 Jenna M. Antonucci
Jenna M. Antonucci Corine St. Gelais
Corine St. Gelais Li Wu
Li Wu- Center for Retrovirus Research, Department of Veterinary Biosciences, The Ohio State University, Columbus, OH, United States
The innate immune response constitutes the first cellular line of defense against initial HIV-1 infection. Immune cells sense invading virus and trigger signaling cascades that induce antiviral defenses to control or eliminate infection. Professional antigen-presenting cells located in mucosal tissues, including dendritic cells and macrophages, are critical for recognizing HIV-1 at the site of initial exposure. These cells are less permissive to HIV-1 infection compared to activated CD4+ T-cells, which is mainly due to host restriction factors that serve an immediate role in controlling the establishment or spread of viral infection. However, HIV-1 can exploit innate immune cells and their cellular factors to avoid detection and clearance by the host immune system. Sterile alpha motif and HD-domain containing protein 1 (SAMHD1) is the mammalian deoxynucleoside triphosphate triphosphohydrolase responsible for regulating intracellular dNTP pools and restricting the replication of HIV-1 in non-dividing myeloid cells and quiescent CD4+ T-cells. Here, we review and analyze the latest literature on the antiviral function of SAMHD1, including the mechanism of HIV-1 restriction and the ability of SAMHD1 to regulate the innate immune response to viral infection. We also provide an overview of the dynamic interplay between HIV-1, SAMHD1, and the cell-intrinsic antiviral response to elucidate how SAMHD1 modulates HIV-1 infection in non-dividing immune cells. A more complete understanding of SAMHD1’s role in the innate immune response to HIV-1 infection may help develop stratagems to enhance its antiviral effects and to more efficiently block HIV-1 replication and avoid the pathogenic result of viral infection.
Introduction
Innate immunity is the cell-intrinsic defense mechanism that senses incoming pathogens and is characterized by type-I interferon (IFN-I) induction and the release of inflammatory cytokines that upregulate antiviral IFN-stimulated genes (ISGs) (1, 2). The activation of the innate response to pathogens is dependent on cellular pattern recognition receptors (PRRs) that detect pathogen-associated molecular patterns (PAMPs), including viral structures or nucleic acids. Interferon-inducible protein IFI16 and cyclic GMP-AMP synthase (cGAS) are cytosolic sensors of HIV-1 that detect viral DNA (3, 4). Recognition of PAMPs results in induction of IFN-I and ISGs to control initial infection and spread, while the concomitant induction of the inflammatory response and cytokines can initiate adaptive immune responses (5, 6). Modulation of IFN-I activation is essential for viral clearance. However, overstimulation of IFN pathways can lead to inflammatory autoimmune disease (7).
HIV-1 is sensitive to ISGs and the IFN-induced antiviral response; so it is not surprising that HIV-1 is a poor inducer of IFN (8). HIV-1 benefits from evading innate immune activation and utilizes a variety of tactics to escape detection (9, 10). Professional antigen-presenting cells located in mucosal tissues, including dendritic cells (DCs) and macrophages, are critical for recognizing HIV-1 at the site of initial exposure. However, these cells are less permissive to HIV-1 infection compared to activated CD4+ T-cells, mainly due to host restriction factors that control the establishment or spread of viral infection. Several host proteins can restrict HIV-1 at various points in the viral lifecycle, including APOBEC proteins, TRIM5α, and tetherin (11–13). However, HIV-1 can exploit innate immune cells and their cellular factors to avoid detection and clearance by the host immune system (13).
