 Stephanie A. Amici
Stephanie A. Amici Joycelyn Dong1,2
Joycelyn Dong1,2 Mireia Guerau-de-Arellano
Mireia Guerau-de-Arellano- 1School of Health and Rehabilitation Sciences, Division of Medical Laboratory Science, College of Medicine, Wexner Medical Center, The Ohio State University, Columbus, OH, United States
- 2McCormick School of Engineering, Division of Biomedical Engineering, Northwestern University, Evanston, IL, United States
- 3Institute for Behavioral Medicine Research, The Ohio State University, Columbus, OH, United States
- 4Department of Microbial Infection and Immunity, The Ohio State University, Columbus, OH, United States
- 5Department of Neuroscience, The Ohio State University, Columbus, OH, United States
Macrophages and microglia play crucial roles during central nervous system development, homeostasis and acute events such as infection or injury. The diverse functions of tissue macrophages and microglia are mirrored by equally diverse phenotypes. A model of inflammatory/M1 versus a resolution phase/M2 macrophages has been widely used. However, the complexity of macrophage function can only be achieved by the existence of varied, plastic and tridimensional macrophage phenotypes. Understanding how tissue macrophages integrate environmental signals via molecular programs to define pathogen/injury inflammatory responses provides an opportunity to better understand the multilayered nature of macrophages, as well as target and modulate cellular programs to control excessive inflammation. This is particularly important in MS and other neuroinflammatory diseases, where chronic inflammatory macrophage and microglial responses may contribute to pathology. Here, we perform a comprehensive review of our current understanding of how molecular pathways modulate tissue macrophage phenotype, covering both classic pathways and the emerging role of microRNAs, receptor-tyrosine kinases and metabolism in macrophage phenotype. In addition, we discuss pathway parallels in microglia, novel markers helpful in the identification of peripheral macrophages versus microglia and markers linked to their phenotype.
Introduction
Macrophages in the central nervous system (CNS) play important homeostatic and immune defense roles (1). While microglia originate from early yolk sac myeloid progenitors and become self-regenerating CNS-resident cells (2–4), macrophages originate from peripheral blood monocytes. Microglia are essential for appropriate synaptic pruning during development (1). During steady state condition, microglia also facilitate learning and memory and remove cellular or other debris. Upon CNS infections and injury, microglial activation and peripheral macrophage recruitment and activation occur. Both macrophages and microglia have the capacity to recognize pathogens or injured cells, activating phagocytic, antigen-presenting and cytokine/chemokine secretion functions that modulate immunity and mediate pathogen or cellular debris elimination (1). Macrophages and microglia also contribute to resolution stages of inflammation and tissue regeneration via switching to anti-inflammatory cytokine patterns, promoting intercellular matrix synthesis and angiogenesis. The complexity of macrophage function is mirrored by the existence of varied, plastic and multilayered macrophage phenotypes in vivo (5). However, for simplicity, a model of inflammatory/classical M1 and resolution/alternatively activated M2 macrophages has been widely used.
Understanding the molecular programs that define inflammatory versus resolution phenotypes provides the opportunity to target and modulate these cellular programs to control the excessive inflammation typical of chronic inflammatory CNS conditions such as Multiple Sclerosis (MS) and CNS injury. In recent years, our understanding of how environmental signals are integrated into macrophage phenotype has greatly advanced. The classic roles of NOTCH, PI3K/AKT, MYC, PPAR, and interferon regulatory factors (IRFs) in macrophage polarization have been further established while prominent roles for metabolism, microRNAs (miRNAs) and receptor-tyrosine kinases (RTKs) are now clear. The ability to distinguish microglia from CNS macrophages and inflammatory vs. resolution macrophages has greatly advanced with the discovery of new markers. In this review, we discuss these findings and present the current understanding in the field of molecular mechanisms and markers of inflammatory versus resolution macrophages, as well as therapeutic implications of macrophage modulation for the CNS autoimmune disease MS.
Macrophages and Microglia: Similar but Not the Same (Developmental Origin, Functions, and Markers)
Microglia and macrophages have many functions in common. They both help to maintain homeostasis during embryogenesis and into adulthood (6–8). Additionally, both cells are sentinels in their respective environments, scanning for foreign invaders and pathogens (9–12). Both cell types also differentiate into a spectrum of proinflammatory to proregenerative subsets in response to injury or insult (13, 14). In addition to the roles microglia play in fighting infection and clearing debris via phagocytosis, microglia are also important in neuronal proliferation and differentiation and the formation and pruning of synaptic connections in neuronal networks (15, 16). Based on the specific genes expressed in microglia and the subset of functions unique to microglia, one can postulate that other tissue-specific macrophages have roles exclusive to their tissue that monocyte-derived macrophages cannot replace.
Monocyte-derived CNS macrophages and microglia have similar morphologies and phagocytic functions but their origins are distinct. Until a short time ago it was believed that solely circulating monocytes replenish tissue macrophage populations, including those in the CNS, but this view is now rejected based on new reports in the literature (17, 18). Although bone marrow derived monocytes can enter tissues such as the CNS and differentiate into macrophages, microglia and other tissue macrophages are now thought to originate most exclusively from earlier embryonic progenitors (19). Embryonic hematopoiesis consists of three main waves, namely primitive, transient definitive and definitive hematopoiesis. Primitive hematopoiesis originates from yolk sac blood islands around embryonic day (E)7, yielding progenitors as early as E7.5 (19). The transient definitive hematopoiesis wave starts around E8 when hemogenic endothelium develops, producing erythromyeloid precursors (EMPs) (19). Upon establishment of circulation starting at E8.5, EMPs migrate to the fetal liver where they support definitive hematopoiesis (19). EMPs will also eventually migrate and support bone marrow hematopoiesis in the adult.
Three models of fetal microglia and tissue macrophage ontogeny have been proposed (19). Two models favor the view that most microglia but few of other tissue macrophages derive from the early wave of primitive hematopoiesis in the yolk sac (20, 21). The remaining model instead supports the view that EMPs from transient definitive hematopoiesis give rise to most microglia and other tissue macrophages (22). All these models are all in agreement on the embryonic origin of microglia, with little or no contribution from monocytes. Sublethally irradiated C56BL/6 CD45.2+ newborn mice reconstituted with hematopoietic cells isolated from CD45.1+ congenic mice had 95% microglia were of host origin (CD45.2+ cells gated on CD11b+CD45int, then Ly6G-F4/80+) 3 months after transplant, while over 30% of circulating leukocytes were of donor origin (2). These data support the idea that microglia are a distinct population not populated/replenished by circulating monocytes. Kierdorf and colleagues added to our knowledge by identifying the earliest yolk sac progenitors with the potential to become microglia to be CD45− c-Kit+ erythromyeloid precursors (EMPs), and these differentiated into Iba-1+ Cx3cr1− cells with microglial-like morphology (3). Two transcription factors important in driving EMPs to differentiate into microglia and CNS macrophages are PU.1 and IRF-8. Pu.1 gene deficient animals lacked microglia completely, while mice-lacking the Irf8 gene had significantly reduced numbers of microglia (3). Upon analysis of the yolk sac progenitors, they found that PU.1 is necessary for the initial transition from EMPs (c-KIT+) to early microglial precursors (CD45+ c-KITlo CX3CR1−); whereas IRF-8 acts downstream of PU.1 and plays a role in the transition from early to mid-stage microgliogenesis (CD45+ c-KIT− CX3CR1+) (3). Another molecule important in shaping microglial development is negative regulator of reactive oxygen species (NRROS, aka LRRC33). Nrros gene deficient mice lack normal CD11bhiCD45lo microglia, and CX3CR1-driven deletion of Nrros leads to impaired expression of Sall1 (lineage-specific transcription factor important for maintenance in adult microglia) and other microglial genes needed for microglial development and function (23). Interestingly, Nrros−/− mice have normal numbers of myeloid progenitor cells in the CNS at E10.5, but the CD11bhiCD45lo microglial population was largely absent by E14.5, suggesting NRROS is important in early microglial development.
Can Microglia Be Differentiated from Peripheral Origin Macrophages?
Discriminating between peripheral macrophages and microglia has been a difficult technical issue. Microglia and macrophages share many markers such as CD11b, F4/80, CX3CR1 and IBA1 (13). High levels of CD45 expression (CD45hi) have long been used to discern peripheral macrophages from microglia, which express lower levels of CD45 (24). However, peripheral macrophages may downregulate CD45 once in the CNS or in response to injury (25). CX3CR1 (aka fractalkine receptor) is expressed by microglia throughout development and into adulthood (26). Since it is not expressed by other CNS-origin cells (27), CX3CR1 can be used to detect microglia in naive tissues. During inflammation, however, peripheral macrophages, monocytes and T cells also express CX3CR1 and infiltrate the CNS (28). The use of irradiated Cx3cr1–green fluorescent protein (GFP) knock-in mice (27, 28) as recipients of WT bone marrow yields a model in which only microglia express GFP and peripheral macrophages can be detected by use of donor markers. Another model, a tamoxifen-inducible Cre mouse line crossed with a red fluorescent protein (RFP) Cre reporter mouse line (Cx3cr1YFP-CreER/wt:R26RFP), can differentially label microglia and recruited macrophages by pulsing mice with tamoxifen and then following the YFP+RFP+ labeled cells (29). Macrophages will turnover quickly and lose RFP expression, while microglia will retain RFP expression because they are long-lived. Besides a marker and chemoattraction role, CX3CR1 has an essential role in promoting a resting microglial phenotype and neuroprotection (30). Cx3cr1−/− mice had worsened neurologic dysfunction in the EAE model (31). In contrast, post-spinal cord injury (SCI) recovery was enhanced in Cx3cr1−/− mice (32), suggesting context and CX3CR1 expression on cells beyond microglia and macrophages influence disease outcomes. Soluble CD163, which is cleaved from CD163 on macrophage/microglia membranes, may also be a marker for MS or for inflammation in general (33–35). In addition, many markers co-expressed by peripheral macrophages and monocytes are present on activated microglia as well. For example, CD169 is a marker for macrophages (13) that was recently identified on early activated microglia in MS and EAE lesions (36). MERTK is another common marker for many tissue specific macrophages including microglia (13).
The most prominent difference between microglia and macrophages appears to be their developmental origin. One marker specific to microglia that does not stain infiltrating peripheral immune cells is TMEM119 (25, 37). TMEM119 protein is expressed on all microglia by postnatal day 14 (P14) and remains expressed in post-sciatic nerve crush injury, LPS injection and optic nerve crush injury (25). Specifically, Ccr2RFP/+ mice (in which RFP is only expressed in infiltrating monocytes) showed IBA1+TMEM119− cells were mostly RFP+ and RFP+ cells were never TMEM119+, suggesting TMEM119 is a stable resident microglia marker that does not recognize infiltrating macrophages. Importantly, TMEM119 is a marker for both mouse and human microglia (25) and is maintained in MS lesional tissue (38). The availability of this marker has revealed that many microglial markers are induced, while macrophage markers are suppressed, in peripheral macrophages that infiltrate the CNS (38). FCRLS is another highly expressed gene specific to murine microglia, but there is no ortholog in humans (39). Another microglial-specific marker P2RY12, a purinergic receptor associated with homeostatic microglia not detected on lymphatic tissue (38), is highly expressed in normal white matter of MS patients. However, as microglia become stimulated in active MS lesions, P2RY12 is sometimes downregulated, while proinflammatory genes such as iNOS and CD86 are upregulated (38), confounding the distinction of microglia from peripheral cells during inflammation. Since another report found that P2RY12 remains elevated after EAE induction (39), this may be a human/mouse model difference. TMEM119 seems to be the most discriminatory of the new markers and has allowed to sort microglia based on their developmental origin. These studies have revealed other genes specific of this population that may lead to additional methods for microglial detection (25).
Macrophage/Microglia Phenotypes, Function, and Nomenclature
The dual role of macrophages and microglia in promoting inflammation vs. resolution is mediated by distinct gene expression programs and macrophage phenotypes. This inflammatory phenotype is induced by ligation of pathogen recognition receptors (PRRs), such as Toll-like receptors (TLRs), on macrophages to pathogen- or danger-associated molecular patterns (PAMPs or DAMPs) from microbes or damaged/dying cells (40). These signals may be combined with inflammatory cytokines produced by Th1 cells, such as IFN-γ. In vitro, this phenotype has been modeled by stimulation of bone marrow-derived macrophages or microglia with LPS (+IFN-γ). Macrophages activated in this manner have long been known as classically activated or M1 macrophages (41). In 2014, in an effort to reach consistency and clarity in the field, novel nomenclature that follows the letter M by a parenthesis enclosing the stimuli used for activation was proposed (42). For example, M1 macrophages stimulated with LPS and IFN-γ are indicated as M(LPS + IFN-γ) while macrophages stimulated with LPS alone would be labeled M(LPS). Macrophages differentiated with GM-CSF, or M(GM-CSF) macrophages, have also been described to have a proinflammatory phenotype (43). This nomenclature is providing an extremely useful standardized tool to communicate macrophage experimental data. In this review, we use this nomenclature when the specific stimulation is known, while the simple M1 vs. M2 notation is used when referring to a general inflammatory vs. resolution/alternatively activated phenotype of macrophages.
