encompassing inhibitory and activating members and monitoring HLA-I, (iii) the C-type lectins with activating natural killer group 2D (NKG2D) and the heterodimers NKG2A-CD94 and NKG2C-CD94, and (iv) the FcγRIIIa receptor (CD16), which can bind to the Fc-region of IgG antibodies. Critical activating signals can also be delivered by other coreceptors including 2B4, DNAM-1, or CD2 (37, 38). Differential expression of activating and inhibitory receptors allows for a certain degree of specificity and shaping of NK-cell function in response to different stimuli. Ultimately, the stochastic expression of receptors on each NK cell leads to substantial NK-cell diversity and determines the differential response to target cells (39, 40).
HIV-1–infected cells can become vulnerable to NK cell-mediated killing by upregulation of stress signals recognized by activating NK-cell receptors and/or by downregulation of inhibitory NK-cell-receptor ligands. Of note, signaling via the FcγRIIIa receptor (CD16), which mediates antibody-dependent cellular cytotoxicity (ADCC), is sufficient to induce NK-cell activation on its own (37). However, the strength of CD16-mediated activation is dependent on tuning of NK-cell responsiveness through inhibitory interactions of KIR or NKG2A with HLA class I (41, 42). Stress ligands upregulated on HIV-1–infected cells are the major histocompatibility complex (MHC) class-I-chain-related proteins (MIC-) A and -B, the UL16-binding proteins (ULBPs) 1–3, which are the ligands for the activating NKG2D receptors (43, 44), and a yet unknown ligand for NKp44 (45, 46). In turn, HIV-1 encodes for multiple accessory proteins with pleiotropic functions to overcome host restriction factors and host immune responses (47–49). The upregulation of stress ligands such as ULBPs and MIC-A/B is counteracted via HIV-1 Nef (50) and the ligands for coactivating receptors such as NTB-A and DNAM-1 are downregulated via HIV-1 Vpu and partially Nef (51–53). The impact of HIV-1 Nef and Vpu on HLA class I expression will be discussed later. In this review, we will focus on the recent progress in understanding the interplay of HLA-I with HLA-I binding NK-cell receptors, and how this interaction either limits HIV-1 replication or is exploited by the virus to enhance pathogenesis.
KIR–HLA Interactions in HIV-1 Disease Progression and Acquisition
Classical and non-classical HLA-I genes (also known as HLA-Ia and HLA-Ib, respectively) are located within the MHC region p21.3 on chromosome 6, the most polymorphic region of the human genome. An extensive amount of allelic variation occurs within the region encoding for classical HLA-I genes (54). In contrast, non-classical HLA-I alleles display varying degrees of oligomorphism. To date, the classical HLA-A, HLA-B, and HLA-C loci comprise >10,000 alleles encoding for 8,662 distinct proteins, whereas the non-classical HLA-E, HLA-F, and HLA-G loci combined encode for 101 alleles and only 30 proteins (The Immuno Polymorphism Database, as of July 2017) (55, 56).
Classical HLA-I is ubiquitously expressed on nucleated cells. Given that the primary function of HLA-I is to present peptides derived from degradation of intracellular proteins (57), it is not surprising that variations mainly occur in regions surrounding the peptide-binding groove (58) so as to maximize diversity of peptides presented across different gene and allele products. Under pathologic conditions such as malignant transformation or infection with intracellular pathogens, HLA-I presents antigenic peptides and thereby can elicit an immune response via HLA-I restricted cytotoxic CD8+-T cells. Historically, it was thought that NK cells only respond to changes in surface levels of classical HLA class I [to missing-self (59)], but there is increasing evidence that KIR can bind differentially depending on the HLA-class I presented peptide (60–65).
In 2007, the first genome-wide association studies reported three protective single-nucleotide polymorphisms (SNPs) in HIV-1 disease (2). The presence of these SNPs was associated with lower viral set point in chronically HIV-1–infected subjects and together explained almost 15% of interindividual disease variability. Strikingly, all three SNPs were located in the MHC region of chromosome 6, emphasizing the crucial role of HLA class I in HIV-1 infection. The first SNP is in high linkage disequilibrium with HLA-B*57, a second SNP was located 35bp upstream of the HLA-C locus, and results in higher HLA-C expression levels. The last SNP was linked to an RNA polymerase subunit, ZNRD1 and affected the time to AIDS progression. Subsequent genome-wide association studies confirmed the first two SNPs and identified six additional SNPs associated with HIV-1 disease control in two different ethnic cohorts. Again, all SNPs were concentrated around the HLA-I region (1). Accordingly, the strongest HLA class I protective effects so far are reported for HLA-B*57 (66, 67) and HLA-B*27 (4, 68); two HLA class I alleles carrying the serologically defined Bw4 motif (determined by the amino acids 77–83). There is a strong association of HLA-Bw4 homozygosity with the ability to suppress viral replication of HIV-1 and with delayed time to AIDS progression (69).
The genes encoding for KIRs are located within the leukocyte receptor cluster on Chromosome 19q13.4, which additionally encodes Ig-like transcripts (ILTs) [also termed leukocyte Ig-like receptors (LIRs)], and leukocyte-associated inhibitory receptors (70). The KIR locus exhibits substantial polymorphism, in its degree only second to the MHC region in the human genome (71). KIRs can be subdivided into two different classes: KIRs with two extracellular Ig-like domains (KIR2Ds) and those with three domains (KIR3Ds). These Ig-like domains are classified as D0, D1, or D2. Type 1 KIR2Ds contain a D1 domain distal to a D2 domain, type 2 KIR2Ds (KIR2DL4 and KIR2DL5) have a D0–D2 domain organization, and KIR3Ds have all three domains as D0–D1–D2. In general, KIR2Ds bind to HLA-C and KIR3D bind to HLA-A and B-ligands (72, 73). Regarding signaling capacity, a long cytoplasmic tail (KIR-L) renders the KIR inhibitory as it contains immune tyrosine inhibitory motifs (ITIMs), whereas a short cytoplasmic tail (KIR-S) associates to adaptor molecules such as DAP12 and delivers activating signals (74). An exception to this is KIR2DL4, which holds an ITIM in its long cytoplasmic tail, but also associates with activating adaptor elements (73). KIRs are a major receptor family on NK cells, but are also expressed on CD4+ and CD8+ T cells (both αβ and γδ T cells) (75–80). Of note, expression of inhibitory KIR on T cells is increased following chronic immune activation, as was observed in the case of CMV reactivation in a posttransplantation setting (81, 82). Increased KIR expression on bulk CD8+ T cells in HIV-1 infection has been reported, but barely detectable KIR expression was described, when investigating HIV-specific CD8+ T cells (83, 84). Overall, little is known about a modulation of KIR-expression on T cells with or without CMV reactivation in HIV-1–infected subjects.
KIR3DS1/KIR3DL1 and HLA-Bw4I80
The first study associating KIRs to HIV-1 control came from the laboratory of Mary Carrington in 2002. This study showed that possessing KIR3DS1 and an HLA-B allele with a Bw4 motif and an isoleucine at position 80 (HLA-Bw4I80) was associated with slower progression to AIDS, when compared to patients having only one or none of these alleles (22). A follow-up analysis by the same group reported a protective effect of combined KIR3DS1 and HLA-Bw4I80 against development of certain opportunistic infections in HIV-1–infected patients, also after controlling for presence of protective (e.g., HLA-B*57 and HLA-B*27) and deleterious (HLA-B*35) alleles (85). The KIR3DS1/KIR3DL1 locus is unique in that it encodes functionally divergent alleles (86). The inhibitory KIR3DL1 binds to HLA-I allotypes that possess a Bw4 motif (HLA-Bw4, which can derive from HLA-A or HLA-B alleles). Polymorphisms in position 80 of these HLA-Bw4 molecules have been shown to modulate the strength of binding to KIR3DL1 (87, 88). In addition, the interaction of KIR3DL1 with HLA-Bw4 is sensitive to the sequence of the HLA-Bw4–presented peptide (61). Contrary to KIR3DL1, a ligand for its activating counterpart, KIR3DS1, remained initially unknown.
In a cohort of recently infected individuals, Barbour et al. did not detect a synergistic protective effect of KIR3DS1 and HLA-Bw4I80 assessing viral load and CD4+ T cell loss. Nonetheless, encoding for at least one KIR3DS1 allele was associated with higher CD4+ T cell counts and encoding for HLA-Bw4I80 alleles correlated with lower viral load, suggesting a protective, but independent effect of KIR3DS1 and HLA-Bw4I80 (89). A further epidemiologic study reported that HIV-1 viral load at set point correlated positively with the number of KIR3DS1 gene copies in the presence of HLA-B Bw4I80 ligands. Higher copy numbers of the KIR3DL1 gene also correlated with lower viral set point in the presence of HLA-Bw4I80 and at least one copy of KIR3DS1 (90). In addition, a study by Jiang et al. (91) in a Chinese cohort showed that KIR3DS1/KIR3DL1 heterozygotes were enriched in HLA-Bw4I80–bearing long-term non-progressors with higher CD4+ T cell counts and decreased viral loads as compared to KIR3DL1 homozygotes or individuals without HLA-Bw4I80 (91).
As KIRs are predominantly expressed on NK cells, Martin et al.’s first report associating a KIR to an outcome in HIV-1 infection (22) triggered multiple studies on NK-cell functionality attempting to elucidate the underlying protective mechanism of KIR3DS1 in combination with HLA-Bw4I80 in HIV-1 disease. In line with the epidemiological data, functional studies reported that NK cells derived from donors possessing KIR3DS1 combined with HLA-Bw4I80 inhibited viral replication in infected autologous CD4+ T cells more potently than NK cells from donors having either or neither allele. Sorted KIR3DS1+ NK cells degranulated significantly more in response to HIV-1–infected HLA-Bw4–expressing CD4+ T cells compared to infected HLA-Bw6+ CD4+ T cells (92). A second study showed that NK cells from individuals encoding for KIR3DS1 displayed enhanced cytotoxic function compared to NK cells from individuals without KIR3DS1, but this was independent of the presence of HLA-Bw4I80 (93). Also, in acutely HIV-1–infected subjects a preferential expansion of KIR3DS1+ NK cells—and to a lesser extent KIR3DL1+ NK cells—was observed, which persisted only in subjects bearing HLA-Bw4I80 (94). Morvan et al. reported an expansion of KIR3DS1+ NK cells in response to various non-specific stimuli, but KIR3DS1+ NK-cell function was not influenced by the presence of HLA-Bw4 in this setting. Nonetheless, the frequency of KIR3DS1+ NK cells and KIR3DS1 expression levels on NK cells were higher in healthy subjects with HLA-Bw4I80 than in those without HLA-Bw4I80 (95). Furthermore, HIV-1 viral inhibition assays demonstrated that in individuals encoding HLA-Bw4, having one copy of KIR3DS1 and one or more copies of KIR3DL1 resulted in increased antiviral capacity of bulk NK cells compared to individuals containing either KIR3DS1 or KIR3DL1 alone, which displayed the lowest amounts of viral inhibition (90). No differences were seen in HLA-Bw6 homozygous donors, whose NK cells had poor antiviral capacity. Having increasing copy numbers of KIR3DL1 was correlated with elevated KIR3DS1 transcript and frequency of KIR3DS1 expression on NK cells. Interestingly, this hinted at a KIR3DL1-related mechanism regulating the peripheral expansion and functionality of KIR3DS1+ NK cells (90). A more recent study reported that NK cells from KIR3DS1 and HLA-Bw4I80 cocarriers produced higher levels of chemokines after cell contact with infected CD4+ T cells than NK cells derived from HLA-Bw6 homozygous donors, leading to superior inhibition of viral replication (96).
Understanding the mechanistic basis of the protective effect of KIR3DS1 has proven difficult, as multiple attempts had failed to demonstrate a functional interaction of KIR3DS1 with its putative HLA-Bw4 ligand (74, 97) or for that matter, an interaction with any ligand. To add an additional layer of complexity, the combined genotype of high expressing KIR3DL1*h alleles and HLA-Bw4I80 (in particular HLA-B*57) conferred strong protection toward HIV-1 disease progression (23, 98). Indeed, increased target cell cytotoxicity was observed in NK cells derived from elite controllers with protective KIR3DL1*h/*y receptor genotypes along with its HLA-Bw4I80 ligand (99). As KIR3DS1 homozygosity is rare, in the majority of studies investigating KIR3DS1 and HLA-Bw4 epistasis, KIR3DS1-bearing subjects possessed also KIR3DL1 as a potential confounding variable.
Protection by an inhibitory KIR in HIV-1 disease seems counterintuitive, but might be mediated through a process called NK-cell licensing or education. Expression of an inhibitory KIR during NK-cell development provides strong inhibitory signals in response to its specific HLA-I ligand, ensuring self-tolerance. This allows NK cells to acquire enhanced cytotoxic function, which becomes apparent once exposed to missing or altered self (100). KIR3DL1 allotypes indeed differ in their inhibition of NK-cell function, with an overall trend toward increasing inhibitory capacity in high-expressing KIR3DL1 allotypes (101). Thus, a potential explanation is that presence of high-expression KIR3DL1*h alleles together with HLA-Bw4I80 determines the increased cytotoxicity of KIR3DL1+ NK cells toward HIV-1–infected targets (taking into account that HLA-B is downregulated via actions of the HIV-1 accessory protein Nef) (23). Indeed, a study in slow progressors to AIDS reported increased polyfunctionality of NK cells from donors carrying the KIR3DL1*h/*y allele together with its HLA-B*57 ligand compared to HLA-Bw6 homozygous donors (102). Boudreau et al. recently demonstrated functionally that killing of HIV-1–infected targets via KIR3DL1+ NK-cells was dependent on the strength of NK-cell education via distinct combinations of KIR3DL1 and HLA-Bw4, with highest cytotoxicity mediated by high-expressing KIR3DL1 and HLA-Bw4I80 interactions (103). Moreover, NK cell education not only leads to enhanced functionality (104), but signaling through inhibitory KIRs on NK cells can additionally promote NK-cell survival (105), potentially leading to accumulation of educated NK cells expressing inhibitory receptors in chronic viral infection.
Supplementary evidence comes from studies in highly exposed HIV-1 seronegative individuals. One study reported a significant overrepresentation of KIR3DS1 homozygosity in high-risk uninfected individuals compared to seroconverted individuals, independent of HLA-Bw4I80 (106). This group also reported an association of the KIR3DL1*h/*y-HLA-B*57 combined genotype with protection from HIV-1 acquisition (107). Another study showed enrichment of the HLA-Bw4 carrier–KIR3DS1 homozygous genotype in HIV-1-exposed seronegative subjects (108). In summary, whereas the results from epidemiological studies are not clear-cut, these studies point toward a potential dual effect of KIR3DS1 (with or without HLA-Bw4I80) on both the course of HIV-1 infection and HIV-1 acquisition.
HLA-C and KIR2Ds
Genome-wide association studies have clearly implicated the HLA-C locus in HIV-1 control, identifying a protective SNP associated with higher HLA-C expression levels (1, 2). Interestingly, HLA-C surface expression levels are only 10% of surface levels of HLA-A and -B (109), and HLA-C alleles demonstrate less polymorphism compared to HLA-B (56). Nonetheless, individuals with high HLA-C expression levels have been shown to have a higher likelihood of mounting an HLA-C-restricted CD8+ T-cell response (110) and exhibit higher mutation rates in HLA-C–presented HIV-1 epitopes, indicating CD8+ T-cell pressure via HLA-C (111). However, given that virtually all individuals encode for KIRs (i.e., KIR2Ds) able to recognize cognate HLA-C molecules, it was proposed that NK cells might play an additional role in mediating the protective effect of higher HLA-C expression. Inhibitory KIR2DL1 binds to HLA-C group 2 allotypes (HLA-C2, which contain Asn77 and Lys80), whereas inhibitory KIR2DL2 and KIR2DL3, which are allelic products of the same KIR2DL2/3 locus, bind to HLA-C group 1 allotypes (HLA-C1, which contain Ser77 and Asn80). Notably, KIR2DL3 also recognizes HLA-B*46:01 due to an intergenic miniconversion between HLA-B*15:01 and HLA-C*01:02 (65). It was long believed that while HIV-1 Nef downregulated HLA-A and HLA-B surface expression to avoid recognition by cytotoxic CD8+ T cells (112), it spared HLA-C surface expression to ensure inhibition of NK cells via engagement of KIR2DL. This paradigm—which initially arose from studies performed with lab-adapted HIV-1 strains—was recently revised, when Apps et al. demonstrated that HLA-C is downregulated by HIV-1 Vpu variants derived from most primary HIV-1 isolates. HIV-1 Vpu-mediated downregulation of HLA-C was shown to subsequently impair the ability of HLA-C–restricted CD8+ T cells to inhibit viral replication (113). Regarding NK-cell function, it was reported earlier that expression of HLA-C (and HLA-E) on activated, HIV-1–infected CD4+ T cells impaired NK-cell killing, whereas blocking the HLA-C interaction with KIR2D enhanced NK-cell cytotoxicity toward HIV-1–infected CD4+ T-cell blasts (114, 115). During primary HIV-1 infection, KIR2DL+ NK-cell frequencies increased with the presence of their cognate HLA-C ligand (e.g., KIR2DL1+ NK cells expanded in HLA-C2 homozygous individuals) and exhibited more polyfunctional responses, presumably due to a licensing effect (116). Downmodulation of HLA-C by various HIV-1 strains resulted in reduced binding of KIR2Ds to HIV-1–infected cells. Moreover, NK cells were able to sense alterations in HLA-C expression as measured by differing degrees of HIV-1-replication inhibition. Yet, remaining HLA-C surface levels were sufficient to inhibit antiviral function of licensed KIR2DL+ NK cells (encountering their cognate HLA-C ligand) compared to unlicensed NK cells (117). Thus, although NK cells licensed through inhibitory KIR2D exhibit increased functionality against HLA-I–deficient target cells, first reports indicate that this subset does not have superior antiviral function against HIV-1–infected targets expressing self-HLA-C.
The Role of HIV-1 Peptides in KIR:HLA-I Interactions
HIV-1 exhibits an extraordinary ability to adapt to and evade host immune responses. The constant battle of the immune system attacking the virus and the virus evading leads to an extremely rapid accumulation of HIV-1 variants and quasispecies that, at least partially, escape from immune pressure (118, 119). Analyzing the major mechanisms of HIV-1 evasion and sites of sequence mutations provides direct insights into where the human immune system is able to apply critical pressure on the virus. A particular example is the rapid increase in HIV-1 mutations in HLA-I presented epitopes recognized by cytotoxic CD8+ T cells (CTL), which allows the virus to overcome adaptive immune pressure. These mutations can abrogate CTL recognition, but sometimes also impair viral replication (120). By now, a substantial body of evidence from structural (62, 63) and functional studies (60, 61, 65, 121–123) shows that KIR binding is modulated by the sequence of HLA-I–presented peptides, and in particular, C-terminal residues of these peptides. Unlike T cells, NK cells have germ-line encoded receptors that do not undergo recombination nor are they “specific” at discriminating self from non-self peptides (27). Instead, they have a moderate degree of peptide “sensitivity,” mediated in large part by KIR:HLA-I interactions, which allows NK cells to monitor for changes in the peptide repertoire expressed by target cells. In fact, common HIV-1 sequence variants can modulate binding of inhibitory KIR to HLA-I, and by this means modulate NK-cell function (61, 124, 125), which has also been demonstrated in the case of SIV (121). Alternatively, NK cells may respond to altered MHC-I peptide processing following induction of the immunoproteasome in response to viral infection. IFN-γ stimulation results in increased cleavage of peptides after hydrophobic and basic residues. Thereby, it alters the C-terminus of available peptides for HLA class I presentation [reviewed in Ref. (126)], which may ultimately affect KIR binding to HLA-I:peptide complexes presented on the cell surface of stressed cells.