SAMHD1 is host protein capable of blocking replication of retroviruses and several DNA viruses in cells (14–18). SAMHD1 is constitutively expressed at various levels in all cell types and highly expressed in myeloid lineage and resting CD4+ T-cells (14, 15, 19). IFN-I treatment increases SAMHD1 expression in certain cell types with low endogenous SAMHD1 levels (20, 21). SAMHD1 has been implicated as a negative regulator of the IFN-I inflammatory response (22–24), however, the underlying mechanism is not fully understood. While HIV-2 encodes the SAMHD1 antagonist Vpx, the more pathogenic HIV-1 does not. It was hypothesized that HIV-1 lacks a countermeasure against SAMHD1 because it is beneficial for infection. In this review, we will discuss the contributions of SAMHD1 to both the direct restriction of HIV-1 and to the modulation of the antiviral innate response and to analyze the hypothesis that HIV-1 restriction by SAMHD1 leads to a diminished induction of innate immunity.
Innate Immune Sensing of HIV-1
Although HIV-1 can be sensed by the innate immune system, the prevailing theory is that HIV-1 avoids immune surveillance through poor replication in immune cells causing ineffective triggering of innate cytosolic sensors (25). Several studies have identified the molecular basis of cytosolic sensors important for targeting viral pathogens. Here, we focus on HIV-1 DNA as a trigger of the innate antiviral response. After sensing viral DNA, cGAS generates the second messenger, cyclic guanosine monophosphate-adenosine monophosphate (26–28), that activates the stimulator of IFN genes (STING) (6). STING activation leads to phosphorylation of TANK-binding kinase 1 (TBK1) and the subsequent phosphorylation and dimerization of IFN-regulatory transcription factors IRF3 and IRF7. Nuclear translocation of the IRF3/IRF7 homo-or-hetero dimers will activate IFN-I gene expression (Figure 1). This signaling cascade results in an upregulation of IFN-I and ISGs as a defense against viral infection (29, 30). Reverse transcribed HIV-1 DNA was identified as the trigger to the cGAS-STING pathway (3). Although cGAS is the primary sensor of cytosolic viral DNA, IFI16 can also act as a sensor of HIV-1 single-stranded DNA that induces an IFN-β response in macrophages by a cGAS-STING-dependent pathway (4).
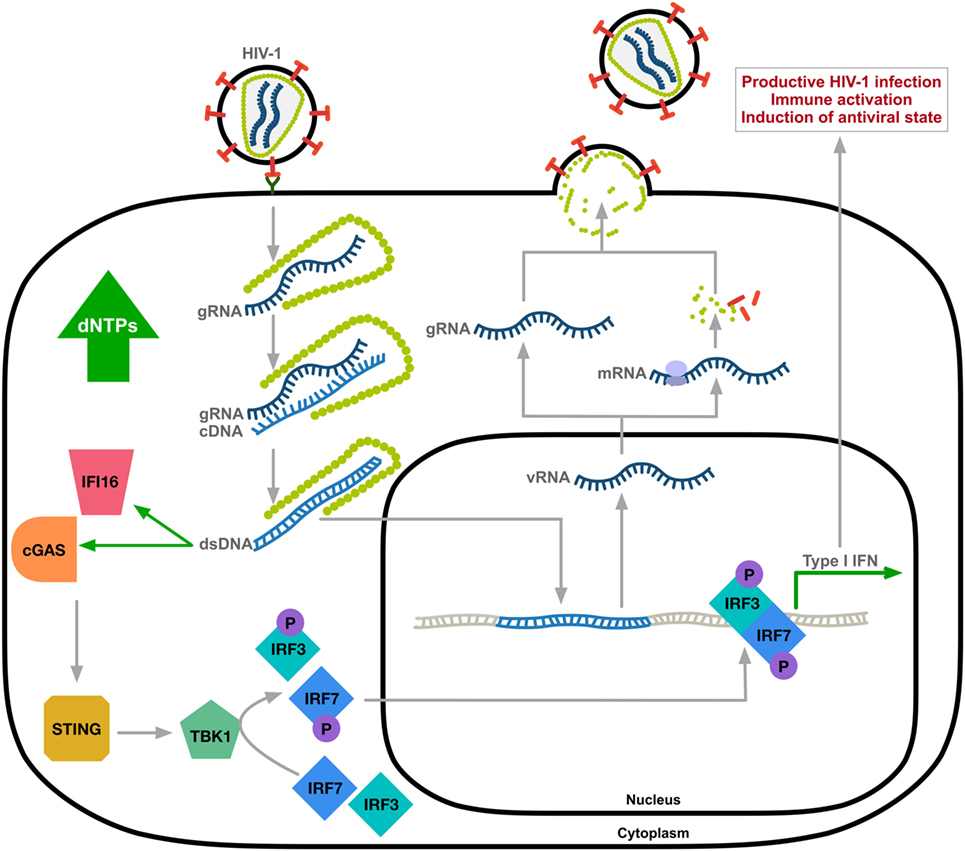
Figure 1. Innate immune sensing of HIV-1 DNA. HIV-1 undergoes uncoating through the interaction between viral capsid and host factors (31, 32). Reverse transcribed HIV-1 DNA, mainly abortive transcripts, activates cytosolic DNA sensors IFI16 and cyclic GMP-AMP synthase (cGAS) resulting in TANK-binding kinase 1 (TBK1)-mediated phosphorylation and nuclear translocation of hetero-or-homo dimers of interferon regulatory factor-3 (IRF3) and IRF7 and induction of type-I IFN response. Expression of ISGs allows for immune activation and the induction of an antiviral state of the cell. gRNA, HIV-1 genomic RNA; cDNA, complementary DNA; vRNA, viral RNA; dsDNA, double-stranded DNA; STING, stimulator of IFN genes; the letter P indicates phosphorylation.