Functionally, M1 macrophages are responsible for fighting bacterial infections and adopt a phenotype characterized by microbicidal, antigen-presenting and immune potentiating abilities. This is accomplished by induction of inducible nitric oxide synthase (iNOS, encoded by the Nos2 gene), which synthesizes microbicidal nitric oxide (NO) in most rodent models (44, 45). It is important to note, however, that iNOS induction does not occur in human macrophages. In addition, M1 macrophages recruit additional cells to the site of infection and bridge innate and adaptive immunity. This is accomplished by induction of chemokines and inflammatory cytokines interleukin (IL)-6, IL-12, IL-1β, IL-23, and TNF-α that recruit immune cells to sites of infection and polarize them to type I responses and by CD80 and CD86 costimulatory molecule expression to prime T cells (42, 46). Our lab has recently characterized CD38 as a marker that is increased in inflammatory murine bone marrow-derived M(LPS + IFN-γ) macrophages and decreased in M2 macrophages compared to untreated M0 macrophages (47). CD38 upregulation is also observed in a sepsis model (47) as well as Experimental Autoimmune Encephalomyelitis (EAE), the mouse model of MS (48). Given that CD38 is a surface marker that allows live cell sorting for downstream applications, it provides an advantage over intracellular markers such as iNOS. Although CD38 is known to be an ectoenzyme that catalyzes conversion of NAD to ADP-ribose and induces calcium signaling inside the cell (49), its exact role in inflammatory phenotype is unknown. However, it appears to play an important role, as CD38 induction by LXR and NAD depletion is necessary to limit bacterial uptake and inflammatory cytokine production (50). Future studies will be necessary to determine whether CD38 plays a similar role in human macrophages. For a listing of current inflammatory phenotype markers in macrophage/microglia, see Table 1. Like macrophages, microglia secrete inflammatory cytokines IL-1β, IL-6, IL-12, and TNF-α when exposed to LPS (+IFN-γ). Similarly, they upregulate iNOS and CD38 (51–56). Although most studies have been done in murine microglia, LPS + IFN-γ also induces M1 phenotype in primary human microglia (57).
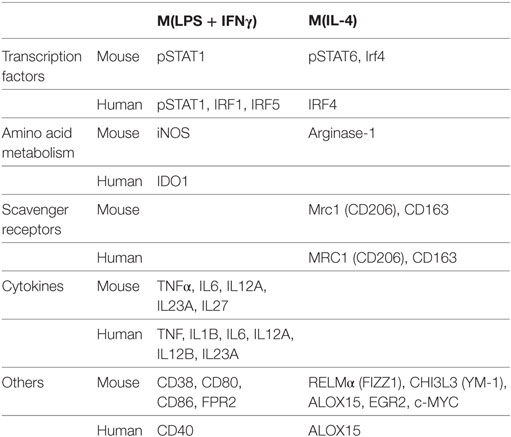
Table 1. Inflammatory phenotype macrophage/microglia markers.
The evolution from acute inflammation to a resolution phase occurs as initial neutrophils undergo apoptosis and monocytes, which will switch to a resolution/M2 phenotype, predominate in the tissue (58). Key lipid mediators in promoting the resolution phase include classical eicosanoids, phospholipids and sphingolipids, endocannabinoids (eCBs) and specialized proresolving mediators (SPMs) (59). The classical eicosanoids thromboxanes (TX) and prostacyclins antagonize inflammation while phospholipids and sphingolipids such as phosphatidylserine (PtdlSer), when recognized by macrophages, promote M2 switch (60). eCBs such as N-arachidonoylethanolamine (AEA) and N-palmitoylethanolamine (PEA) have immunomodulatory roles, particularly in neuroinflammation (61). Last, but not least, SPMs have major proresolution activities. Main SPMs include lipoxins (LX)A4 and LXB4, resolvins (Rv) RvD1-6 derived from docosahexanoic acid (DHA), RvE1-3 derived from eicosapentanoic acid (EPA), protectin D1 and maresins. In particular, maresins have been shown to shift to a resolving macrophage phenotype (62), which can also be induced by exposure to Th2 cytokines like IL-4 and IL-13, parasites, fungal cells, apoptotic cells, immune complexes, adenosine, or transforming growth factor (TGF)-β (63). In vitro, four M2 type macrophages were initially described, corresponding to macrophages stimulated with IL-4 or IL-13 (M2a), IL-1R ligands or immune complexes (M2b), IL-10, TGF-β or glucocorticoid (M2c) and IL-6 and adenosine (M2d) (64). This notation has now been replaced with the M(stimulus) nomenclature that clearly defines the inducing stimulus (42). For, example, macrophages stimulated with IL-4 are indicated as M(IL-4) and pro-M2 macrophages differentiated with M-CSF are called M(M-CSF).
It is currently being debated whether resolution/wound healing macrophages are an evolution of initial inflammatory macrophages under the changing local environment or, rather, they originate from newly recruited peripheral monocytes. Similarly, resolution spectrum macrophages may revert to an inflammatory phenotype if new inflammatory stimuli are encountered (65). Resolution macrophages suppress IL-12 secretion and may secrete anti-inflammatory mediators IL-10, TGF-β, IL-1R antagonist (IL-1RA), and decoy IL-1R II (66). In addition, these macrophages express arginase-1 instead of iNOS, switching arginine metabolism from production of NO to ornithine and polyamines for collagen and extracellular matrix synthesis (67). M2 markers arginase-1, resistin-like alpha (RELMα/FIZZ1), and chitinase 3-like protein 3 (CHI3L3/YM1) are detected in murine but not human macrophages (68–70), although it should be noted that a portion of murine M1 stimulated cells also upregulates arginase-1 (42, 47). Murine M(IL-4), but not M(LPS + IFN-γ), bone marrow derived macrophages (BMDMs) can be identified by flow cytometry detection of the intracellular transcription factor EGR2 (47), which is related to the M2 transcription factor C-MYC. C-MYC (47, 71) and CD206 (41) are M2 markers common to murine and human macrophages. CD169 [aka sialic acid binding Ig-like lectin-1 (SIGLEC-1)] in certain in vivo macrophage populations (72) and tyrosine-protein kinase Mer (MERTK) in M2c macrophages (73) have also been identified as markers useful in flow cytometry (13, 74). CD169 and MERTK are also expressed in microglia.
Does macrophage and microglial phenotype modulate neuroinflammation and MS? The hypothesis that an inflammatory phenotype in macrophages or microglia is damaging to the CNS while a resolving phenotype contributes to neuroregeneration was introduced by the Popovich group in the late 2000s (75). They found that while inflammatory macrophage responses cause neurotoxicity, resolving macrophage responses instead promote neuron viability and regenerative growth toward repair (75). Evidence supporting this hypothesis, which has important therapeutic implications, has since accumulated in multiple neuroinflammatory paradigms. We will discuss below the latest evidence for a role of macrophage and microglial phenotype on modulation of CNS neuroinflammation and remyelination in multiple sclerosis and its animal model EAE.
Activated microglia are one of the first observations very early in MS while both monocyte-derived macrophages and activated P2RY12−/lo microglia are found later in active lesions (38). Large numbers of macrophages and microglia, coinciding with myelin breakdown, are a hallmark of the acute MS lesion (76). High oxidative activity and expression of IL-1β and IL-23p19, all characteristic of inflammatory macrophages and microglia, are observed in these lesions. Exacerbated and fast-progressing EAE occurs in mice with a microglial-specific Nr4a1 deficiency which results in increased microglial activation and NO production, consistent with the damaging M1 responses. Neurodegenerative microglia characterized by a TREM2-APOE pathway signature, are generated after neuron phagocytosis, further establishing a neurodegenerative cycle (77). Inflammatory microglia do in turn induce a subtype of inflammatory astrocytes, termed A1, which can no longer sustain neurons and induce neuron and oligodendrocyte cell death. A1 astrocytes are abundant in the CNS of various neurodegenerative diseases, including MS lesions (78).
So, what about resolution? It is unclear what exactly makes an active MS lesion evolve toward resolution. PtdlSer, a phospholipid present in myelin, may play a role. PtdlSer liposomes suppress NO and inflammatory cytokine production in macrophages and in vivo treatment ameliorates EAE (79). The early resolving lesion contains instead lipid-laden, aka foamy, macrophages or microglia spread throughout the lesion and forming a layer at the lesion edge. There is no evidence of remyelination in early resolving lesions but examples of remyelination in late resolving lesions are fairly common (80). Interestingly, foamy macrophages or microglia abound within these remyelinated areas (80, 81), as if consistent with a reparative role. Consistent with a beneficial role of resolution macrophages, mice deficient in the M2-promoting factor SOCS3 suffer from chronic and more severe EAE while adoptive transfer or promotion of resolution spectrum macrophages suppresses EAE disease (82–84). Besides being less neurotoxic and chemotactic, M2 macrophages may play an active proregeneration role. Consistent with the latter, a shift from inflammatory to resolving phenotype in microglia and infiltrating macrophages is observed during remyelination (81). The shift to M2 phenotype drives oligodendrocyte differentiation in an activin A-dependent manner (81). Taken together, these findings provide a framework for the importance of inflammatory macrophage and microglial phenotype in driving MS neuroinflammation and the therapeutic promise of promoting opposing M2 responses.
Classic Molecular Mechanisms Modulating Macrophage Phenotype
The distinct phenotypic features of inflammatory vs. wound-healing macrophages are controlled by a network of molecular pathways that relay environmental signals via signaling cascades to impact gene expression and cellular metabolism (see Figure 1 for a summary of molecular pathways that modulate macrophage phenotype). PI3K/AKT, NOTCH, PPARs, MYC, and IRFs have been known to modulate macrophage phenotype. Novel data demonstrating crucial roles for metabolism, RTKs, miRNA and epigenetic modifications will be discussed in a subsequent section.
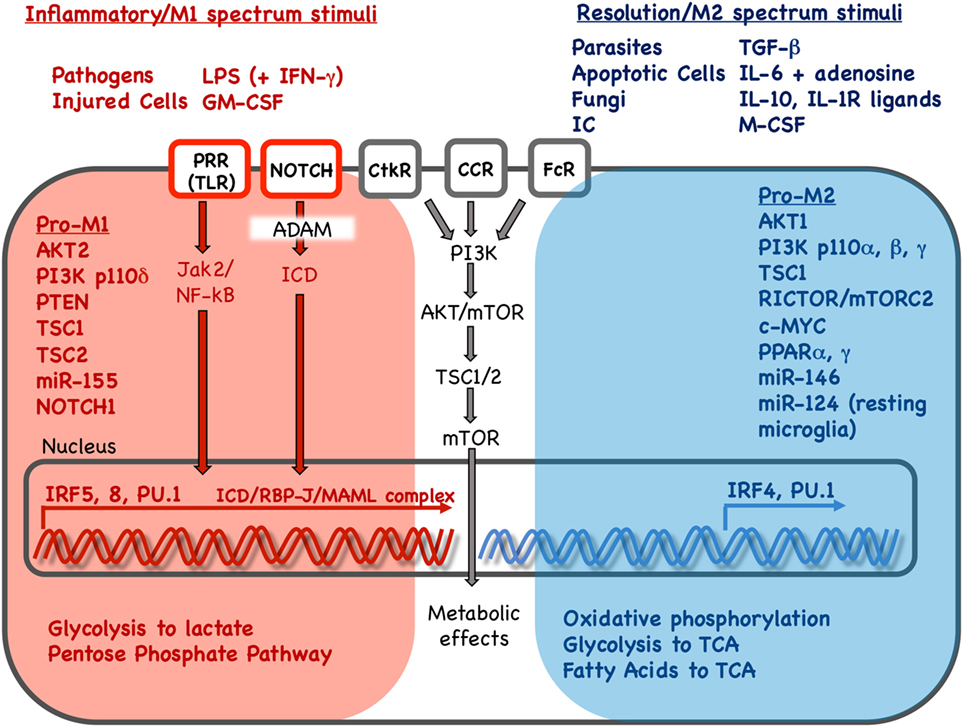
Figure 1. Key stimuli and molecular pathways in inflammatory vs. resolution phenotype in mononuclear phagocytes. Inflammatory and resolution macrophage phenotype results as external stimuli are integrated via signaling pathways to drive phenotype-supporting transcriptional programs and cellular metabolism. Red and blue color indicates pathways, stimuli, transcription factors and metabolic processes associated with inflammatory or resolution phenotype, respectively. Inflammatory phenotype is induced or promoted by pathogens, injured cells and in vitro stimuli. In contrast, resolution spectrum phenotypes are induced or promoted by parasites, fungi, apoptotic cells, immune complexes and other cytokine/growth factor stimuli. Pathogen or injury signals sensed via pathogen-recognition receptors (PRR) such as Toll-like receptors (TLR) result in Janus activated kinase (Jak)2 and nuclear factor kappa B (NF-κB) activation. Signals received via Notch receptors, cytokine receptor (CtkR), chemokine receptor (CCR), and Fc receptor (FcR) stimulation are also integrated, defining gene expression and downstream metabolic reprogramming. Interferon regulatory factors (IRFs) 5 and 8 promote inflammatory gene expression while IRF 4 promotes resolution phenotype genes. Gene expression promotes changes in nutrient uptake and metabolic pathways that support inflammatory or resolution macrophage phenotype. LPS, lipopolysaccharide; IFN-γ: interferon-γ; GM-CSF, granulocyte monocyte colony stimulation factor; IC, immune complexes; TGF-β, transforming growth factor-β; IL, interleukin; IL-1R, interleukin 1 receptor; M-CSF, monocyte colony stimulation factor; PI3K, phosphoinositide 3 kinase; AKT, serine threonine kinase; mTOR, mammalian target of rapamycin; PTEN, phosphatase and tensin homolog; TSC, tuberous sclerosis complex; c-MYC, PPAR, peroxisome proliferator activated receptor; ADAM, A disintegrin and metalloproteinase; RBP-J, recombination signal binding protein for immunoglobulin kappa J region; MAML, mastermind-like; Rictor, rapamycin-insensitive companion of mTOR; TCA, tricarboxylic acid/Krebs cycle.
