 Kevin D. Pavelko
Kevin D. Pavelko Michael P. Bell1
Michael P. Bell1 Haidong Dong
Haidong Dong- 1Department of Immunology, College of Medicine, Mayo Clinic, Rochester, MN, United States
- 2Department of Urology, College of Medicine, Mayo Clinic, Rochester, MN, United States
Therapies that target the PD-1/B7-H1 axis have revolutionized cancer treatment, yet precise knowledge of how this pathway provides benefit continues to evolve. Here, we report a novel role for the immune checkpoint ligand B7-H1 in the accumulation of tissue-resident memory CD8+ T-cells (TRM). After intracranial infection, Theiler’s murine encephalomyelitis virus (TMEV) generates TRM that are maintained in the central nervous system (CNS) tissues of B7-H1WT animals. Although no differences in acute T-cell responses between B7-H1WT and B7-H1KO are observed, at long-term periods post-infection the maintenance of CD8+ TRM is diminished in B7-H1KO animals. This is accompanied by redistribution of the resident CD8+ population from primarily CD103+ TRM to a diminished population of TRM and a preponderance of non-specified PD-1+ CD103− CD8+ T-cells. T-cell transfer studies demonstrate that host B7-H1 is necessary for maintaining TRM and limiting accumulation of PD-1+ CD103− CD8+ T-cells. The lack of host B7-H1 results in compromised control of a heterologous virus re-challenge demonstrating a functional defect in TRM mediated virus control. This study reveals a new role for B7-H1 in TRM and pro-inflammatory PD-1+ CD103− CD8+ T-cell accumulation in the CNS and gives insight for using B7-H1/PD-1 blockade in modulating long-term T-cell protection.
Introduction
The development of immunologic memory is a fundamental process in response to vaccination and is critical for protection from secondary exposure to viral pathogens. However, many questions regarding the establishment and maintenance of immunologic memory remain unanswered. Acute viral infections are often accompanied by the recruitment and expansion of cytotoxic effector CD8+ T-cells that target MHC class I restricted viral antigens presented on the surface of infected cells. This targeting promotes killing of the infected cell and clearance of the virus from invaded tissues. Upon viral clearance, the CD8+ T-cell population contracts in a regulated process to avoid continuous immune mediated pathology and to restore tissue homeostasis. Although there is a substantial reduction of the effector CD8+ T-cell population, the CD8+ T-cells that remain persist and become long-term memory CD8+ T-cells (TM). Upon re-exposure to pathogens, TM respond with increased potency and efficiency and rapidly clear secondary infections. These TM can be classified into subgroups based on their location, cell surface biomarker expression, and transcriptional profiles (1). CD8+ central memory cells (TCM) and effector memory cells (TEM) remain within the lymphoid organs or in circulation and provide a rapid response that is activated upon antigen re-exposure (2). In contrast, tissue resident memory CD8+ T-cells (TRM) remain at sites of previous infection and provide tissue-specific protection from secondary exposure (3). TRM have been identified in a variety of tissues, including lung, skin, intestine, and brain (4–7). The integrin CD103 has been shown to promote the retention of TRM in brain tissues (7) and interactions with ligands expressed in the brain may provide support for their maintenance and retention within the CNS. Although emphasis continues to be placed on the mechanisms involved in TCM and TEM development in lymphoid organs and circulating blood, more recently interest has grown in understanding tissue-specific TRM development, regulation, and maintenance (8–12).
The expression of the immune checkpoint molecule B7-H1 (PD-L1) and interactions with its receptor programmed death-1 (PD-1) constitute a fundamental mechanism in regulating the magnitude of effector T-cell responses. This response is particularly critical after inflammatory viral infections and activation of CD8+ cytotoxic lymphocytes in vital host tissues. The upregulation of PD-1 on T-cells after activation and subsequent interaction with B7-H1 in host tissues ensures that a regulated process of T-cell contraction and elimination of unnecessary inflammation occurs (13). In the presence of chronic antigen exposure, some CD8+ T-cells convert to an exhausted state, which coincides with their upregulation of PD-1 and the gradual loss of their ability to eliminate their intended targets (14). These findings have formed the basis for the development of immunotherapeutic interventions aimed at targeting immune checkpoint molecules for the treatment of cancers (15). Some effector CD8+ T-cells that ultimately eliminate virus from infected tissues are downregulated yet are maintained as TRM for protection from subsequent re-exposure to pathogens. Although both exhausted CD8+ cells and TRM develop after viral infection, it is unclear how specific tissue interactions regulate the development of either of these CD8 T-cell phenotypes. Several factors unique to the responsive T-cell population, the host tissues or the virus have potential roles in the development of these persisting tissue specific T-cell populations.
Intracranial infection with the mouse pathogen Theiler’s murine encephalomyelitis virus (TMEV) is often used as a model of persistent infection and neuropathology in mice expressing susceptible MHC class I alleles (16). However, in mice expressing the H-2Db allele acute encephalitis and virus infection is cleared by an immunodominant CD8+ T-cell response to the virus antigen VP2121–130 (17). Upon viral clearance, immune-mediated pathology subsides and replicating virus is no longer detected in the central nervous system (CNS). This clearance is preferentially associated with the H-2Db allele in C57BL/6 mice and H-2Db provides exclusive protection in FVB transgenic mice (18). Although acute CD8+ T-cell responses to VP2121–130 comprise up to 70% of the central nervous system infiltrating lymphocyte (CNS-IL) pool of CD8+ T-cells, little is known about the development of brain-specific TRM after infection with this virus. Previously, others have shown a role for immune checkpoint molecule blockade in the protection of the CNS from persistent virus infection and chronic inflammation (19). However, it is unclear what role B7-H1 may play in the generation of TRM after acute encephalitis and viral clearance. To that end, we examined the continued presence of TRM in the CNS at time points greater than 30 days post-TMEV infection using both B7-H1WT and B7-H1KO animals. We found that after the resolution of acute infection, T-cells responsive to the viral peptide VP2121–130 and to the embedded peptide antigen OVA257–264 developed into TRM and persisted within the CNS for longer than 200 days post-infection in the absence of replicating virus. Further, we identified a role for host B7-H1 in enhancing the accumulation of TRM in the CNS and demonstrated that, as a consequence of that role B7-H1 is required for controlling a secondary virus challenge. These findings have implications for the design and use of immune checkpoint blockade therapies and extend our understanding of the role B7-H1 plays in resolving CNS inflammation and of its contribution to the development of TRM.
Materials and Methods
Animals and Cell Lines
C57BL/6 (B7-H1WT) and C57BL/6 CD45.1 (Ly5.1) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). OT-1 TCR (CD90.1+) transgenic mice were provided by T. Tian (Harvard University, Boston, MA, USA). B7-H1KO C57BL/6 mice were provided by L. Chen (Yale University, New Haven, CT, USA). All animals were housed in the Mayo Clinic Department of Comparative Medicine and cared for according to the Mayo Clinic Institutional Animal Care and Use Committee and NIH guidelines for animal use and care. All biologic agents were used in accordance with the policies and guidelines designated by the Mayo Clinic Institutional Biosafety Committee.
Antibodies and Tetramers
The antibodies used for flow cytometry were Brilliant Violet 421™ anti-mouse CD8α (Biolegend; Clone 53-6.7), APC/Cy7 anti-mouse CD8β Antibody (Biolegend; YTS156.7.7), Brilliant Violet 605™ anti-mouse CD45 (Biolegend; 30-F11), PerCP/Cy5.5 anti-mouse CD103 (Biolegend; 2E7), BV421 anti-Mouse CD103 (BD Biosciences; M290), PE-Cyanine7 anti-human/mouse CD44 (Tonbo; IM7), PE anti-mouse CD279/PD-1 (Tonbo; J43.l), FITC anti-mouse CD279/PD-1 (eBioscience; RMP1-30), FITC, anti-mouse CD69 (BD Biosciences; H1.2F3), and FITC anti-mouse CD90.1 (eBioscience; HIS51). Allophycocyanin labeled H-2Db/VP2121–130, H-2Kb/SIYR, and H-2Kb/OVA257–264 tetramers were kindly provided by Dr. Aaron Johnson (Mayo Clinic, Rochester, MN, USA) and were generated using previously described methods (17, 20). Anti-PD1 hamster monoclonal antibody (G4) was purified and dosed as previously described (21). Animals were treated with 200 µg of anti-PD-1 or isotype IgG on day three post-infection with TMEV-OVA8. The antibody used for in vivo intravascular labeling of CD8+ T-cells was PE anti-mouse CD8α (BD Biosciences; 53-6.7). Intravascular labeling of peripheral blood lymphocytes and CNS-IL was performed using previously described methods (22).