Viral variants arising due to CTL-mediated pressure can in turn impact KIR recognition by (i) impairing binding to inhibitory KIRs (61, 127), (ii) reducing HLA-C surface levels (128), or (iii) enhancing binding to inhibitory KIRs directly, a mechanism termed as “double-escape” (129). Furthermore, several amino acid polymorphisms in the viral genome, which showed a significant enrichment in subjects having a specific KIR gene, have been identified (130). As one example, a polymorphism in the overlapping reading frame of vpu and env was associated with the presence of KIR2DL2 in HIV-1–infected subjects. Antiviral activity of KIR2DL2+ NK cells against this viral variant was reduced in vitro (130). However, a role for HLA-I in this process could not be determined due to small sample size. A subsequent study in a larger cohort of HIV-1 clade C–infected individuals identified two viral sequence variants, that were significantly enriched in individuals in the presence of the combined KIR2DL3–HLA-C*03:04 genotype. One of the variants (Tgag303V) was contained within a CTL epitope and located at the C-terminal end of the nonamer (YVDRFFKVL), but did not mediate escape from recognition by HLA-C*03:04-restricted CTLs compared to the wild-type sequence (131). This viral variant, however, enhanced binding to KIR2DL3 and inhibited KIR2DL3+ NK cells in vitro (132). Overall, these studies support the concept of KIR-mediated selection pressure on HIV-1 as an additional source driving viral evolution. Furthermore, a recent report showed that binding of KIR2DL2/3 to HLA-C1 allotypes is more selective to presented peptides than KIR2DL1 binding to HLA-C2 (60), further enhancing our mechanistic understanding of KIR:peptide:HLA-I interactions. Moreover, this study showed that certain peptides (including an HIV-1 Gag peptide) allow for binding of KIR2DLs to non-canonical HLA-C molecules (60). Taken together, whereas NK cells are not able to distinguish between self- and non-self peptides, KIR binding to HLA-I is certainly sensitive to changes in the peptide sequence presented on HLA-I molecules. This may in turn facilitate recognition of HIV-1–infected cells, potentially not only via presentation of viral peptides but also due to stressed-induced changes in the HLA-I-presented peptide repertoire.
Of note, the majority of studies evaluating the peptide sensitivity of KIR:HLA-I interactions to date have relied on external labeling with peptides, but overall, the abundance of viral peptides eluted from HLA-I compared to self-peptides is low (133, 134). Yet, antagonist peptides (i.e., peptides that are presented by HLA-I but abrogate KIR binding) can significantly interfere with KIR clustering at immune synapses and override NK-cell inhibition (135, 136). Therefore, HIV-1 infection may lead to NK-cell activation by causing a shift between antagonist and agonist peptides. Consequently, further investigations on how HIV-1 infection changes the HLA-I–presented peptide repertoire and how this impacts NK-cell function are needed. Nonetheless, CTL pressure on viral sequence appears to be dominant, as the first escape mutations arise after peak viremia and following expansion of HIV-1–specific CTLs (9). KIR are also expressed on T cells and can modulate CTL activity (75, 83); therefore, a potential role of KIR+ T cells in explaining KIR/HLA disease associations has to be considered. Overall, studies suggest a complex interplay between innate and adaptive immune pressures in driving HIV-1 sequence evolution, with HLA-I being central to the interaction with KIRs and TCRs.
KIR3DS1 and the Non-Classical HLA-F—A Non-Classical Stress Ligand?
Genetic evidence and functional data not only implicate KIR3DS1 in HIV-1 disease but indicate a widespread effect of KIR3DS1 in autoimmunity, transplantation, cancer, and other viral infections (137). Yet, for years, a definite ligand for this receptor that could account for these effects remained elusive. Only recently, we and others discovered that KIR3DS1 can bind open conformers (OCs) of HLA-F, a non-classical HLA-I molecule (138, 139). This was confirmed via surface plasmon resonance (SPR), pull-down experiments, HLA-F tetramer binding studies, as well as KIR3DS1+ reporter cell assays (138, 139). Functionally, HLA-F OCs led to degranulation of KIR3DS1+ NK cells, as well as cytokine production in response to HLA-F (138). HLA-F is a non-classical HLA-I molecule with a unique combination of features. It is (i) highly conserved with one dominant allele (140) [similar to KIR3DS1 (71)], (ii) displays tight tissue specific regulation, with a mostly intracellular localization (141, 142), and (iii) is expressed on the cell surface of activated lymphocytes (143). HLA-F is known to bind to inhibitory KIR3DL2 (138, 144), as well as inhibitory LILRBs (145, 146), whereas the results on binding of HLA-F to KIRDS4 are conflicting (138, 139, 144, 146). In HLA-F, 5 of 10 residues, which are highly conserved in other HLA class I molecules, are substituted, resulting in an altered peptide groove (146). To date, there has been no structural data published describing the OC of HLA-F. Given that KIR3DS1, KIR3DL2 and KIR3DL1 (albeit weaker), bind to HLA-F, one could imagine a role of the D0 domain in contacting HLA-F OC as the D0 domain enhances KIR3DL binding to HLA class I and mediates binding to a non-HLA class I ligand (72, 147). Nevertheless, the contact residues of KIR3DS1 to OCs of HLA-F conferring specificity and high-affinity of the interaction are entirely unknown to date. SPR data suggest that KIR3DS1 additionally binds to OCs of classical HLA class I, but so far, the functionality of this binding remains to be demonstrated (139).
We previously demonstrated that HIV-1 infection causes upregulation of HLA-F at a transcriptional level in stimulated CD4+ T cells. Therefore, KIR3DS1 binding to HLA-F expressed as a “stressed self” signal on HIV-1–infected cells might explain the superior ability of KIR3DS1+ NK cells to inhibit viral replication in autologous CD4+ T cells (92, 138). Thus, the interaction between KIR3DS1 and HLA-F upregulated on HIV-1–infected cells may have similarities to the well-reported upregulation of stress ligands such as ULPBs and MIC-A/MIC-B in HIV-1 infection, which are in turn recognized by the activating NK-cell receptor NKG2D (43).
Given the identification of HLA-F as a KIR3DS1 ligand, the following question remains unsolved: why is the strong genetic protective effect of KIR3DS1 observed preferentially in combination with HLA-Bw4I80 in HIV-I infection? We can conceive four potential models that are not mutually exclusive and may explain this phenomenon (Figure 1):
(A) KIR3DS1 binds to HLA-B*57:01 expressing particular HIV-1 peptides: Only six residues differ in the extracellular domain of KIR3DS1 and KIR3DL1 (*013 versus *001 allele products, respectively) and one of these substitutions (L166R) abolishes binding to HLA-B*57:01. Nonetheless, one recent modeling study identified two HIV-1 derived peptides that can overcome the steric hindrance of R166 with HLA-B*57:01 R83 and allow for binding of KIR3DS1 to the HLA-B*57:01–peptide complex. Binding was of sufficient avidity to activate KIR3DS1+ Jurkat reporter cells (148). Thus, a change in the peptide repertoire resulting from HIV-1 infection might therefore allow KIR3DS1 to engage certain HLA-Bw4I80 molecules and trigger KIR3DS1+ NK-cell cytotoxicity. However, further studies are needed to confirm this and assess the functional relevance in natural HIV-1 infection.
(B) HLA-Bw4I80enhances HLA-F expression at the cell surface of HIV-1–infected cells: HLA-I gene products differ in their ability to form homodimers on the cell surface. In particular, the protective HLA-B*27:05 allotype is commonly expressed as a β2m-free disulfide-bonded homodimer (149, 150). Formation of HLA-I dimers in turn can affect recognition by immune receptors (151–153). HLA-F was reported to bind to OCs of other HLA-I to varying degrees and form heterodimers (154). Goodridge et al. discuss that the varying potential of different HLA-I gene products to interact as OCs with HLA-F may modulate HLA-F surface expression levels (144). Protective HLA-B allotypes (e.g., HLA-B*57:01) indeed demonstrate a higher degree of tapasin-dependent assembly and less stability as an OC compared to HLA-B allotypes associated with rapid progression (e.g., HLA-B*35: 03) (155). Thus, protective allotypes might differ from susceptible allotypes in their ability to interact as HLA-I OCs with HLA-F in a setting of HIV-1 infection, in turn enhancing or diminishing recognition by KIR3DS1+ NK cells. This would indicate a KIR3DS1: HLA-Bw4I80:HLA-F protective axis in HIV-1 infection that is independent of KIR3DL1.
(C) KIR3DS1:HLA-F and KIR3DL1:HLA-Bw4I80interactions are independently, but synergistically protective: Martin et al. identified the protective effect of combined KIR3DS1 and HLA-Bw4I80, but the vast majority of individuals bearing KIR3DS1 in this study were heterozygous and thus also encoded for KIR3DL1 (22). Furthermore, Jiang et al. demonstrated that KIR3DS1/KIR3DL1 heterozygosity in HLA-Bw4I80–carrying individuals conferred superior HIV-1 disease control. Therefore, it might be the heterozygous state of KIR3DS1/KIR3DL1 in the context of HLA-Bw4I80 that confers protection in HIV-1 infection, rather than KIR3DS1 alone with HLA-Bw4I80 (91). Thus, protection could derive from a synergistic but independent effect of KIR3DS1–HLA-F and KIR3DL1–HLA-Bw4I80 interactions. Long et al. showed that possessing KIR3DS1 confers greater NK-cell functionality, also in absence of HLA-Bw4I80 (93). Under this model, the most effective NK cells against HIV-1–infected target cells would express both KIR3DS1 and KIR3DL1 and undergo activation via KIR3DS1-mediated engagement of HLA-F and KIR3DL1-dependent loss of inhibition due to HLA-B downregulation.
Yet, there is evidence that KIR3DS1 expression and function is not completely independent from KIR3DL1, as KIR3DS1 mRNA, and KIR3DS1+ NK-cell frequency increases with more gene copies of KIR3DL1 (90). Additionally, KIR3DS1/KIR3DL1 individuals display superior viral inhibition activity than individuals with either KIR alone in the presence of HLA-Bw4 (90). Thus, there is a possibility of KIR3DL1-mediated epistatic regulation of KIR3DS1 expression and function. However, the existence of a KIR3DS1+KIR3DL1+ coexpressing NK-cell subset has not yet been definitively proven due to the limitations of current anti-KIR antibody cross-reactivity.
(D) KIR3DL1:HLA-Bw4I80interactions are necessary to limit KIR3DS1-HLA-F-mediated immune activation: As chronic viral infections can drive inflammatory processes resulting from persistent immune activation (156), downmodulation of the immune response is important for host homeostasis and preventing immunopathology; especially in HIV-1 infection where immune activation can accelerate disease progression (157). Thus, it is conceivable that inhibition of NK cells via KIR3DL1:HLA-Bw4 interactions may be important to counteract an exuberant immune response mediated by KIR3DS1+ NK cells recognizing HLA-F on “stressed”/infected cells. Moreover, education through inhibitory KIRs has been shown to promote increased survival of iKIR+ NK cells (105). Increased survival of educated KIR3DL1+ NK cells might counteract chronic immune activation that can result in disease progression. In line with this, the study of Martin et al. showed that KIR3DS1 homozygosity without HLA-Bw4I80 was modestly associated with rapid progression to AIDS (22). Therefore, as supported by mouse models that implicate NK cells as “rheostats” in chronic viral infections (158), combined stimulatory and inhibitory signaling may result in a tunable antiviral response that confers optimal HIV-1 disease control without causing immunopathology.
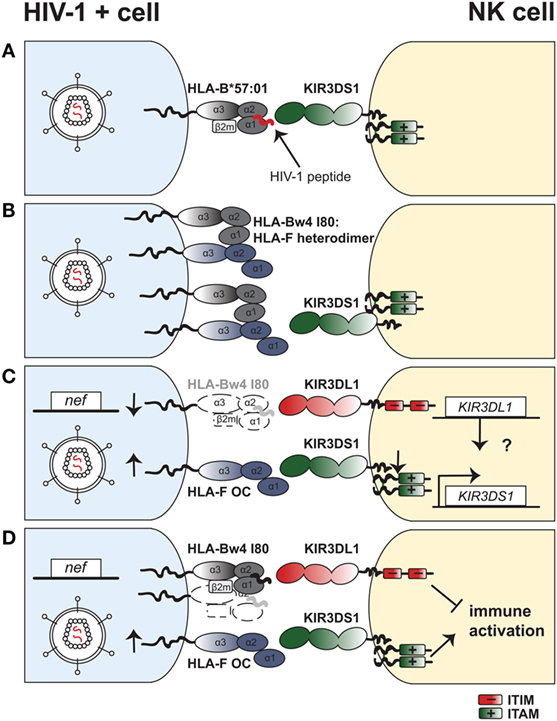
Figure 1. Four models with potential mechanisms to explain the underlying protective effect of the combined KIR3DS1–HLA-Bw4 genotype in HIV-1 infection. (A) Viral peptides allow for KIR3DS1 binding to HLA-B*57:01 and trigger natural killer (NK)-cell activation. Presentation of viral peptides (in red) on HLA-B*57:01 upon HIV-1 infection of target cells (blue) enables binding of KIR3DS1 on NK cells (yellow). The short cytoplasmic tail of KIR3DS1 associates to the adaptor molecule DAP12, which bears two ITAMs. (B) HLA-Bw4 enhances human leukocyte antigen F (HLA-F) expression at the cell surface of HIV-1–infected cells. Open conformers of HLA-F exist as heterodimers with open conformers of HLA-Bw4 on the cell surface of HIV-1–infected cells. This enhances binding and triggering via KIR3DS1 on NK cells. (C) KIR3DS1:HLA-F and KIR3DL1:HLA-Bw4 interactions have independent but synergistic protective effects in HIV-1 infection. HIV-1 infection of target cells leads to downregulation of HLA-Bw4 from the cell surface via action of the accessory protein Nef. Loss of HLA-Bw4 on the infected cells leads to loss of inhibition via KIR3DL1. Simultaneously, cell stress induced by HIV-1 infection leads to upregulation of open conformers of HLA-F, which bind to KIR3DS1 and trigger NK-cell activation. A potential epistatic regulation of KIR3DS1 gene expression via the KIR3DL1 gene is depicted. (D) KIR3DL1:HLA-Bw4I80 interactions limit KIR3DS1:HLA-F-mediated immune activation. HIV-1 infection directly (and indirectly) causes cellular stress, which in turn upregulates surface expression of HLA-F open conformers on CD4+ T cells and other cell types. OCs of HLA-F bind to KIR3DS1 and trigger NK-cell activation. On the other hand, KIR3DL1 binds to HLA-Bw4 molecules, which are present on HIV-1–infected cells, although at low levels due to HIV-1 Nef-mediated downregulation. Inhibitory signaling via KIR3DL1 limits NK-cell activation and inflammatory cytokine production, thus limiting activation via KIR3DS1.
In summary, our mechanistic understanding of how protection in HIV-1 disease is mediated in the context of combined KIR3DS1 and HLA-Bw4 is still limited and requires further study. Although we focus on NK cells, a potential role for HLA-Bw4I80-restricted CD8+ T cells expressing KIR3DS1 has also to be considered (84). So far, genetic studies of disease susceptibility have been extremely resourceful in guiding our understanding of the mechanisms involved in HIV-1 control. Therefore, HIV-1 disease association studies that are able to tease out the effect of KIR3DS1 homozygosity in the context of HLA-Bw4I80 would be of great utility, but will require large sample sizes.
The Role of Peptide:HLA-F Complex
Major histocompatibility complex class I exists in two biologically relevant conformations on the cell surface: (i) as a membrane-bound heavy chain lacking peptide and β2-microglobulin (β2m) termed open conformer (OC) or (ii) as a trimeric heavy chain:β2m:peptide complex (159). Recently, thermal denaturation assays demonstrated that OCs of HLA-F are more stable (146) than OCs of other HLA-I gene products (160). Earlier findings assessing stability after cold treatment suggested an increased stability of HLA-F OCs compared to open conformers of classical HLA-I (141, 142). This—and the fact that no canonical peptides could be eluted from HLA-F—supported the notion that HLA-F is mainly expressed as an OC devoid of peptide (142, 154).
Recently, the crystal structure of HLA-F (in complex with β2m and peptide) was solved, shedding first light onto the molecular structure of HLA-F (146). Surprisingly, this work showed that HLA-F has a unique peptide-binding grove that resembles the groove of classical HLA-I but does not anchor peptides at their N-terminus, allowing for binding of longer peptides. Indeed, peptides eluted from HLA-F and characterized by mass spectrometry had an extended length distribution compared to classical HLA-I molecules, peaking at 12 amino acids and with peptides up to 30 amino acids observed. This unconventional length rather resembles the length of HLA class II-presented peptides.
Moreover, new insights into the structure and docking mode of LILRB1 interacting with the HLA-F:β2m:peptide complex were gained. The LILR family (also termed LIR, ILT, or CD85) are encoded on chromosome 19 within the leukocyte receptor complex along with the KIR locus. In total, 13 different LILRs have been identified. Similar to KIRs, LILRs can provide an either inhibitory (LILRB) or activating (LILRA) signal, depending on the presence of an ITIM or the association to ITAM-containing adaptor molecules, but also depending on the cellular context (161). LILRB2 is not expressed on NK cells and its implications in HIV-1 disease are reviewed elsewhere (24). LILRB1 recognizes most classical and non-classical HLA-I molecules, except for HLA-E (162–164), given that it binds to the conserved α3 domain of the HLA-I heavy chain as well as β2m (165, 166). Intriguingly, the affinity of LILRB1 to peptide-bound HLA-F:β2m is the highest observed so far compared to other HLA-I ligands (146, 167). LILRB1 is expressed on NK cells in varying percentages (0–50% with high interindividual variability), as well as on T cells and professional antigen presenting cells such as DCs, monocytes/macrophages, and B cells (146, 168). Engagement of LILRB1 in vitro leads to inhibition of cytotoxicity and cytokine production in a subset of NK cells (151, 169, 170), but interestingly, LILRB1+ (but not LILRB1–) NK cells are able to markedly suppress HIV-1 replication in infected monocyte-derived DCs in a manner independent of classical HLA-I (171), hinting at a possible role of HLA-F.