PI3K/AKT
The PI3K/AKT pathway is activated in response to environmental stimuli such as PAMPs, cytokines/chemokines and hormones to regulate cell survival, proliferation, and differentiation. This pathway plays a pivotal role in the activation phenotype of macrophages [for a thorough review, see Vergadi et al. (85)]. PI3K activates downstream kinase AKT that may exist as three different isoforms, namely AKT1, AKT2, and AKT3. AKT signaling is considered to be an activation dampening signal that controls NO and inflammatory cytokine production after TLR signaling (86–88) and promotes anti-inflammatory cytokines such as IL-10 (89, 90). However, AKT signaling is also required for normal M1 responses (91–93). The AKT1 and AKT2 isoforms play opposing roles in macrophage polarization. AKT1 KO macrophages show enhanced iNOS and IL-12 production and bacterial clearance (91, 94, 95). These effects were mediated by induction of the pro-M1 factor miR-155 (96, 97) that suppresses the target CCAAT/enhancer-binding protein beta CEBPβ (91, 95), a pro-M2 factor (98). In contrast, AKT2 deficiency has the opposite effect, resulting in macrophages that express CEBPβ and signature M2 markers such as arginase-1, YM1, REMLα, and the regulatory cytokine IL-10 (91, 99). AKT2 KO macrophages appear to adopt this phenotype via high levels of miR-146 (93), which has been associated with dampening of inflammatory responses via targeting of IRAK1, TRAF6, and IRF5 leading to suppression of TLR signaling in macrophages and microglia (100–103). Consistent with AKT signaling dampening inflammation, Akt3 gene deficient mice suffered more severe disease in the murine EAE model of MS, an effect mediated by both peripheral macrophages and microglia (104).
Downstream, the PI3K/AKT pathway regulates cellular metabolism via the tuberous sclerosis (TSC)/mammalian target of rapamycin (mTOR) pathway. This is interesting in light of the prominent role metabolism plays in determining macrophage polarization (see metabolism section below). Some data suggest that mTOR signaling inhibits M1 and promotes M2 polarization (105–108). In contrast, other results are more consistent with mTOR signaling promoting M1 polarization (105, 106, 109–112). However, further clarity on the precise roles of TSC and mTORC1 and mTORC2 gene isoforms on metabolism and phenotype is required.
In summary, AKT signaling relays diverse extracellular signals to engage the metabolic regulator mTOR pathway. While AKT signaling can activate inflammatory responses, it is essential for promoting a dampening response, thereby promoting resolution.
Notch
Notch signaling controls embryonic development and differentiation in multiple tissues and organs. Notch receptors NOTCH 1-4 are expressed on the cell surface, where they bind Jagged (JAGGED1, JAGGED2) or Delta-like (DLL1, 3, 4) family ligands on neighboring cells. Binding triggers A disintegrin and metalloproteinase (ADAM)/γ-secretase-catalyzed release of the Notch receptors intracellular domain (ICD), allowing ICD translocation to the nucleus, where it heterodimerizes with recombination signal binding protein for immunoglobulin kappa J region (RBP-J). While RBP-J normally acts as a corepressor recruiter, the ICD/RBP-J complex promotes gene expression via recruitment of mastermind-like coactivator (MAML) (113, 114). In murine macrophages, increased expression of NOTCH1, NOTCH2, and Notch ligands DLL4 and JAGGED1 has been observed in response to inflammatory cues such as LPS, IFN-γ, or IL-1β (115, 116). NOTCH/RBP-J signaling in macrophages results in enhanced NF-κB signaling and induction of pro-M1 transcription factors IRF1 and IRF8 that in turn drive expression of multiple classical activation genes (115–121). Accordingly, reduced levels of inflammatory cytokines IL-6, IL-12, and IFN-γ are observed in response to LPS + IFN-γ in macrophages deficient in Notch1 or treated with the γ-secretase inhibitor DAPT (122, 123). Notch signaling also increases M1 phenotype by modulating glucose flux to the tricarboxylic acid (TCA) cycle, respiratory chain components and reactive oxygen species (ROS) generation (124). Microglial responses are similarly impacted by NOTCH signaling (125–127). Macrophage-specific Notch gene deficiency or γ-secretase inhibitor DAPT treatment suppress clinical disease in in vivo disease models, including EAE (126, 128, 129). Overall, NOTCH signaling appears to have a pivotal role in the development of pathogenic macrophage responses and therapeutic strategies that target NOTCH signaling may be beneficial in inflammatory diseases, including MS.
Peroxisome Proliferator Activated Receptors
Peroxisome proliferator activated receptors (PPARs) are nuclear hormone receptors that act as transcription factors and play important roles in development, differentiation and metabolic regulation (130). PPAR ligands include fatty acids, prostaglandins (PG) such as PG J2 and leukotrienes (LT) such as LT B4 (131). PPARs heterodimerize with the retinoid X receptor (RXR) and bind DNA, modulating target gene transcription. There are three PPAR receptors, PPAR α, β/δ, and γ. Stimulation of macrophages with M2 stimuli such as IL-4 and IL-13 induces PPARγ and PPARβ/δ, which are necessary to stabilize M2 phenotype (132, 133).
PPARα activation has been linked to anti-inflammatory innate immune responses in macrophages. PPARα agonists have therapeutic activity in several inflammatory disease models [reviewed in Ref. (134), including EAE (135)]. Similarly, PPARα deficient mice suffer from worsened EAE disease (136). PPARα activation has also been shown to promote regulatory macrophages and mediate microbiota/gut homeostasis (137).
PPARγ activation has long been known to promote M2 polarization and suppress inflammatory cytokines in mouse and human macrophages (138–140). Activation of PPARγ with the flavonoid apigenin suppresses M1 macrophage inflammatory cytokine IL-1β and iNOS expression and promotes expression of alternatively activated phenotype markers by modulating NF-κB signaling (141). Similar suppression of LPS-induced inflammatory microglia has been reported via increased PPARγ signals (142). Accordingly PPARγ agonists suppress CNS neuroinflammation in the EAE model (143).
PPARβ/delta is thought to play an anti-inflammatory role, although immune activating effects have also been reported. For example, induction of M2 polarization by IL-4 and IL-13 is dependent on PPARβ/δ (144, 145). In addition, PPARβ/δ agonists suppress intestinal inflammation and EAE (146–148). However, studies in human monocyte-derived macrophages have shown that PPARβ/δ agonists both suppress inflammatory cytokines and suppress cytotoxic T cell inhibitory molecules PD-1L and IDO (149).
In summary, PPAR nuclear receptors are activated by resolution phase lipid mediators, promoting CNS macrophage and microglia phenotype switching toward resolution. Therefore, PPARs stand out as potential therapeutic targets in neuroinflammatory disease.
c-Myc
c-Myc is a transcription factor that modulates cellular survival and proliferation and metabolism, with important roles in angiogenesis, tumorigenesis, and immune responses (71, 150). c-MYC was found first to be induced by M2 stimuli such as IL-4 and IL-13 (71). In human macrophages, c-MYC translocates to the nucleus and controls the expression of half of the M2-associated genes (71). Human M2 markers SCARB, ALOX15, and MRC1 are directly promoted by c-MYC while others are indirectly induced (71). C-MYC also promotes STAT6 and PPAR-γ expression, further stabilizing M2 phenotype (71). In mice, c-Myc has also been found in human tumor-associated macrophages (TAM) (151) and transcriptional profiling of murine BMDM has demonstrated that c-Myc is also a selective marker of murine M2 macrophages (47). c-Myc expression correlated with detectable Egr2 protein, specifically labeling M(IL-4) but not M (LPS + IFN-γ) BMDM macrophages (47).
The exact role of c-MYC in M2 macrophages is not entirely clear. c-MYC may influence macrophage proliferation, which is consistent with the loss of proliferation observed during M1 macrophage stimulation (152). A pivotal role of c-MYC may be to metabolically program the macrophage. While HIF-1α induction in M1 macrophages promotes use of glucose via aerobic glycolysis to yield lactate and produce ROS, c-MYC expression in M2 macrophages may allow glycolytic activity necessary for M2 differentiation (153), providing additional sources of fuel for TCA cycle/oxidative phosphorylation (152). To summarize, c-MYC is gaining recognition as a mouse to human-conserved pro-M2 transcription factor. While much research is needed to understand c-MYC’s mechanistic actions, its connections to metabolic regulation of phenotype provide an intriguing area for exploration.
Interferon Regulatory Factors
Interferon regulatory factors are transcription factors that are activated in response to cytokines, via JAK/STAT signaling, and/or PAMPS and play important roles in innate and adaptive immunity. There are nine IRF family members, named IRF1-9 (154), which modulate macrophage phenotype. Our current understanding is that IRF1, 5 and 8 promote classical activation while IRF4 promotes alternative activation (155).
IRF5 is strongly induced by LPS and IFN-γ and GM-CSF stimuli and plays a prominent role in M1 activation. IRF5 interacts with RelA to bind target gene loci (156), resulting in enhanced IL-12 and IL-23 (157). IRF5 also promotes M1 polarization by association to MyD88 (158). IRF5 variants have been linked to MS (159), possibly via enhanced inflammatory activation. Suppression of IRF5 in EAE, via inhibition of the Aurora Kinase A, reduced inflammatory cytokines and improved clinical disease (160). Another IRF, IRF1, also contributes to M1 phenotype via induction of iNOS and IL-12 (161). IFN-γ stimulation of macrophages induces Batf2, which was shown be an M1-specific factor than interacts with IRF1 to induce Nos2, Tnf-α and Ccl5 (162). IRF8 contributes to M1 phenotype by activating IL-12 transcription in cooperation with IRF1 (163). The clinical relevance of IRF8 is highlighted by the link between IRF8 variants and MS (164, 165). Mice with a myeloid-specific deletion of IRF8 are resistant to EAE. IRF8 activates microglia and drives an IL-12 and IL-23 rich environment that promotes Th1 and Th17 responses (166). In contrast, IRF4 is instead a major mediator of M2 polarization (167). IRF4 inhibits pathogen sensing via suppression of MyD88 signaling (167, 168) and collaborates with histone deacetylase Jumanji D3 (Jmjd3) to promote expression of M2 genes Arg1, CD206, Ym1, and Fizz1.
Overall, IRFs essentially link cytokine and PAMP extracellular stimuli to signaling that enhances (IRF1, 5, 8) or suppresses (IRF4) inflammatory transcriptional programs. The association of IRFs to MS risk and EAE disease by impacting macrophages and microglia highlight the importance of IRFs and their potential as therapeutic targets.
Emerging Pathways
MicroRNA
MicroRNA are small (~22 nucleotides) RNAs that are regulated in response to inflammatory signals, modulating macrophage and microglia activation and phenotype. The biogenesis of miRNA starts with transcription of a primary (pri-miRNA) transcript that undergoes several processing steps (169). The first involves Drosha/DCGR8 complex cleavage to generate a double-stranded hairpin precursor termed pre-miRNA. This is followed by pre-miRNA export to the cytoplasm, where Dicer eliminates the hairpin yielding a miRNA–miRNA duplex. One miRNA strand is then loaded onto the RISC complex for binding to target mRNA transcript. miRNA generally suppresses target gene expression via either induction of mRNA degradation or inhibition of translation (170).
In the context of inflammatory stimuli, miRNA modulate their expression and macrophage polarization (see Table 2). The importance of miRNA in macrophage/microglia polarization in now well documented. miRNA such as miR-155, miR-146, miR-101, miR-21 and let-7 family are induced in response to inflammatory stimuli while miR-223, miR-125b, and the miR-23/27a/24-2 cluster are instead downregulated. Among these miRNAs, miR-155 stands out as a miRNA necessary for inflammatory phenotype. miR-146 is instead induced by inflammatory stimuli to dampen the inflammatory response. These two miRNAs and the pathways they control are therefore discussed in detail below, together with a summary of the contributions of other miRNAs to pathways that modulate macrophage and microglia phenotype.