CNS-IL Isolation and FACS Analysis
Isolation of CNS-ILs was performed using previously described methods (23). Briefly, at designated time points, post-infection mice were euthanized with CO2 prior to collection of brain and spinal cord into 5 mL of 4°C RPMI. Animals were perfused with 50 mL of PBS prior to tissue harvest to exclude the possibility of contamination by blood-derived rather than tissue-derived cells. Tissues were then transferred to a Pyrex Ten Broeck homogenizer (Corning 7 mL, 0.15 mm gap) and homogenized until complete tissue dissociation is attained (5–7 strokes). The CNS homogenate was then sieved through a Corning™ 100 µm strainer (Fisher Scientific; Cat. No. 08-771-19) followed by addition of 5 mL RPMI. The homogenate was then brought to 70% Percoll prepared in PBS in a final volume of 30 mL prior to centrifugation in at 7,840 g for 25 min at 4°C. After centrifugation a top myelin debris layer was removed and isolated cells were resuspended in a total volume of 50 mL RPMI before pelleting at 800 g. Red blood cells were lysed with ACK (ammonium chloride potassium) lysing buffer, washed with RPMI, and resuspended in FACS buffer before addition of appropriate fluorochrome labeled antibodies or tetramers and analysis by FACS. Final stained cell populations were spiked with 50 µL of CountBright™ absolute counting beads prior to FACS to determine absolute cell numbers. Cells were run on a BD LSRII flow cytometer (BD Bioscience), and data were analyzed using FloJo Software version 7.6.5 (Tree Star, Ashland, OR, USA). For reinfection experiments mice were depleted of peripheral CD8+ cells by injection of 0.5 mg of anti-mouse CD8 antibody (Lyt2.43) on day 7, 5, and 3 prior to reinfection with GD7-KS1 as previously described (24).
Virus, Infection, and Quantitation
Molecularly defined viruses derived from the two TMEV subgroup viruses were used for these experiments. The XhoI-OVA8 virus is derived from the Daniel’s strain which belongs to the less neurovirulent subgroup TO. The TMEV-GD7-KS1 virus is derived from the GDVII substrain that belongs to the highly neurovirulent subgroup GDVII (25). The XhoI-OVA8 (TMEV-OVA8) and GD7-KS1 (TMEV-GD7) viruses were described previously (26, 27). Mice were infected intraperitoneally with 2 × 106 or intracranially with 2 × 105 PFU of TMEV-OVA8. Virus titers were determined by plaque assay using NCTC clone 929 (L-929) cells (ATCC, Manassas, VA, USA) using a protocol described previously (28). Semi-quantitative RT-PCR targeting the VP2 capsid protein of TMEV-OVA8 was used to determine virus RNA levels and has been described (27).
In Vivo Killing Assay
A modified version of a previously described technique was used to test in vivo killing by cytotoxic lymphocyte responses induced with TMEV-OVA8 (29). On day 6 after intraperitoneal infection of B7-H1WT or B7-H1KO mice, three peptide-pulsed target cell populations were prepared from C57BL/6 CD45.1 donor splenocytes. Two concentrations of carboxyfluorescein succinimidyl ester (CFSE; Excitation/Emission 490 nm/520 nm) were used to label the no peptide population (CFSELow) and the virus peptide VP2121–130 (FHAGSLLVFM; CFSEHigh). Chicken ovalbumen257–264 (SIINFEKL) pulsed splenocytes were labeled with a second dye PKH26 (Ex/Em; 551 nm/567 nm). The three populations were mixed at equal numbers before challenge of TMEV infected mice by intravenous injection. 2 × 107 total cells were injected per mouse. Percent killing was determined by relative number of cells recovered from the splenocytes of infected and naïve animals. Splenocytes were assessed by FACS for the number of cells having the CD45.1 marker and the distribution of the three labeled populations. Percent killing was determined using the following equation: % specific lysis = 1−[rnaive/rinfected] × 100 where: r = %CFSElow cells/%CFSEhigh or PKH26 labeled cells.
Adoptive Transfer of OT-1 T-Cells and Virus Infection
CD8+ T-cells were purified from the spleens of OT-1 (Thy1.1 or Thy1.2) TCR transgenic mice or from OT-1B7-H1KO mice (30) using magnetic bead separation (Miltinyi Biotec, Auburn, CA, USA). After isolation, 2 × 106 CD8+ cells were injected intravenously into B7-H1WT or B7-H1KO mice. On the day of transfer mice were infected intracranially with 2 × 105 PFU of TMEV-OVA8. At designated times, post-infection CNS tissues and spleens were harvested and processed for lymphocyte isolation prior to immunophenotyping by FACS.
Statistics
Mean and SD values were calculated using Excel 2010. Statistical analysis was performed using Sigmaplot for Windows version 11.0. All parametric data were analyzed by t-test. Non-parametric data were analyzed by Rank-sum assay. Significance was determined by P < 0.05.
Results
Intracranial Infection with an Attenuated TMEV Virus Results in the Accumulation and Maintenance of Tissue Resident Memory CD8+ T-Cells in the CNS
Theiler’s murine encephalomyelitis virus infection of the CNS generates an acute encephalomyelitis that is ultimately cleared by day 14 due to an immunodominant CD8+ T-cell response to the viral peptide VP2121–130 (18, 31). The generation of this response and the activation of cytotoxic perforin expressing T-cells promotes viral clearance and subsequently inflammation resolves. To determine whether infection with TMEV generates CNS TRM, we infected C57BL/6 mice intraperitoneally or intracranially with TMEV-OVA8 and examined the accumulation of virus-specific memory CD8+ T-cells 98 days post-infection. This attenuated TMEV virus generates strong CD8+ T-cell responses to a virus genome embedded OVA8 peptide as well as the endogenous immunodominant virus antigen VP2121–130 (27). At 98 days post-infection, we isolated CNS-ILs and splenocytes from infected animals and found that virus specific H-2Kb-OVA8+ CD8+ T-cells (OVA8+) were present in the CNS after intracranial infection, but not after intraperitoneal infection (Figure 1A). Recovered splenocytes from both intracranial and intraperitoneal infection generated a population of virus specific OVA8+ T-cells, verifying that infection via both routes promotes the generation of antigen-specific (VP2+) CD8+ T cells (Figure 1B). Phenotypic analysis of total CD8+ T-cells recovered from the CNS of infected animals revealed that all CD8+ T-cells expressed high levels of CD44 (effector/memory T-cell marker) on day 6 but dimmer levels of CD44 at day 98, while CD44 levels were comparable between CD8+ T-cells recovered from the spleen (Figure 1C). Further analysis of OVA8+ T-cells recovered from both the CNS and spleen demonstrated that the CNS derived virus specific CD8+ T-cells expressed high levels of CD69 (T-cell activation marker) and CD103 (tissue-resident memory T-cell marker) compared to spleen derived OVA8+ cells (Figure 1D). These findings demonstrate that intracranial TMEV infection results in the development and maintenance of a long lived CNS CD103+ CD69+ CD8+ TRM population.
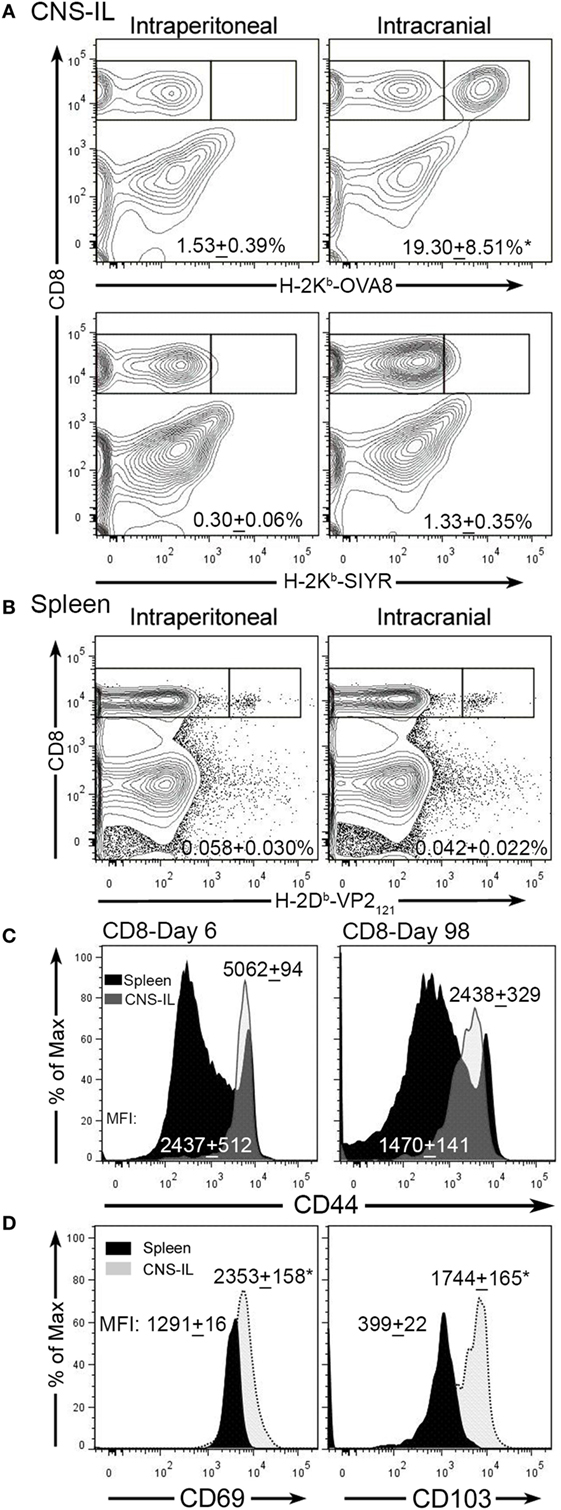
Figure 1. Intracranial infection with Theiler’s murine encephalomyelitis virus (TMEV)-OVA8 generates long lived TRM. (A) Central nervous system (CNS) infiltrating lymphocytes from intraperitoneally or intracranially infected C57BL/6 mice were analyzed 140 days post-infection for antigen specific CD8+ T-cell responses using the virus specific tetramer H-2Kb-OVA8 or the non-specific control tetramer H-2Kb-SIYR (n = 3 for each group). (B) Splenocytes from C57BL/6 infected mice infected with TMEV-OVA8 were analyzed with the virus specific tetramer H-2Db-VP2121–130 to determine the frequency of virus specific CD8+ T-cells. (C) Analysis of CD8+ cells recovered from the CNS and spleens of C57BL/6 mice for CD44 expression on 6 day 6 and 98 post infection with TMEV-OVA8. Numbers are mean fluorescence intensity (MFI; mean + SD, n = 5 per time point). (D) CD69 and CD103 expression on H-2Kb-OVA8+ CD8+ T-cells isolated from the CNS of C57BL/6 mice intracranially infected with TMEV-OVA8 on day 140 post-infection (n = 3 for each group). Data are expressed as mean + SD.