Looking at the binding footprint of LILRB1 on HLA-F, it is improbable that the interaction is sensitive to the nature of the presented peptide—in contrast to certain KIRs. In the case of KIR3DS1, it was shown that KIR3DS1+ reporter cells responded to HLA-F OCs, but were not triggered by peptide-bound HLA-F complexes (146). This could be due to peptide-induced conformational changes in HLA-F structure or direct steric inhibition by the bound peptides. Furthermore, inhibitory KIR3DL2 recognizes OCs of HLA-F or HLA-I and posssibly heterodimers of HLA-F with HLA-I heavy chains, with the latter also being increasingly expressed on activated lymphocytes (144). This raises interesting possibilities for a cell-stress induced conformational change in HLA-F allowing binding to activating receptors, such as KIR3DS1, while abrogating binding to inhibitory receptors, such as LILRB1. Thus, although HLA-F is not expressed on the surface of lymphocytes in a resting state (143), it potentially can exist in various conformations on stressed cells (154) with differential impact on NK-cell function (Figure 2).
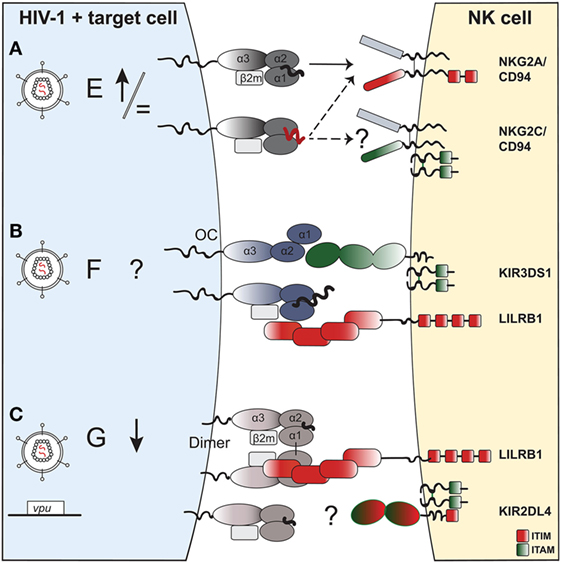
Figure 2. The potential impact of HIV-1 infection on expression of non-classical human leukocyte antigen class I (HLA-I) molecules on a target cell and effect on natural killer (NK)-cell receptor binding. (A) HLA-E. HIV-1 infection of target cells leaves HLA-E surface levels either unchanged or slightly upregulated. HLA-E can present self-peptides (in black) that bind to the NKG2A:CD94 receptor complex, and inhibit NK-cell function. In the context of HIV-1 infection, HLA-E can present viral or “stress”-self-peptides (in red) that abrogate (or reinforce) binding to NKG2A:CD94 and modulate NK-cell activity. The potential role of viral or “stress” peptides presented on HLA-E that could trigger the activating NKG2C:CD94 receptor complex on NK cells is currently unknown. NKG2C associates to DAP12, an adaptor molecule containing two ITAMs. (B) HLA-F. The exact impact of HIV-1 infection on HLA-F surface expression in different cell types needs yet to be established. In general, HLA-F is expressed on activated or stressed cells, potentially in distinct functionally relevant conformations: (i) an open conformer that binds to the activating NK-cell receptor KIR3DS1 or (ii) a β2m-bound complex presenting peptides of unusually long length for HLA-I, which allows binding of the inhibitory receptor LILRB1. (C) HLA-G: one study showed downregulation of HLA-G in monocyte-derived macrophages, potentially via HIV-1 Vpu, although this has not yet been confirmed in primary cells. Moreover, the functional relevance of HLA-G downregulation in antiviral immune responses has not been established to date, although HLA-G is thought to play a predominantly immunoregulatory role given its interaction with inhibitory receptors. HLA-G can form dimers on the cell surface via an interchain α1 disulfide bond, which enhances recognition by inhibitory LILRB1 on NK cells. KIR2DL4 binding to HLA-G remains controversial. KIR2DL4 has a dual activating and inhibitory effect on NK cells, given that its cytoplasmic tail contains an ITIM and it associates to ITAM-bearing FcRγ.
The Non-Classical HLA-G—An Immune Modulator?
Human leukocyte antigen-G is a non-classical HLA-I that displays a high degree of tissue restriction. It was first discovered in extravillous trophoblast cells in the fetal placenta (172), where HLA-G protein is abundant (173) and since then has been extensively studied in the context of reproduction. Further studies showed that under healthy conditions, HLA-G is expressed in other immune-privileged sites including the cornea (174), thymus (175), nail matrix, and on mesenchymal stem cells (176, 177). Under inflammatory conditions such as CMV infection or within a tumor microenvironment, HLA-G can be expressed on DCs and monocytes/macrophages (178, 179), and is reported to be upregulated in monocytes treated with IFN-γ (a potent inducer of HLA-I and -II expression) as well as IL-10 (180–182). Also, an increasing number of studies shows aberrant HLA-G mRNA expression by tumors (183, 184) [reviewed in Ref. (185)], but some of these findings remain controversial as in other studies no HLA-G protein was identified (179, 186) [reviewed in Ref. (187)]. Overall, there is evidence that HLA-G expression is induced on various immune cells under inflammatory conditions resulting from infections, allergies, or allogeneic stimulation following transplantation (188–191).
As a result of a premature stop codon in exon 6 (192), the cytoplasmic tail of HLA-G is truncated and the heavy chain has a molecular weight of only 39 kDa, compared to the 45 kDa weight of classical HLA class I heavy chain. In total, seven splicing variants of HLA-G have been described (193, 194). The predominant splice variant in vivo is HLA-G1, which encodes for the full-length, membrane-bound HLA-G protein (195, 196). Alternatively, soluble HLA-G (sHLA-G) can be generated from three splice variants or via proteolysis of the HLA-G1 isoform (197). Interestingly, sHLA-G can confer a protective effect to cells normally permissive to NK-cell killing (197). Apart from HLA-G1, three other alternatively spliced transcripts encode membrane-bound HLA-G, albeit in a truncated form: HLA-G2 lacks the α2 domain, HLA-G4 lacks the α3 domain, and HLA-G3 lacks both the α2 and α3 domains (198). These transcripts were reported to inhibit NK-cell function, although it remains unclear through which NK-cell receptors this occurs (198–200). Moreover, it was suggested that isoforms HLA-G2 and -G3 are expressed in individuals homozygous for the HLA-G*0105N null allele (201), possibly explaining the existence of healthy adults lacking full length HLA-G1 (201). Of note, all splicing variants encode the leader sequence enabling HLA-E expression (202) and thereby their expression in target cells can indirectly inhibit NK cells via NKG2A:CD94 (163).
To date, 18 distinct functional proteins of HLA-G have been identified, with the HLA-G gene encoding a total of 54 HLA-G alleles (including two HLA-G null alleles) (56). While most of the polymorphism of classical HLA-I genes lies in the α1 and α2 domains that bear the peptide-binding groove (203), HLA-G has a relatively conserved peptide-binding groove and has allelic variability occurring within the 3′UTR, which is important for posttranscriptional regulation of HLA-G (203, 204). Peptides eluted from HLA-G thus far appear to be derived from a restricted number of proteins (205) and a crystal structure demonstrates that these presented peptides are buried deep within the peptide-binding groove (206). The induction of an HLA-G-restricted CD8+ T cell response against a human cytomegalovirus peptide in mice was described, but the cytolytic capacity of these T cells was limited (207). Overall, it seems that the immune modulatory functions of HLA-G mediated through binding of inhibitory receptors expressed on a variety of immune cells dominates over a potential role in presenting peptides.
NK-Cell Receptors Recognizing HLA-G
Human leukocyte antigen-G is recognized by LILRs with greater affinity than HLA-A, -B, or -C molecules (208). In addition, HLA-G is unique in possessing a cysteine at position 42 of its α1 domain, which allows for an unusual conformation of HLA-G as a homodimer of two β2m-associated HLA-G complexes (152, 209, 210) (Figure 2C). This conformation dramatically enhances recognition and signaling of LILRB1 (151) and has been demonstrated to occur naturally on trophoblasts (173). Indeed, inhibition of LILRB1+ NK-cell function is sensitive to the conformation of HLA-G, as the heavy chain of HLA-G alone does not inhibit LILRB1+ NK cells (211). Studies measuring inhibition of LILRB1+ NK-cell cytolytic function via HLA-G have to account for HLA-E expression as it is upregulated through the HLA-G leader peptide—an exception being the K562 cell line, which does not express HLA-E (212). Independent of HLA-E, HLA-G interferes with immunological synapse formation and inhibits NK-cell cytotoxicity (212, 213). Additionally, Riteau et al. demonstrated that HLA-G expression has a major inhibitory effect on NK cell lysis through LILRB1, also when coexpressed with other HLA-I ligands (214). Besides the inhibitory effect of HLA-G expression on NK-cell-effector function itself, HLA-G can impair NK-DC crosstalk. Pretreatment of DCs with sHLA-G leads to reduced activation and IFN-γ production by NK cells (215), while IFN-γ in turn triggers HLA-G surface expression (180, 181). This again supports the notion that HLA-G has tolerogenic properties.
In addition to LILRB1, HLA-G has been proposed to modulate NK-cell function via binding KIR2DL4. KIR2DL4 is a framework gene within the KIR locus and thus is present in virtually all haplotypes, but there is a high frequency of alleles lacking the transmembrane domain or having truncated cytoplasmic tails (216). In peripheral blood, expression of KIR2DL4 is weak and restricted to the CD56bright subset, but can be induced on NK cells in vitro with stimulation (217, 218). KIR2DL4 has unique functional properties compared to other receptors of this family. A positively basic arginine residue in the transmembrane domain allows for association with the activating Fc receptor gamma protein (219), while the long cytoplasmic tail contains one immunoreceptor tyrosine-based inhibitory motif (ITIM). This results in mixed activating and inhibitory signaling, which has been shown to occur in vitro (219–222). In line with this, crosslinking of KIR2DL4 on peripheral blood NK cells induces IFN-γ production, and (albeit weaker) NK-cell cytotoxicity (217, 218, 223).
Newer reports provide conflicting evidence regarding the interaction of HLA-G with KIR2DL4 (224, 225). Although several groups reported binding using various techniques including cellular transfectants, SPR, and functional assays (169, 226–230), others have failed to reproduce KIR2DL4 binding via SPR, tetramers, or functional IFN-γ responses to sHLA-G (210, 231, 232). The crystal structure of the extracellular domains of KIR2DL4 solved by Moradi et al. (233) demonstrated oligomerization of KIR2DL4, uncharacteristic of other KIRs. In this study, no binding of KIR2DL4 to HLA-G was detected via SPR (233). An explanation might be that signaling via KIR2DL4 only occurs upon concentration of the ligand in endosomes [as discussed in Ref. (187)], since sHLA-G endocytosed into KIR2DL4-containing compartments was shown to induce cytokine secretion of NK cells (229, 234). Regardless of its binding to HLA-G, higher copy numbers of KIR2DL4 have been linked to better survival of CD4+ T cells and increased IFN-γ responses from NK cells during acute SIV infection in rhesus macaques (235).
HLA-G Expression in HIV-1
Only a low percentage of immune cells in healthy subjects expresses HLA-G, whereas in HIV-1 infection a substantial upregulation of HLA-G has been observed in both peripheral blood monocytes and T-cell subsets (236). This was later partly attributed to antiretroviral treatment, as frequencies of HLA-G+ monocytes decreased after treatment interruption (237). In fact, nucleoside reverse transcriptase inhibitors were found to increase HLA-G expression, whereas protease inhibitors did not (238). A role for HLA-G+ HIV-1–restricted CD8+ T cells has furthermore been described in HIV-1–infected subjects (239). Contrary to in vivo studies of high HLA-G expression levels on monocytes of patients undergoing HAART (236–238), one study showed downregulation of HLA-G1 surface expression in HIV-1–infected monocyte-derived macrophages in vitro (240). This downregulation was suggested to be mediated via HIV-1 Vpu (240), given that the truncated tail of HLA-G renders it resistant to HIV-1 Nef-mediated downregulation (241). Yet, this needs to be confirmed in primary cells. Overall, how HIV-1 directly impacts HLA-G expression in different cell types remains unclear.
In addition to inhibiting DC function via LILRB2 (242) and regulating CD4+ T-cell proliferation (243), sHLA-G can inhibit NK-cell killing in vitro and may therefore suppress NK-cell function in vivo (197). sHLA-G plasma levels change during the course of HIV-1 infection and treatment, as two groups reported high sHLA-G levels in early infection (244) with a significant decrease after treatment initiation (245). In rapid progressors, however, levels of sHLA-G were persistently elevated even despite treatment initiation, while this was not the case for untreated normal progressors and long-term non-progressors (244). Furthermore, sHLA-G levels were higher in patients with opportunistic infections, indicating a potential role of sHLA-G as a surrogate marker of disease progression (246). In a cohort of female commercial sex workers from Benin, HIV-1–infected subjects were reported to have lower levels of sHLA-G in plasma (247) but higher levels of sHLA-G in the genital mucosa (248). Of note, levels of sHLA-G are also in part determined genetically by distinct HLA-G alleles (249). Thus, data on sHLA-G levels in HIV-1 infection need to be carefully controlled for confounding factors such as HAART (237, 238), HLA-G genetic background (249), sampling site (247, 248), or coinfections (246, 248). In summary, it is not known whether higher sHLA-G levels have direct functional consequences on HIV-1 disease progression via modulation of NK and other immune cells, or whether sHLA-G levels are rather a reflection of viremia and the antiviral immune response.
Genetic Evidence for a Role of HLA-G in HIV-1 Infection
Although HLA-G polymorphisms are limited, certain HLA-G alleles have been suggested to be involved in susceptibility to HIV-1 infection. In 2004, Matte et al. reported that the HLA-G*0105N allele, a null variant which does not encode functional HLA-G1, was protective in HIV-1 acquisition, whereas the HLA-G*01:01:08 allele encoding for full-length HLA-G increased the risk of HIV-1 infection. They formulated the hypothesis that non-functional HLA-G proteins may allow for better NK-cell killing of HIV-1–infected cells (250). This observation was not consistent with findings of subsequent studies, which reported either enrichment of HLA-G*0105N in HIV-1–positive women (251) or did not identify HLA-G*0105N allele as a disease modifying factor (252). Other HLA-G alleles identified were HLA-G*01:04:04, which associated with susceptibility to HIV-1 infection, and HLA-G*01:01:01, which was enriched in HIV-1–resistant women (252). One study states that these conflicting findings may be explained by variation of HLA-G polymorphisms among different ethnic populations and reports no association of HLA-G polymorphisms to HIV-1 susceptibility except in African-American cohorts (253).
As HLA-G is an important player involved in maternal-fetal tolerance, HLA-G polymorphisms have been studied in the context of vertical HIV-1 transmission from mother-to-child. Mothers bearing the HLA-G*01:03 allele were less likely to perinatally transmit HIV-1 (254). Upregulation of the functional isoform HLA-G1 mRNA in the placenta has been associated with increased risk of HIV-1 mother-to-child transmission (255). Further studies have assessed the risk of variants in the 5′ and 3′UTR of HLA-G, and in particular, the impact of the 14-bp insertion/deletion in the 3′UTR of HLA-G on mother-to-child transmission. In healthy subjects, the 14-bp insertion genotype (ins/ins) correlates with lower plasma levels of sHLA-G (256). In vitro, transfection of the 14-bp ins/ins HLA-G into K562 cells resulted in increased levels of membrane-bound HLA-G1 expression with higher mRNA stability and lower sHLA-G1 ratio (257). However, studies on the impact of the 14-bp insertion on HIV-1 vertical transmission risk report conflicting results (258–260). In horizontal transmission, the frequency of the 14-bp ins/ins genotype was enriched in HIV-1–infected patients in African (but not European) subjects (261). Overall, population studies attempting to shed light on the question whether functional versus non-functional HLA-G alleles are associated with HIV-1 susceptibility have painted an inconsistent picture. Moreover, posttranscriptional regulation of the HLA-G gene through variations in the 3′ and 5′ LTR and alternative splicing has to be considered as an important genetic factor modulating HLA-G expression levels in these studies.
The Oligomorphic Interaction between HLA-E and NKG2:CD94—A Contrast to the Diversified HLA-KIR System
Inhibition of NK cells can be achieved either through highly diversified KIR:HLA-I interactions or through a second inhibitory system indirectly monitoring the level of overall HLA-I expression. This latter inhibitory mechanism is achieved via the well-conserved NK-cell receptor–ligand interaction of NKG2A/CD94 with HLA-E (262). Contrary to other non-classical HLA-I gene products, HLA-E is ubiquitously expressed (263), but at substantially lower levels as compared to classical HLA-A, -B, and -C (264). Its expression is dependent on the expression of other HLA-I, as it presents a nonamer peptide derived from the signal sequence of several HLA-A, -B, and -C gene products as well as HLA-G. HLA-F and HLA-E itself lack an HLA-E–presented leader peptide (265).
Human leukocyte antigen-E has restricted polymorphism with to date only 25 known alleles (56), of which two—HLA-E*01:01 and *01:03—are the most frequent in the human population and are believed to be in balancing selection (266, 267). HLA-E*01:01 encodes for an arginine at position 107 (HLA-ER), whereas HLA-E*01:03 encodes for a glycine at this position (HLA-EG). This substitution leads to higher surface expression levels of the latter, despite similar intracellular protein levels (160). HLA-E is highly relevant to innate immune responses due to its interaction with heterodimeric NKG2/CD94 type II transmembrane-anchored receptors, which are expressed on a large proportion of NK cells as well as on a subset of CD4+ and CD8+ T cells (268–270).
Natural killer group 2 receptors are a family of C-type lectin receptors encoded within the NK gene complex on chromosome 12p12-13 (271). Almost all NKG2 gene products heterodimerize with CD94, a non-signaling invariant glycoprotein also encoded within the NK gene complex. These include NKG2A [which produces NKG2A and NKG2B gene products via alternative splicing (272)], NKG2C, NKG2E, NKG2F, and NKG2H. The NKG2D gene is also located within the NK gene complex, but its gene product has low sequence homology to other NKG2 receptors and forms an NKG2D:NKG2D homodimer (without CD94) that binds to the stress ligands MIC-A, MIC-B, and ULBPs, but not to HLA-E (273). Unlike KIR genes, NKG2 genes exhibit limited polymorphism (262, 274). Aside from being expressed widely on NK cells, they can also be expressed on subsets of T cells (275). Here, we focus on NKG2A:CD94 and NKG2C:CD94 receptor complexes, both of which bind HLA-E but have opposite effects on NK-cell function. While NKG2A signaling inhibits NK-cell cytotoxicity via two ITIMs in its cytoplasmic tail (276), NKG2C delivers activating signals through its associated adaptor molecule DAP12 (277).