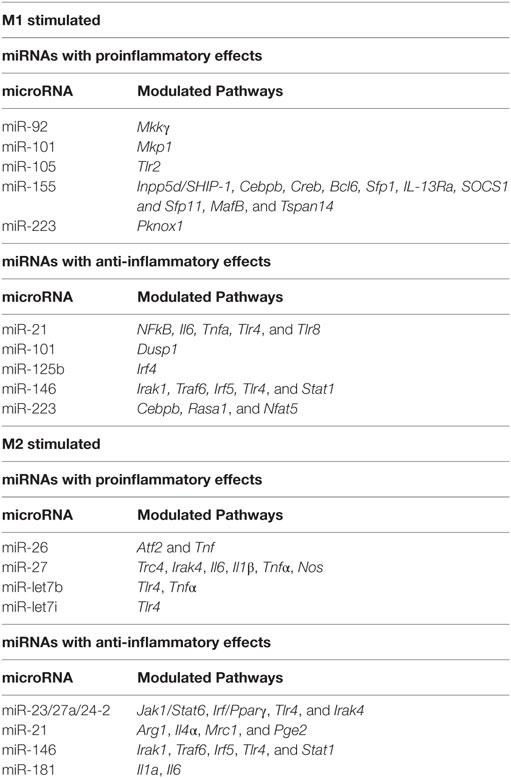
Table 2. Summary of miRNAs linked with M1 or M2 phenotype.
miR-155 is robustly induced in CNS inflammatory conditions such as MS and spinal cord injury (171–174). Several cells in the CNS, including macrophages (175), microglia (176), astrocytes (177), and neurons (178) may express miR-155. miR-155 is the most highly upregulated miRNA after exposure to a range of inflammatory stimuli including M [LPS + (IFN-γ)] conditions in both murine/human macrophages (96, 173, 179–183) and microglia (173, 176, 184). In contrast, exposure to alternatively activating stimuli such as IL-4 does not induce miR-155 (96, 185). The quick and swift induction of miR-155 suggests that miR-155 plays a crucial role in determining the classically activated macrophage phenotype. In support of this hypothesis, miR-155 delivery into macrophages or microglia via exosomes enhances inflammatory gene expression, including IL-6 and IL-12 (186). miR-155 in microglia also modulates phenotype via suppression of SOCS-1 and enhancement of NO and cytokine production (176). So, what are the global effects of miR-155 on M1 phenotype? Transcriptional profiling in miR-155 KO macrophages exposed to M(LPS + IFN-γ) conditions reveals that approximately half of the M(LPS + IFN-γ) signature is miR-155 dependent (96). These results indicate that miR-155 is required for full expression of inflammatory macrophage signature. Among the most impacted genes, inflammatory cytokines such as IL-1β, IL-6 and IL-12, inflammatory enzymes such as iNOS and costimulatory molecules such as CD86 and CD40 and adhesion and migration molecules such as CD49E and CCR7 stand out (96, 97). These data are consistent with clinical improvement of EAE, SCI and stroke in animals deficient in miR-155 or treated with miR-155 antisense oligonucleotide inhibitors (171, 187). Since miRNAs typically suppress targets rather than promote them, the inflammatory gene activation effects of miR-155 are expected to be mediated by suppression of deactivating genes. Validated miR-155 targets include transcripts of the Inpp5d, Cebpβ, Creb, Bcl6, Sfp1, IL-13Rα, Socs1, Sfp11, MafB, and Tspan14 genes. We found that the transcripts of target genes Inpp5d, Ptprj, MafB and Tspan14 inversely correlated with miR-155 (96). Targeting of Inpp5d by miR-155 promotes AKT signaling (188) in murine macrophages. In addition, CEPBβ has been shown to promote alternatively activated genes IL-10, IL-13Ra, arginase-1, RELMα (98) and targeting by miR-155 may suppress M2 phenotype. This is in contrast to the later finding that CEBPβ targeting by miR-223 is essential to prevent inflammatory macrophage development and colitis (189). MAF suppresses IL-12 and promotes IL-10 production in macrophages (190, 191) and its inactivation by miR-155 may be required to initiate the inflammatory gene expression program.
Although miR-146a/b are coinduced with miR-155 in response to M1 stimuli (192), they have opposite effects. While miR-155’s role is to release the brake on inflammation, miR-146 instead sets off a series of events that will eventually dampen inflammation. miR-146 targeting of TRAF6, IRAK1, TLR4, and STAT1 appear to mediate some of these effects by limiting responsiveness to inflammatory stimuli (100–102). Similar results have been observed in microglia, where miR-146 has been shown to promote M2 phenotype by targeting IRAK1/TRAF6 (103).
Other miRNAs modulated with macrophage/microglia phenotype include miR-124, miR-125b, miR-223, miR-101, miR-21, the let-7 family and the miR-23a/27a/24-2 cluster. Effects on phenotype appear to be achieved through targeting to JAK/STAT, NF-kB, or MAPK pathways and CEBP, PPAR, or IRF family transcription factors as further described below or in Table 2.
The CEBP family of transcription factors is targeted by miR-124, miR-223, and let-7 family miRNAs in macrophages and microglia. miR-124 and miR-223 target CEBPβ, which promotes macrophage development (98) and both inflammatory (193–196) and alternatively activating cytokines (197, 198). miRNA-124 is expressed in microglia, promoting a resting phenotype, but not in peripheral origin macrophages/monocytes (199). miR-124 treatment suppresses microglia and macrophage activation, T cell infiltration and clinical disease in EAE and other CNS inflammatory models, suggesting it may beneficial in MS (199, 200). miR-223 maintains a deactivated state through targeting of CEBPβ (189). miR-let-7c is associated with less inflammatory GM-CSF induced macrophages, where it suppresses the pro-inflammatory TF CEPBδ (201). miR-let-7i (202) and miR-let-7b (203) have also been shown to dampen inflammation by suppression of TLR4 expression.
Many miRNAs modulate macrophage phenotype by dampening or promoting the NF-kB pathway that proinflammatory stimuli induce. For example, miR-125b promotes activated microglia phenotype by suppressing A20/NF-κB signaling (204). miR-let-7f also targets the NF-κB negative regulator A20, resulting in enhanced IL-1β and TNF-α (205). In addition, let-7b also acts as a TLR7 agonist in microglia, activating TLR signaling and downstream NF-kB activity and leading to inflammatory microglia and neurodegeneration (206). Consistent with this, let-7b correlates with TNF-α production in tumor-associated macrophages (207).
In summary, miRNAs are now established as important regulators of macrophage and microglia that modulate neuroinflammation and neurodegeneration. miR-155 and miR-146 have robust pro- and anti-inflammatory roles, respectively, that may be therapeutically harnessed in MS and other neuroinflammatory diseases.
Metabolism
Metabolism is taking center stage in our understanding of pathways that modulate macrophage phenotype. While it was understood that inflammatory macrophages necessitate metabolic adaptations to offset high energy requirements, the realization that metabolism in fact determines inflammatory or regulatory phenotype is possibly a paradigm shift. Since metabolic pathway choice depends on enzyme activity, it provides interesting new (or repurposed) therapeutic strategies for inflammatory disease, further discussed in section 8. For a summary of how metabolic pathways differ in M1 and M2 macrophages, refer to Figure 2.
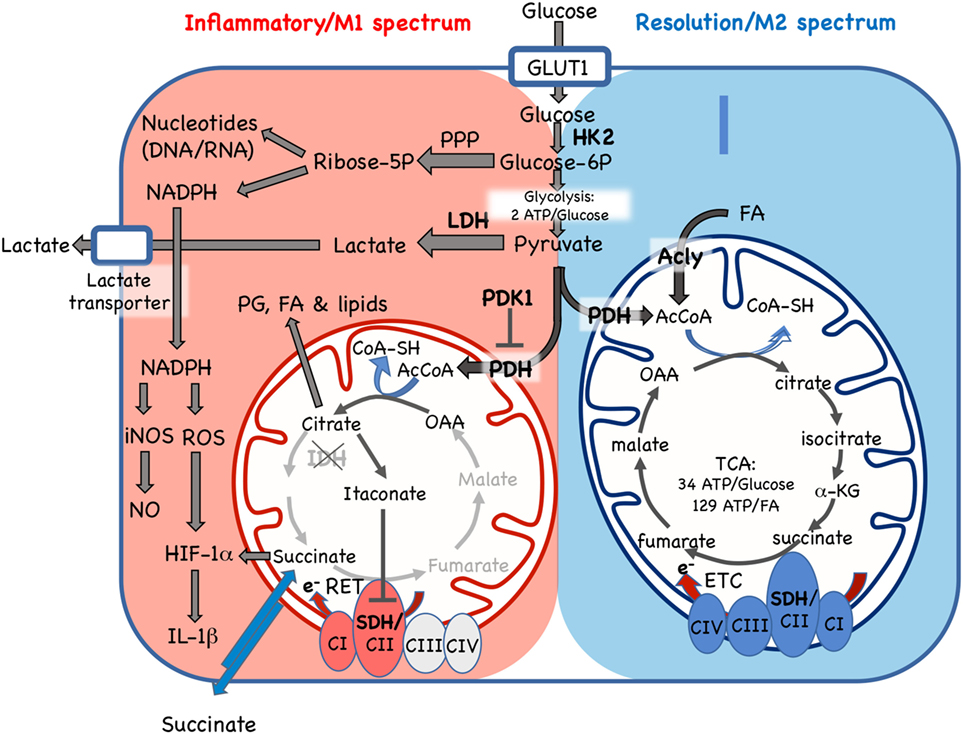
Figure 2. Cellular metabolic pathways driving mononuclear phagocyte inflammatory vs. resolution phenotype. Glucose is essential for both inflammatory and resolution macrophage phenotypes. In inflammatory macrophages, glucose is largely processed to yield lactate via aerobic glycolysis. Another major pathway in M1 spectrum macrophages is conversion to ribose-5P via the pentose phosphate pathway (PPP) for synthesis of nucleotides and NADPH, which supports nitric oxide (NO), reactive oxygen species (ROS), and IL-1β production. In resolution macrophages, the major fate of glucose is the TCA/Krebs cycle via pyruvate dehydrogenase (PDH)-catalyzed conversion of pyruvate to AcetylCoA (AcCoA). The TCA cycle in resolution macrophages is also fed by fatty acids (FA) via the ATP-citrate lyase (Acly) enzyme and promotes forward electron transport chain (ETC), from C1 to CIV, for ATP generation. In contrast, while some glucose enters the mitochondria in inflammatory macrophages, where it is converted to citrate, the TCA cycle is broken, with stops at the isocitrate dehydrogenase (IDH) and succinate dehydrogenase (SHD) steps. Citrate accumulation results in itaconate production, which inhibits SDH, and also promotes prostaglandin (PG), lipid and FA synthesis. In inflammatory macrophages, these blocks promote reverse electron transport chain (RET) from CII to CI. GLUT1, glucose transporter 1/SLC2A1; HK2, hexokinase 2; Glucose-6P, glucose-6-phosphate; LDH, lactose dehydrogenase; PHK1, pyruvate dehydrogenase kinase 1; AcCoa, acetyl coenzyme A; TCA, tricarboxylic acid/Krebs cycle; a-KG, a-ketoglutarate; CI-IV, ETC complexes I–IV, SDH, succinate dehydrogenase; OAA, oxaloacetate; e-, electrons; RET, reverse electron transport chain; iNOS, inducible nitric oxide synthase; HIF-1α, hypoxia inducible factor-1α. Enzymes are indicated by bold font.
Differences in amino acid metabolism have long been observed among macrophage phenotypes (208, 209). While M1 macrophages upregulate iNOS to convert arginine to NO for microbial killing, M2 macrophages induce arginase-1 and catabolize arginine to produce polyamines and proline for collagen biosynthesis. Additional differences in ATP generation (glycolysis vs. mitochondrial oxidative phosphorylation), pentose phosphate pathway activity, and TCA use have recently been demonstrated (210).
• ATP generation. Most tissues, including M2 macrophages, use mitochondrial oxidative phosphorylation (34 ATP/glucose or 129/palmitic acid) as a source of energy. In contrast, M1 macrophages rely on aerobic glycolysis to lactate (2 ATP/glucose) for energy generation (211). Aerobic glycolysis is also known as Warburg metabolism and this phenotype has also been observed in cancer cells (212).
Why a lower ATP output pathway would be chosen by M1 macrophages is intriguing. Glycolysis may provide rapid ATP production. However, new evidence that NO production in M1 macrophages strongly inhibits oxidative phosphorylation by impairing the electron transport chain (213) provides an alternative explanation for the need to use an alternative pathway for ATP generation.
While M1 macrophages have increased glycolytic activity as compared to M2 macrophages, M2 macrophages also depend to some extent on access to glucose and its oxidation via glycolysis. Although the TCA cycle in M2 macrophages was thought to be mostly fueled by fatty acids (210), recent work by has shown that an active glycolysis pathway is essential for TCA/oxidative phosphorylation and M2 phenotype (153).
• Pentose phosphate pathway (PPP). The PPP directs some glucose-6P away from the glycolysis pathway and into generation of ribose-6P and derivatives. This pathway yields nucleotides to support DNA replication and RNA transcription and NADPH for ROS and NO generation. In addition to increased glycolysis, high PPP activity is characteristic of M1 macrophages (214).