B7-H1 Does Not Regulate Acute Cytotoxic T-Cell Function or Alter Virus Clearance
To determine the role that B7-H1 plays in the development of virus specific cytotoxic T-cell responses we infected B7-H1 wild-type (B7-H1WT) and B7-H1 knockout mice (B7-H1KO) with TMEV-OVA8 for 6 days to determine whether immune responses induced by infection can kill virus specific target cells in an in vivo CTL assay. We found that the effector T-cells generated by B7-H1WT or B7-H1KO mice equivalently killed both VP2121–130 and OVA257–264 target cells (Figure 2A). In addition, intracranial infection of B7-H1WT and B7-H1KO mice for 6 or 98 days demonstrated no difference in the level of TMEV RNA obtained from CNS tissues (Figure 2B). A further analysis of CNS homogenates demonstrated that no replicating virus remains in the CNS of C57BL/6 mice after 34 days as assessed by plaque assay, a finding consistent with attenuation of this strain (27) and with previous investigations using intracranial infection with the DA strain of TMEV in C57BL/6 (H-2b) mice (32).
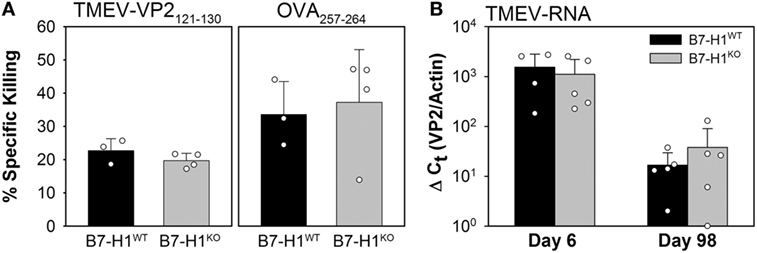
Figure 2. Acute in vivo cytotoxic T-cell responses and virus control in the absence of B7-H1. (A) In vivo killing of the virus-specific peptide [OVA8 and Theiler’s murine encephalomyelitis virus (TMEV)-VP2121–130] pulsed target cells recovered from the spleens of B7-H1 competent and B7-H1 deficient mice 6 days post TMEV-OVA8 intraperitoneal challenge. Data are presented as the percent specific killing of virus antigen specific targets recovered and analyzed by FACS relative to unpulsed targets and uninfected control mice. (B) RNA purified from the CNS of mice infected intracranially for 6 and 98 days was assessed by semi-quantitative RT-PCR for the presence of TMEV specific transcripts.
B7-H1 Promotes the Retention of Long-term Virus-Specific T-Cells in the CNS
We next investigated the role of B7-H1 in the development and maintenance of virus specific CD8+ T cell responses in the CNS after intracranial infection with TMEV-OVA8. On day 6 post-infection, intracranial infection of either B7-H1WT or B7-H1KO mice revealed no difference in the percentage or absolute numbers of virus specific OVA8+ or H-2Db-VP2121–130+ (VP2+) cells recovered from the CNS. Although there was a modest increase in total CD8+ T-cells recovered from B7-H1KO mice at this acute time point this difference was not significant (p = 0.112) (Figures 3A–C).
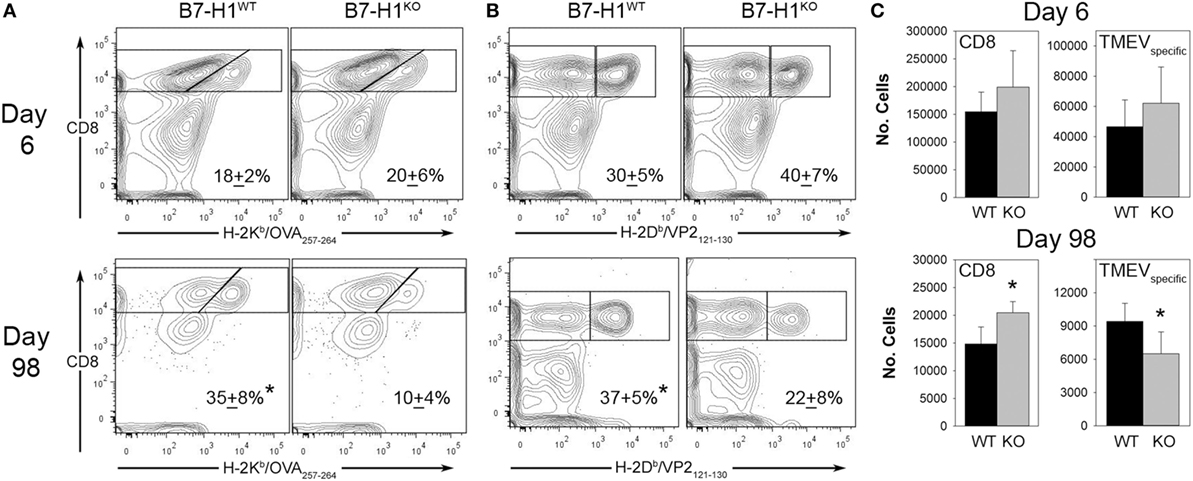
Figure 3. The accumulation of viral antigen specific CD8+ T-cells in the CNS of B7-H1KO mice post Theiler’s murine encephalomyelitis virus (TMEV) infection. (A) CD45+ cells recovered from the CNS were analyzed for percent of total OVA8+ CD8+ T-cells in B7-H1WT and B7-H1KO mice on day 6 and day 98 post intracranial TMEV-OVA8 infection (n = 5 per group; mean + SD percent of total CD8+ cells). (B) CD45+ gated cells in (A) were analyzed to determine the percent of B7-H1WT and B7-H1KO CD8+ T-cells that were specific to the TMEV peptide antigen VP2121–130 on day 6 and day 98. (C) Absolute numbers of total CD8+ tetramer positive OVA8+ and VP2+ CD8+ T-cells recovered from the CNS of B7-H1WT and B7-H1KO mice on day 6 and day 98 post TMEV-OVA8 intracranial challenge. *denotes significance of p < 0.05 by t-test when comparing B7-H1WT and B7-H1KO.
Long-term post-infection time points were further investigated to determine the role of B7-H1 in promoting the accumulation of virus-specific CD8+ T-cells. We found that the quantity of CD8+ T-cells remaining in the CNS at 98 days post-infection was approximately 10% of that seen during acute infection in both B7-H1WT and B7-H1KO mice (Figure 3C). To further characterize the residual CD8+ T-cell population, we determined the percent and quantity of T-cells that were specific for the known virus specific epitopes. We found that the percentage of OVA8+ and VP2+ T-cells recovered from the CNS of mice infected for 98 days was decreased in B7-H1KO mice when compared to B7-H1WT mice (Figures 3A,B). In addition, a decrease in the absolute number of virus specific T-cells identified by both tetramers was observed for the CD8+ T-cell population in B7-H1KO mice (Figure 3C). These findings suggest that B7-H1 plays a role in the long-term accumulation of TRM cells after intracranial virus infection.
B7-H1 Influences the Accumulation of PD-1+ CD103− CD8+ T-Cells in the CNS
Although our finding suggests that B7-H1 plays a role in promoting CNS-TRM after virus infection, the absolute number of total CD8+ T-cells that remain in the CNS of mice infected for 98 days was increased in B7-H1KO mice compared to B7-H1WT (Figure 3C), suggesting that non-specified CD8+ T-cell populations fail to contract or are maintained in the CNS at this extended time point post-infection. To determine if phenotypic differences between non-specified and virus specific CD8+ T-cells existed, we examined the phenotype of the CNS resident CD8+ T-cell populations during acute infection and at 98 days post-infection in B7-H1WT and B7-H1KO mice. First, we assessed the expression levels of CD103 on CD8+ T-cells recovered from the CNS of TMEV-OVA8 infected animals, a marker previously used to identify CNS TRM (7, 33). During the acute inflammatory phase of infection, very few of the total CD8+ T-cells or the virus-specific VP2+ cells expressed the TRM marker CD103, and no differences in this marker were observed between B7-H1WT and B7-H1KO mice (Figure 4A). On day 98 post-infection, a majority of the total CD8+ T-cells recovered from the CNS of B7-H1WT animals expressed high levels of CD103. In contrast, this population in B7-H1KO animals was decreased and coincided with an enhanced population of CD8+ T-cells that expressed lower levels of CD103 (Figure 4A). Since the phenotypic expression of CD103 in the virus-specific CD8+ T-cells was comparable between B7-H1WT and B7-H1KO, it seems that the generation of virus-specific TRM was not necessarily influenced by B7-H1 expression. However, the decrease of virus-specific CD8+ T-cells in B7-H1KO CNS-IL compared to the CD103− CD8+ T-cell population (Figure 3C) suggests that the quality of the virus-specific TRM response and their accumulation in the CNS could be regulated directly by B7-H1 or indirectly by competing cells present in the absence of B7-H1 (Figure 4A).