Introduction to SAMHD1
Human SAMHD1 is a 626-amino acid protein containing an N-terminal nuclear localization signal followed by a sterile-alpha motif and histidine/aspartic acid (HD) domain. SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase (dNTPase) (33, 34) that converts dNTPs into the constituent deoxynucleoside and inorganic triphosphate upon stimulation by dGTP or GTP (33–35). SAMHD1 and ribonuclease reductase, the enzyme responsible for de novo dNTP synthesis through the conversion of ribonucleotide diphosphates to deoxyribonucleotides (36), are allosterically regulated to achieve balanced intracellular dNTP levels in a cell-cycle-dependent manner (37). During G1 to S-phase transition in actively proliferating cells, ribonuclease reductase expression increases, leading to expansion of the dNTP pool to facilitate DNA synthesis (38, 39). The activity of SAMHD1 is activated by high dNTP levels, and degradation of nucleic acids in the absence of DNA replication protects the cell from innate immune activation and cancer development (40, 41). Mutations in SAMHD1 that affect its enzyme activity are associated with Aicardi-Goutières syndrome (AGS), an encephalopathic autoimmune disease characterized by symptoms mimicking chronic viral infection (22). The accumulation of intracellular dNTPs caused by mutations in the genes encoding proteins involved in nucleic acid metabolism, including SAMHD1 and TREX1 (42), are sensed by PRRs, resulting in aberrant production of IFN-I (43). AGS patients present with increased production of IFN-α, the chemokine most characteristic of congenital virus infection. AGS patients with SAMHD1 mutations can present with signs of lupus erythematosus, with many symptoms mimicking those of HIV-1 infection (22, 44). Furthermore, cells isolated from AGS patients with homozygous SAMHD1 mutation revealed that SAMHD1-deficient monocytes supported productive infection by HIV-1 (20), suggesting a link between SAMHD1 function in both autoimmunity and HIV-1 restriction.
Long interspersed element 1 (LINE-1) is the only autonomous and active human retroelement capable of producing new genomic insertions through its endogenous endonuclease and reverse transcriptase activities (45, 46). A study on AGS-related SAMHD1 mutations indicate that all disease-related mutations reduced LINE-1 inhibition in dividing cells (47). Recent work suggests that SAMHD1 potently blocks LINE-1 transposition in cycling cells by triggering the sequestration of LINE-1 ORF1p into stress granules (48). Impaired inhibition of LINE-1 retrotransposition may lead to triggering of the autoimmune response by stimulating toll-like receptors (TLRs) (49), although this has not been confirmed. Impaired dNTPase activity and LINE-1 suppression by mutant SAMHD1 could explain the chronic inflammatory response characteristics of AGS disease. These studies outlining the pathogenic effect of SAMHD1 deficiency on autoimmune disease implicate SAMHD1 as a negative regulator of the innate immune system.