• Krebs/TCA cycle. M2 macrophages rely almost exclusively on ATP generation via oxidative phosphorylation coupled to an intact TCA cycle (213, 215, 216). In contrast, in M1 macrophages the TCA cycle is “broken” at two steps: citrate to α-ketoglutarate and succinate to fumarate (217, 218). Reduced isocitrate dehydrogenase activity leads to citrate accumulation in M1 macrophages. Citrate supports M1 phenotype by promoting FAS and membrane biosynthesis, prostaglandin and itaconate production. Itaconate inhibits succinate dehydrogenase causing the second break in the cycle and succinate accumulation (219). Succinate stabilizes HIF-1α and promotes IL-1β production in LPS stimulated macrophages (217, 218). The second break in the TCA cycle is linked to a reversal in electron transport chain direction that fuels an increase in mitochondrial membrane potential and supports classic M1 NO and ROS generation (217).
In summary, M1 and M2 macrophages use opposing metabolic pathways to fulfill energy and biosynthetic requirements. M1 macrophages favor glycolysis to lactate and PPP pathways to provide energy and support NO and ROS generation. M2 macrophages instead rely on TCA cycle for ATP generation. This metabolic dichotomy is not a consequence of M1 or M2 phenotype, but rather a requirement for either phenotype, therefore providing exciting opportunities for therapeutic targeting.
Receptor-Tyrosine Kinases
Receptor-tyrosine kinases have been proposed to fine-tune macrophage function in immunity and tissue homeostasis. Macrophages are known to express RTKs within three families of RTKs, namely platelet-derived growth factor receptor (PDGFR), the AXL/TYRO3/MERTK family, and the RON superfamily. Colony stimulation factor receptor 1 (CSF1R) is involved in macrophage development and is a member of the PDGFR superfamily (220). The PDGFR family receptors are characterized by 5 Immunoglobulin (Ig)-family domains and a kinase domain. The TAM (TYRO3/AXL/MERTK) family is instead characterized by two Ig-like domains, two fibronectin III repeats and a Kinase domain (221, 222). Finally, the human RON receptor (STK in mouse) is a member of the MET family of RTKs (223, 224). Ligation of RTKs to ligands, such as M-CSF to CSF1R, apoptotic cell phosphatidyl serine (PtdSer) via grown arrest specific 6 (Gas6) and Protein S bridging to TAM family RTKs and macrophage-stimulating protein (MSP) to STK results in activation of kinase activity (225–227).
The active form of STK ligand MSP is generated via the coagulation cascade (228) and has a crucial role in the response of macrophages to inflammatory cytokines and LPS. MSP dampens NO and PGE2 production via suppression of iNOS and COX-2 expression (229–231). TAM receptors mediate apoptotic cell removal after PtdSer is recognized via Gas6 or Protein S, activating MerTK and reducing TNF-α and LPS responsiveness (232). NO activity, IL-12 production and MHCII expression are also controlled by MSP: MSP exposure prior to LPS + IFN-γ activation inhibits these signature M1 factors via arginase-1 induction (233). Consistent with the proresolving role of RTKs, macrophages from Tyro3, MerTK, and Axl triple KO mice display enhanced IL-12, MHCII, and costimulatory molecules in response to LPS (234).
The AXL RTK is also induced in mouse and human macrophages by type I IFNs and TLR3 stimulation (235). This induction may signal enhanced apoptotic cell removal needs during inflammation. The Rothlin group added a relevant layer to the physiologic role of TAM RTKs AXL and MERTK in resolution of immune responses to infection and promotion of tissue repair (60). They found that including apoptotic cell ligands for TAM RTKs strongly enhanced the expression of anti-inflammatory and tissue repair genes, including RELMα, CHI3L3, FN1, and EAR2 in response M2 (IL-4) stimuli (60) in vitro. Such signaling was essential to dampen inflammation and allow tissue repair in thioglycollate and helminth infection models. Their results implicate that apoptotic cell sensing by AXL and MERTK in the presence of IL-4 responses drives anti-inflammatory and tissue repair programs in macrophages. It is interesting to speculate whether the failure of most interventions to enhance M2 phenotype during injury (236, 237) may stem from deficiencies in these pathways. If so, small molecule-mediated stimulation of these pathways during chronic CNS inflammation may be a promising therapeutic strategy to promote proregenerative macrophages/microglia.
In summary, RTK activity is largely stimulated by resolution mediators, such as MSP and apoptotic neutrophil PtdlSer in late stages of acute inflammation. These signals effectively suppress inflammatory responses and promote and amplify a resolution macrophage phenotype switch. These findings highlight a physiologically relevant pathway for inflammation resolution that may be therapeutically harnessed in neuroinflammatory disease.
Therapeutic Strategies for Macrophage/Microglia Phenotype Reprogramming
Increased understanding of the molecular pathways that promote inflammatory and resolution phenotypes in macrophages and microglia provides therapeutic targets for inflammatory diseases, including the autoimmune disease MS. Due to the increasing importance of metabolic reprogramming in macrophage phenotype, we will focus our discussion on the current understanding of how available metabolic reprogramming drugs may impact macrophage phenotype and MS.
Dichloroacetate (DCA) is an inhibitor of PDK1, a kinase that in turn suppresses PDH. DCA treatment therefore increased PDH activity, shifting cellular metabolism toward the TCA cycle/oxidative phosphorylation and promoting a proregenerative M2 resolution phenotype (238). Consistent with a shift from M1 to M2 phenotype in macrophages and microglia, treatment with DCA suppressed clinical disease scores and T cell infiltration in the EAE model of MS (239).
Dimethylfumarate (DMF) is an approved oral drug for MS treatment that has been shown to efficiently reduce relapse rate and disease progression (240). DMF is thought to exert its therapeutic effects via activation of the Nrf2 pathway and induction of the antioxidant response. Could DMF’s metabolite monomethyl fumarate enter the TCA cycle at the fumarate step, thereby modulating oxidative phosphorylation? Consistent with this scenario, increased TCA cycle intermediates malate, fumarate and succinate are observed in DMF-treated oligodendrocytes (241). However, it is unknown whether similar effects occur in macrophages and microglia. Microglia and myeloid cell pretreatment with DMF does however reduce NO and inflammatory cytokine production (242, 243), although these effects have not been recapitulated in DMF-treated EAE mice (244). Similarly, increased superoxide generation has been observed in fumarate-treated monocytes (245), indicating that DMF’s effects may depend on timing and context.
Many of the current drugs targeting metabolic pathways are currently approved for type II diabetes and metabolic syndrome therapy. One of the first drugs for type II diabetes, the biguanide family metformin, inhibits AMPK and complex I of the electron transport chain (ETC) (246). Treatment of LPS-activated macrophages with metformin suppresses NO and IL-1β production while increasing IL-10 (247). This effect was due to inhibition of complex I, which is necessary for respiratory electron chain in M1 macrophages (247). In vivo metformin treatment reduced inflammatory cytokine production, reduced Th17 responses and enhanced Tregs, overall ameliorating EAE disease (248, 249). The common use of metformin for metabolic syndrome has allowed to test whether it is beneficial in MS. In an open-label study of 50 obese MS patients, the 20 patients who received metformin had decreased new or enlarging T2 and gadolinium-enhancing lesions (250, 251). These clinical effects were accompanied by significant decreases in inflammatory Th1 and Th17 cells but significant increases in Tregs in metformin-treated patients (250).
Pioglitazone and rosiglitazone are antidiabetic drugs from the thiazolidinedione family that act as full agonists of PPARγ, modulating multiple metabolic processes, particularly lipid and glucose metabolism (130, 252). A smaller number of patients in the metformin study above were treated with pioglitazone and similarly benefited from reduced lesions and shifts from inflammatory to regulatory T cell responses (250). These clinical effects are consistent with previously reported suppression of microglial activation and clinical disease in the EAE model (253, 254).
Concluding Remarks
Recent studies have provided us with a more thorough and insightful understanding about how steady-state and acute environmental signals are integrated by macrophages and their microglial counterparts to maintain optimal neurologic function, eliminate infection and resolve injury or inflammation. Besides PRR-induced Jak2/NF-κB signaling, Notch receptor signaling and PI3K/AKT2/miR-155 signaling contributes to M1 phenotype. In contrast, PI3K/AKT1/miR-146 and apoptotic cell-induced RTK signaling promotes M2 phenotype. These effects are achieved through IRF-mediated control of gene expression, leading to changes in how cellular nutrients are used via metabolic pathways. M1 macrophages turn to aerobic glycolysis/Warburg metabolism and high PPP activity for energy production and simultaneously promote NO, ROS, and IL-1β expression. Interestingly, NO inhibits oxidative phosphorylation, which may explain the need for TCA-independent sources of energy in M1 macrophages. In contrast, M2 macrophage polarization and function is entirely dependent on TCA/oxidative phosphorylation, although glucose is also essential for M2 phenotype. Links between classic pathways known to affect macrophage phenotype and metabolism are starting to emerge. The PI3K/AKT pathway activates mTOR signaling, a major regulator of glucose metabolic pathways in the cell. Similarly, PPARγ is another major regulator of macrophage phenotype and lipid/glucose metabolism. Finally, c-MYC is an important mediator of M2 phenotype in murine and human macrophages that modulates glucose fuel utilization. The requirement of active metabolic pathways for specific macrophage phenotypes constitutes a paradigm shift in the field, away from a mere supporting role. Perhaps more importantly, it provides an opportunity for therapeutic modulation at the step where signaling pathways converge to determine phenotype. Novel and repurposed metabolic reprogramming drugs may provide promising alternative therapeutic strategies in MS and other neuroinflammatory disorders. So far, a study has shown excellent clinical MS responsiveness using metformin and pioglitazone in obese MS patients. Further studies are required to evaluate the efficacy of these treatments in the general MS population and understand how they impact macrophages and microglia phenotype. The field is ripe to address these questions and exciting basic knowledge and therapeutic opportunities lie ahead.
Author Contributions
All authors: conception of idea, review of the literature, manuscript writing and editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MA is supported by NIH NIAID R01AI121405, NIH NIAID 1R21AI127354, and Ohio State University College of Medicine start-up funds. JD is supported by the Undergraduate Research Grant Program, administered by Northwestern University’s Office of Undergraduate Research.
References
1. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid cells in the central nervous system. Immunity (2017) 46:943–56. doi:10.1016/j.immuni.2017.06.007
2. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi:10.1126/science.1194637
3. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci (2013) 16:273–80. doi:10.1038/nn.3318
4. Gomez Perdiguero E, Schulz C, Geissmann F. Development and homeostasis of “resident” myeloid cells: the case of the microglia. Glia (2013) 61:112–20. doi:10.1002/glia.22393
5. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643
6. Gow DJ, Sester DP, Hume DA. CSF-1, IGF-1, and the control of postnatal growth and development. J Leukoc Biol (2010) 88:475–81. doi:10.1189/jlb.0310158
7. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol (2009) 9:259–70. doi:10.1038/nri2528
8. Waisman A, Ginhoux F, Greter M, Bruttger J. Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol (2015) 36:625–36. doi:10.1016/j.it.2015.08.005
9. Benoit M, Desnues B, Mege J-L. Macrophage polarization in bacterial infections. J Immunol (2008) 181:3733–9. doi:10.4049/jimmunol.181.6.3733
10. Andersson U, Wang H, Palmblad K, Aveberger A-C, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (Hmg-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med (2000) 192:565–70. doi:10.1084/jem.192.4.565
11. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci (2005) 8:752–8. doi:10.1038/nn1472
12. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308:1314–8. doi:10.1126/science.1110647
13. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13:1118–28. doi:10.1038/ni.2419
14. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi:10.1016/j.immuni.2014.01.006
15. Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol (2015) 36:605–13. doi:10.1016/j.it.2015.08.008
16. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333:1456–8. doi:10.1126/science.1202529
17. Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology (2015) 144:541–8. doi:10.1111/imm.12451
18. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity (2013) 39:599–610. doi:10.1016/j.immuni.2013.08.007
19. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44:439–49. doi:10.1016/j.immuni.2016.02.024
20. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi:10.1016/j.immuni.2015.03.011
21. Sheng J, Ruedl C, Karjalainen K. Fetal HSCs versus EMP2s. Immunity (2015) 43:1025. doi:10.1016/j.immuni.2015.11.023
22. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51. doi:10.1038/nature13989
23. Wong K, Noubade R, Manzanillo P, Ota N, Foreman O, Hackney JA, et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol (2017) 18:633–41. doi:10.1038/ni.3743
24. Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol (1995) 154:4309–21.
25. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A (2016) 113:E1738–46. doi:10.1073/pnas.1525528113
26. Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol (2012) 188:29–36. doi:10.4049/jimmunol.1100421
27. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci (2006) 9:917–24. doi:10.1038/nn1715
28. Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol (2000) 20:4106–14. doi:10.1128/MCB.20.11.4106-4114.2000
29. O’Koren EG, Mathew R, Saban DR. Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci Rep (2016) 6:20636. doi:10.1038/srep20636
30. Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal ‘On’ and “Off” signals control microglia. Trends Neurosci (2007) 30:596–602. doi:10.1016/j.tins.2007.08.007
31. Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, et al. Regulation of adaptive immunity by the fractalkine receptor during autoimmune inflammation. J Immunol (2013) 191:1063–72. doi:10.4049/jimmunol.1300040
32. Freria CM, Hall JCE, Wei P, Guan Z, McTigue DM, Popovich PG. Deletion of the fractalkine receptor, CX3CR1, improves endogenous repair, axon sprouting, and synaptogenesis after spinal cord injury in mice. J Neurosci (2017) 37:3568–87. doi:10.1523/JNEUROSCI.2841-16.2017
33. Fabriek BO, Møller HJ, Vloet RPM, van Winsen LM, Hanemaaijer R, Teunissen CE, et al. Proteolytic shedding of the macrophage scavenger receptor CD163 in multiple sclerosis. J Neuroimmunol (2007) 187:179–86. doi:10.1016/j.jneuroim.2007.04.016
34. Stilund M, Reuschlein A-K, Christensen T, Møller HJ, Rasmussen PV, Petersen T. Soluble CD163 as a marker of macrophage activity in newly diagnosed patients with multiple sclerosis. PLoS One (2014) 9:e98588. doi:10.1371/journal.pone.0098588
35. Stilund M, Gjelstrup MC, Petersen T, Møller HJ, Rasmussen PV, Christensen T. Biomarkers of inflammation and axonal degeneration/damage in patients with newly diagnosed multiple sclerosis: contributions of the soluble CD163 CSF/serum ratio to a biomarker panel. PLoS One (2015) 10:e0119681. doi:10.1371/journal.pone.0119681
36. Bogie JF, Boelen E, Louagie E, Delputte P, Elewaut D, van Horssen J, et al. CD169 is a marker for highly pathogenic phagocytes in multiple sclerosis. Mult Scler (2017) 101:1352458517698759. doi:10.1177/1352458517698759
37. Satoh J-I, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology (2016) 36:39–49. doi:10.1111/neup.12235
38. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain (2017) 140:1900–13. doi:10.1093/brain/awx113
39. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci (2014) 17:131–43. doi:10.1038/nn.3599
40. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81:1–5. doi:10.1189/jlb.0306164
41. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
42. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
43. Jaguin M, Houlbert N, Fardel O, Lecureur V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol (2013) 281:51–61. doi:10.1016/j.cellimm.2013.01.010
44. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A (1997) 94:5243–8. doi:10.1073/pnas.94.10.5243
45. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology (2014) 141:96–110. doi:10.1111/imm.12173
46. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
47. Jablonski KA, Amici SA, Webb LM, de Dios Ruiz-Rosado J, Popovich PG, Partida-Sánchez S, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS One (2015) 10:e0145342. doi:10.1371/journal.pone.0145342
48. Herrmann MM, Barth S, Greve B, Schumann KM, Bartels A, Weissert R. Identification of gene expression patterns crucially involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Dis Model Mech (2016) 9:1211–20. doi:10.1242/dmm.025536
49. Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med (2001) 7:1209–16. doi:10.1038/nm1101-1209
50. Matalonga J, Glaria E, Bresque M, Escande C, Carbó JM, Kiefer K, et al. The nuclear receptor LXR limits bacterial infection of host macrophages through a mechanism that impacts cellular NAD metabolism. Cell Rep (2017) 18:1241–55. doi:10.1016/j.celrep.2017.01.007
51. Hanisch U-K, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci (2007) 10:1387–94. doi:10.1038/nn1997
52. Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol (2009) 4:389–98. doi:10.1007/s11481-009-9171-5
53. Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets (2010) 9:174–91. doi:10.2174/187152710791012053
54. Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) (2012) 60:251–66. doi:10.1007/s00005-012-0181-2
55. Boche D, Perry VH, Nicoll JAR. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol (2013) 39:3–18. doi:10.1111/nan.12011
56. Mayo L, Jacob-Hirsch J, Amariglio N, Rechavi G, Moutin M-J, Lund FE, et al. Dual role of CD38 in microglial activation and activation-induced cell death. J Immunol (2008) 181:92–103. doi:10.4049/jimmunol.181.1.92
57. Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot M-C, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia (2012) 60:717–27. doi:10.1002/glia.22298
58. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature (2014) 510:92–101. doi:10.1038/nature13479
59. Chiurchiù V, Maccarrone M. Bioactive lipids as modulators of immunity, inflammation and emotions. Curr Opin Pharmacol (2016) 29:54–62. doi:10.1016/j.coph.2016.06.005
60. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science (2017) 356:1072–6. doi:10.1126/science.aai8132
61. Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia (2010) 58:1017–30. doi:10.1002/glia.20983
62. Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggström JZ, Petasis NA, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J (2013) 27:2573–83. doi:10.1096/fj.13-227728
63. Koh TJ, DiPietro LA. Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med (2011) 13:e23. doi:10.1017/S1462399411001943
64. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi:10.1016/j.it.2004.09.015
65. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio (2013) 4:e264–213. doi:10.1128/mBio.00264-13
66. Brancato SK, Albina JE. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am J Pathol (2011) 178:19–25. doi:10.1016/j.ajpath.2010.08.003
67. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32:593–604. doi:10.1016/j.immuni.2010.05.007
68. Raes G, De Baetselier P, Noël W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol (2002) 71:597–602.
69. Duluc D, Delneste Y, Tan F, Moles M-P, Grimaud L, Lenoir J, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood (2007) 110:4319–30. doi:10.1182/blood-2007-02-072587
70. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm (2015) 2015:816460. doi:10.1155/2015/816460
71. Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood (2012) 119:411–21. doi:10.1182/blood-2011-02-339911
72. Chávez-Galán L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol (2015) 6:263. doi:10.3389/fimmu.2015.00263
73. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol (2012) 189:3508–20. doi:10.4049/jimmunol.1200662
74. Yu Y-RA, Hotten DF, Malakhau Y, Volker E, Ghio AJ, Noble PW, et al. Flow cytometric analysis of myeloid cells in human blood, bronchoalveolar lavage, and lung tissues. Am J Respir Cell Mol Biol (2016) 54:13–24. doi:10.1165/rcmb.2015-0146OC
75. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci (2009) 29:13435–44. doi:10.1523/JNEUROSCI.3257-09.2009
76. Barnett MH, Henderson APD, Prineas JW. The macrophage in MS: just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult Scler (2006) 12:121–32. doi:10.1191/135248506ms1304rr
77. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, Fatimy El R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity (2017) 47:566–81.e9. doi:10.1016/j.immuni.2017.08.008
78. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi:10.1038/nature21029
79. Bogie JFJ, Jorissen W, Mailleux J, Nijland PG, Zelcer N, Vanmierlo T, et al. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol Commun (2013) 1:43. doi:10.1186/2051-5960-1-43
80. Raine CS. Multiple sclerosis: the resolving lesion revealed. J Neuroimmunol (2017) 304:2–6. doi:10.1016/j.jneuroim.2016.05.021
81. Miron VE, Boyd A, Zhao J-W, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci (2013) 16:1211–8. doi:10.1038/nn.3469
82. Tierney JB, Kharkrang M, La Flamme AC. Type II-activated macrophages suppress the development of experimental autoimmune encephalomyelitis. Immunol Cell Biol (2009) 87:235–40. doi:10.1038/icb.2008.99
83. Jiang H-R, Milovanović M, Allan D, Niedbala W, Besnard A-G, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur J Immunol (2012) 42:1804–14. doi:10.1002/eji.201141947
84. Terrazas C, de Dios Ruiz-Rosado J, Amici SA, Jablonski KA, Martinez-Saucedo D, Webb LM, et al. Helminth-induced Ly6C(hi) monocyte-derived alternatively activated macrophages suppress experimental autoimmune encephalomyelitis. Sci Rep (2017) 7:40814. doi:10.1038/srep40814
85. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198:1006–14. doi:10.4049/jimmunol.1601515
86. López-Peláez M, Soria-Castro I, Boscá L, Fernández M, Alemany S. Cot/tpl2 activity is required for TLR-induced activation of the Akt p70 S6k pathway in macrophages: implications for NO synthase 2 expression. Eur J Immunol (2011) 41:1733–41. doi:10.1002/eji.201041101
87. Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol (2008) 180:4218–26. doi:10.4049/jimmunol.180.6.4218
88. Díaz-Guerra MJ, Castrillo A, Martín-Sanz P, Boscá L. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Immunol (1999) 162:6184–90.
89. Polumuri SK, Toshchakov VY, Vogel SN. Role of phosphatidylinositol-3 kinase in transcriptional regulation of TLR-induced IL-12 and IL-10 by Fc gamma receptor ligation in murine macrophages. J Immunol (2007) 179:236–46. doi:10.4049/jimmunol.179.1.236
90. Pengal RA, Ganesan LP, Wei G, Fang H, Ostrowski MC, Tridandapani S. Lipopolysaccharide-induced production of interleukin-10 is promoted by the serine/threonine kinase Akt. Mol Immunol (2006) 43:1557–64. doi:10.1016/j.molimm.2005.09.022
91. Arranz A, Doxaki C, Vergadi E, la Torre de YM, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A (2012) 109:9517–22. doi:10.1073/pnas.1119038109
92. Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, et al. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A (2008) 105:8292–7. doi:10.1073/pnas.0707761105
93. Vergadi E, Vaporidi K, Theodorakis EE, Doxaki C, Lagoudaki E, Ieronymaki E, et al. Akt2 deficiency protects from acute lung injury via alternative macrophage activation and miR-146a induction in mice. J Immunol (2014) 192:394–406. doi:10.4049/jimmunol.1300959
94. Kuijl C, Savage NDL, Marsman M, Tuin AW, Janssen L, Egan DA, et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature (2007) 450:725–30. doi:10.1038/nature06345
95. Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity (2009) 31:220–31. doi:10.1016/j.immuni.2009.06.024
96. Jablonski KA, Gaudet AD, Amici SA, Popovich PG, Guerau-de-Arellano M. Control of the inflammatory macrophage transcriptional signature by miR-155. PLoS One (2016) 11:e0159724. doi:10.1371/journal.pone.0159724
97. Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang C-Y, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol (2012) 4:341–3. doi:10.1093/jmcb/mjs044
98. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A (2009) 106:17475–80. doi:10.1073/pnas.0908641106
99. Nandan D, Camargo de Oliveira C, Moeenrezakhanlou A, Lopez M, Silverman JM, Subek J, et al. Myeloid cell IL-10 production in response to Leishmania involves inactivation of glycogen synthase kinase-3β downstream of phosphatidylinositol-3 kinase. J Immunol (2012) 188:367–78. doi:10.4049/jimmunol.1100076
100. Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, et al. Attenuation of cardiac dysfunction in polymicrobial sepsis by microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J Immunol (2015) 195:672–82. doi:10.4049/jimmunol.1403155
101. Li X, Ji Z, Li S, Sun Y-N, Liu J, Liu Y, et al. miR-146a-5p antagonized AGEs- and P.g-LPS-induced ABCA1 and ABCG1 dysregulation in macrophages via IRAK-1 downregulation. Inflammation (2015) 38:1761–8. doi:10.1007/s10753-015-0153-x
102. He X, Tang R, Sun Y, Wang Y-G, Zhen K-Y, Zhang D-M, et al. MicroR-146 blocks the activation of M1 macrophage by targeting signal transducer and activator of transcription 1 in hepatic schistosomiasis. EBioMedicine (2016) 13:339–47. doi:10.1016/j.ebiom.2016.10.024
103. Sharma N, Verma R, Kumawat KL, Basu A, Singh SK. miR-146a suppresses cellular immune response during Japanese encephalitis virus JaOArS982 strain infection in human microglial cells. J Neuroinflammation (2015) 12:30. doi:10.1186/s12974-015-0249-0
104. Tsiperson V, Gruber RC, Goldberg MF, Jordan A, Weinger JG, Macian F, et al. Suppression of inflammatory responses during myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis is regulated by AKT3 signaling. J Immunol (2013) 190:1528–39. doi:10.4049/jimmunol.1201387
105. Fang C, Yu J, Luo Y, Chen S, Wang W, Zhao C, et al. Tsc1 is a critical regulator of macrophage survival and function. Cell Physiol Biochem (2015) 36:1406–18. doi:10.1159/000430306
106. Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity (2008) 29:565–77. doi:10.1016/j.immuni.2008.08.012
107. Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem (2011) 286:44295–305. doi:10.1074/jbc.M111.258053
108. Festuccia WT, Pouliot P, Bakan I, Sabatini DM, Laplante M. Myeloid-specific Rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS One (2014) 9:e95432. doi:10.1371/journal.pone.0095432
109. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun (2013) 4:2834. doi:10.1038/ncomms3834
110. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife (2016) 5:R106. doi:10.7554/eLife.11612
111. Pan H, O’Brien TF, Zhang P, Zhong X-P. The role of tuberous sclerosis complex 1 in regulating innate immunity. J Immunol (2012) 188:3658–66. doi:10.4049/jimmunol.1102187
112. Mercalli A, Calavita I, Dugnani E, Citro A, Cantarelli E, Nano R, et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology (2013) 140:179–90. doi:10.1111/imm.12126
113. Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol (2013) 13:427–37. doi:10.1038/nri3445
114. Nakano T, Fukuda D, Koga J-I, Aikawa M. Delta-like ligand 4-notch signaling in macrophage activation. Arterioscler Thromb Vasc Biol (2016) 36:2038–47. doi:10.1161/ATVBAHA.116.306926