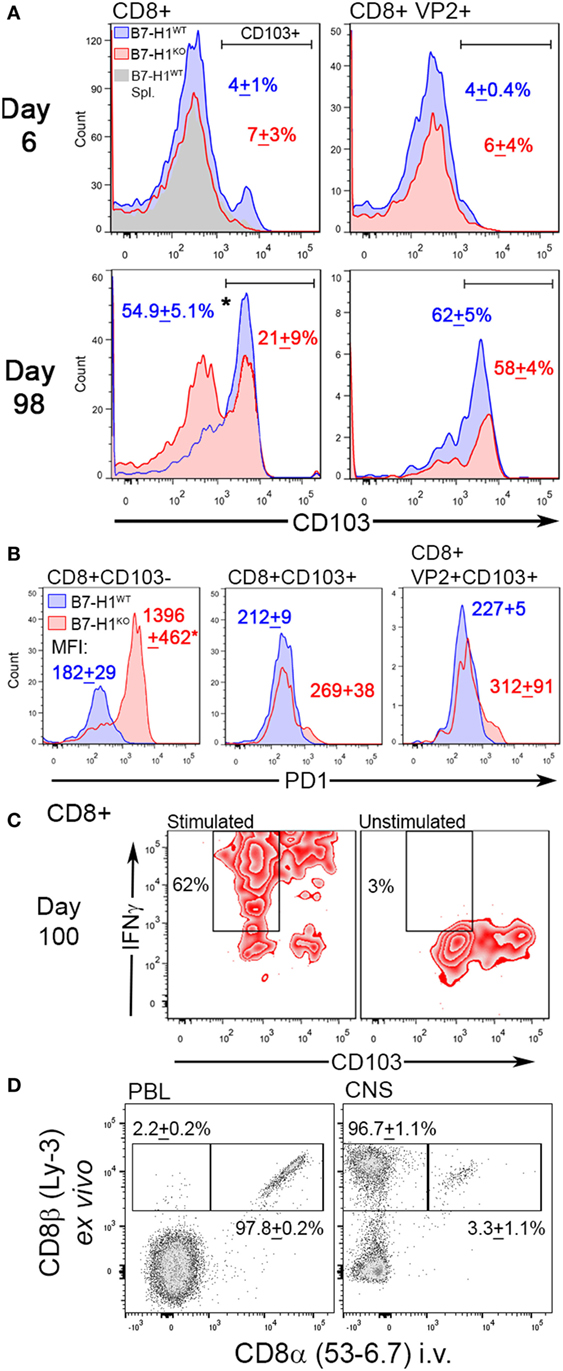
Figure 4. The TRM marker CD103 and the immune checkpoint molecule programmed death-1 (PD-1) distinguish CNS resident CD8+ T-cells. (A) Expression of CD103 in total CD8+ and virus-specific VP2+ CD8+ CNS-IL recovered from B7-H1WT and B7-H1KO mice on day 6 and 98 days post Theiler’s murine encephalomyelitis virus (TMEV)-OVA8 challenge (n = 5 per group; mean percent CD103+ SD). Gray histogram—B7-H1WT CD8+ splenocyte control. (B) PD-1 expression levels in total CD8+ and VP2+ CD8+ CNS-IL recovered from B7-H1WT and B7-H1KO mice on day 99 post-infection. CD103 was used to discriminate TRM and non-TRM populations. (Data are presented as mean fluorescence intensity + SD; n = 3.) (C) IFNγ secretion relative to CD103 expression in CD8+ T-cells recovered from the CNS of B7-H1KO mice infected with TMEV-OVA8 for 100 days. Pooled cells (n = 6) were stimulated with PMA/Ionomycin overnight and are compared to unstimulated cells from the same pool. (D) Expression of ex vivo labeled CD8β and in vivo labeled CD8α in B7-H1KO mice infected intracranially with TMEV-OVA8 greater than 90 days prior to analysis. Peripheral blood lymphocytes and CNS-IL were analyzed. (Data are presented as percent CD8β+ only and percent CD8β+ CD8α+ + SD; n = 3).
To investigate potential mechanisms for retention of CD103− CD8+ T-cells in the absence of B7-H1, we measured their expression of PD-1, a ligand of B7-H1 identified as a marker for both regulatory T-cells and exhausted T-cells. Of interest, the CD103− CD8+ T-cell population that accumulates in the CNS of B7-H1KO mice expressed significantly higher levels of PD-1 compared to that of B7-H1WT mice (Figure 4B). In contrast, total CD103+ CD8+ T-cells and virus-specific VP2+ CD103+ TRM derived from the CNS and spleen of B7-H1WT and B7-H1KO mice expressed comparable but low levels of PD-1 (Figure 4B; Figure S1A in Supplementary Material). Since PD-1high CD103− CD8+ T-cells dramatically increased in the CNS of B7-H1KO mice, it is possible that their accumulation is directly under the control of B7-H1 when present.
Since CD103− CD8+ T-cells represent a population of T-cells that persist in the CNS from early acute infection through time points well beyond virus clearance, we investigated whether CD103− CD8+ T-cells were pro-inflammatory cells that persist in the absence of B7-H1. We isolated CNS-IL from B7-H1KO mice and restimulated them in vitro to assess the potential for secreting the pro-inflammatory cytokine IFNγ. We found that the non-specific CD103− CD8+ T-cells derived from the CNS of B7-H1KO mice secreted IFNγ in response to polyclonal activation of T-cells using PMA/ionomycin (Figure 4C). Similar to a population of CD103− CD8+ T-cells present in the spleen of previously infected B7-H1KO mice (Figure S1B in Supplementary Material). Taken together, our findings suggest that B7-H1 promotes the maintenance of PD-1low CD103+ TRM by limiting the accumulation of pro-inflammatory PD-1high CD103− CD8+ T-cells.
To verify that peripheral blood cells and intravascular lymphocytes do not contribute to the pool of CD8+ T-cells that are recovered from the CNS, we performed in vivo intravascular staining to discriminate circulating cells from tissue-resident lymphocytes in B7-H1KO mice infected with TMEV-OVA8 greater than 90 days prior to testing. After a 3-min pulse with in vivo anti-CD8 antibody, whole peripheral blood and CNS-IL were harvested and analyzed by flow cytometry (Figure 4D). We found that 97.8% of peripheral blood circulating CD8+ cells were labeled with the intravascular CD8 antibody, whereas the CNS-IL-derived tissue resident CD8+ cells had very few cells labeled by this approach (3.3%). This provides further evidence that the CD8+ cells derived by these methods are indeed resident T cells embedded within CNS tissues.
Host Specific B7-H1 Influences TRM Maintenance in the CNS
Previously, we have shown that expression of B7-H1 on tissue or on T-cells can modulate the function of CD8+ T-cells (13, 34). To determine the role of host tissue-specific B7-H1 expression on the regulation of CD8+ TRM, we transferred congenically marked wild-type OT-1 T-cells into B7-H1WT or B7-H1KO host animals prior to infection with TMEV-OVA8 (Figure 5A). After 6 days of infection, the percent and number of transferred OT-1 T-cells that infiltrated the CNS were comparable between B7-H1WT and B7-H1KO host mice (Figures 5B,C). In addition, the endogenous virus-specific TRM cell percentages were comparable to those observed previously in B7-H1WT and B7-H1KO animals without adoptive transfer (Figures S2A,B in Supplementary Material). On day 33 post-infection, the percentage and number of recovered OT-1 CD8+ T-cells were lower in the CNS of B7-H1KO mice compared to B7-H1WT mice (Figure 5B). To verify the conversion of transferred OT-1 T-cells to TRM, we measured the levels of CD103 expression on OT-1 T-cells on day 6 and 33 post-infection. We found that the majority of OT-1 T-cells shifted from CD103− at day 6 to CD103+ at day 33 (Figure 5C). In addition, the absolute number of CD103+ OT-1 TRM cells decreased in the absence of B7-H1, from 27,982 + 8,723 cells (mean + SD) in B7-H1WT to 9,602 + 10,647 cells in B7-H1KO (t-test, p = 0.017). Endogenous host CD8+ T-cells increased in B7-H1KO mice compared to B7-H1WT (Figure 5B) which was due to an increased number of non-specified CD103− CD8+ T-cells (Figures S2A,B in Supplementary Material). These results suggest that host-specific B7-H1 influences the accumulation of CD103+ TRM in the CNS post-TMEV infection.