SAMHD1-Mediated HIV-1 Restriction
HIV-1 replicates inefficiently in non-diving cells, such as quiescent CD4+ T-cells, DCs, and monocytes. HIV-1 infection can be enhanced in these cells by Vpx, an accessory protein encoded by HIV-2 and certain lineages of simian immunodeficiency viruses (SIVs) (50, 51). This hinted at the existence of a cellular restriction factor counteracted by Vpx (50). SAMHD1 was identified as the mystery HIV-1 restriction factor by a mass spectrometry analysis of cellular proteins immunoprecipitated from cells expressing Vpx (14, 15). Vpx interacts with the C-terminal domain of SAMHD1, thereby initiating proteasomal degradation by an E3 ubiquitin ligase complex, and relieving SAMHD1-mediated lentiviral restriction (14, 15, 52, 53).
The mechanism and modulation of SAMHD1-mediated HIV-1 restriction is an area of intense scrutiny (Figure 2). Overexpression of SAMHD1 in PMA-treated monocytic U937 cells results in a depletion of dNTP levels (54). It was later confirmed that SAMHD1 restricts the replication of retroviruses and several DNA viruses by depleting the concentration of intracellular dNTPs to levels insufficient to support viral DNA synthesis (14–18, 54, 55). Structural studies strengthened a model of nucleotide-dependent tetramer assembly of SAMHD1 (56–58), where GTP binds to guanine-specific allosteric sites and dNTP binds to non-specific activator sites, initiating the formation of enzymatically active tetramers with the catalytic core of the HD domain (33, 34, 37, 59). Moreover, binding of single-stranded nucleic acids (ssNAs) to the dimer–dimer interface of SAMHD1 inhibits the formation of the catalytically active tetramer (60).
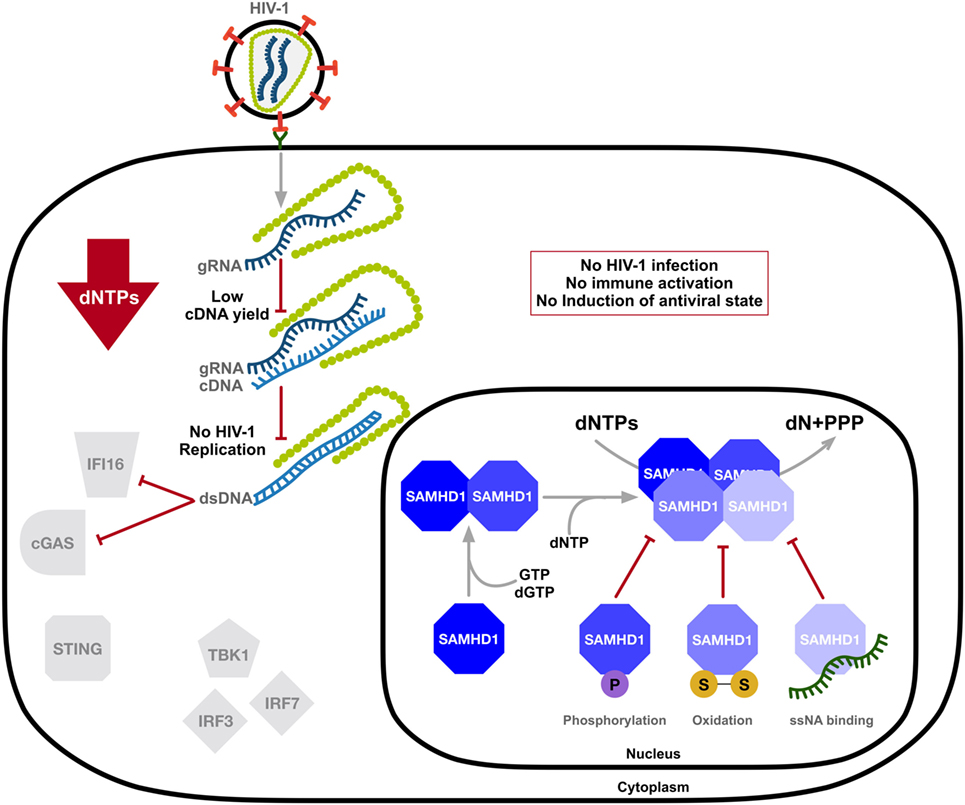
Figure 2. SAMHD1 negatively regulates the innate immune sensing of HIV-1 DNA. SAMHD1 blocks HIV-1 infection through intracellular dNTP depletion, thus preventing the accumulation of viral DNA accessible to sensing by IFI16 and cyclic GMP-AMP synthase (cGAS) and the activation of the type-I interferon (IFN-I) response. The dNTPase activity of SAMHD1 is structurally regulated. Consecutive binding of dGTP/GTP and any dNTP to two allosteric sites provokes formation of the catalytically active tetramer, which can be destabilized by phosphorylation, oxidation, or the binding of single-stranded nucleic acids (ssNAs). dN, deoxynucleosides; PPPs, triphosphate; two linked letters S indicate the disulfide bond.
As SAMHD1 is also highly expressed in activated CD4+ T-cells that support productive infection, several studies demonstrated posttranslational modification as a means of mechanistic regulation of SAMHD1 function in restricting HIV-1. SAMHD1 is phosphorylated at several residues; however, phosphorylation of threonine 592 was identified as essential for the negative modulation of its HIV-1 restriction activity (61–65) and tetramer