115. Fung E, Tang S-MT, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation (2007) 115:2948–56. doi:10.1161/CIRCULATIONAHA.106.675462
116. Monsalve E, Ruiz-García A, Baladrón V, Ruiz-Hidalgo MJ, Sánchez-Solana B, Rivero S, et al. Notch1 upregulates LPS-induced macrophage activation by increasing NF-kappaB activity. Eur J Immunol (2009) 39:2556–70. doi:10.1002/eji.200838722
117. Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol (2012) 13:642–50. doi:10.1038/ni.2304
118. Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SHE, et al. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol (2008) 38:174–83. doi:10.1002/eji.200636999
119. Monsalve E, Pérez MA, Rubio A, Ruiz-Hidalgo MJ, Baladrón V, García-Ramírez JJ, et al. Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol (2006) 176:5362–73. doi:10.4049/jimmunol.176.9.5362
120. Boonyatecha N, Sangphech N, Wongchana W, Kueanjinda P, Palaga T. Involvement of Notch signaling pathway in regulating IL-12 expression via c-Rel in activated macrophages. Mol Immunol (2012) 51:255–62. doi:10.1016/j.molimm.2012.03.017
121. Wang Y, Huang J-W, Calses P, Kemp CJ, Taniguchi T. miR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res (2012) 72:4037–46. doi:10.1158/0008-5472.CAN-12-0103
122. Outtz HH, Wu JK, Wang X, Kitajewski J. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol (2010) 185:4363–73. doi:10.4049/jimmunol.1000720
123. Singla RD, Wang J, Singla DK. Regulation of Notch 1 signaling in THP-1 cells enhances M2 macrophage differentiation. Am J Physiol Heart Circ Physiol (2014) 307:H1634–42. doi:10.1152/ajpheart.00896.2013
124. Xu J, Chi F, Guo T, Punj V, Lee WNP, French SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest (2015) 125:1579–90. doi:10.1172/JCI76468
125. Cao Q, Li P, Lu J, Dheen ST, Kaur C, Ling E-A. Nuclear factor-κB/p65 responds to changes in the Notch signaling pathway in murine BV-2 cells and in amoeboid microglia in postnatal rats treated with the γ-secretase complex blocker DAPT. J Neurosci Res (2010) 88:2701–14. doi:10.1002/jnr.22429
126. Wei Z, Chigurupati S, Arumugam TV, Jo D-G, Li H, Chan SL. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke (2011) 42:2589–94. doi:10.1161/STROKEAHA.111.614834
127. Yao L, Kan EM, Kaur C, Dheen ST, Hao A, Lu J, et al. Notch-1 signaling regulates microglia activation via NF-κB pathway after hypoxic exposure in vivo and in vitro. PLoS One (2013) 8:e78439. doi:10.1371/journal.pone.0078439
128. Wongchana W, Lawlor RG, Osborne BA, Palaga T. Impact of Notch1 deletion in macrophages on proinflammatory cytokine production and the outcome of experimental autoimmune encephalomyelitis. J Immunol (2015) 195:5337–46. doi:10.4049/jimmunol.1401770
129. Wang CYY, Wei Q, Han I, Sato S, Ghanbari-Azarnier R, Whetstone H, et al. Hedgehog and Notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer Res (2012) 72:1013–22. doi:10.1158/0008-5472.CAN-11-2531
130. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International union of pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev (2006) 58:726–41. doi:10.1124/pr.58.4.5
131. Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res (2000) 49:497–505. doi:10.1007/s000110050622
132. Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, et al. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature (1999) 400:378–82. doi:10.1038/22572
133. Coste A, Dubourdeau M, Linas MD, Cassaing S, Lepert J-C, Balard P, et al. PPARgamma promotes mannose receptor gene expression in murine macrophages and contributes to the induction of this receptor by IL-13. Immunity (2003) 19:329–39. doi:10.1016/S1074-7613(03)00229-2
134. Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol (2007) 28:551–8. doi:10.1016/j.it.2007.09.003
135. Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, et al. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J Immunol (2004) 172:5790–8. doi:10.4049/jimmunol.172.9.5790
136. Dunn SE, Ousman SS, Sobel RA, Zuniga L, Baranzini SE, Youssef S, et al. Peroxisome proliferator-activated receptor (PPAR)alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J Exp Med (2007) 204:321–30. doi:10.1084/jem.20061839
137. Manoharan I, Suryawanshi A, Hong Y, Ranganathan P, Shanmugam A, Ahmad S, et al. Homeostatic PPARα signaling limits inflammatory responses to commensal microbiota in the intestine. J Immunol (2016) 196:4739–49. doi:10.4049/jimmunol.1501489
138. Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab (2007) 6:137–43. doi:10.1016/j.cmet.2007.06.010
139. Dasu MR, Park S, Devaraj S, Jialal I. Pioglitazone inhibits toll-like receptor expression and activity in human monocytes and db/db mice. Endocrinology (2009) 150:3457–64. doi:10.1210/en.2008-1757
140. Yang W-S, Jeng C-Y, Wu T-J, Tanaka S, Funahashi T, Matsuzawa Y, et al. Synthetic peroxisome proliferator-activated receptor-gamma agonist, rosiglitazone, increases plasma levels of adiponectin in type 2 diabetic patients. Diabetes Care (2002) 25:376–80. doi:10.2337/diacare.25.2.376
141. Feng X, Weng D, Zhou F, Owen YD, Qin H, Zhao J, et al. Activation of PPARγ by a natural flavonoid modulator, apigenin ameliorates obesity-related inflammation via regulation of macrophage polarization. EBioMedicine (2016) 9:61–76. doi:10.1016/j.ebiom.2016.06.017
142. Han Q, Yuan Q, Meng X, Huo J, Bao Y, Xie G. 6-Shogaol attenuates LPS-induced inflammation in BV2 microglia cells by activating PPAR-γ. Oncotarget (2017) 8:42001–6. doi:10.18632/oncotarget.16719
143. Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res (2003) 71:315–25. doi:10.1002/jnr.10501
144. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab (2008) 7:485–95. doi:10.1016/j.cmet.2008.04.002
145. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab (2008) 7:496–507. doi:10.1016/j.cmet.2008.04.003
146. Polak PE, Kalinin S, Russo Dello C, Gavrilyuk V, Sharp A, Peters JM, et al. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol (2005) 168:65–75. doi:10.1016/j.jneuroim.2005.07.006
147. Bassaganya-Riera J, DiGuardo M, Climent M, Vives C, Carbo A, Jouni ZE, et al. Activation of PPARγ and δ by dietary punicic acid ameliorates intestinal inflammation in mice. Br J Nutr (2011) 106:878–86. doi:10.1017/S0007114511001188
148. Kanakasabai S, Chearwae W, Walline CC, Iams W, Adams SM, Bright JJ. Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology (2010) 130:572–88. doi:10.1111/j.1365-2567.2010.03261.x
149. Adhikary T, Wortmann A, Schumann T, Finkernagel F, Lieber S, Roth K, et al. The transcriptional PPARβ/δ network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res (2015) 43:5033–51. doi:10.1093/nar/gkv331
150. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med (2013) 3:a014217. doi:10.1101/cshperspect.a014217
151. Pello OM, Chèvre R, Laoui D, De Juan A, Lolo F, Andrés-Manzano MJ, et al. In vivo inhibition of c-MYC in myeloid cells impairs tumor-associated macrophage maturation and pro-tumoral activities. PLoS One (2012) 7:e45399. doi:10.1371/journal.pone.0045399
152. Liu L, Lu Y, Martinez J, Bi Y, Lian G, Wang T, et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc Natl Acad Sci U S A (2016) 113:1564–9. doi:10.1073/pnas.1518000113
153. Huang SC-C, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity (2016) 45:817–30. doi:10.1016/j.immuni.2016.09.016
154. Takaoka A, Tamura T, Taniguchi T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci (2008) 99:467–78. doi:10.1111/j.1349-7006.2007.00720.x
155. Günthner R, Anders H-J. Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm (2013) 2013:731023–8. doi:10.1155/2013/731023
156. Saliba DG, Heger A, Eames HL, Oikonomopoulos S, Teixeira A, Blazek K, et al. IRF5:RelA interaction targets inflammatory genes in macrophages. Cell Rep (2014) 8:1308–17. doi:10.1016/j.celrep.2014.07.034
157. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol (2011) 12:231–8. doi:10.1038/ni.1990
158. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by toll-like receptors. Nature (2005) 434:243–9. doi:10.1038/nature03308
159. Kristjansdottir G, Sandling JK, Bonetti A, Roos IM, Milani L, Wang C, et al. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J Med Genet (2008) 45:362–9. doi:10.1136/jmg.2007.055012
160. Ding L, Gu H, Gao X, Xiong S, Zheng B. Aurora kinase a regulates m1 macrophage polarization and plays a role in experimental autoimmune encephalomyelitis. Inflammation (2015) 38:800–11. doi:10.1007/s10753-014-9990-2
161. Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in toll-like receptor-dependent gene induction program. Proc Natl Acad Sci U S A (2006) 103:15136–41. doi:10.1073/pnas.0607181103
162. Roy S, Guler R, Parihar SP, Schmeier S, Kaczkowski B, Nishimura H, et al. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J Immunol (2015) 194:6035–44. doi:10.4049/jimmunol.1402521
163. Masumi A, Tamaoki S, Wang IM, Ozato K, Komuro K. IRF-8/ICSBP and IRF-1 cooperatively stimulate mouse IL-12 promoter activity in macrophages. FEBS Lett (2002) 531:348–53. doi:10.1016/S0014-5793(02)03556-1
164. International Multiple Sclerosis Genetics Consortium (IMSGC)Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet (2013) 45:1353–60. doi:10.1038/ng.2770
165. Disanto G, Sandve GK, Berlanga-Taylor AJ, Morahan JM, Dobson R, Giovannoni G, et al. Genomic regions associated with multiple sclerosis are active in B cells. PLoS One (2012) 7:e32281. doi:10.1371/journal.pone.0032281
166. Yoshida Y, Yoshimi R, Yoshii H, Kim D, Dey A, Xiong H, et al. The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation. Immunity (2014) 40:187–98. doi:10.1016/j.immuni.2013.11.022
167. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol (2010) 11:936–44. doi:10.1038/ni.1920
168. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A (2005) 102:15989–94. doi:10.1073/pnas.0508327102
169. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116:281–97. doi:10.1016/S0092-8674(04)00045-5
170. Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions – beyond repression of gene expression. Nat Rev Genet (2014) 15:599–612. doi:10.1038/nrg3765
171. Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol (2011) 187:2213–21. doi:10.4049/jimmunol.1003952
172. Paraboschi EM, Soldà G, Gemmati D, Orioli E, Zeri G, Benedetti MD, et al. Genetic association and altered gene expression of mir-155 in multiple sclerosis patients. Int J Mol Sci (2011) 12:8695–712. doi:10.3390/ijms12128695
173. Moore CS, Rao VTS, Durafourt BA, Bedell BJ, Ludwin SK, Bar-Or A, et al. miR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Ann Neurol (2013) 74:709–20. doi:10.1002/ana.23967
174. Gaudet AD, Mandrekar-Colucci S, Hall JCE, Sweet DR, Schmitt PJ, Xu X, et al. miR-155 deletion in mice overcomes neuron-intrinsic and neuron-extrinsic barriers to spinal cord repair. J Neurosci (2016) 36:8516–32. doi:10.1523/JNEUROSCI.0735-16.2016
175. Tili E, Michaille J-J, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol (2007) 179:5082–9. doi:10.4049/jimmunol.179.8.5082
176. Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology (2012) 135:73–88. doi:10.1111/j.1365-2567.2011.03514.x
177. Tarassishin L, Loudig O, Bauman A, Shafit-Zagardo B, Suh H-S, Lee SC. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155*. Glia (2011) 59:1911–22. doi:10.1002/glia.21233
178. Herai RR, Stefanacci L, Hrvoj-Mihic B, Chailangkarn T, Hanson K, Semendeferi K, et al. Micro RNA detection in long-term fixed tissue of cortical glutamatergic pyramidal neurons after targeted laser-capture neuroanatomical microdissection. J Neurosci Methods (2014) 235:76–82. doi:10.1016/j.jneumeth.2014.06.028
179. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A (2007) 104:1604–9. doi:10.1073/pnas.0610731104
180. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A (2009) 106:2735–40. doi:10.1073/pnas.0811073106
181. Cobos Jiménez V, Bradley EJ, Willemsen AM, van Kampen AHC, Baas F, Kootstra NA. Next-generation sequencing of microRNAs uncovers expression signatures in polarized macrophages. Physiol Genomics (2013) 46:91–103. doi:10.1152/physiolgenomics.00140.2013
182. Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem (2012) 287:21816–25. doi:10.1074/jbc.M111.327031
183. Woodbury ME, Freilich RW, Cheng CJ, Asai H, Ikezu S, Boucher JD, et al. miR-155 is essential for inflammation-induced hippocampal neurogenic dysfunction. J Neurosci (2015) 35:9764–81. doi:10.1523/JNEUROSCI.4790-14.2015
184. Freilich RW, Woodbury ME, Ikezu T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS One (2013) 8:e79416. doi:10.1371/journal.pone.0079416
185. Zhang Y, Zhang M, Zhong M, Suo Q, Lv K. Expression profiles of miRNAs in polarized macrophages. Int J Mol Med (2013) 31:797–802. doi:10.3892/ijmm.2013.1260
186. Alexander M, Hu R, Runtsch MC, Kagele DA, Mosbruger TL, Tolmachova T, et al. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun (2015) 6:7321. doi:10.1038/ncomms8321
187. Caballero-Garrido E, Pena-Philippides JC, Lordkipanidze T, Bragin D, Yang Y, Erhardt EB, et al. In vivo inhibition of miR-155 promotes recovery after experimental mouse stroke. J Neurosci (2015) 35:12446–64. doi:10.1523/JNEUROSCI.1641-15.2015
188. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A (2009) 106:7113–8. doi:10.1073/pnas.0902636106
189. Zhou H, Xiao J, Wu N, Liu C, Xu J, Liu F, et al. MicroRNA-223 regulates the differentiation and function of intestinal dendritic cells and macrophages by targeting C/EBPβ. Cell Rep (2015) 13:1149–60. doi:10.1016/j.celrep.2015.09.073
190. Cao S, Liu J, Song L, Ma X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol (2005) 174:3484–92. doi:10.4049/jimmunol.174.6.3484
191. Cao S, Liu J, Chesi M, Bergsagel PL, Ho I-C, Donnelly RP, et al. Differential regulation of IL-12 and IL-10 gene expression in macrophages by the basic leucine zipper transcription factor c-Maf fibrosarcoma. J Immunol (2002) 169:5715–25. doi:10.4049/jimmunol.169.10.5715
192. Schulte LN, Westermann AJ, Vogel J. Differential activation and functional specialization of miR-146 and miR-155 in innate immune sensing. Nucleic Acids Res (2013) 41:542–53. doi:10.1093/nar/gks1030
193. Natsuka S, Akira S, Nishio Y, Hashimoto S, Sugita T, Isshiki H, et al. Macrophage differentiation-specific expression of NF-IL6, a transcription factor for interleukin-6. Blood (1992) 79:460–6.