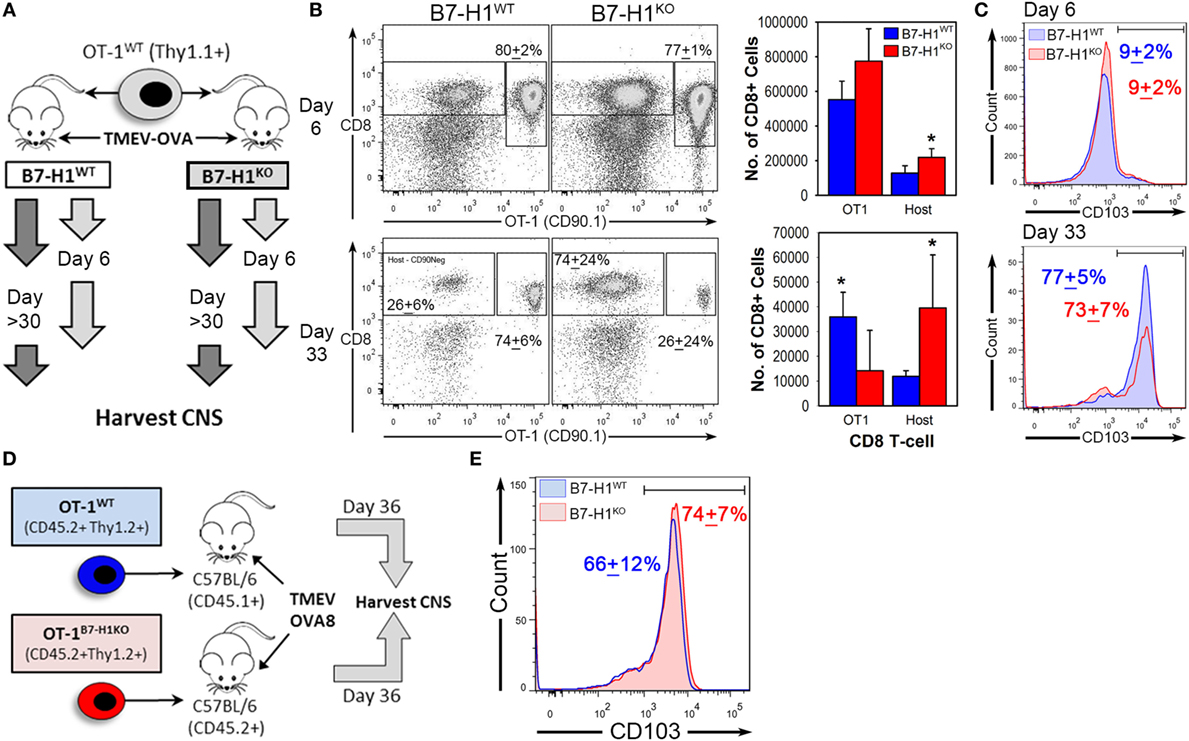
Figure 5. Host-specific B7-H1 promotes central nervous system (CNS) retention of transferred virus specific TRM after intracranial virus infection. (A) Adoptive transfer strategy to determine the consequences of B7-H1 expression on T-cells versus host tissues. B7-H1WT CD90.1+ OT-1 T-cells were transferred into B7-H1WT CD90.2+ hosts (C57BL/6) or into B7-H1KO CD90.2+ hosts before intracranial infection with Theiler’s murine encephalomyelitis virus (TMEV)-OVA8. Tissues were harvested on day 6 and day 33 post-infection. (B) Total CD45+ cells were analyzed for frequency and absolute numbers of recovered CD90.1+ CD8+ OT-1 and host CD8+ T-cells (Host) on day 6 and 33 post TMEV-OVA8 infection from the CNS infected animals. (Percent of total recovered CD90.1+ CD8+ T-cells + SD.) (C) CD103 expression of recovered CD90.1+ OT-1 T-cells recovered from the CNS of B7-H1WT and B7-H1KO mice on day 6 and day 33 post intracranial infection with TMEV-OVA8. (D) Adoptive transfer strategy and TMEV-OVA8 infection of C57BL/6 recipient hosts of B7-H1WT and B7-H1KO CD8+ OT-1 T-cells. (E) CD103 expression of B7-H1WT and B7-H1KO CD8+ OT-1 T-cells recovered from the CNS of C57BL/6 hosts on day 36 post intracranial infection. *Significant by t-test p < 0.05; B7-H1WT versus B7-H1KO.
To determine whether T-cell specific B7-H1 expression would affect the accumulation of TRM, we transferred OT-1 wild-type T-cells (OT-1WT) or B7-H1 deficient OT-1 CD8+ T-cells (OT-1B7-H1KO) into wild-type mice before intracranial infection with TMEV-OVA8 (Figure 5D). The frequencies of OT-1WT and OT-1B7-H1KO TRM were comparable on day 36 post-infection (Figure 5E), demonstrating that B7-H1 expressed by the CD8+ OT-1 T-cells does not contribute to the accumulation of TRM in the CNS. Together, our data suggest that host specific B7-H1, but not T-cell B7-H1 promotes accumulation of CD103+ TRM in the CNS.
B7-H1 Is Required for Virus Control upon Secondary Brain Infection
Since B7-H1 promotes the maintenance of TRM, we asked whether lack of B7-H1 would affect virus clearance in the CNS after secondary infection. To address this question, we used the highly pathogenic TMEV variant GD7-KS1 (TMEV-GD7) as a heterologous secondary CNS infection (26), this TMEV variant contains genetic determinants known to increase neuropathology and is attributed to high pathogenicity and lethality even at low doses when injected directly into the CNS (25). Although few cross neutralizing antibody epitopes have been defined, the neutralization profiles are unique (35). Importantly, the immunodominant CD8 T-cell epitope VP2121–130 is conserved between TMEV-OVA8 (primary infection) and TMEV-GD7 (secondary infection), allowing direct assessment of virus specific TRM function in the CNS. We first infected B7-H1WT and B7-H1KO mice with TMEV-OVA8. Then to evaluate the potential protection from TRM on day 277 post-infection, we eliminated circulating CD8+ T-cells using an anti-CD8 depleting antibody. After CD8 T-cell depletion, the previously infected mice were re-infected intracranially with TMEV-GD7 (Figure 6A). Three days post-challenge CNS-IL and splenocytes were isolated from rechallenged mice to verify peripheral CD8 depletion and to determine the phenotype of CNS derived cells. 8 days after the final dose of CD8 depleting antibody and 3 days after virus rechallenge CD8 depletion was maintained with a 91.4 + 1.3% reduction in total CD8+ T-cells in the spleen of B7-H1WT mice and an 87 + 4.2% reduction in B7-H1KO mice compared to non-depleted control (Figure 6B). CNS-IL were analyzed by flow cytometry to determine the phenotype of resident CD8+ T-cells. Consistent with previous experiments, this cohort of B7-H1KO mice had a reduced number of CD8+ TRM compared to the B7-H1WT group (Figure 6C).
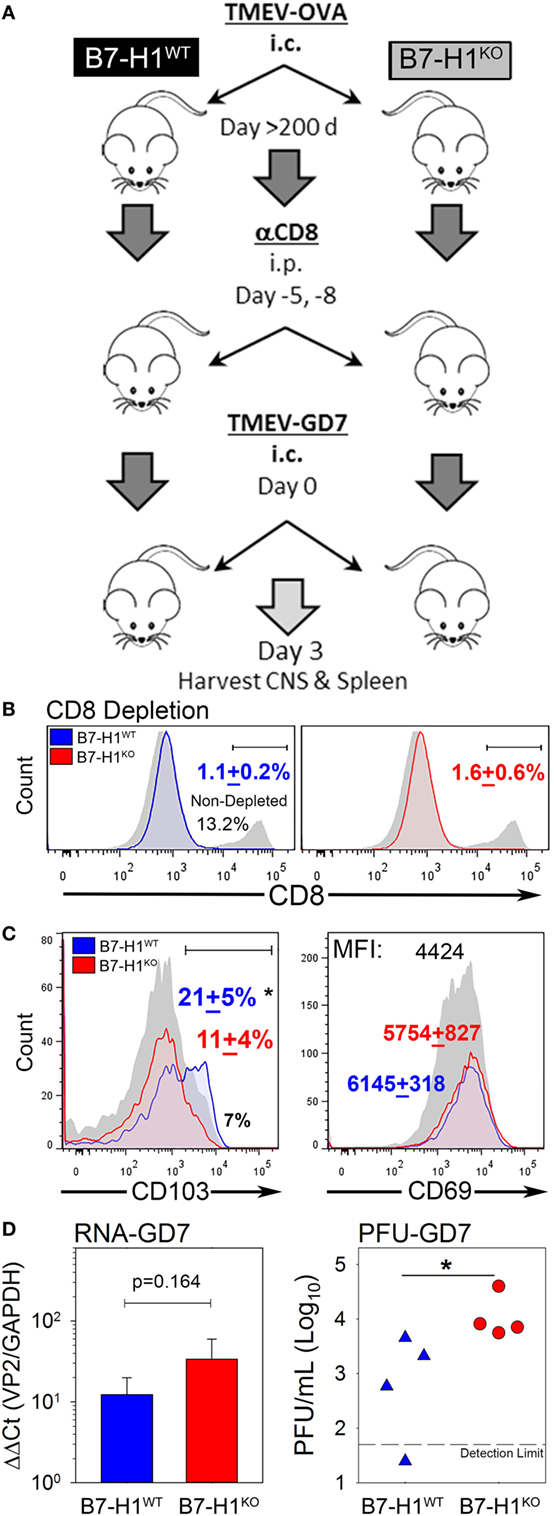
Figure 6. B7-H1 promotes tissue resident memory CD8+ T-cell protection from heterologous secondary infection. (A) Virus re-challenge scheme to determine the role of B7-H1 in protecting mice from a secondary tissue-specific re-challenge with the Theiler’s murine encephalomyelitis virus (TMEV) variant GD7. (B) Splenocytes were analyzed for CD8 expression in B7-H1WT and B7-H1KO mice depleted with CD8 depleting antibody for verify depletion of peripheral CD8+ T-cells. Splenocytes were gated on forward-side scatter and CD45 subsequent to analysis of the percent of CD8+ cells (n = 4 per group). One non-CD8 depleted animal infected with TMEV-OVA8 is used as a reference to determine relative depletion (gray/black). (C) CD103 and CD69 expression on CD8+ T-cells recovered from the CNS on day 3 after TMEV-GD7 re-challenge. (D) Semi-quantitative RT-PCR analysis for TMEV-GD7 specific transcripts from the CNS of B7-H1WT and B7-H1KO mice given a re-challenge with TMEV-GD7. (Right) Plaque assay performed on CNS homogenates recovered from re-infected B7-H1WT and B7-H1KO mice (PFU, plaque-forming units). *Significant by t-test p < 0.05; B7-H1WT versus B7-H1KO.