formation (66, 67). SAMHD1 is phosphorylated by cyclin-dependent kinase 1 (CDK1) and CDK2 in complex with cell cycle regulatory protein cyclin A. This regulation of SAMHD1 function is associated with the cell cycle, as CDK1 and cyclin A are highly expressed in dividing cells. Furthermore, S-phase requires elevated dNTP levels, indicating modulation of the dNTPase activity of SAMHD1 during the cell cycle (64). SAMHD1 protein levels may be altered during various stages of the cell cycle depending on different cell types (68, 69). Interestingly, proliferation-induced oxidation of SAMHD1 by hydrogen peroxide reversibly inhibits its dNTPase activity through the formation of tetramer-inhibiting disulfide bonds (70), suggesting a dynamic structure-based regulatory mechanism of SAMHD1’s dNTPase activity that is influenced by the cell cycle (Figure 2).
Although the accepted consensus is that SAMHD1 restricts HIV-1 infection through the depletion of intracellular dNTPs, several studies suggested the existence of an additional yet-undiscovered mechanism of SAMHD1-mediated retroviral restriction. This undefined antiviral activity appears to be dependent on phosphorylation (61, 63, 65) and is not fully dependent on low dNTP levels (71). SAMHD1 acts as a ssNA binding protein that degrades single-stranded DNA and RNA via a metal-dependent 3′–5′ exonuclease activity in vitro (72–74). It has been suggested that SAMHD1 utilizes its nucleic acid binding potential to exert a ribonuclease activity against incoming HIV-1 genomic RNA in a phosphorylation-dependent manner (75). SAMHD1 was shown to restrict retroviruses though degradation of HIV-1 RNA in human monocyte-derived macrophages (MDMs), monocytes, and CD4+ T-cells (75, 76). It was proposed that SAMHD1 degrades incoming HIV-1 gRNA, thereby restricting infection and preventing innate immune sensing of viral nucleic acids. However, recent studies have been unable to confirm the controversial findings (55, 77–79). As a nuclear-localized protein (80), incoming viral genomic RNA would be inaccessible by SAMHD1 for hydrolysis. Additional studies showed that dNTPase inactive SAMHD1 mutant retained exonuclease activities in vitro, indicating the exonuclease activity could not be attributed to the known dNTP-binding active site (77). Seamon et al. (77) suggested that the nuclease activity attributed to SAMHD1 was due to contamination during purification. Cell-based assays also failed to recapitulate the findings, thereby confirming the lack of SAMHD1 RNase activity to restrict HIV-1 in infected cells (55, 78). Ryoo et al. suggested that the differences in experimental conditions are responsible for the conflicting results, including a shorter infection time and the use of RNaseH-defective reverse transcriptase (81). They further identified SAMHD1 as a phosphorolytic not hydrolytic ribonuclease (82).