Despite their similarity, the two major alleles of HLA-E differ in the subset of peptides they present (278). An example is the HLA-B*27–derived leader peptide, which stabilizes HLA-EG, but does not bind detectably to HLA-ER (160, 279). Similar to KIR:HLA interactions, binding of the NKG2:CD94 heterodimer to HLA-E is sensitive to the presented peptide (279, 280). The crystal structures of NKG2A:CD94 and NKG2C:CD94 receptor complexes bound to HLA-E presenting the HLA-G leader peptide (VMAPRTLFL; VL9) illustrate that both subunits (NKG2 and CD94) intimately interact with the peptide-binding domains (α1 and α2) of HLA-E. Interestingly, CD94 occupied the majority of the binding site, yet despite this, the NKG2A:CD94 complex had six times stronger binding affinity to HLA-E:VL9 than NKG2C:CD94 (280). Consequently, it is believed that CD94 is the main driver of HLA-E binding and peptide sensitivity, while the NKG2 subunit modulates affinity (and possibly sensitivity to some extent). Leader peptides of classical HLA-I presented on HLA-E do not trigger NK-cell activation through NKG2C, whereas NKG2A+ NK cells are potently inhibited by a wide range of different HLA-I–derived leader peptides (281). Therefore, the NKG2A:CD94–HLA-E interaction allows NK cells to indirectly monitor for changes in overall HLA-I expression without causing aberrant immune activation through NKG2C:CD94. An exception to this is HLA-E in complex with the HLA-G leader peptide, which can engage NKG2C:CD94 and trigger activation (279, 281). As HLA-G displays high tissue-specific restriction, this nonetheless allows for tight regulation of NKG2C triggering. The amount of surface stabilization of HLA-E by various leader peptides does not strictly correlate with the level of inhibition through NKG2A:CD94, which emphasizes the role of specific peptides in the binding of NKG2A:CD94 to HLA-E (282).
Peptide Presentation by HLA-E in the Context of Viral Infections
Like classical HLA-I, HLA-E can also present virus- or “stress”-derived peptides. The leader sequences of heat shock protein 60 (HSP60), which is induced under stress conditions (283), stabilizes HLA-E, but disrupts binding to NKG2A:CD94 and thus disinhibits NK-cell function (284). HLA-E can also be the target of viral immune evasion. CMV, for example, encodes for a sequence identical to the HLA-C*03 leader peptide that can increase HLA-E expression and inhibit NK-cell cytotoxicity (285). Additionally, an HCV-derived epitope (HCV Core35-44) stabilizes HLA-E and inhibits NK-cell lysis (286). Cheent et al. showed that viral- or heat shock protein-derived peptides in isolation did not inhibit NK-cell lysis. However, these peptides enhanced inhibition in the presence of HLA-E–presented leader peptides and therefore were termed “synergistic peptides.” Confocal microscopy has shown that these synergistic peptides act by recruiting non-signaling CD94 (without NKG2A) to the immunological synapse (262). Similar to peptide antagonism in KIR-HLA interactions (135, 136), this adds an additional layer of complexity to peptide-dependent modulation of NK-cell-effector function.
For HIV-1, the capsid-derived p24 aa14–22 epitope AISPRTLNA (AA9) has been described to stabilize HLA-E. One study by Natterman et al. demonstrated that AA9 inhibited NK cell-mediated cytolysis of peptide-pulsed HLA-E–transfected K562 cells (287), and that NK-cell killing could be restored via antibody blockade of either HLA-E or NKG2A. Contrary to this study, however, Davis et al. reported that HLA-E:AA9 tetramers did not bind to NKG2A+ CD56bright NK cells (while HLA-E:VL9 tetramers did). Thus, the authors suggest a potential role for the AA9 peptide in abrogating HLA-E binding to NKG2A:CD94 on NK cells, explaining enhanced degranulation of NKG2A+ NK cells against HIV-1–infected cells as compared to NKG2A− NK cells (288). In line with a role of HLA-E in HIV-1 infection, a genetic study in a cohort of Zimbabwean women demonstrated a four-fold reduced risk of HIV-1 acquisition in individuals homozygous for HLA-E*01:03 (HLA-EG) alleles compared to heterozygous or HLA-E*01:01 homozygous individuals. Given that HLA-EG is a high-expression allele, the authors speculated that increased presentation of HIV-1 peptides by HLA-E enhances NK cell cytotoxicity against HIV-1–infected target cells during the initial stages of infection (289).
Besides the role of HLA-E in innate immunity, increasing evidence demonstrates that HLA-E presentation of viral peptides derived from CMV, EBV, and HCV can elicit HLA-E–restricted CD8+ T-cell responses (290–292). Furthermore, Hansen et al. (293) showed that inoculation of rhesus CMV-based SIVgag vectors leads to presentation of surprisingly diverse epitopes on MHC-E, inducing a broadly directed and protective CD8+ T cell response in rhesus macaques (293). So far, HIV-1–specific HLA-E–restricted CD8+ T cells have not been shown in humans (294), but the conserved nature of HLA-E alleles among different populations, its ability to present viral peptides, and its dual role in innate and adaptive immunity renders HLA-E an important target for future research.
NKG2A+ NK Cells—A Subset with Enhanced (Not Reduced) Antiviral Capacity in HIV-1
Chronic HIV-1 viremia leads to a decrease in the proportion of NK cells expressing NKG2A (32, 295–297), and normal NKG2A levels are restored only after prolonged times of antiretroviral therapy (297). Subset analyses show, however, that NKG2A+ cell frequency increases within the CD56dimCD16bright NK-cell subset over the course of HIV-1 disease progression, whereas NKG2A+ cell frequency is decreased in the dysfunctional CD56− NK cell subset (298). Given that this highly dysfunctional CD56− NK cell subset with poor cytotoxic capacity expands in viremic subjects (33, 34), bulk NKG2A+ NK-cell frequencies are reduced (298). Presence of viremia in patients with low CD4+ T-cell counts correlated with significantly higher NKG2A+ frequencies on CD56dimCD16bright NK cells compared to aviremic patients with low CD4+ T cell counts (298), which may suggest a potential effect of long-term HIV-1 exposure itself on modulating NKG2A expression.
On the other side of the equation, HLA-E levels on CD4+ T cells from HIV-1–infected patients increase with declining CD4+ T cell counts in vivo (299). Upon HIV-1 infection or reactivation in vitro, HLA-E surface levels remain unchanged (36, 114, 288) or increase (287, 299). Functionally, blocking of the inhibitory NKG2A:CD94 interaction with HLA-E increases the ability of NK cells to kill HIV-1–infected CD4+ T cells in vitro (114, 287). Similarly, blocking of NKG2A enhances ADCC of NK cells toward antibody-coated HIV-1–infected CD4+ T cell blasts (115). Although these initial data implied the notion that HLA-E–NKG2A:CD94 interactions were inhibitory and detrimental to elimination of HIV-1–infected cells, recent experimental data demonstrated a superior ability of the NKG2A+ NK-cell subset to degranulate in response to HIV-1–infected CD4+ T-cell blast compared to NKG2A− subsets (288). Moreover, NKG2A+ NK cells showed the highest polyfunctional responses with increased IFN-γ and MIP-1β, as well as higher CD107a expression against HIV-1–infected CD4+ T cell blasts (300). This suggests that HLA-E-mediated inhibition of NK-cell function via engagement of NKG2A:CD94 is incomplete, potentially due to a skewed peptide repertoire in infected cells (288) (Figure 2A). Moreover, the increased functionality of NKG2A+ NK cells highlights the concept that the inhibitory NKG2A:CD94 receptor is important in NK-cell education (301), as described later in more detail. Taken together, the factors driving an overall decline in NK-cell function in HIV-1–infected individuals are not entirely clear, although decreased frequency of NKG2A+ NK cells may play a role.
NKG2C+ NK Cells—A Role in HIV-1 Independent (or Dependent) of CMV?
It is conceivable that ligation of activating NKG2C:CD94 via HLA-E may enhance cytotoxicity toward HIV-1–infected cells, but this has not been demonstrated. In healthy subjects, NKG2C is expressed only at low-to-moderate frequencies depending on NKG2C zygosity and CMV status (288, 302). In HIV-1–infected subjects, an increased frequency of NKG2C+ NK cells can be detected (295), independent of HIV-1 disease stage or presence of viremia (298), leading to a reversed NKG2A+-to-NKG2C+ NK-cell ratio in HIV-1–infected subjects compared to healthy controls (296). Additionally, NKG2C+ NK cells form part of the dysfunctional CD56−CD16+ NK-cell population in HIV-1–positive viremic patients (303). Thus, NKG2C expression appears to be modulated by HIV-1 infection, but differences in NKG2C+ NK-cell activity toward HIV-1–infected cells have not been demonstrated (Figure 2A).
It is important to note that CMV infection substantially skews the NK-cell repertoire toward NKG2C-expressing NK cells (304, 305). Furthermore, NK cells of CMV seropositive patients display enhanced cytotoxicity against target cells expressing HLA-E, which can be blocked by anti-NKG2C (306). Therefore, coinfection of CMV in HIV-1–infected patients is a highly relevant confounding factor when assessing NKG2C+ frequencies and function on NK cells. In a cohort of HIV-1–positive aviremic individuals, the association between increased NKG2C expression and HIV-1 infection disappeared when accounting for CMV seropositivity (307). Furthermore, Brunetta et al. showed that NKG2C+ NK-cell frequencies are higher in CMV seropositive individuals with HIV-1 infection compared to CMV seropositive HIV-1–negative subjects (297). Overall, the leading notion is that HIV-1 infection may render individuals more susceptible to CMV reactivation and impair immune control of CMV, potentially explaining the higher degree of CMV-driven expansion of NKG2C+ NK-cell subsets in HIV-1–infected subjects (308, 309). Additional evidence for a potential role of NKG2C comes from HIV-1 disease association studies, where homozygous deletion of NKG2C in a cohort of HIV-1–infected subjects was associated with increased risk of HIV-1 infection. Moreover, a genotype with two functional copies of NKG2C was significantly enriched in long-term non-progressors compared to normal progressors. This indicates a functional role for the NKG2C receptor in HIV-1 infection (310), which remains to be established experimentally.
CMV-driven expansion of NKG2C+ NK cells has received great interest as it has been implied in conferring adaptive, memory-like functions to NK cells (311). Briefly, first evidence came from a study in hematopoietic stem cell transplantation (HSCT), where infusing NK cells from CMV-seropositive donors into CMV-seropositive HSCT recipients led to expansion of donor NKG2C+ NK cells and production of increased amounts of IFN-γ in comparison to donor NKG2C+ NK cells infused into CMV seronegative HSCT recipients (312). This hinted at a previous priming of donor NKG2C+ NK cells leading to an enhanced antiviral response upon re-challenge with CMV in the CMV seropositive HSCT recipient (312). Additional evidence of adaptive NK-cell function in a rhesus macaque model demonstrated that splenic NK cells derived from previously SIV-infected macaques specifically lysed DCs pulsed with SIV Gag or Env in vitro. Remarkably, antigen-specific NK-cell cytotoxicity against Gag- or Env-pulsed DCs was reduced by blocking NKG2A and NKG2C, which suggests a potential role of these receptors in NK-cell memory (313). Taken together, in humans, the role of activating NKG2C:CD94 receptors in HIV-1 infection, either for increased recognition of HIV-1–infected target cells via HLA-E (independent of CMV) or for a potential HIV-1 specific NK-cell response remains to be further investigated.
HLA-E Is Affected by Dimorphism in the Leader Peptide of HLA-B
An additional factor impacting HLA-E surface expression is a dimorphism in the leader peptide of HLA-A, -B, and -C. HLA-A and HLA-C alleles encode for a methionine at position 2 of the leader sequence, whereas HLA-B alleles can either encode for methionine (-21M) or threonine (-21T) at this position. Leader sequences with threonine at P2 do not allow for stable induction of HLA-E surface levels and consequently fail to confer protection from NK cells through engagement of inhibitory NKG2A:CD94 (279). In HIV-1 infection, HLA-B alleles containing a Bw4 motif are associated with protection from AIDS (69) and all HLA-Bw4 alleles (with the exception of HLA-B*38:01) encode for the −21T polymorphism (42), whereas HLA-Bw6 alleles encode for either −21T or −21M. In a large cohort of serodiscordant Zambian couples, Merino et al. aimed to elucidate the impact of HLA-B leader peptide dimorphism independent of the Bw4 motif. Compound carriage of either Bw6/−21T or Bw4/−21T alleles displayed similar levels of protection in comparison to Bw6/−21M alleles, which were associated with increased risk of seroconversion. This indicates an independent protective effect of the −21T dimorphism on HIV-1 acquisition (314). Moreover, NK cells lysed HIV-1–infected CD4+ T cells or HIV-1–infected monocyte-derived macrophages preferentially when target cells encoded for −21T/T over a range of various HIV-1 strains. Antibody-mediated blockade of HLA-E on −21M/M target cells increased NK-cell cytotoxicity, whereas no change was observed for −21T/T target cells. Surprisingly, in this study mean fluorescence intensity of HLA-E surface expression did not differ between the −21T/T, T/M, or M/M subsets (315). However, recent analyses employing mass cytometry revealed that donors with at least one copy of −21M displayed increased surface HLA-E levels compared to −21T homozygous donors. NK cells of −21M donors displayed reduced amounts and frequencies of NKG2A:CD94, but a higher phenotypic diversity (42). In this study, Horowitz et al. additionally showed that the increased availability of HLA-E peptides in −21M donors is important for NK-cell functionality (42).
Shaping of NK cell function via self-reactive inhibitory NK-cell receptors is a well-described process termed licensing or education (316). It is governed by two independent systems, the well-conserved interaction of NKG2A with HLA-E and the diversified interaction of HLA-I and inhibitory KIR (301). The presence of −21M leader peptides available for HLA-E stabilization indeed correlated with an increased polyfunctional NK-cell response in terms of ADCC, IFN-γ production and degranulation against the missing-self K562 target cell line compared to NK cells derived from donors with a −21T/T genotype (42). Based on the dimorphism in the HLA-B leader peptide, Horowitz et al. described the evolution of two distinct HLA-I haplotypes, which can be distinguished by the inhibitory receptor system operating in NK-cell education. The first, more ancient haplotype encoding the HLA-E permissive −21M and HLA-C1 alleles is skewed toward the supply of ligands for NKG2A:CD94, whereas the second haplotype encodes the non-HLA-E–permissive −21T dimorphism, HLA-B with a Bw4 motif and HLA-C2/C1 allotypes, hence being more skewed toward encoding strong KIR ligands (42). Studying the two potential routes of licensing, Bernard et al. reported that the NKG2A+ NK cell subset mounted the highest polyfunctional response against infected CD4+ T cells, without further modulation through coexpression of inhibitory KIR3DL1 (300). An earlier study reported a dual effect of NKG2A and inhibitory KIR coexpression in promoting NK cell education as well as survival (105). Coexpression of inhibitory KIR (with the presence of cognate ligand) with the activating NKG2C receptor following CMV reactivation after hematopoietic cell transplantation was required for robust cytokine production by NK cells (317). This raises the question of which combination of KIR and NKG2 receptors results in best possible NK-cell functionality and survival in combating HIV-1 infection. Overall, HLA-E—aside from presenting peptides—has clearly an additional function in NK-cell education through NKG2A. This in turn may explain the antiviral capacity of NKG2A+ NK cells as observed in vitro following HIV-1 infection.
Concluding Remarks
Studies from the preantiretroviral treatment era suggest that early events in acute HIV-1 infection influence the rate of HIV-1 disease progression. NK cells, as first-responding innate effector cells, have been shown to expand in early HIV-1 infection and kill HIV-1–infected cells, with genetic studies robustly linking variants in NK-cell receptors to HIV-1 acquisition and disease progression. Additionally, according to mouse models of chronic viral infections, NK cells have the potential to regulate adaptive immune responses, possibly even impairing an effective adaptive response (158, 318). Notably, the antiviral effector potential of NK cells is closely linked to HLA-I. HLA-I allows not only for effective NK-cell education, but also modulates NK-cell activity toward HIV-1-infected cells via changes in HLA-I surface expression and peptide presentation. While numerous studies have established a role for the KIR interaction with classical HLA-I in HIV-1, recent advances have increased our understanding of non-classical HLA-E, -F, and -G in HIV-1 infection. First, HLA-F was identified as a ligand for KIR3DS1, which is prominently associated with HIV-1 disease control. HLA-F may serve as a “stress” signal on HIV-1–infected cells, at best enhancing KIR3DS1+ NK-cell killing of infected cells, and at worst mediating HIV-1–associated immunopathology (Figure 2B). Second, HLA-E expression levels are not downregulated in HIV-1, which is important as HLA-E is capable of presenting viral peptides. Moreover, HLA-E can tune NK-cell function through NKG2A in virtually all individuals, and is linked to a superior antiviral capacity of NKG2A+ NK cells (Figure 2A). Third, HLA-G has predominantly immunomodulatory properties (rather than a peptide-presenting function), and although genetic studies are teasing apart the link between HLA-G polymorphisms and HIV-1 disease, the impact of HLA-G on NK-cell function in HIV-1 has yet to be determined (Figure 2C). Eventually, the unique properties of these non-classical HLA-I molecules and their conservation between individuals renders them an ideal target for new approaches aimed at harnessing innate immunity against HIV-1.
Author Contributions
AH wrote the first draft of the manuscript, WG-B and MA have made substantial, direct, and intellectual contributions to the work and all authors approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Charles L. Dulberger for his helpful discussion and critical remarks on the manuscript and Gloria Martrus Zapater for her valuable input on the figures.
Funding
AH was supported by the German Center for Infection Research (DZIF) through a MD/PhD Stipend (TI 07.002) and via the Clinician Scientist Program of the Faculty of Medicine, University Medical Centre Hamburg-Eppendorf, Hamburg, Germany.