Further, TMEV load was assessed from the CNS of B7-H1WT and B7-H1KO animals by analysis of viral RNA and replicating virus. The levels of TMEV RNA were comparable between the CNS of B7-H1KO and B7-H1WT mice (p = 0.164), suggesting viral RNA was present as either encapsidated functional virions or as residual viral transcripts present after virus inactivation. However, more plaque forming units were identified in the CNS of B7-H1KO mice compared to B7-H1WT mice (Figure 6D), suggesting that host B7-H1 is required to promote protection from secondary CNS virus infection by promoting the accumulation of virus-specific CD8+ TRM in the CNS.
Systemic Delivery of PD-1 Blockade during Acute Virus Infection Fails to Modulate CNS TRM
Since B7-H1 influences the accumulation of PD-1+ CD103− CD8+ T-cells in the CNS, we hypothesized that targeting PD-1 on these cells may potentially restore the TRM population in the CNS by eliminating their presence in the CNS. To address this, we infected B7-H1KO mice intracranially with TMEV-OVA8 and treated mice with control IgG or anti-PD-1 antibody on day 3 post-infection and assessed the distribution of CD8+ and CD4+ T-cells in the CNS and spleen. At 6 days and 35 days post-challenge, CD45+ CNS-IL and spleen cells were analyzed by flow cytometry. On day 6 post-infection, the percentage of CD8+ CNS-IL and the absolute number of CD8+ cells in the spleen was decreased in the PD-1 blockade group (Figure 7A). By day 35; however, no difference in the percentage or absolute numbers of CD8+ or CD4+ cells were detected in the CNS of B7-H1KO mice when comparing anti-PD-1-treated versus control IgG-treated animals. Decreases in the absolute numbers of CD8+ and CD4+ cells were observed in the spleen at day 35 in groups treated with anti-PD-1 compared to control IgG-treated group (Figure 7A).
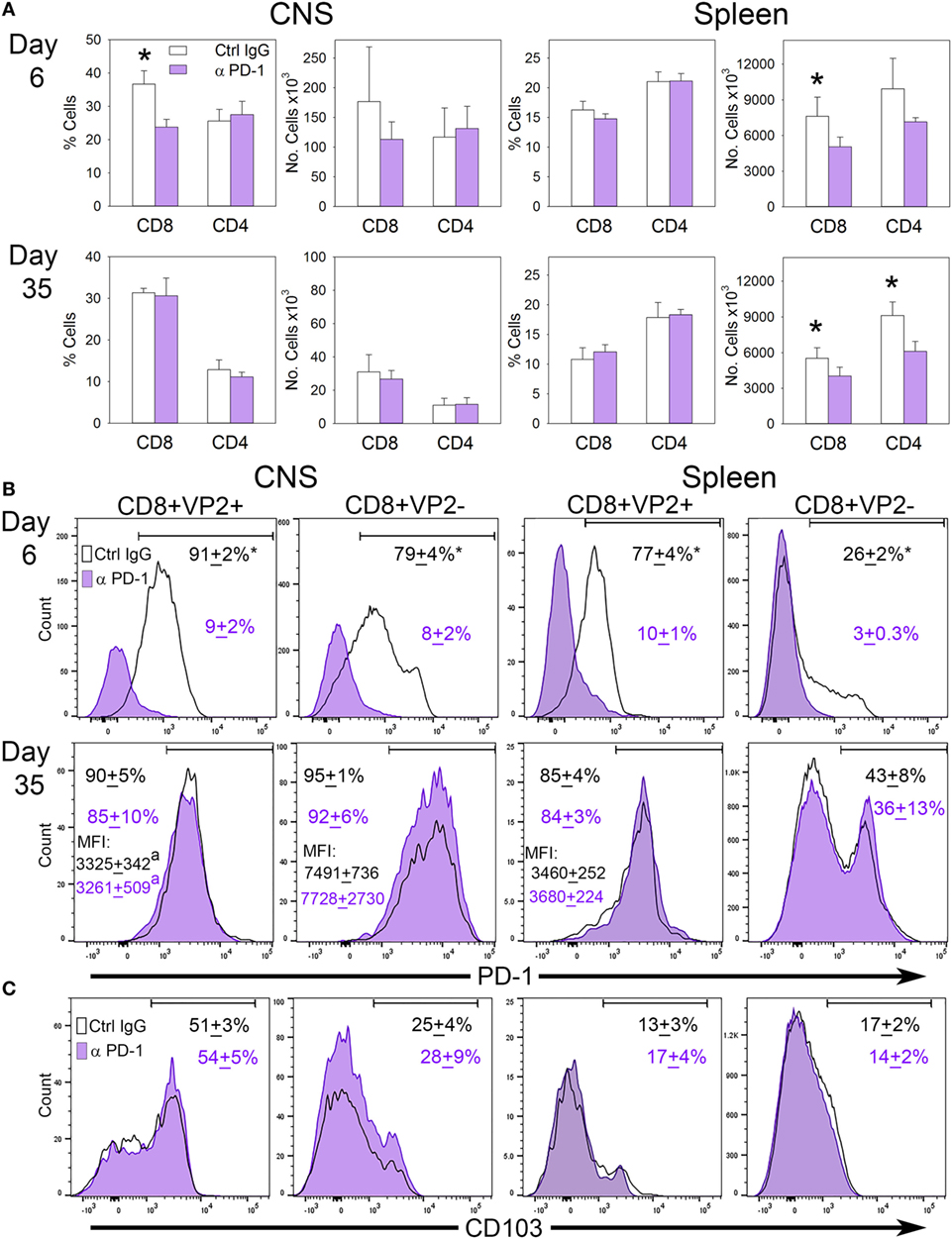
Figure 7. The influence of programmed death-1 (PD-1) blockade on the development of virus specific TRM in B7-H1KO mice after CNS infection with Theiler’s murine encephalomyelitis virus (TMEV)-OVA8. (A) Percent and number of CD8+ and CD4+ cells recovered from the CNS and spleen from B7-H1KO mice infected intracranially and then treated with control poly IgG antibody (black outline) or anti-PD-1 (G4; purple) on day 3 post-infection. CNS infiltrating lymphocytes and spleen cells were analyzed by flow cytometry on day 6 (Ctrl. IgG n = 4, anti-PD-1 n = 5) and day 35 (Ctrl. IgG n = 5, anti-PD-1 n = 5) post-infection. (B) CD8+ cells in (A) assessed for VP2+ specificity and PD-1 expression and (C) the TRM marker CD103. Significant by t-test p < 0.05 *Control IgG versus anti-PD-1 or CD8+ VP2+ versus CD8+ VP2−.
To further interrogate the CD8+ memory populations, we analyzed CD8+ VP2+ cells by flow cytometry to determine how PD-1 blockade influences the accumulation of virus-specific and non-specific CD8+ PD-1+ cells in the CNS and spleen. We found that the populations of PD-1+ cells in the CD8+ VP2+ and the CD8+ VP2− subsets were significantly reduced using anti-PD-1 antibody demonstrating that PD-1 blockade reduces the CD8+ PD-1+ population in both the CNS and the spleen at the 6 day acute time point. After 35 days, the differences between the anti-PD-1 and control IgG groups resolve in the CNS and spleen (Figure 7B). Furthermore, the expression of the TRM marker CD103 is consistent with the retention of a CD103− population of cells in the CNS of B7-H1KO animals that is not influenced by systemic treatment with PD-1 blockade (Figure 7C). Of interest, the expression levels of PD-1 in the VP2− populations in both treatment groups at day 35 were increased in relation to the VP2+ group suggesting that the accumulation of the CD8+ CD103− PD-1high population in the CNS was not influenced by PD-1 blockade (Figures 7B,C). We hypothesize that the inability of the anti-PD-1 antibody to access the CNS through the blood–brain barrier may prevent therapeutic targeting of PD-1 expressing cells in the CNS.
Discussion
Although much attention has been focused on the generation of tissue resident memory T-cells (TRM) in protection from infection or cancer, less is known about how these cells accumulate in distinct niches throughout the body. In this study, we identified a novel role for the immune checkpoint ligand B7-H1 in the promotion of TRM accumulation in the CNS after intracranial viral infection with TMEV. We find that B7-H1 expression in host tissues is important for restricting the inflammatory PD-1high CD103− CD8+ T-cells that limit TRM in the CNS. We found a deficit in the quality and quantity of virus-specific TRM in the CNS of B7-H1 deficient hosts after intracranial infection with TMEV, a virus that is ultimately cleared by CD8+ T-cell responses to virus antigens. Similarly, others have reported a reduction of TRM in the brain of B7-H1KO mice following infection with the herpes virus murine cytomegalovirus (MCMV) (36). Our findings suggest a new role for B7-H1 within the CNS which includes promotion of TRM maintenance through restriction of PD-1high CD103− CD8+ T-cells that accumulate in the absence of B7-H1.