194. Yan C, Zhu M, Staiger J, Johnson PF, Gao H. C5a-regulated CCAAT/enhancer-binding proteins β and δ are essential in Fcγ receptor-mediated inflammatory cytokine and chemokine production in macrophages. J Biol Chem (2012) 287:3217–30. doi:10.1074/jbc.M111.280834
195. Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, et al. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. EMBO J (1995) 14:1932–41.
196. Gorgoni B, Maritano D, Marthyn P, Righi M, Poli V. C/EBP beta gene inactivation causes both impaired and enhanced gene expression and inverse regulation of IL-12 p40 and p35 mRNAs in macrophages. J Immunol (2002) 168:4055–62. doi:10.4049/jimmunol.168.8.4055
197. Davydov IV, Krammer PH, Li-Weber M. Nuclear factor-IL6 activates the human IL-4 promoter in T cells. J Immunol (1995) 155:5273–9.
198. van Dijk TB, Baltus B, Raaijmakers JA, Lammers JW, Koenderman L, de Groot RP. A composite C/EBP binding site is essential for the activity of the promoter of the IL-3/IL-5/granulocyte-macrophage colony-stimulating factor receptor beta c gene. J Immunol (1999) 163:2674–80.
199. Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat Med (2011) 17:64–70. doi:10.1038/nm.2266
200. Hamzei Taj S, Kho W, Aswendt M, Collmann FM, Green C, Adamczak J, et al. Dynamic modulation of microglia/macrophage polarization by miR-124 after focal cerebral ischemia. J Neuroimmune Pharmacol (2016) 11:733–48. doi:10.1007/s11481-016-9700-y
201. Banerjee S, Xie N, Cui H, Tan Z, Yang S, Icyuz M, et al. MicroRNA let-7c regulates macrophage polarization. J Immunol (2013) 190:6542–9. doi:10.4049/jimmunol.1202496
202. Chen X-M, Splinter PL, O’Hara SP, LaRusso NF. A cellular micro-RNA, let-7i, regulates toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem (2007) 282:28929–38. doi:10.1074/jbc.M702633200
203. Sharma BS, Abo-Ismail MK, Schenkel FS, You Q, Verschoor CP, Pant SD, et al. Association of TLR4 polymorphisms with Mycobacterium avium subspecies paratuberculosis infection status in Canadian Holsteins. Anim Genet (2015) 46:560–5. doi:10.1111/age.12333
204. Parisi C, Napoli G, Amadio S, Spalloni A, Apolloni S, Longone P, et al. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ (2016) 23:531–41. doi:10.1038/cdd.2015.153
205. Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, et al. MicroRNA let-7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-κB pathway. Cell Host Microbe (2015) 17:345–56. doi:10.1016/j.chom.2015.01.007
206. Lehmann SM, Krüger C, Park B, Derkow K, Rosenberger K, Baumgart J, et al. An unconventional role for miRNA: let-7 activates toll-like receptor 7 and causes neurodegeneration. Nat Neurosci (2012) 15:827–35. doi:10.1038/nn.3113
207. Wang Z, Xu L, Hu Y, Huang Y, Zhang Y, Zheng X, et al. miRNA let-7b modulates macrophage polarization and enhances tumor-associated macrophages to promote angiogenesis and mobility in prostate cancer. Sci Rep (2016) 6:25602. doi:10.1038/srep25602
208. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun (1995) 206:667–73. doi:10.1006/bbrc.1995.1094
209. Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol (1998) 160:5347–54.
210. Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol (2017) 38:395–406. doi:10.1016/j.it.2017.03.001
211. Newsholme P, Gordon S, Newsholme EA. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J (1987) 242:631–6. doi:10.1042/bj2420631
212. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8:519–30. doi:10.1085/jgp.8.6.519
213. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep (2016) 17:684–96. doi:10.1016/j.celrep.2016.09.008
214. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15:813–26. doi:10.1016/j.cmet.2012.04.023
215. Van den Bossche J, Baardman J, de Winther MPJ. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J Vis Exp (2015) 105:e53424. doi:10.3791/53424
216. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab (2006) 4:13–24. doi:10.1016/j.cmet.2006.05.011
217. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167:457–70.e13. doi:10.1016/j.cell.2016.08.064
218. O’Neill LAJ. A metabolic roadblock in inflammatory macrophages. Cell Rep (2016) 17:625–6. doi:10.1016/j.celrep.2016.09.085
219. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem (2016) 291:14274–84. doi:10.1074/jbc.M115.685792
220. Dai X-M, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99:111–20. doi:10.1182/blood.V99.1.111
221. Graham DK, Dawson TL, Mullaney DL, Snodgrass HR, Earp HS. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ (1994) 5:647–57.
222. Graham DK, Bowman GW, Dawson TL, Stanford WL, Earp HS, Snodgrass HR. Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene (1995) 10:2349–59.
223. Iwama A, Okano K, Sudo T, Matsuda Y, Suda T. Molecular cloning of a novel receptor tyrosine kinase gene, STK, derived from enriched hematopoietic stem cells. Blood (1994) 83:3160–9.
224. Ronsin C, Muscatelli F, Mattei MG, Breathnach R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene (1993) 8:1195–202.
225. Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature (1995) 373:623–6. doi:10.1038/373623a0
226. Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem (1996) 271:30022–7. doi:10.1074/jbc.271.47.30022
227. Wang MH, Ronsin C, Gesnel MC, Coupey L, Skeel A, Leonard EJ, et al. Identification of the ron gene product as the receptor for the human macrophage stimulating protein. Science (1994) 266:117–9. doi:10.1126/science.7939629
228. Wang MH, Yoshimura T, Skeel A, Leonard EJ. Proteolytic conversion of single chain precursor macrophage-stimulating protein to a biologically active heterodimer by contact enzymes of the coagulation cascade. J Biol Chem (1994) 269:3436–40.
229. Wang MH, Cox GW, Yoshimura T, Sheffler LA, Skeel A, Leonard EJ. Macrophage-stimulating protein inhibits induction of nitric oxide production by endotoxin- or cytokine-stimulated mouse macrophages. J Biol Chem (1994) 269:14027–31.
230. Liu QP, Fruit K, Ward J, Correll PH. Negative regulation of macrophage activation in response to IFN-gamma and lipopolysaccharide by the STK/RON receptor tyrosine kinase. J Immunol (1999) 163:6606–13.
231. Zhou Y-Q, Chen Y-Q, Fisher JH, Wang M-H. Activation of the RON receptor tyrosine kinase by macrophage-stimulating protein inhibits inducible cyclooxygenase-2 expression in murine macrophages. J Biol Chem (2002) 277:38104–10. doi:10.1074/jbc.M206167200
232. Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol (1999) 162:3498–503.
233. Morrison AC, Wilson CB, Ray M, Correll PH. Macrophage-stimulating protein, the ligand for the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase, inhibits IL-12 production by primary peritoneal macrophages stimulated with IFN-gamma and lipopolysaccharide. J Immunol (2004) 172:1825–32. doi:10.4049/jimmunol.172.3.1825
234. Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science (2001) 293:306–11. doi:10.1126/science.1061663
235. Fujimori T, Grabiec AM, Kaur M, Bell TJ, Fujino N, Cook PC, et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol (2015) 8:1021–30. doi:10.1038/mi.2014.129
236. Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, Stappenbeck TS. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci U S A (2009) 106:256–61. doi:10.1073/pnas.0803343106
237. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204:3037–47. doi:10.1084/jem.20070885
238. Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure (2007) 15:992–1004. doi:10.1016/j.str.2007.07.001
239. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125:194–207. doi:10.1172/JCI76012
240. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med (2012) 367:1098–107. doi:10.1056/NEJMoa1114287
241. Huang H, Taraboletti A, Shriver LP. Dimethyl fumarate modulates antioxidant and lipid metabolism in oligodendrocytes. Redox Biol (2015) 5:169–75. doi:10.1016/j.redox.2015.04.011
242. Asadullah K, Schmid H, Friedrich M, Randow F, Volk HD, Sterry W, et al. Influence of monomethylfumarate on monocytic cytokine formation – explanation for adverse and therapeutic effects in psoriasis? Arch Dermatol Res (1997) 289:623–30. doi:10.1007/s004030050251
243. Peng H, Guerau-de-Arellano M, Mehta VB, Yang Y, Huss DJ, Papenfuss TL, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF-κB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem (2012) 287:28017–26. doi:10.1074/jbc.M112.383380
244. Linker RA, Lee D-H, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain (2011) 134:678–92. doi:10.1093/brain/awq386
245. Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation (2010) 7:30. doi:10.1186/1742-2094-7-30
246. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (2012) 122:253–70. doi:10.1042/CS20110386
247. Kelly B, Tannahill GM, Murphy MP, O’Neill LAJ. Metformin inhibits the production of reactive oxygen species from NADH:Ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem (2015) 290:20348–59. doi:10.1074/jbc.M115.662114
248. Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol (2009) 182:8005–14. doi:10.4049/jimmunol.0803563
249. Sun Y, Tian T, Gao J, Liu X, Hou H, Cao R, et al. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J Neuroimmunol (2016) 292:58–67. doi:10.1016/j.jneuroim.2016.01.014
250. Negrotto L, Farez MF, Correale J. Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol (2016) 73:520–8. doi:10.1001/jamaneurol.2015.4807
251. Haghikia A, Gold R. Positive effect on multiple sclerosis with treatment of metabolic syndrome. JAMA Neurol (2016) 73:499–501. doi:10.1001/jamaneurol.2015.5050
252. Al-Majed A, Bakheit AHH, Abdel Aziz HA, Alharbi H, Al-Jenoobi FI. Pioglitazone. Profiles Drug Subst Excip Relat Methodol (2016) 41:379–438. doi:10.1016/bs.podrm.2015.11.002
253. Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol (2002) 51:694–702. doi:10.1002/ana.10206
Keywords: macrophages, microglia, central nervous system, inflammation, molecular, microRNA, metabolism
Citation: Amici SA, Dong J and Guerau-de-Arellano M (2017) Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 8:1520. doi: 10.3389/fimmu.2017.01520
Received: 18 August 2017; Accepted: 26 October 2017;
Published: 10 November 2017
Edited by:
Valentin A. Pavlov, Northwell Health, United StatesReviewed by:
Valerio Chiurchiù, Università Campus Bio-Medico, ItalySilvia Brunelli, Università degli studi di Milano Bicocca, Italy
Caroline Jefferies, Cedars-Sinai Medical Center, United States
Copyright: © 2017 Amici, Dong and Guerau-de-Arellano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mireia Guerau-de-Arellano, bWlyZWlhLmd1ZXJhdUBvc3VtYy5lZHU=