References
1. Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PIW, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science (2010) 330(6010):1551–7. doi:10.1126/science.1195271
2. Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A whole-genome association study of major determinants for host control of HIV-1. Science (2007) 317(5840):944–7. doi:10.1126/science.1143767
3. Kuniholm MH, Gao X, Xue X, Kovacs A, Anastos K, Marti D, et al. Human leukocyte antigen genotype and risk of HIV disease progression before and after initiation of antiretroviral therapy. J Virol (2011) 85(20):10826–33. doi:10.1128/JVI.00804-11
4. Kaslow RA, Carrington M, Apple R, Park L, Muñoz A, Saah AJ, et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med (1996) 2(4):405–11. doi:10.1038/nm0496-405
5. Martinez-picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol (2006) 80(7):3617–23. doi:10.1128/JVI.80.7.3617-3623.2006
6. Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature (2009) 458(7238):641–5. doi:10.1038/nature07746
7. Frater AJ, Brown H, Oxenius A, Günthard HF, Hirschel B, Robinson N, et al. Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J Virol (2007) 81(12):6742–51. doi:10.1128/JVI.00022-07
8. Altfeld M, Allen TM. Hitting HIV where it hurts: an alternative approach to HIV vaccine design. Trends Immunol (2006) 27(11):504–10. doi:10.1016/j.it.2006.09.007
9. McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol (2010) 10(1):11–23. doi:10.1038/nri2674
10. Lichterfeld M, Kaufmann DE, Yu XG, Mui SK, Addo MM, Johnston MN, et al. Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J Exp Med (2004) 200(6):701–12. doi:10.1084/jem.20041270
11. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol (2002) 3(11):1061–8. doi:10.1038/ni845
12. Thobakgale CF, Streeck H, Mkhwanazi N, Mncube Z, Maphumulo L, Chonco F, et al. Short communication: CD8+ T cell polyfunctionality profiles in progressive and nonprogressive pediatric HIV type 1 infection. AIDS Res Hum Retroviruses (2011) 27(9):1005–12. doi:10.1089/aid.2010.0227
13. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood (2006) 107(12):4781–9. doi:10.1182/blood-2005-12-4818
14. Altfeld M, Gale M. Innate immunity against HIV-1 infection. Nat Immunol (2015) 16(6):554–62. doi:10.1038/ni.3157
15. Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell (1996) 86(3):367–77. doi:10.1016/S0092-8674(00)80110-5
16. Martin MP, Dean M, Smith MW, Winkler C, Gerrard B, Michael NL, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science (1998) 282(5395):1907–11. doi:10.1126/science.282.5395.1907
17. Lambotte O, Ferrari G, Moog C, Yates NL, Liao H-X, Parks RJ, et al. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS (2009) 23(8):897–906. doi:10.1097/QAD.0b013e328329f97d
18. Vieillard V, Fausther-Bovendo H, Samri A, Debré P, French Asymptomatiques à Long Terme (ALT) ANRS-CO15 Study Group. Specific phenotypic and functional features of natural killer cells from HIV-infected long-term nonprogressors and HIV controllers. J Acquir Immune Defic Syndr (2010) 53(5):564–73. doi:10.1097/QAI.0b013e3181d0c5b4
19. Kottilil S, Chun TW, Moir S, Liu S, McLaughlin M, Hallahan CW, et al. Innate immunity in human immunodeficiency virus infection: effect of viremia on natural killer cell function. J Infect Dis (2003) 187(7):1038–45. doi:10.1086/368222
20. Parham P. Influence of KIR diversity on human immunity. Adv Exp Med Biol (2005) 560:47–50. doi:10.1007/0-387-24180-9_6
21. Borges L, Cosman D. LIRs/ILTs/MIRs, inhibitory and stimulatory Ig-superfamily receptors expressed in myeloid and lymphoid cells. Cytokine Growth Factor Rev (2000) 11(3):209–17. doi:10.1016/S1359-6101(00)00007-1
22. Martin MP, Gao X, Lee J-H, Nelson GW, Detels R, Goedert JJ, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet (2002) 31(4):429–34. doi:10.1038/ng934
23. Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet (2007) 39(6):733–40. doi:10.1038/ng2035
24. Lichterfeld M, Yu XG. The emerging role of leukocyte immunoglobulin-like receptors (LILRs) in HIV-1 infection. J Leukoc Biol (2012) 91(1):27–33. doi:10.1189/jlb.0811442
25. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22(11):633–40. doi:10.1016/S1471-4906(01)02060-9
26. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med (1989) 320(26):1731–5. doi:10.1056/NEJM198906293202605
27. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi:10.1146/annurev-immunol-020711-075005
28. Lodoen MB, Lanier LL. Viral modulation of NK cell immunity. Nat Rev Microbiol (2005) 3(1):59–69. doi:10.1038/nrmicro1066
29. Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol (2013) 31:163–94. doi:10.1146/annurev-immunol-032712-100001
30. Lifson JD, Nowak MA, Goldstein S, Rossio JL, Kinter A, Vasquez G, et al. The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. J Virol (1997) 71(12):9508–14.
31. Alter G, Teigen N, Ahern R, Streeck H, Meier A, Rosenberg ES, et al. Evolution of innate and adaptive effector cell functions during acute HIV-1 infection. J Infect Dis (2007) 195(10):1452–60. doi:10.1086/513878
32. Mavilio D, Benjamin J, Daucher M, Lombardo G, Kottilil S, Planta MA, et al. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci U S A (2003) 100(25):15011–6. doi:10.1073/pnas.2336091100
33. Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, et al. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci U S A (2005) 102(8):2886–91. doi:10.1073/pnas.0409872102
34. Alter G, Teigen N, Davis BT, Addo MM, Suscovich TJ, Waring MT, et al. Sequential deregulation of NK cell subset distribution and function starting in acute HIV-1 infection. Blood (2005) 106(10):3366–9. doi:10.1182/blood-2005-03-1100
35. Alter G, Malenfant JM, Delabre RM, Burgett NC, Yu XG, Lichterfeld M, et al. Increased natural killer cell activity in viremic HIV-1 infection. J Immunol (2004) 173(8):5305–11. doi:10.4049/jimmunol.173.8.5305
36. Fogli M, Mavilio D, Brunetta E, Varchetta S, Ata K, Roby G, et al. Lysis of endogenously infected CD4+ T cell blasts by rIL-2 activated autologous natural killer cells from HIV-infected viremic individuals. PLoS Pathog (2008) 4(7):e1000101. doi:10.1371/journal.ppat.1000101
37. Bryceson YT, March ME, Ljunggren H, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107(1):159–66. doi:10.1182/blood-2005-04-1351
38. Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood (2009) 114(13):2657–66. doi:10.1182/blood-2009-01-201632
39. Valiante NM, Uhrberg M, Shilling HG, Lienert-Weidenbach K, Arnett KL, D’Andrea A, et al. Functionally and structurally distinct NK cell receptor repertoires in the peripheral blood of two human donors. Immunity (1997) 7(6):739–51. doi:10.1016/S1074-7613(00)80393-3
40. Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med (2013) 5(208):208ra145. doi:10.1126/scitranslmed.3006702
41. Anfossi N, André P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity (2006) 25(2):331–42. doi:10.1016/j.immuni.2006.06.013
42. Horowitz A, Djaoud Z, Nemat-Gorgani N, Blokhuis J, Hilton HG, Béziat V, et al. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci Immunol (2016) 1(3):eaag1672. doi:10.1126/sciimmunol.aag1672
43. Richard J, Sindhu S, Pham TNQ, Belzile J-P, Cohen EA. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood (2010) 115(7):1354–63. doi:10.1182/blood-2009-08-237370
44. Ward J, Davis Z, Dehart J, Zimmerman E, Bosque A, Brunetta E, et al. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog (2009) 5(10):e1000613. doi:10.1371/journal.ppat.1000613
45. Vieillard V, Strominger JL, Debre P. NK cytotoxicity against CD4+ T cells during HIV-1 infection:a gp41 peptide induces the expression of an NKp44 ligand. Proc Natl Acad Sci U S A (2005) 102(31):10981–6. doi:10.1073/pnas.0504315102
46. Ward J, Bonaparte M, Sacks J, Guterman J, Fogli M, Mavilio D, et al. HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood (2007) 110(4):1207–14. doi:10.1182/blood-2006-06-028175
47. Sandberg JK, Andersson SK, Bächle SM, Nixon DF, Moll M. HIV-1 Vpu interference with innate cell-mediated immune mechanisms. Curr HIV Res (2012) 10(4):327–33. doi:10.2174/157016212800792513
48. Collins DR, Collins KL. HIV-1 accessory proteins adapt cellular adaptors to facilitate immune evasion. PLoS Pathog (2014) 10(1):e1003851. doi:10.1371/journal.ppat.1003851
49. Wonderlich ER, Leonard JA, Collins KL. HIV immune evasion disruption of antigen presentation by the HIV Nef protein. Adv Virus Res (2011) 80(1–2):103–27. doi:10.1016/B978-0-12-385987-7.00005-1
50. Cerboni C, Neri F, Casartelli N, Zingoni A, Cosman D, Rossi P, et al. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J Gen Virol (2007) 88(Pt 1):242–50. doi:10.1099/vir.0.82125-0
51. Matusali G, Potesta M, Santoni A, Cerboni C, Doria M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J Virol (2012) 86(8):4496–504. doi:10.1128/JVI.05788-11
52. Bolduan S, Reif T, Schindler M, Schubert U. HIV-1 Vpu mediated downregulation of CD155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology (2014) 464–465:375–84. doi:10.1016/j.virol.2014.07.034
53. Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell M, Planelles V, et al. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe (2010) 8(5):397–409. doi:10.1016/j.chom.2010.10.008
54. Parham P, Lomen CE, Lawlor DA, Ways JP, Holmes N, Coppin HL, et al. Nature of polymorphism in HLA-A, -B, and -C molecules. Proc Natl Acad Sci U S A (1988) 85:4005–9. doi:10.1073/pnas.85.11.4005
55. Robinson J, Guethlein LA, Cereb N, Yang SY, Norman PJ, Marsh SGE, et al. Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLoS Genet (2017) 13(6):e1006862. doi:10.1371/journal.pgen.1006862
56. Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res (2015) 43(D1):D423–31. doi:10.1093/nar/gku1161
57. Rock KL, York IA, Goldberg AL. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat Immunol (2004) 5(7):670–7. doi:10.1038/ni1089
58. Martin MP, Carrington M. Immunogenetics of HIV disease. Immunol Rev (2013) 254(1):245–64. doi:10.1111/imr.12071
59. Sentman CL, Olsson MY, Kärre K. Missing self recognition by natural killer cells in MHC class I transgenic mice. A “receptor calibration” model for how effector cells adapt to self. Semin Immunol (1995) 7(2):109–19. doi:10.1006/smim.1995.0015
60. Sim MJW, Malaker SA, Khan A, Stowell JM, Shabanowitz J, Peterson M, et al. Canonical and cross-reactive binding of NK cell inhibitory receptors to HLA-C allotypes is dictated by peptides bound to HLA-C. Front Immunol (2017) 8:1–16. doi:10.3389/fimmu.2017.00193
61. Fadda L, O’Connor GM, Kumar S, Piechocka-Trocha A, Gardiner CM, Carrington M, et al. Common HIV-1 peptide variants mediate differential binding of KIR3DL1 to HLA-Bw4 molecules. J Virol (2011) 85(12):5970–4. doi:10.1128/JVI.00412-11
62. Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature (2000) 405(6786):537–43. doi:10.1038/35014520
63. Stewart-Jones GBE, di Gleria K, Kollnberger S, McMichael AJ, Jones EY, Bowness P. Crystal structures and KIR3DL1 recognition of three immunodominant viral peptides complexed to HLA-B*2705. Eur J Immunol (2005) 35(2):341–51. doi:10.1002/eji.200425724
64. Lunemann S, Martrus G, Hölzemer A, Chapel A, Ziegler M, Körner C, et al. Sequence variations in HCV core-derived epitopes alter binding of KIR2DL3 to HLA-C*03:04 and modulate NK cell function. J Hepatol (2016) 65(2):252–8. doi:10.1016/j.jhep.2016.03.016
65. Hilton HG, McMurtrey CP, Han AS, Djaoud Z, Guethlein LA, Blokhuis JH, et al. The intergenic recombinant HLA-B*46:01 has a distinctive peptidome that includes KIR2DL3 ligands. Cell Rep (2017) 19(7):1394–405. doi:10.1016/j.celrep.2017.04.059
66. Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature (2004) 432(7018):769–75. doi:10.1038/nature03113
67. Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A (2000) 97(6):2709–14. doi:10.1073/pnas.050567397
68. Magierowska M, Theodorou I, Debré P, Sanson F, Autran B, Rivière Y, et al. Combined genotypes of CCR5, CCR2, SDF1, and HLA genes can predict the long-term nonprogressor status in human immunodeficiency virus-1-infected individuals. Blood (1999) 93(3):936–41.
69. Flores-Villanueva PO, Yunis EJ, Delgado-Galvan J, Vittinghoff E, Buchbinder S, Leung JY, et al. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci U S A (2001) 98(9):5140–5. doi:10.1073/pnas.071548198
70. Wilson MJ, Torkar M, Haude A, Milne S, Jones T, Sheer D, et al. Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci U S A (2000) 97(9):4778–83. doi:10.1073/pnas.080588597
71. Parham P, Norman PJ, Abi-Rached L, Guethlein LA. Variable NK cell receptors exemplified by human KIR3DL1/S1. J Immunol (2011) 187(1):11–9. doi:10.4049/jimmunol.0902332
72. Khakoo SI, Geller R, Shin S, Jenkins JA, Parham P. The D0 domain of KIR3D acts as a major histocompatibility complex class I binding enhancer. J Exp Med (2002) 196(7):911–21. doi:10.1084/jem.20020304
73. Carrington M, Norman P. The KIR gene cluster [Internet]. Bethesda, MD: National Center for Biotechnology Information (US) (2003). Available from: https://www.ncbi.nlm.nih.gov/books/NBK10135/
74. Carr WH, Rosen DB, Arase H, Nixon DF, Michaelsson J, Lanier LL. Cutting edge: KIR3DS1, a gene implicated in resistance to progression to AIDS, encodes a DAP12-associated receptor expressed on NK cells that triggers NK cell activation. J Immunol (2007) 178(2):647–51. doi:10.4049/jimmunol.178.2.647
75. Arlettaz L, Degermann S, De Rham C, Roosnek E, Huard B. Expression of inhibitory KIR is confined to CD8+ effector T cells and limits their proliferative capacity. Eur J Immunol (2004) 34(12):3413–22. doi:10.1002/eji.200324756
76. van der Veken LT, Diez Campelo M, van der Hoorn MAWG, Hagedoorn RS, van Egmond HME, van Bergen J, et al. Functional analysis of killer Ig-like receptor-expressing cytomegalovirus-specific CD8+ T cells. J Immunol (2009) 182(1):92–101. doi:10.4049/jimmunol.182.1.92
77. Björkström NK, Béziat V, Cichocki F, Liu LL, Levine J, Larsson S, et al. CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood (2012) 120(17):3455–65. doi:10.1182/blood-2012-03-416867
78. van Bergen J, Thompson A, van der Slik A, Ottenhoff THM, Gussekloo J, Koning F. Phenotypic and functional characterization of CD4 T cells expressing killer Ig-like receptors. J Immunol (2004) 173(11):6719–26. doi:10.4049/jimmunol.173.11.6719
79. Lafarge X, Pitard V, Ravet S, Roumanes D, Halary F, Dromer C, et al. Expression of MHC class I receptors confers functional intraclonal heterogeneity to a reactive expansion of γδ T cells. Eur J Immunol (2005) 35(6):1896–905. doi:10.1002/eji.200425837
80. Uhrberg M, Valiante NM, Young NT, Lanier LL, Phillips JH, Parham P. The repertoire of killer cell Ig-like receptor and CD94:NKG2A receptors in T cells: clones sharing identical alpha beta TCR rearrangement express highly diverse killer cell Ig-like receptor patterns. J Immunol (2001) 166(38):3923–32. doi:10.4049/jimmunol.166.6.3923
81. Horowitz A, Guethlein LA, Nemat-Gorgani N, Norman PJ, Cooley S, Miller JS, et al. Regulation of adaptive NK cells and CD8 T cells by HLA-C correlates with allogeneic hematopoietic cell transplantation and with cytomegalovirus reactivation. J Immunol (2015) 195(9):4524–36. doi:10.4049/jimmunol.1401990
82. Chan WK, Rujkijyanont P, Neale G, Yang J, Bari R, Das Gupta N, et al. Multiplex and genome-wide analyses reveal distinctive properties of KIR+ and CD56+ T cells in human blood. J Immunol (2013) 191(4):1625–36. doi:10.4049/jimmunol.1300111
83. Anfossi N, Doisne J-M, Peyrat M-A, Ugolini S, Bonnaud O, Bossy D, et al. Coordinated expression of Ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol (2004) 173(12):7223–9. doi:10.4049/jimmunol.173.12.7223
84. Zipperlen K, Gallant M, Stapleton S, Heath J, Barrett L, Grant M. Protective genotypes in HIV infection reflect superior function of KIR3DS1+ over KIR3DL1+ CD8+ T cells. Immunol Cell Biol (2015) 93(1):67–76. doi:10.1038/icb.2014.68
85. Qi Y, Martin MP, Gao X, Jacobson L, Goedert JJ, Buchbinder S, et al. KIR/HLA pleiotropism: protection against both HIV and opportunistic infections. PLoS Pathog (2006) 2(8):e79. doi:10.1371/journal.ppat.0020079
86. Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol (2013) 13(2):133–44. doi:10.1038/nri3370
87. Cella M, Longo A, Ferrara GB, Strominger JL, Colonna M. NK3-specific natural killer cells are selectively inhibited by Bw4-positive HLA alleles with isoleucine 80. J Exp Med (1994) 180(4):1235–42. doi:10.1084/jem.180.4.1235
88. Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med (1995) 181(3):1133–44. doi:10.1084/jem.181.3.1133
89. Barbour JD, Sriram U, Caillier SJ, Levy JA, Hecht FM, Oksenberg JR. Synergy or independence? Deciphering the interaction of HLA Class I and NK cell KIR alleles in early HIV-1 disease progression. PLoS Pathog (2007) 3(4):e43. doi:10.1371/journal.ppat.0030043
90. Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ, et al. Copy number variation of KIR genes influences HIV-1 control. PLoS Biol (2011) 9(11):e1001208. doi:10.1371/journal.pbio.1001208
91. Jiang Y, Chen O, Cui C, Zhao B, Han X, Zhang Z, et al. KIR3DS1/L1 and HLA-Bw4-80I are associated with HIV disease progression among HIV typical progressors and long-term nonprogressors. BMC Infect Dis (2013) 13(1):405. doi:10.1186/1471-2334-13-405
92. Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med (2007) 204(12):3027–36. doi:10.1084/jem.20070695
93. Long BR, Ndhlovu LC, Oksenberg JR, Lanier LL, Hecht FM, Nixon DF, et al. Conferral of enhanced natural killer cell function by KIR3DS1 in early human immunodeficiency virus type 1 infection. J Virol (2008) 82(10):4785–92. doi:10.1128/JVI.02449-07
94. Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol (2009) 83(13):6798–805. doi:10.1128/JVI.00256-09
95. Morvan M, Willem C, Gagne K, Kerdudou N, David G, Sébille V, et al. Phenotypic and functional analyses of KIR3DL1+ and KIR3DS1+ NK cell subsets demonstrate differential regulation by Bw4 molecules and induced KIR3DS1 expression on stimulated NK cells. J Immunol (2009) 182(11):6727–35. doi:10.4049/jimmunol.0900212