The interaction of cell surface PD-1 expressed by CD8+ T-cells with the PD-1 ligand B7-H1 is important for resolving CD8+ T-cell responses and preventing sustained immunopathology. One mechanism by which T cell responses are resolved is through the induction of T-cell apoptosis induced by PD-1 ligation in tissues that express B7-H1 (13). Consistent with this hypothesis, we find that B7-H1 may be involved in the depletion of non-specified inflammatory PD-1high CD8+ T-cells in the CNS following viral infection. One hallmark of these T-cells is high expression of PD-1, suggesting that in the absence of ligand B7-H1, inflammatory CD8+ T-cells with high levels of PD-1 would compete for long-term residence with virus-specific TRM in the CNS post-infection. Importantly, these PD-1high CD8+ T-cells may regulate the accumulation of virus-specific TRM. One potential hypothesis is that B7-H1 limits the accumulation and survival of low-affinity or antigen non-specific CD8+ T-cells following clearance of infections in order to provide space for tissue resident T-cells that are protective against secondary infections, a finding consistent with an associated regulatory function of PD-1 expressing cells (37).
We found that the increased population of CD103− CD8+ T-cells observed in B7-H1KO mice produced IFNγ upon stimulation. It is plausible that IFNγ produced by CD103− pro-inflammatory T-cells suppressed the accumulation of TRM in B7-H1KO mice. The role of IFNγ in suppression of memory CD8+ T-cells has been reported in lung following influenza infection (38). Although IFNγ does not directly inhibit T-cell proliferation, it suppresses memory T-cell generation through two processes, including the induction of activation-induced cell death of effector cells (39, 40) and through activation of T-bet and its subsequent inhibition of IL-7 receptor expression (41). A consequence of IL-7 receptor signaling in T-cells is reduced levels of bcl-2, a pro-survival molecule needed for memory T-cell survival. Further, low expression of bcl-2 was observed in CD103+ TRM cells in the brain of B7-H1KO mice following MCMV infection (36). Together, these studies suggest that IFNγ produced by PD-1high CD103− pro-inflammatory T-cells may contribute to the decreased number of TRM in the CNS of B7-H1KO mice. Future studies are warranted to determine how this unique population of PD-1high inflammatory CD8+ T-cells modulates virus antigen-specific TRM.
Previous studies have demonstrated that intracranial virus infection results in the accumulation and maintenance of a persistent population of activated CD8+ T-cells that persist in the absence of continued virus replication (42). Bergmann et al. found that both antigen specific and non-specific cells accumulate in the CNS after virus clearance with limited cytolytic and pro-inflammatory activity (43). Similarly, we identified two unique populations of CNS antigen-specific and -non-specific CD8+ T-cells that can be discriminated by their expression of PD-1. We find that both inflammatory non-specific PD-1high CD8 T-cells and PD-1low antigen-specific TRM were sensitive to the presence of B7-H1. Further, we found that the TRM within B7-H1WT or B7-H1KO CNS tissues express a low level of PD-1 compared to inflammatory PD-1high CD103− CD8+ T-cells. This finding suggests that the low level PD-1 expression may prevent TRM in the CNS from a strong engagement with B7-H1 that would lead to T-cell apoptosis or exhaustion. It is not clear why PD-1 levels are different in these two T-cell subsets, but cues within the tissue microenvironment along with TCR signals may differentially regulate PD-1 expression (44, 45). It should be noted that stimulation through a strong T-cell receptor interaction may not be the only signal promoting PD-1 expression, since antigen-specific TRM express low levels of PD-1, while antigen-non-specific CD103− CD8+ T-cells expressed higher levels of PD-1 compared to TRM after viral infection.
Although PD-1 has been proposed as a biomarker for exhausted CD8+ T-cells that develop during chronic viral infections (14, 46, 47), PD-1 expression per se does not always lead to T cell exhaustion and its absence is not necessary for the prevention of exhaustion (48). Since the inflammatory PD-1high CD103− CD8+ T-cells can persist in the CNS for extended periods post-infection in the absence of B7-H1, suggesting that the expression of PD-1, even at high levels, did not affect their residence in the CNS. A possible scenario following viral infection in the CNS is that host B7-H1 eliminates inflammatory PD-1high CD103− CD8+ T-cells in order to preserve the virus specific TRM that are necessary for protection from secondary infection. It is also possible that the PD-1high inflammatory CD8+ T cells represent a new subset of regulatory CD8+ T-cells that increases in B7-H1 deficient environments (49). The similarities observed between the inflammatory CD8+ T-cells here and regulatory CD8+ T-cells warrants further investigation.
The development of immunity in vital organs must be carefully regulated to provide sufficient control from long lasting pathology and protection of tissues with limited renewal capacity. The regulation of B7-H1 expression in the brain and how it can affect the long-term accumulation of CD8+ T-cells has implications for studies of immune-mediated neurologic disease. Although baseline B7-H1 expression levels are thought to be low in the CNS, it is often upregulated after injury or infection (50–52). Our study demonstrates that the level of B7-H1 expression in host tissues can determine the quality of the CD8+ T-cell population that occupies the CNS. Previous evidence demonstrates that brain resident CD8+ T-cells accumulate with age (53). However, it is unclear under what consequences these CD8+ T-cells reach the brain and accumulate. One could speculate that over a lifetime, clinical or subclinical CNS infections and their associated inflammatory cascades could lead to an accumulation of CD8+ TRM in the brain and spinal cord. However, aging is also associated with deterioration of the blood brain barrier and an increase in immunosurvellance that allows CD8+ T-cells to enter the CNS under non-inflammatory conditions (54). Nevertheless, both inflammatory and non-inflammatory mechanisms can promote the accumulation of TRM. Our findings here suggest that the level of B7-H1 expressed by host tissues can have consequences for the quality and quantity of the CD8+ T-cell subsets that accumulate within CNS tissues.
The discovery of immune checkpoints and the use of reagents that target these pathways have revolutionized cancer immunotherapy. However, their development and use in clinical trials for chronic viral infections or for long-term tumor control has been met with mixed success (55, 56). Similarly here we find that PD-1antibody fails to deplete or influence the PD-1high expressing CNS resident population of CD8+ cells or to enhance TRM accumulation. The major difference between targeting B7-H1/PD-1 pathway in cancer and in viral infection is the cellular sources of B7-H1 or PD-1. In the tumor tissues, the blockade of PD-1/B7-H1 is mainly aimed at blocking the interaction between PD-1+ effector T cells and B7-H1+ tumor cells. However, in the infected tissues, since both effector and regulatory cells express PD-1, the net outcome of the blockade of PD-1/B7-H1 would be determined by a balance of the functions of effector cells and immune regulatory cells. To that end, our study identified a unique population of PD-1high CD103− CD8+ T-cells as a potential new cellular target of B7-H1/PD-1 therapy in chronic viral infections. However, since unique barriers exist in protected tissues like the CNS, the use of antibody based therapies will be challenging (57). A prolonged blockade of B7-H1 or PD-1 may provide a risk for the accumulation of the inflammatory PD-1high CD8+ T cells that eventually would impair the maintenance of protective TRM in particular tissues. To reduce the possibility of compromising long-term protection mediated by tissue resident CD8+ T cells, the timing and location of B7-H1/PD-1 blockade as a therapy for virus vaccines or infection should be carefully optimized in order to support the generation and maintenance of a pool of antigen specific CD8+ TRM.
In summary, our study reveals a new function of B7-H1 for the promotion of TRM maintenance by limiting the accumulation of inflammatory PD-1high CD103− CD8+ T cells in the CNS. This finding extends our understanding of the maintenance of TRM in the brain and provides new insights for optimizing B7-H1/PD-1 blockade to promote long-term protection from cancer or virus infection.
Ethics Statement
This study was carried out in accordance with the recommendations of the National Institute of Health and the Mayo Clinic Department of Comparative Medicine. The protocol was approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Author Note
One Sentence Summary: The immune checkpoint molecule B7-H1 modulates the accumulation of tissue resident CD8 T-cells.
Author Contributions
HD and KP conceived the project and were responsible for research design, data analysis, and drafting the manuscript. KP and MB performed the experiments and data acquisition. SH helped with the animal models.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the National Institute of Allergy and Infectious Disease grant R01-AI095239 (HD).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01532/full#supplementary-material.
References
1. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol (2013) 31:137–61. doi:10.1146/annurev-immunol-032712-095954
2. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi:10.1038/44385
3. Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science (2001) 291:2413–7. doi:10.1126/science.1058867
4. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10:524–30. doi:10.1038/ni.1718
5. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med (2010) 207:553–64. doi:10.1084/jem.20090858
6. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol (2011) 187:5510–4. doi:10.4049/jimmunol.1102243
7. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107:17872–9. doi:10.1073/pnas.1010201107
8. Iijima N, Iwasaki A. Tissue instruction for migration and retention of TRM cells. Trends Immunol (2015) 36:556–64. doi:10.1016/j.it.2015.07.002
9. Carbone FR. Tissue-resident memory T cells and fixed immune surveillance in nonlymphoid organs. J Immunol (2015) 195:17–22. doi:10.4049/jimmunol.1500515
10. Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol (2017) 17(3):179–94. doi:10.1038/nri.2016.144
11. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol (2015) 16:406–14. doi:10.1038/ni.3108
12. Schenkel JM, Fraser KA, Casey KA, Beura LK, Pauken KE, Vezys V, et al. IL-15-independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J Immunol (2016) 196:3920–6. doi:10.4049/jimmunol.1502337
13. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity (2004) 20:327–36. doi:10.1016/S1074-7613(04)00050-0
14. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. doi:10.1038/nature05115
15. Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer (2007) 7:95–106. doi:10.1038/nrc2051
16. Rodriguez M, Leibowitz J, David CS. Susceptibility to Theiler’s virus-induced demyelination. Mapping of the gene within the H-2D region. J Exp Med (1986) 163:620–31. doi:10.1084/jem.163.3.620
17. Johnson AJ, Njenga MK, Hansen MJ, Kuhns ST, Chen L, Rodriguez M, et al. Prevalent class I-restricted T-cell response to the Theiler’s virus epitope Db:VP2121-130 in the absence of endogenous CD4 help, tumor necrosis factor alpha, gamma interferon, perforin, or costimulation through CD28. J Virol (1999) 73:3702–8.
18. Mendez-Fernandez YV, Johnson AJ, Rodriguez M, Pease LR. Clearance of Theiler’s virus infection depends on the ability to generate a CD8+ T cell response against a single immunodominant viral peptide. Eur J Immunol (2003) 33:2501–10. doi:10.1002/eji.200324007
19. Duncan DS, Miller SD. CNS expression of B7-H1 regulates pro-inflammatory cytokine production and alters severity of Theiler’s virus-induced demyelinating disease. PLoS One (2011) 6:e18548. doi:10.1371/journal.pone.0018548
20. Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (1996) 274:94–6. doi:10.1126/science.274.5284.94
21. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res (2005) 65:1089–96.
22. Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc (2014) 9:209–22. doi:10.1038/nprot.2014.005
23. Cumba Garcia LM, Huseby Kelcher AM, Malo CS, Johnson AJ. Superior isolation of antigen-specific brain infiltrating T cells using manual homogenization technique. J Immunol Methods (2016) 439:23–8. doi:10.1016/j.jim.2016.09.002
24. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med (2016) 213:1571–87. doi:10.1084/jem.20151916
25. Fu JL, Stein S, Rosenstein L, Bodwell T, Routbort M, Semler BL, et al. Neurovirulence determinants of genetically engineered Theiler viruses. Proc Natl Acad Sci U S A (1990) 87:4125–9. doi:10.1073/pnas.87.11.4125
26. Bell MP, Pavelko KD. Enhancing the tumor selectivity of a picornavirus virotherapy promotes tumor regression and the accumulation of infiltrating CD8+ T cells. Mol Cancer Ther (2016) 15:523–30. doi:10.1158/1535-7163.MCT-15-0459
27. Pavelko KD, Bell MP, Karyampudi L, Hansen MJ, Allen KS, Knutson KL, et al. The epitope integration site for vaccine antigens determines virus control while maintaining efficacy in an engineered cancer vaccine. Mol Ther (2013) 21:1087–95. doi:10.1038/mt.2013.52
28. Rodriguez M, Leibowitz JL, Powell HC, Lampert PW. Neonatal infection with the Daniels strain of Theiler’s murine encephalomyelitis virus. Lab Invest (1983) 49:672–9.
29. Wonderlich J, Shearer G, Livingstone A, Brooks A. Induction and measurement of cytotoxic T lymphocyte activity. Curr Protoc Immunol (2006) Chapter 3:Unit 3.11. doi:10.1002/0471142735.im0311s72
30. Pulko V, Harris KJ, Liu X, Gibbons RM, Harrington SM, Krco CJ, et al. B7-H1 expressed by activated CD8 T cells is essential for their survival. J Immunol (2011) 187:5606–14. doi:10.4049/jimmunol.1003976
31. Rodriguez M, David CS. Demyelination induced by Theiler’s virus: influence of the H-2 haplotype. J Immunol (1985) 135:2145–8.
32. Myoung J, Hou W, Kang B, Lyman MA, Kang JA, Kim BS. The immunodominant CD8+ T cell epitope region of Theiler’s virus in resistant C57BL/6 mice is critical for anti-viral immune responses, viral persistence, and binding to the host cells. Virology (2007) 360:159–71. doi:10.1016/j.virol.2006.09.045
33. Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol (2012) 189:3462–71. doi:10.4049/jimmunol.1201305
34. Gibbons RM, Liu X, Pulko V, Harrington SM, Krco CJ, Kwon ED, et al. B7-H1 limits the entry of effector CD8(+) T cells to the memory pool by upregulating Bim. Oncoimmunology (2012) 1:1061–73. doi:10.4161/onci.20850
35. Nitayaphan S, Toth MM, Roos RP. Neutralizing monoclonal antibodies to Theiler’s murine encephalomyelitis viruses. J Virol (1985) 53:651–7.
36. Prasad S, Hu S, Sheng WS, Chauhan P, Singh A, Lokensgard JR. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J Neuroinflammation (2017) 14:82. doi:10.1186/s12974-017-0860-3
37. Dai H, Wan N, Zhang S, Moore Y, Wan F, Dai Z. Cutting edge: programmed death-1 defines CD8+CD122+ T cells as regulatory versus memory T cells. J Immunol (2010) 185:803–7. doi:10.4049/jimmunol.1000661
38. Prabhu N, Ho AW, Wong KH, Hutchinson PE, Chua YL, Kandasamy M, et al. Gamma interferon regulates contraction of the influenza virus-specific CD8 T cell response and limits the size of the memory population. J Virol (2013) 87:12510–22. doi:10.1128/JVI.01776-13
39. Liu Y, Janeway CA Jr. Interferon gamma plays a critical role in induced cell death of effector T cell: a possible third mechanism of self-tolerance. J Exp Med (1990) 172:1735–9. doi:10.1084/jem.172.6.1735
40. Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med (2002) 196:999–1005. doi:10.1084/jem.20020666
41. Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, et al. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med (2007) 204:2015–21. doi:10.1084/jem.20070841
42. Hawke S, Stevenson PG, Freeman S, Bangham CR. Long-term persistence of activated cytotoxic T lymphocytes after viral infection of the central nervous system. J Exp Med (1998) 187:1575–82. doi:10.1084/jem.187.10.1575
43. Bergmann CC, Altman JD, Hinton D, Stohlman SA. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J Immunol (1999) 163:3379–87.
44. Blackburn SD, Crawford A, Shin H, Polley A, Freeman GJ, Wherry EJ. Tissue-specific differences in PD-1 and PD-L1 expression during chronic viral infection: implications for CD8 T-cell exhaustion. J Virol (2010) 84:2078–89. doi:10.1128/JVI.01579-09
45. Vargas-Inchaustegui DA, Xiao P, Hogg AE, Demberg T, McKinnon K, Venzon D, et al. Immune targeting of PD-1(hi) expressing cells during and after antiretroviral therapy in SIV-infected rhesus macaques. Virology (2013) 447:274–84. doi:10.1016/j.virol.2013.09.015
46. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi:10.1038/nature04444
47. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol (2007) 81:4215–25. doi:10.1128/JVI.02844-06
48. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med (2015) 212:1125–37. doi:10.1084/jem.20142237
49. Zozulya AL, Ortler S, Fabry Z, Sandor M, Wiendl H. The level of B7 homologue 1 expression on brain DC is decisive for CD8 Treg cell recruitment into the CNS during EAE. Eur J Immunol (2009) 39:1536–43. doi:10.1002/eji.200839165
50. Phares TW, Ramakrishna C, Parra GI, Epstein A, Chen L, Atkinson R, et al. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J Immunol (2009) 182:5430–8. doi:10.4049/jimmunol.0803557
51. Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med (2003) 198:71–8. doi:10.1084/jem.20022119
52. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. PD-L1 enhances CNS inflammation and infarct volume following experimental stroke in mice in opposition to PD-1. J Neuroinflammation (2013) 10:111. doi:10.1186/1742-2094-10-111
53. Ritzel RM, Crapser J, Patel AR, Verma R, Grenier JM, Chauhan A, et al. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol (2016) 196:3318–30. doi:10.4049/jimmunol.1502021
54. Gemechu JM, Bentivoglio M. T cell recruitment in the brain during normal aging. Front Cell Neurosci (2012) 6:38. doi:10.3389/fncel.2012.00038
55. Barouch DH, Deeks SG. Immunologic strategies for HIV-1 remission and eradication. Science (2014) 345:169–74. doi:10.1126/science.1255512
56. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med (2016) 8:328rv324. doi:10.1126/scitranslmed.aad7118
Keywords: Theiler’s murine encephalomyelitis virus, B7-H1, programmed death-1, CD8, tissue resident memory, picornaviruses, immune checkpoint, CTL, viruses
Citation: Pavelko KD, Bell MP, Harrington SM and Dong H (2017) B7-H1 Influences the Accumulation of Virus-Specific Tissue Resident Memory T Cells in the Central Nervous System. Front. Immunol. 8:1532. doi: 10.3389/fimmu.2017.01532
Received: 24 August 2017; Accepted: 27 October 2017;
Published: 09 November 2017
Edited by:
Kamal M. Khanna, NYU Langone Medical Center, United StatesReviewed by:
Kimberly Sue Schluns, University of Texas MD Anderson Cancer Center, United StatesJason Kyle Whitmire, University of North Carolina at Chapel Hill, United States
Copyright: © 2017 Pavelko, Bell, Harrington and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin D. Pavelko, cGF2ZWxrby5rZXZpbkBtYXlvLmVkdQ==;
Haidong Dong, ZG9uZy5oYWlkb25nQG1heW8uZWR1