96. Song R, Lisovsky I, Lebouché B, Routy JP, Bruneau J, Bernard NF. HIV protective KIR3DL1/S1-HLA-B genotypes influence NK cell-mediated inhibition of HIV replication in autologous CD4 targets. PLoS Pathog (2014) 10(1):e1003867. doi:10.1371/journal.ppat.1003867
97. O’Connor GM, Guinan KJ, Cunningham RT, Middleton D, Parham P, Gardiner CM. Functional polymorphism of the KIR3DL1/S1 receptor on human NK cells. J Immunol (2007) 178(1):235–41. doi:10.4049/jimmunol.178.1.235
98. López-Vázquez A, Miña-Blanco A, Martínez-Borra J, Njobvu PD, Suárez-Alvarez B, Blanco-Gelaz MA, et al. Interaction between KIR3DL1 and HLA-B*57 supertype alleles influences the progression of HIV-1 infection in a Zambian population. Hum Immunol (2005) 66(3):285–9. doi:10.1016/j.humimm.2005.01.001
99. Tomescu C, Duh F-M, Hoh R, Viviani A, Harvill K, Martin MP, et al. Impact of protective killer inhibitory receptor/human leukocyte antigen genotypes on natural killer cell and T-cell function in HIV-1-infected controllers. AIDS (2012) 26(15):1869–78. doi:10.1097/QAD.0b013e32835861b0
100. Jonsson AH, Yokoyama WM. Natural killer cell tolerance licensing and other mechanisms. Adv Immunol (2009) 101:27–79. doi:10.1016/S0065-2776(08)01002-X
101. Yawata M, Yawata N, Draghi M, Little A-M, Partheniou F, Parham P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med (2006) 203(3):633–45. doi:10.1084/jem.20051884
102. Kamya P, Boulet S, Tsoukas CM, Routy J-P, Thomas R, Côté P, et al. Receptor-ligand requirements for increased NK cell polyfunctional potential in slow progressors infected with HIV-1 coexpressing KIR3DL1*h/*y and HLA-B*57. J Virol (2011) 85(12):5949–60. doi:10.1128/JVI.02652-10
103. Boudreau JE, Mulrooney TJ, Le Luduec J-B, Barker E, Hsu KC. KIR3DL1 and HLA-B density and binding calibrate NK education and response to HIV. J Immunol (2016) 196(8):3398–410. doi:10.4049/jimmunol.1502469
104. Brodin P, Lakshmikanth T, Johansson S, Karre K, Hoglund P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood (2009) 113(11):2434–41. doi:10.1182/blood-2008-05-156836
105. Felices M, Lenvik TR, Ankarlo DEM, Foley B, Curtsinger J, Luo X, et al. Functional NK cell repertoires are maintained through IL-2Rα and Fas ligand. J Immunol (2014) 192(8):3889–97. doi:10.4049/jimmunol.1302601
106. Boulet S, Sharafi S, Simic N, Bruneau J, Routy J-P, Tsoukas CM, et al. Increased proportion of KIR3DS1 homozygotes in HIV-exposed uninfected individuals. AIDS (2008) 22(5):595–9. doi:10.1097/QAD.0b013e3282f56b23
107. Boulet S, Kleyman M, Kim JY, Kamya P, Sharafi S, Simic N, et al. A combined genotype of KIR3DL1 high expressing alleles and HLA-B*57 is associated with a reduced risk of HIV infection. AIDS (2008) 22(12):1487–91. doi:10.1097/QAD.0b013e3282ffde7e
108. Guerini FR, Caputo SL, Gori A, Bandera A, Mazzotta F, Uglietti A, et al. Under representation of the inhibitory KIR3DL1 molecule and the KIR3DL1+/BW4+ complex in HIV exposed seronegative individuals. J Infect Dis (2011) 203(9):1235–9. doi:10.1093/infdis/jir020
109. Zemmour J, Parham P. Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C by Jacqueline Zemmour and Peter Parham. J Exp Med (1992) 176:937–50. doi:10.1084/jem.176.4.937
110. Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, et al. Influence of HLA-C expression level on HIV control. Science (2013) 340(6128):87–91. doi:10.1126/science.1232685
111. Blais M-E, Zhang Y, Rostron T, Griffin H, Taylor S, Xu K, et al. High frequency of HIV mutations associated with HLA-C suggests enhanced HLA-C-restricted CTL selective pressure associated with an AIDS-protective polymorphism. J Immunol (2012) 188(9):4663–70. doi:10.4049/jimmunol.1103472
112. Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity (1999) 10(6):661–71. doi:10.1016/S1074-7613(00)80065-5
113. Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, et al. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe (2016) 19(5):686–95. doi:10.1016/j.chom.2016.04.005
114. Bonaparte MI, Barker E. Killing of human immunodeficiency virus – infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood (2004) 104(7):2087–94. doi:10.1182/blood-2004-02-0696
115. Ward JP, Bonaparte MI, Barker E. HLA-C and HLA-E reduce antibody-dependent natural killer cell-mediated cytotoxicity of HIV-infected primary T cell blasts. AIDS (2004) 18(13):1769–79. doi:10.1097/00002030-200409030-00005
116. Körner C, Granoff ME, Amero MA, Sirignano MN, Vaidya SA, Jost S, et al. Increased frequency and function of KIR2DL1-3+ NK cells in primary HIV-1 infection are determined by HLA-C group haplotypes. Eur J Immunol (2014) 44(10):2938–48. doi:10.1002/eji.201444751
117. Körner C, Simoneau CR, Schommers P, Granoff M, Ziegler M, Hölzemer A, et al. HIV-1-mediated downmodulation of HLA-C impacts target cell recognition and antiviral activity of NK cells. Cell Host Microbe (2017) 22(1):111.e–9.e. doi:10.1016/j.chom.2017.06.008
118. Kløverpris HN, Leslie A, Goulder P. Role of HLA adaptation in HIV evolution. Front Immunol (2016) 6:665. doi:10.3389/fimmu.2015.00665
119. Goulder PJR, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol (2008) 8(8):619–30. doi:10.1038/nri2357
120. Goulder PJR, Watkins DI. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol (2004) 4(8):630–40. doi:10.1038/nri1417
121. Schafer JL, Ries M, Guha N, Connole M, Colantonio AD, Wiertz EJ, et al. Suppression of a natural killer cell response by simian immunodeficiency virus peptides. PLoS Pathog (2015) 11(9):e1005145. doi:10.1371/journal.ppat.1005145
122. Colantonio AD, Bimber BN, Neidermyer WJ, Reeves RK, Alter G, Altfeld M, et al. KIR polymorphisms modulate peptide-dependent binding to an MHC class I ligand with a Bw6 motif. PLoS Pathog (2011) 7(3):e1001316. doi:10.1371/journal.ppat.1001316
123. Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, et al. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur J Immunol (2004) 34(6):1673–9. doi:10.1002/eji.200425089
124. Fadda L, Körner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw*0102-restricted HIV-1 p24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS Pathog (2012) 8(7):e1002805. doi:10.1371/journal.ppat.1002805
125. Van Teijlingen NH, Hölzemer A, Körner C, García-Beltrán WF, Schafer JL, Fadda L, et al. Sequence variations in HIV-1 p24 Gag-derived epitopes can alter binding of KIR2DL2 to HLA-C*03: 04 and modulate primary natural killer cell function. AIDS (2014) 28(10):1399–408. doi:10.1097/QAD.0000000000000284
126. McCarthy MK, Weinberg JB. The immunoproteasome and viral infection: a complex regulator of inflammation. Front Microbiol (2015) 6:21. doi:10.3389/fmicb.2015.00021
127. Brackenridge S, Evans EJ, Toebes M, Goonetilleke N, Liu MKP, Gleria K, et al. An Early HIV mutation within an HLA-B * 57-restricted T cell epitope abrogates binding to the killer inhibitory receptor 3DL1†. J Virol (2011) 85(11):5415–22. doi:10.1128/JVI.00238-11
128. Lin Z, Kuroki K, Kuse N, Sun X, Akahoshi T, Chikata T, et al. HIV-1 control by NK cells via reduced interaction between KIR2DL2 and HLAC*12:02/C*14:03. Cell Rep (2016) 17(9):2210–20. doi:10.1016/j.celrep.2016.10.075
129. Thananchai H, Makadzange T, Maenaka K, Kuroki K, Peng Y, Conlon C, et al. Reciprocal recognition of an HLA-Cw4-restricted HIV-1 gp120 epitope by CD8+ T cells and NK cells. AIDS (2009) 23(2):189–93. doi:10.1097/QAD.0b013e32831fb55a
130. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature (2011) 476(7358):96–100. doi:10.1038/nature10237
131. Honeyborne I, Codoñer FM, Leslie A, Tudor-Williams G, Luzzi G, Ndung’u T, et al. HLA-Cw*03-restricted CD8+ T-cell responses targeting the HIV-1 gag major homology region drive virus immune escape and fitness constraints compensated for by intracodon variation. J Virol (2010) 84(21):11279–88. doi:10.1128/JVI.01144-10
132. Hölzemer A, Thobakgale CF, Garcia-Beltran CAJ, Garcia-Beltran WF, Carlson JM, van Teijlingen NH, et al. Selection of an HLA-C *03: 04-restricted HIV-1 p24 Gag sequence variant is associated with viral escape from KIR2DL3+ Natural Killer Cells: data from an observational cohort in South Africa. PLoS Med (2015) 12(11):e1001900. doi:10.1371/journal.pmed.1001900
133. Rucevic M, Kourjian G, Boucau J, Blatnik R, Garcia Bertran W, Berberich MJ, et al. MHC-bound HIV peptides identified from various cell types reveal common nested peptides and novel T cell responses. J Virol (2016) 90(19):8605–20. doi:10.1128/JVI.00599-16
134. Ternette N, Yang H, Partridge T, Llano A, Cedeño S, Fischer R, et al. Defining the HLA class I-associated viral antigen repertoire from HIV-1-infected human cells. Eur J Immunol (2016) 46(1):60–9. doi:10.1002/eji.201545890
135. Borhis G, Ahmed PS, Mbiribindi B, Naiyer MM, Davis DM, Purbhoo MA, et al. A peptide antagonist disrupts NK cell inhibitory synapse formation. J Immunol (2013) 190(6):2924–30. doi:10.4049/jimmunol.1201032
136. Fadda L, Borhis G, Ahmed P, Cheent K, Pageon SV, Cazaly A, et al. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci U S A (2010) 107(22):10160–5. doi:10.1073/pnas.0913745107
137. Körner C, Altfeld M. Role of KIR3DS1 in human diseases. Front Immunol (2012) 3:326. doi:10.3389/fimmu.2012.00326
138. Garcia-Beltran WF, Hölzemer A, Martrus G, Chung AW, Pacheco Y, Simoneau CR, et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat Immunol (2016) 17(9):1067–74. doi:10.1038/ni.3513
139. Burian A, Wang KL, Finton KAK, Lee N, Ishitani A, Strong RK, et al. HLA-F and MHC-I open conformers bind natural killer cell ig-like receptor KIR3DS1. PLoS One (2016) 11(9):e0163297. doi:10.1371/journal.pone.0163297
140. Pan FH, Liu XX, Tian W. Characterization of HLA-F polymorphism in four distinct populations in Mainland China. Int J Immunogenet (2013) 40(5):369–76. doi:10.1111/iji.12047
141. Lee N, Geraghty DE. HLA-F surface expression on B cell and monocyte cell lines is partially independent from tapasin and completely independent from TAP. J Immunol (2003) 171(10):5264–71. doi:10.4049/jimmunol.171.10.5264
142. Wainwright SD, Biro PA, Holmes CH. HLA-F is a predominantly empty, intracellular, TAP-associated MHC class Ib protein with a restricted expression pattern. J Immunol (2000) 164(1):319–28. doi:10.4049/jimmunol.164.1.319
143. Lee N, Ishitani A, Geraghty DE. HLA-F is a surface marker on activated lymphocytes. Eur J Immunol (2010) 40(8):2308–18. doi:10.1002/eji.201040348
144. Goodridge JP, Burian A, Lee N, Geraghty DE. HLA-F and MHC class I open conformers are ligands for NK cell Ig-like receptors. J Immunol (2013) 191(7):3553–62. doi:10.4049/jimmunol.1300081
145. Lepin EJM, Bastin JM, Allan DSJ, Roncador G, Braud VM, Mason DY, et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol (2000) 30(12):3552–61. doi:10.1002/1521-4141(200012)30:12<3552::AID-IMMU3552>3.0.CO;2-L
146. Dulberger CL, McMurtrey CP, Hölzemer A, Neu KE, Liu V, Steinbach AM, et al. Human leukocyte antigen F presents peptides and regulates immunity through interactions with NK cell receptors. Immunity (2017) 46(6):1018.e–29.e. doi:10.1016/j.immuni.2017.06.002
147. Sivori S, Falco M, Carlomagno S, Romeo E, Soldani C, Bensussan A, et al. A novel KIR-associated function: evidence that CpG DNA uptake and shuttling to early endosomes is mediated by KIR3DL2. Blood (2010) 116(10):1637–47. doi:10.1182/blood-2009-12-256586
148. O’Connor GM, Vivian JP, Gostick E, Pymm P, Lafont BAP, Price DA, et al. Peptide-dependent recognition of HLA-B*57:01 by KIR3DS1. J Virol (2015) 89(10):5213–21. doi:10.1128/JVI.03586-14
149. Campbell EC, Antoniou AN, Powis SJ. The multi-faceted nature of HLA class I dimer molecules. Immunology (2012) 136(4):380–4. doi:10.1111/j.1365-2567.2012.03593.x
150. Bird LA, Peh CA, Kollnbeger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol (2003) 33(3):748–59. doi:10.1002/eji.200323678
151. Gonen-Gross T, Achdout H, Gazit R, Hanna J, Mizrahi S, Markel G, et al. Complexes of HLA-G protein on the cell surface are important for leukocyte Ig-like receptor-1 function. J Immunol (2003) 171(3):1343–51. doi:10.4049/jimmunol.171.3.1343
152. Shiroishi M, Kuroki K, Rasubala L, Tsumoto K, Kumagai I, Kurimoto E, et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci U S A (2006) 103(44):16412–7. doi:10.1073/pnas.0605228103
153. Kollnberger S, Chan A, Sun M, Chen LY, Wright C, Gleria K, et al. Interaction of HLA-B27 homodimers with KIR3DL1 and KIR3DL2, unlike HLA-B27 heterotrimers, is independent of the sequence of bound peptide. Eur J Immunol (2007) 37(5):1313–22. doi:10.1002/eji.200635997
154. Goodridge JP, Burian A, Lee N, Geraghty DE. HLA-F complex without peptide binds to MHC class I protein in the open conformer form. J Immunol (2010) 184(11):6199–208. doi:10.4049/jimmunol.1000078
155. Rizvi SM, Salam N, Geng J, Qi Y, Bream JH, Duggal P, et al. Distinct assembly profiles of HLA-B molecules. J Immunol (2014) 192(11):4967–76. doi:10.4049/jimmunol.1301670
156. Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol (2010) 10(7):514–26. doi:10.1038/nri2802
157. Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol (2008) 214(2):231–41. doi:10.1002/path.2276
158. Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature (2012) 481(7381):394–8. doi:10.1038/nature10624
159. Arosa FA, Santos SG, Powis SJ. Open conformers: the hidden face of MHC-I molecules. Trends Immunol (2007) 28(3):115–23. doi:10.1016/j.it.2007.01.002
160. Strong RK, Holmes MA, Li P, Braun L, Lee N, Geraghty DE. HLA-E allelic variants: correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J Biol Chem (2003) 278(7):5082–90. doi:10.1074/jbc.M208268200
161. Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol (2006) 36(7):1646–53. doi:10.1002/eji.200636195
162. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, López-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A (1998) 95(9):5199–204. doi:10.1073/pnas.95.9.5199
163. Navarro F, Llano M, Bellón T, Colonna M, Geraghty DE, López-Botet M. The ILT2(LIR1) and CD94/NKG2A NK cell receptors respectively recognize HLA-G1 and HLA-E molecules co-expressed on target cells. Eur J Immunol (1999) 29(1):277–83. doi:10.1002/(SICI)1521-4141(199901)29:01<277::AID-IMMU277>3.0.CO;2-4
164. Achdout H, Arnon TI, Markel G, Gonen-Gross T, Katz G, Lieberman N, et al. Enhanced recognition of human NK receptors after influenza virus infection. J Immunol (2003) 171(2):915–23. doi:10.4049/jimmunol.171.2.915
165. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol (2003) 4(9):913–9. doi:10.1038/ni961
166. Clements CS, Kjer-Nielsen L, Kostenko L, Hoare HL, Dunstone MA, Moses E, et al. Crystal structure of HLA-G: a nonclassical MHC class I molecule expressed at the fetal-maternal interface. Proc Natl Acad Sci U S A (2005) 102(9):3360–5. doi:10.1073/pnas.0409676102
167. Chapman TL, Heikema AP, Bjorkman PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity (1999) 11(5):603–13. doi:10.1016/S1074-7613(00)80135-1
168. Fanger NA, Cosman D, Peterson L, Braddy SC, Maliszewski CR, Borges L. The MHC class I binding proteins LIR-1 and LIR-2 inhibit Fe receptor-mediated signaling in monocytes. Eur J Immunol (1998) 28(11):3423–34. doi:10.1002/(SICI)1521-4141(199811)28:11<3423::AID-IMMU3423>3.0.CO;2-2
169. Ponte M, Cantoni C, Biassoni R, Tradori-Cappai A, Bentivoglio G, Vitale C, et al. Inhibitory receptors sensing HLA-G1 molecules in pregnancy: decidua-associated natural killer cells express LIR-1 and CD94/NKG2A and acquire p49, an HLA-G1-specific receptor. Proc Natl Acad Sci U S A (1999) 96(10):5674–9. doi:10.1073/pnas.96.10.5674
170. Rouas-Freiss N, Gonçalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci U S A (1997) 94(21):11520–5. doi:10.1073/pnas.94.21.11520
171. Scott-Algara D, Arnold V, Didier C, Kattan T, Pirozzi G, Barré-Sinoussi F, et al. The CD85j+ NK cell subset potently controls HIV-1 replication in autologous dendritic cells. PLoS One (2008) 3(4):e1975. doi:10.1371/journal.pone.0001975
172. Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A Class I antigen, HLA-G, expressed in human trophoblasts. Science (1990) 248(4952):220–3. doi:10.1126/science.2326636
173. Apps R, Gardner L, Sharkey AM, Holmes N, Moffett A. A homodimeric complex of HLA-G on normal trophoblast cells modulates antigen-presenting cells via LILRB1. Eur J Immunol (2007) 37(7):1924–37. doi:10.1002/eji.200737089
174. Le Discorde M, Moreau P, Sabatier P, Legeais JM, Carosella ED. Expression of HLA-G in human cornea, an immune-privileged tissue. Hum Immunol (2003) 64(11):1039–44. doi:10.1016/j.humimm.2003.08.346
175. Mallet V, Fournel S, Schmitt C, Campan A, Lenfant F, Le Bouteiller P. Primary cultured human thymic epithelial cells express both membrane-bound and soluble HLA-G translated products. J Reprod Immunol (1999) 43(2):225–34. doi:10.1016/S0165-0378(99)00036-4
176. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells (2008) 26(1):212–22. doi:10.1634/stemcells.2007-0554
177. Ito T, Ito N, Saathoff M, Stampachiacchiere B, Bettermann A, Bulfone-Paus S, et al. Immunology of the human nail apparatus: the nail matrix is a site of relative immune privilege. J Invest Dermatol (2005) 125(6):1139–48. doi:10.1111/j.0022-202X.2005.23927.x
178. Onno M, Pangault C, Le Friec G, Guilloux V, André P, Fauchet R. Modulation of HLA-G antigens expression by human cytomegalovirus: specific induction in activated macrophages harboring human cytomegalovirus infection. J Immunol (2000) 164(12):6426–34. doi:10.4049/jimmunol.164.12.6426
179. Pangault C, Amiot L, Caulet-Maugendre S, Brasseur F, Burtin F, Guilloux V, et al. HLA-G protein expression is not induced during malignant transformation. Tissue Antigens (1999) 53(4 Pt 1):335–46. doi:10.1034/j.1399-0039.1999.530403.x
180. Yang Y, Chu W, Geraghty DE, Hunt JS. Expression of HLA-G in human mononuclear phagocytes and selective induction by IFN-gamma. J Immunol (1996) 156(11):4224–31.
181. Lefebvre S, Moreau P, Guiard V, Cherif E, Adrian-cabestre F, Menier C, et al. Molecular mechanisms controlling constitutive and IFN- g -inducible HLA-G expression in various cell types. J Reprod Immunol (1999) 43:213–24. doi:10.1016/S0165-0378(99)00035-2
182. Moreau P, Adrian-Cabestre F, Menier C, Guiard V, Gourand L, Dausset J, et al. IL-10 selectively induces HLA-G expression in human trophoblasts and monocytes. Int Immunol (1999) 11(5):803–11. doi:10.1093/intimm/11.5.803
183. Bukur J, Rebmann V, Grosse-Wilde H, Luboldt H, Ruebben H, Drexler I, et al. Functional role of human leukocyte antigen-G up-regulation in renal cell carcinoma. Cancer Res (2003) 63(14):4107–11.
184. Davidson B, Elstrand MB, McMaster MT, Berner A, Kurman RJ, Risberg B, et al. HLA-G expression in effusions is a possible marker of tumor susceptibility to chemotherapy in ovarian carcinoma. Gynecol Oncol (2005) 96(1):42–7. doi:10.1016/j.ygyno.2004.09.049
185. Yan W-H. HLA-G expression in cancers: potential role in diagnosis, prognosis and therapy. Endocr Metab Immune Disord Drug Targets (2011) 11(1):76–89. doi:10.2174/187153011794982059
186. Davies B, Hiby S, Gardner L, Loke YW, King A. HLA-G expression by tumors. Am J Reprod Immunol (2001) 45(2):103–7. doi:10.1111/j.8755-8920.2001.450207.x
187. Apps R, Gardner L, Moffett A. A critical look at HLA-G. Trends Immunol (2008) 29(7):313–21. doi:10.1016/j.it.2008.02.012
188. Contini P, Puppo F, Canonica GW, Murdaca G, Ciprandi G. Allergen-driven HLA-G expression and secretion in peripheral blood mononuclear cells from allergic rhinitis patients. Hum Immunol (2016) 77(12):1172–8. doi:10.1016/j.humimm.2016.08.005
189. Saurabh A, Thakral D, Mourya MK, Singh A, Mohan A, Bhatnagar AK, et al. Differential expression of HLA-G and ILT-2 receptor in human tuberculosis: Localized versus disseminated disease. Hum Immunol (2016) 77(9):746–53. doi:10.1016/j.humimm.2016.01.004
190. Le Rond S, Le Maoult J, Créput C, Menier C, Deschamps M, Le Friec G, et al. Alloreactive CD4+ and CD8+ T cells express the immunotolerant HLA-G molecule in mixed lymphocyte reactions: in vivo implications in transplanted patients. Eur J Immunol (2004) 34(3):649–60. doi:10.1002/eji.200324266
191. Amiot L, Vu N, Rauch M, L’Helgoualc’h A, Chalmel F, Gascan H, et al. Expression of HLA-G by mast cells is associated with hepatitis C virus-induced liver fibrosis. J Hepatol (2014) 60(2):245–52. doi:10.1016/j.jhep.2013.09.006
192. Geraghty DE, Koller BH, Orr HT. A human major histocompatibility complex class I gene that encodes a protein with a shortened cytoplasmic segment. Proc Natl Acad Sci U S A (1987) 84(24):9145–9. doi:10.1073/pnas.84.24.9145
193. Paul P, Adrian Cabestre F, Ibrahim EC, Lefebvre S, Khalil-Daher I, Vazeux G, et al. Identification of HLA-G7 as a new splice variant of the HLA-G mRNA and expression of soluble HLA-G5, -G6, and -G7 transcripts in human transfected cells. Hum Immunol (2000) 61(11):1138–49. doi:10.1016/S0198-8859(00)00197-X
194. Ishitani A, Geraghty DE. Alternative splicing of HLA-G transcripts yields proteins with primary structures resembling both class I and class II antigens. Proc Natl Acad Sci U S A (1992) 89(9):3947–51. doi:10.1073/pnas.89.9.3947
195. Hiby SE, King A, Sharkey A, Loke YW. Molecular studies of trophoblast HLA-G: polymorphism, isoforms, imprinting and expression in preimplantation embryo. Tissue Antigens (1999) 53(1):1–13. doi:10.1034/j.1399-0039.1999.530101.x
196. Blaschitz A, Juch H, Volz A, Hutter H, Daxboeck C, Desoye G, et al. The soluble pool of HLA-G produced by human trophoblasts does not include detectable levels of the intron 4-containing HLA-G5 and HLA-G6 isoforms. Mol Hum Reprod (2005) 11(10):699–710. doi:10.1093/molehr/gah185
197. Park GM, Lee S, Park B, Kim E, Shin J, Cho K, et al. Soluble HLA-G generated by proteolytic shedding inhibits NK-mediated cell lysis. Biochem Biophys Res Commun (2004) 313(3):606–11. doi:10.1016/j.bbrc.2003.11.153
198. Riteau B, Rouas-Freiss N, Menier C, Paul P, Dausset J, Carosella ED. HLA-G2, -G3, and -G4 isoforms expressed as nonmature cell surface glycoproteins inhibit NK and antigen-specific CTL cytolysis. J Immunol (2001) 166(8):5018–26. doi:10.4049/jimmunol.166.8.5018
199. Menier C, Riteau B, Dausset J, Carosella ED, Rouas-Freiss N. HLA-G truncated isoforms can substitute for HLA-G1 in fetal survival. Hum Immunol (2000) 61(11):1118–25. doi:10.1016/S0198-8859(00)00194-4
200. Rouas-Freiss N, Marchal RE, Kirszenbaum M, Dausset J, Carosella ED. The a1 domain of HLA-G1 and HLA-G2 inhibits cytotoxicity induced by natural killer cells: Is HLA-G the public ligand for natural killer cell inhibitory receptors? Immunology (1997) 94:5249–54.
201. Le Discorde M, Le Danff C, Moreau P, Rouas-Freiss N, Carosella ED. HLA-G*0105N null allele encodes functional HLA-G isoforms. Biol Reprod (2005) 73(2):280–8. doi:10.1095/biolreprod.104.037986
202. Bainbridge D, Ellis S, Le Bouteiller P, Sargent I. HLA-G remains a mystery. Trends Immunol (2001) 22(10):548–52. doi:10.1016/S1471-4906(01)02031-2
203. Donadi EA, Castelli EC, Arnaiz-Villena A, Roger M, Rey D, Moreau P. Implications of the polymorphism of HLA-G on its function, regulation, evolution and disease association. Cell Mol Life Sci (2011) 68(3):369–95. doi:10.1007/s00018-010-0580-7
204. Ferreira LMR, Meissner TB, Tilburgs T, Strominger JLHLA-G. At the interface of maternal–fetal tolerance. Trends Immunol (2017) 38(4):272–86. doi:10.1016/j.it.2017.01.009
205. Ishitani A, Sageshima N, Lee N, Dorofeeva N, Hatake K, Marquardt H, et al. Protein expression and peptide binding suggest unique and interacting functional roles for HLA-E, F, and G in maternal-placental immune recognition. J Immunol (2003) 171(3):1376–84. doi:10.4049/jimmunol.171.3.1376
206. Clements CS, Kjer-Nielsen L, McCluskey J, Rossjohn J. Structural studies on HLA-G: implications for ligand and receptor binding. Hum Immunol (2007) 68(4):220–6. doi:10.1016/j.humimm.2006.09.003
207. Lenfant F, Pizzato N, Liang S, Davrinche C, Le Bouteiller P, Horuzsko A. Induction of HLA-G-restricted human cytomegalovirus pp65 (UL83)-specific cytotoxic T lymphocytes in HLA-G transgenic mice. J Gen Virol (2003) 84(2):307–17. doi:10.1099/vir.0.18735-0
208. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci U S A (2003) 100(15):8856–61. doi:10.1073/pnas.1431057100
209. Gonen-Gross T, Gazit R, Achdout H, Hanna J, Mizrahi S, Markel G, et al. Special organization of the HLA-G protein on the cell surface. Hum Immunol (2003) 64(11):1011–6. doi:10.1016/j.humimm.2003.08.355
210. Boyson JE, Erskine R, Whitman MC, Chiu M, Lau JM, Koopman LA, et al. Disulfide bond-mediated dimerization of HLA-G on the cell surface. Proc Natl Acad Sci U S A (2002) 99(25):16180–5. doi:10.1073/pnas.212643199
211. Gonen-Gross T, Achdout H, Arnon TI, Gazit R, Stern N, Horejsi V, et al. The CD85J/leukocyte inhibitory receptor-1 distinguishes between conformed and 2-microglobulin-free HLA-G molecules. J Immunol (2005) 175(8):4866–74. doi:10.4049/jimmunol.175.8.4866
212. Khalil-Daher I, Riteau B, Menier C, Sedlik C, Paul P, Dausset J, et al. Role of HLA-G versus HLA-E on NK function: HLA-G is able to inhibit NK cytolysis by itself. J Reprod Immunol (1999) 43(2):175–82. doi:10.1016/S0165-0378(99)00032-7
213. Yu Y, Wang Y, Feng M. Human leukocyte antigen-G1 inhibits natural killer cytotoxicity through blocking the activating signal transduction pathway and formation of activating immunologic synapse. Hum Immunol (2008) 69(1):16–23. doi:10.1016/j.humimm.2007.11.005
214. Riteau B, Menier C, Khalil-Daher I, Martinozzi S, Pla M, Dausset J, et al. HLA-G1 co-expression boosts the HLA class I-mediated NK lysis inhibition. Int Immunol (2001) 13(2):193–201. doi:10.1093/intimm/13.2.193
215. Gros F, Cabillic F, Tourirais O, Le Maux A, Sebti Y, Amiot L. Soluble HLA-G molecules impair natural killer/dendritic cell crosstalk via inhibition of dendritic cells. Eur J Immunol (2008) 38(3):742–9. doi:10.1002/eji.200736918
216. Goodridge JP, Lathbury LJ, Steiner NK, Shulse CN, Pullikotil P, Seidah NG, et al. Three common alleles of KIR2DL4 (CD158d) encode constitutively expressed, inducible and secreted receptors in NK cells. Eur J Immunol (2007) 37(1):199–211. doi:10.1002/eji.200636316
217. Goodridge JP, Witt CS, Christiansen FT, Warren HS. KIR2DL4 (CD158d) genotype influences expression and function in NK cells. J Immunol (2003) 171(4):1768–74. doi:10.4049/jimmunol.171.4.1768
218. Kikuchi-Maki A, Yusa S, Catina TL, Campbell KS. KIR2DL4 is an IL-2-regulated NK cell receptor that exhibits limited expression in humans but triggers strong IFN-gamma production. J Immunol (2003) 171(18):3415–25. doi:10.4049/jimmunol.171.7.3415
219. Kikuchi-Maki A, Catina TL, Campbell KS. Cutting edge: KIR2DL4 transduces signals into human NK cells through association with the Fc receptor gamma protein. J Immunol (2005) 174(7):3859–63. doi:10.4049/jimmunol.174.7.3859
220. Yusa S, Catina TL, Campbell KS. SHP-1- and phosphotyrosine-independent inhibitory signaling by a killer cell Ig-like receptor cytoplasmic domain in human NK cells. J Immunol (2002) 168(10):5047–57. doi:10.4049/jimmunol.168.10.5047
221. Faure M, Long EO. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J Immunol (2002) 168(12):6208–14. doi:10.4049/jimmunol.168.12.6208
222. Rajagopalan S, Long EO. KIR2DL4 (CD158d): an activation receptor for HLA-G. Front Immunol (2012) 3:258. doi:10.3389/fimmu.2012.00258
223. Rajagopalan S, Fu J, Long EO, Alerts E. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J Immunol (2001) 167:1877–81. doi:10.4049/jimmunol.167.4.1877
224. Le Page MEL, Goodridge JP, John E, Christiansen FT, Witt CS. Response to comment on “killer Ig-like receptor 2DL4 does not mediate NK cell IFN-γ responses to soluble HLA-G preparations”. J Immunol (2014) 192(9):4003–4. doi:10.4049/jimmunol.1301748
225. Rajagopalan S, Long EO. Comment on “Killer Ig-like receptor 2DL4 does not mediate NK Cell IFN-γ responses to soluble HLA-G preparations”. J Immunol (2014) 192(9):4003. doi:10.4049/jimmunol.1400492
226. Rajagopalan S, Long EO. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med (1999) 189(7):1093–100. doi:10.1084/jem.189.7.1093
227. Cantoni C, Verdiani S, Falco M, Pessino A, Cilli M, Conte R, et al. p49, a putative HLA class I-specific inhibitory NK receptor belonging to the immunoglobulin superfamily. Eur J Immunol (1998) 28(10):3398. doi:10.1002/(SICI)1521-4141(199806)28:06<1980::AID-IMMU1980>3.0.CO;2-F
228. Yan WH, Fan LA. Residues Met76 and Gln79 in HLA-G alpha1 domain involve in KIR2DL4 recognition. Cell Res (2005) 15(3):176–82. doi:10.1038/sj.cr.7290283
229. Rajagopalan S, Bryceson YT, Kuppusamy SP, Geraghty DE, Van Der Meer A, Joosten I, et al. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol (2006) 4(1):e9. doi:10.1371/journal.pbio.0040009
230. Yu YR, Tian XH, Wang Y, Feng MF. Rapid production of human KIR2DL4 extracellular domain and verification of its interaction with HLA-G. Biochem (2006) 71(Suppl 1):S60–4. doi:10.1134/S0006297906130104
231. Allan DS, Colonna M, Lanier LL, Churakova TD, Abrams JS, Ellis SA, et al. Tetrameric complexes of human histocompatibility leukocyte antigen (HLA)-G bind to peripheral blood myelomonocytic cells. J Exp Med (1999) 189(7):1149–56. doi:10.1084/jem.189.7.1149
232. Le Page MEL, Goodridge JP, John E, Christiansen FT, Witt CS. Killer Ig-like receptor 2DL4 does not mediate NK cell IFN-γ responses to soluble HLA-G preparations. J Immunol (2014) 192(2):732–40. doi:10.4049/jimmunol.1400492
233. Moradi S, Berry R, Pymm P, Hitchen C, Beckham SA, Wilce MCJ, et al. The structure of the atypical killer cell immunoglobulin-like receptor, KIR2DL4. J Biol Chem (2015) 290(16):10460–71. doi:10.1074/jbc.M114.612291
234. Rajagopalan S, Moyle MW, Joosten I, Long EO. DNA-PKcs controls an endosomal signaling pathway for a proinflammatory response by natural killer cells. Sci Signal (2010) 3(110):ra14. doi:10.1126/scisignal.2000467
235. Hellmann I, Letvin NL, Schmitz JE. KIR2DL4 copy number variation is associated with CD4+ T-cell depletion and function of cytokine-producing NK cell subsets in SIV-infected Mamu-A*01-negative rhesus macaques. J Virol (2013) 87(9):5305–10. doi:10.1128/JVI.02949-12
236. Lozano JM, Gonzalez R, Kindelan JM, Rouas-Freiss N, Caballos R, Dausset J, et al. Monocytes and T lymphocytes in HIV-1-positive patients express HLA-G molecule. AIDS (2002) 16(3):347–51. doi:10.1097/00002030-200202150-00005
237. Cabello A, Rivero A, Garcia MJ, Lozano JM, Torre-Cisneros J, González R, et al. HAART induces the expression of HLA-G on peripheral monocytes in HIV-1 infected individuals. Hum Immunol (2003) 64(11):1045–9. doi:10.1016/j.humimm.2003.08.353
238. Rivero A, Lozano JM, González R, García-Jurado G, Camacho A, Torres-Cisneros J, et al. Nucleoside reverse transcriptase inhibitors are able and protease inhibitors unable to induce the tolerogenic molecule HLA-G1 on monocytes from HIV-1 infected patients. Hum Immunol (2007) 68(4):303–6. doi:10.1016/j.humimm.2006.10.014
239. Viganò S, Negrón JJ, Tse S, Chowdhury FZ, Lichterfeld M, Yu XG. HLA-G+ HIV-1-specific CD8 T cells are associated with HIV-1 immune control. AIDS (2017) 31(2):207–12. doi:10.1097/QAD.0000000000001326
240. Derrien M, Pizzato N, Dolcini G, Menu E, Chaouat G, Lenfant F, et al. Human immunodeficiency virus 1 downregulates cell surface expression of the non-classical major histocompatibility class I molecule HLA-G1. J Gen Virol (2004) 85(Pt 7):1945–54. doi:10.1099/vir.0.79867-0
241. Pizzato N, Derrien M, Lenfant F. The short cytoplasmic tail of HLA-G determines its resistance to HIV-1 Nef-mediated cell surface downregulation. Hum Immunol (2004) 65(11):1389–96. doi:10.1016/j.humimm.2004.07.239
242. Huang J, Burke P, Yang Y, Seiss K, Beamon J, Cung T, et al. Soluble HLA-G inhibits myeloid dendritic cell function in HIV-1 infection by interacting with leukocyte immunoglobulin-like receptor B2. J Virol (2010) 84(20):10784–91. doi:10.1128/JVI.01292-10
243. Lila N, Rouas-Freiss N, Dausset J, Carpentier A, Carosella ED. Soluble HLA-G protein secreted by allo-specific CD4+ T cells suppresses the allo-proliferative response: a CD4+ T cell regulatory mechanism. Proc Natl Acad Sci U S A (2001) 98(21):12150–5. doi:10.1073/pnas.201407398
244. Lajoie J, Fontaine J, Tremblay C, Routy J-P, Poudrier J, Roger M. Persistence of high levels of blood soluble human leukocyte antigen-G is associated with rapid progression of HIV infection. AIDS (2009) 23(11):1437–40. doi:10.1097/QAD.0b013e32832d0825
245. Murdaca G, Contini P, Setti M, Cagnati P, Lantieri F, Indiveri F, et al. Behavior of non-classical soluble HLA class G antigens in human immunodeficiency virus 1-infected patients before and after HAART: comparison with classical soluble HLA-A, -B, -C antigens and potential role in immune-reconstitution. Clin Immunol (2009) 133(2):238–44. doi:10.1016/j.clim.2009.08.002
246. Murdaca G, Contini P, Setti M, Cagnati P, Spanò F, Lantieri F, et al. Soluble human leukocyte antigen-G serum levels in patients with acquired immune deficiency syndrome affected by different disease-defining conditions before and after antiretroviral treatment. Hum Immunol (2011) 72(9):712–6. doi:10.1016/j.humimm.2011.05.008
247. Lajoie J, Massinga Loembe M, Poudrier J, Guédou F, Pépin J, Labbé A-C, et al. Blood soluble human leukocyte antigen G levels are associated with human immunodeficiency virus type 1 infection in Beninese commercial sex workers. Hum Immunol (2010) 71(2):182–5. doi:10.1016/j.humimm.2009.11.007
248. Thibodeau V, Lajoie J, Labbé A-C, Zannou MD, Fowke KR, Alary M, et al. High level of soluble HLA-G in the female genital tract of Beninese commercial sex workers is associated with HIV-1 infection. PLoS One (2011) 6(9):e25185. doi:10.1371/journal.pone.0025185
249. Rebmann V, van der Ven K, Pässler M, Pfeiffer K, Krebs D, Grosse-Wilde H. Association of soluble HLA-G plasma levels with HLA-G alleles. Tissue Antigens (2001) 57(1):15–21. doi:10.1034/j.1399-0039.2001.057001015.x
250. Matte C, Lajoie J, Lacaille J, Zijenah LS, Ward BJ, Roger M. Functionally active HLA-G polymorphisms are associated with the risk of heterosexual HIV-1 infection in African women. AIDS (2004) 18(3):427–31. doi:10.1097/00002030-200402200-00008
251. Segat L, Catamo E, Fabris A, Morgutti M, D’Agaro P, Campello C, et al. HLA-G*0105N allele is associated with augmented risk for HIV infection in white female patients. AIDS (2010) 24(12):1961–4. doi:10.1097/QAD.0b013e32833c3324
252. Turk WJR, Kimani J, Bielawny T, Wachihi C, Ball TB, Plummer FA, et al. Associations of human leukocyte antigen-G with resistance and susceptibility to HIV-1 infection in the Pumwani sex worker cohort. AIDS (2013) 27(1):7–15. doi:10.1097/QAD.0b013e32835ab1f2
253. Aghafar MZKA, Witt C, Kamarulzaman A, Ismail R, Lederman MM, Rodriguez B, et al. Genetic variations in loci relevant to natural killer cell function are affected by ethnicity but are generally not correlated with susceptibility to HIV-1. Tissue Antigens (2012) 79(5):367–71. doi:10.1111/j.1399-0039.2012.01843.x
254. Luo M, Czarnecki C, Ramdahin S, Embree J, Plummer FA. HLA-G and mother-child perinatal HIV transmission. Hum Immunol (2013) 74(4):459–63. doi:10.1016/j.humimm.2012.11.023
255. Moodley S, Bobat R. Expression of HLA-G1 at the placental interface of HIV-1 infected pregnant women and vertical transmission of HIV. Placenta (2011) 32(10):778–82. doi:10.1016/j.placenta.2011.07.012
256. Chen XY, Yan WH, Lin A, Xu HH, Zhang JG, Wang XX. The 14 bp deletion polymorphisms in HLA-G gene play an important role in the expression of soluble HLA-G in plasma. Tissue Antigens (2008) 72(4):335–41. doi:10.1111/j.1399-0039.2008.01107.x
257. Svendsen SG, Hantash BM, Zhao L, Faber C, Bzorek M, Nissen MH, et al. The expression and functional activity of membrane-bound human leukocyte antigen-G1 are influenced by the 3’-untranslated region. Hum Immunol (2013) 74(7):818–27. doi:10.1016/j.humimm.2013.03.003
258. Segat L, Zupin L, Kim H-Y, Catamo E, Thea DM, Kankasa C, et al. HLA-G 14 bp deletion/insertion polymorphism and mother-to-child transmission of HIV. Tissue Antigens (2014) 83(3):161–7. doi:10.1111/tan.12296
259. Fabris A, Catamo E, Segat L, Morgutti M, Arraes LC, De Lima-Filho JL, et al. Association between HLA-G 3’UTR 14-bp polymorphism and HIV vertical transmission in Brazilian children. AIDS (2009) 23(2):177–82. doi:10.1097/QAD.0b013e32832027bf
260. Hong HA, Paximadis M, Gray GE, Kuhn L, Tiemessen CT. Maternal human leukocyte antigen-G (HLA-G) genetic variants associate with in utero mother-to-child transmission of HIV-1 in Black South Africans. Infect Genet Evol (2015) 30:147–58. doi:10.1016/j.meegid.2014.12.021
261. da Silva GK, Vianna P, Veit TD, Crovella S, Catamo E, Cordero EA, et al. Influence of HLA-G polymorphisms in human immunodeficiency virus infection and hepatitis C virus co-infection in Brazilian and Italian individuals. Infect Genet Evol (2014) 21:418–23. doi:10.1016/j.meegid.2013.12.013
262. Cheent KS, Jamil KM, Cassidy S, Liu M, Mbiribindi B, Mulder A, et al. Synergistic inhibition of natural killer cells by the nonsignaling molecule CD94. Proc Natl Acad Sci U S A (2013) 110(42):16981–6. doi:10.1073/pnas.1304366110
263. Wei X, Orr HT. Differential expression of HLA-E, HLA-F, and HLA-G transcripts in human tissue. Hum Immunol (1990) 29(2):131–42.
264. Apps R, Meng Z, Del Prete GQ, Lifson JD, Zhou M, Carrington M. Relative expression levels of the HLA class-I proteins in normal and HIV-infected cells. J Immunol (2015) 194(8):3594–600. doi:10.4049/jimmunol.1403234
265. Lee N, Goodlett DR, Ishitani A, Marquardt H, Geraghty DE. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J Immunol (1998) 160(10):4951–60.
266. Grimsley C, Ober C. Population genetic studies of HLA-E: evidence for selection. Hum Immunol (1997) 52(1):33–40. doi:10.1016/S0198-8859(96)00241-8
267. Felício LP, Porto IOP, Mendes-Junior CT, Veiga-Castelli LC, Santos KE, Vianello-Brondani RP, et al. Worldwide HLA-E nucleotide and haplotype variability reveals a conserved gene for coding and 3′ untranslated regions. Tissue Antigens (2014) 83(2):82–93. doi:10.1111/tan.12283
268. Romero P, Ortega C, Palma A, Molina IJ, Peña J, Santamaría M. Expression of CD94 and NKG2 molecules on human CD4(+) T cells in response to CD3-mediated stimulation. J Leukoc Biol (2001) 70:219–24.
269. Braud VM, Aldemir H, Breart B, Ferlin WG. Expression of CD94-NKG2A inhibitory receptor is restricted to a subset of CD8+ T cells. Trends Immunol (2003) 24(4):162–4. doi:10.1016/S1471-4906(03)00064-4
270. Gunturi A, Berg RE, Forman J. The role of CD94/NKG2 in innate and adaptive immunity. Immunol Res (2004) 30(1):29–34. doi:10.1385/IR:30:1:029
271. Renedo M, Arce I, Rodríguez A, Carretero M, Lanier LL, López-Botet M, et al. The human natural killer gene complex is located on chromosome 12p12-p13. Immunogenetics (1997) 46(4):307–11. doi:10.1007/s002510050276
272. Plougastel B, Jones T, Trowsdale J. Genomic structure, chromosome location, and alternative splicing of the human NKG2A gene. Immunogenetics (1996) 44(4):286–91. doi:10.1007/BF02602558
273. Spear P, Wu M-R, Sentman M-L, Sentman CL. NKG2D ligands as therapeutic targets. Cancer Immun (2013) 13:8.
274. Shum BP, Flodin LR, Muir DG, Rajalingam R, Khakoo SI, Cleland S, et al. Conservation and variation in human and common chimpanzee CD94 and NKG2 genes. J Immunol (2002) 168:240–52. doi:10.4049/jimmunol.168.1.240
275. McMahon CW, Raulet DH. Expression and function of NK cell receptors in CD8+ T cells. Curr Opin Immunol (2001) 13(4):465–70. doi:10.1016/S0952-7915(00)00242-9
276. Brooks AG, Posch PE, Scorzelli CJ, Borrego F, Coligan JE. NKG2A complexed with CD94 defines a novel inhibitory natural killer cell receptor. J Exp Med (1997) 185(4):795–800. doi:10.1084/jem.185.4.795
277. Lanier LL, Corliss B, Wu J, Phillips JH. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity (1998) 8(6):693–701. doi:10.1016/S1074-7613(00)80574-9
278. Celik AA, Kraemer T, Huyton T, Blasczyk R, Bade-Döding C. The diversity of the HLA-E-restricted peptide repertoire explains the immunological impact of the Arg107Gly mismatch. Immunogenetics (2016) 68(1):29–41. doi:10.1007/s00251-015-0880-z
279. Llano M, Lee N, Navarro F, García P, Albar JP, Geraghty DE, et al. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonamer. Eur J Immunol (1998) 28(9):2854–63. doi:10.1002/(SICI)1521-4141(199809)28:09<2854::AID-IMMU2854>3.0.CO;2-W
280. Petrie EJ, Clements CS, Lin J, Sullivan LC, Johnson D, Huyton T, et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J Exp Med (2008) 205(3):725–35. doi:10.1084/jem.20072525
281. Lauterbach N, Wieten L, Popeijus HE, Voorter CEM, Tilanus MGJ. HLA-E regulates NKG2C+ natural killer cell function through presentation of a restricted peptide repertoire. Hum Immunol (2015) 76(8):578–86. doi:10.1016/j.humimm.2015.09.003
282. Maier S, Grzeschik M, Weiss EH, Ulbrecht M. Implications of HLA-E allele expression and different HLA-E ligand diversity for the regulation of NK cells. Hum Immunol (2000) 61(11):1059–65. doi:10.1016/S0198-8859(00)00190-7
283. Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell (1998) 92(3):351–66. doi:10.1016/S0092-8674(00)80928-9
284. Michaëlsson J, Teixeira de Matos C, Achour A, Lanier LL, Kärre K, Söderström K. A signal peptide derived from hsp60 Binds HLA-E and interferes with CD94/NKG2A recognition. J Exp Med (2002) 196(11):1403–14. doi:10.1084/jem.20020797
285. Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science (2000) 287(5455):1031–3. doi:10.1126/science.287.5455.1031
286. Nattermann J, Nischalke HD, Hofmeister V, Ahlenstiel G, Zimmermann H, Leifeld L, et al. The HLA-A2 restricted T cell epitope HCV core 35-44 stabilizes HLA-E expression and inhibits cytolysis mediated by natural killer cells. Am J Pathol (2005) 166(2):443–53. doi:10.1016/S0002-9440(10)62267-5
287. Nattermann J, Nischalke HD, Hofmeister V, Kupfer B, Ahlenstiel G, Feldmann G, et al. HIV-1 infection leads to increased HLA-E expression resulting in impaired function of natural killer cells. Antivir Ther (2005) 10(1):95–107.
288. Davis ZB, Cogswell A, Scott H, Mertsching A, Boucau J, Wambua D, et al. A conserved HIV-1-derived peptide presented by HLA-E renders infected T-cells highly susceptible to attack by NKG2A/CD94-bearing natural killer cells. PLoS Pathog (2016) 12(2):e1005421. doi:10.1371/journal.ppat.1005421
289. Lajoie J, Hargrove J, Zijenah LS, Humphrey JH, Ward BJ, Roger M. Genetic variants in nonclassical major histocompatibility complex class I human leukocyte antigen (HLA)-E and HLA-G molecules are associated with susceptibility to heterosexual acquisition of HIV-1. J Infect Dis (2006) 193(2):298–301. doi:10.1086/498877
290. Pietra G, Romagnani C, Mazzarino P, Falco M, Millo E, Moretta A, et al. HLA-E-restricted recognition of cytomegalovirus-derived peptides by human CD8+ cytolytic T lymphocytes. Proc Natl Acad Sci U S A (2003) 100(19):10896–901. doi:10.1073/pnas.1834449100
291. Romagnani C, Pietra G, Falco M, Millo E, Mazzarino P, Biassoni R, et al. Identification of HLA-E-specific alloreactive T lymphocytes: a cell subset that undergoes preferential expansion in mixed lymphocyte culture and displays a broad cytolytic activity against allogeneic cells. Proc Natl Acad Sci U S A (2002) 99(17):11328–33. doi:10.1073/pnas.172369799
292. Schulte D, Vogel M, Langhans B, Kramer B, Korner C, Nischalke HD, et al. The HLA-E(R)/HLA-E(R) genotype affects the natural course of hepatitis C virus (HCV) infection and is associated with HLA-E-restricted recognition of an HCV-derived peptide by interferon-gamma-secreting human CD8(+) T cells. J Infect Dis (2009) 200(9):1397–401. doi:10.1086/605889
293. Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, et al. Broadly targeted CD8+ T cell responses restricted by major histocompatibility complex E. Science (2016) 351(6274):714–20. doi:10.1126/science.aac9475
294. Haynes BF, Shaw GM, Korber B, Kelsoe G, Sodroski J, Hahn BH, et al. HIV-host interactions: implications for vaccine design. Cell Host Microbe (2016) 19(3):292–303. doi:10.1016/j.chom.2016.02.002
295. Fausther-Bovendo H, Wauquier N, Cherfils-Vicini J, Cremer I, Debré P, Vieillard V. NKG2C is a major triggering receptor involved in the Vδ1 T cell-mediated cytotoxicity against HIV-infected CD4 T cells. AIDS (2008) 22(2):217–26. doi:10.1097/QAD.0b013e3282f46e7c
296. Mela CM, Burton CT, Imami N, Nelson M, Steel A, Gazzard BG, et al. Switch from inhibitory to activating NKG2 receptor expression in HIV-1 infection: lack of reversion with highly active antiretroviral therapy. AIDS (2005) 19(16):1761–9. doi:10.1097/01.aids.0000183632.12418.33
297. Brunetta E, Fogli M, Varchetta S, Bozzo L, Hudspeth KL, Marcenaro E, et al. Chronic HIV-1 viremia reverses NKG2A/NKG2C ratio on natural killer cells in patients with human cytomegalovirus co-infection. AIDS (2010) 24(1):27–34. doi:10.1097/QAD.0b013e3283328d1f
298. Zhang R, Xu J, Hong K, Yuan L, Peng H, Tang H, et al. Increased NKG2A found in cytotoxic natural killer subset in HIV-1 patients with advanced clinical status. AIDS (2007) 21(Suppl 8):S9–17. doi:10.1097/01.aids.0000304691.32014.19
299. Martini F, Agrati C, D’Offizi G, Poccia F. HLA-E up-regulation induced by HIV infection may directly contribute to CD94-mediated impairment of NK cells. Int J Immunopathol Pharmacol (2005) 18(2):269–76. doi:10.1177/039463200501800209
300. Lisovsky I, Isitman G, Song R, DaFonseca S, Tremblay-McLean A, Lebouché B, et al. A higher frequency of NKG2A+ than of NKG2A- NK cells responds to autologous HIV-infected CD4 cells irrespective of whether or not they coexpress KIR3DL1. J Virol (2015) 89(19):9909–19. doi:10.1128/JVI.01546-15
301. Yawata M, Yawata N, Draghi M, Partheniou F, Little AM, Parham P. MHC class I specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood (2008) 112(6):2369–80. doi:10.1182/blood-2008-03-143727
302. Muntasell A, López-Montañés M, Vera A, Heredia G, Romo N, Peñafiel J, et al. NKG2C zygosity influences CD94/NKG2C receptor function and the NK-cell compartment redistribution in response to human cytomegalovirus. Eur J Immunol (2013) 43(12):3268–78. doi:10.1002/eji.201343773
303. Gregson JNS, Kuri-Cervantes L, Mela CM, Gazzard BG, Bower M, Goodier MR. Short communication: NKG2C+ NK cells contribute to increases in CD16+CD56- cells in HIV type 1+ individuals with high plasma viral load. AIDS Res Hum Retroviruses (2013) 29(1):84–8. doi:10.1089/AID.2011.0397
304. Béziat V, Liu LL, Malmberg J-A, Ivarsson MA, Sohlberg E, Björklund AT, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood (2013) 121(14):2678–88. doi:10.1182/blood-2012-10-459545
305. Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104(12):3664–71. doi:10.1182/blood-2004-05-2058
306. Bigley AB, Rezvani K, Shah N, Sekine T, Balneger N, Pistillo M, et al. Latent cytomegalovirus infection enhances anti-tumour cytotoxicity through accumulation of NKG2C+ NK cells in healthy humans. Clin Exp Immunol (2016) 185(2):239–51. doi:10.1111/cei.12785
307. Gumá M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J Infect Dis (2006) 194(1):38–41. doi:10.1086/504719
308. Gumá M, López-Botet M. Reply to Mela and Goodier [2]. J Infect Dis (2007) 195(1):159–60. doi:10.1086/509814
309. Mela CM, Goodier MR. The contribution of cytomegalovirus to changes in NK cell receptor expression in HIV-1-infected individuals. J Infect Dis (2007) 195(1):158–9. doi:10.1086/509811
310. Thomas R, Low HZ, Kniesch K, Jacobs R, Schmidt RE, Witte T. NKG2C deletion is a risk factor of HIV infection. AIDS Res Hum Retroviruses (2012) 28(8):844–51. doi:10.1089/aid.2011.0253
311. Della Chiesa M, Sivori S, Carlomagno S, Moretta L, Moretta A. Activating KIRs and NKG2C in viral infections: toward NK cell memory? Front Immunol (2015) 6:573. doi:10.3389/fimmu.2015.00573
312. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol (2012) 189(10):5082–8. doi:10.4049/jimmunol.1201964
313. Reeves RK, Li H, Jost S, Blass E, Li H, Schafer JL, et al. Antigen-specific NK cell memory in rhesus macaques. Nat Immunol (2015) 16(9):927–32. doi:10.1038/ni.3227
314. Merino AM, Song W, He D, Mulenga J, Allen S, Hunter E, et al. HLA-B signal peptide polymorphism influences the rate of HIV-1 acquisition but not viral load. J Infect Dis (2012) 205(12):1797–805. doi:10.1093/infdis/jis275
315. Merino AM, Sabbaj S, Easlick J, Goepfert P, Kaslow RA, Tang J. Dimorphic HLA-B signal peptides differentially influence HLA-E- and natural killer cell-mediated cytolysis of HIV-1-infected target cells. Clin Exp Immunol (2013) 174(3):414–23. doi:10.1111/cei.12187
316. Elliott JM, Yokoyama WM. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol (2011) 32(8):364–72. doi:10.1016/j.it.2011.06.001
317. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C + natural killer cells with potent function. Blood (2012) 119(11):2665–74. doi:10.1182/blood-2011-10-386995
Keywords: HIV-1, innate immunity, natural killer cells, killer cell immunoglobulin-like receptor, human leukocyte antigen class I, human leukocyte antigen-F, human leukocyte antigen-E
Citation: Hölzemer A, Garcia-Beltran WF and Altfeld M (2017) Natural Killer Cell Interactions with Classical and Non-Classical Human Leukocyte Antigen Class I in HIV-1 Infection. Front. Immunol. 8:1496. doi: 10.3389/fimmu.2017.01496
Received: 05 July 2017; Accepted: 24 October 2017;
Published: 14 November 2017
Edited by:
Persephone Borrow, University of Oxford, United KingdomReviewed by:
Amir Horowitz, Icahn School of Medicine at Mount Sinai, United StatesSimon Kollnberger, Cardiff University, United Kingdom
Copyright: © 2017 Hölzemer, Garcia-Beltran and Altfeld. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelique Hölzemer, YW5nZWxpcXVlLmhvZWx6ZW1lckBsZWlibml6LWhwaS5kZQ==