The Intersection of HIV-1, SAMHD1, and the Innate Antiviral Response
SAMHD1 cDNA was originally identified as a ortholog of the mouse IFN-γ-induced gene Mg11 in human DCs (83). A link to the innate immune response was strengthened by the discovery that cytokines, including toll-like agonists and IFNs, can induce SAMHD1 expression (84, 85). Cell lines treated with IFN-I (21, 86) and human primary monocytes treated with IFN-α and IFN-γ (20, 84, 87) show enhanced expression of SAMHD1. While SAMHD1 is highly expressed in MDMs, monocyte-derived dendritic cells (MDDCs), and primary CD4+ T-cells, IFN treatment does not increase SAMHD1 protein levels further (21, 88–90). However, treatment of MDMs and MDDCs with IFN-I results in reduced phosphorylation of SAMHD1 at residue T592 (61), indicating a shift from catalytically inactive to active SAMHD1. Interestingly, the SAMHD1 promoter is a direct target of IRF3. The overexpression and activation of IRF3 enhances SAMHD1 promoter activity in HeLa cells (86).
HIV-1 does not trigger a sterilizing immune response (91) and is a poor activator of inflammatory pathways (8), resulting in an impaired response to HIV-1 and the development of persistent infection. The DC response to HIV-1 infection contributes to this dysfunctional immune response (92). Myeloid cells constantly sample the cellular environment to identify pathogens and send out danger signals in the form of IFN-I. DCs are essential for activating the adaptive immune response to infection, as maturation leads to T-cell responses through antigen priming (91, 93). Interestingly, HIV-1 infects DCs without activating an effective antiviral response. As SAMHD1 limits HIV-1 cDNA synthesis in myeloid cells (14, 54), it was hypothesized that degradation of SAMHD1 by Vpx in DCs would result in productive HIV-1 infection and the synthesis of viral proteins that would directly enter antigen presentation, thereby strengthening the T-cell response to infection (94). This could be why the vpx gene was lost from the ancestor of HIV-1 during the coevolution of primate SAMHD1 and lentiviruses (95).
Vpx-mediated degradation of SAMHD1 in DCs leads to enhanced HIV-1 infection, and studies in primary MDMs and MDDCs indicate that Vpx-mediated SAMHD1 degradation results in cGAS stimulation and IRF3 activation (3). Early work suggested that enhanced infection by SAMHD1 depletion leads to DC maturation (94). A study utilizing coculture of autologous activated CD4+ T lymphocytes with SAMHD1-deficient MDDCs infected with primary clinical HIV-1 isolates indicated enhancement of both infection and IFN response (96). Interestingly, cocultured primary T-lymphocytes, but not HIV-1, trigger a decrease in SAMHD1 expression in MDDCs independent of dNTP levels (96). This study suggests that crosstalk between lymphocytes and DCs induces downregulation of SAMHD1 expression, a requirement for stimulation of HIV-1 production in DCs, thereby inducing the innate sensing of HIV-1 and DC maturation (96).
Conversely, recent work indicates that DC maturation, measured by CD83 and CD86 expression, does not occur in SAMHD1-deficient cells due to additional manipulation of the innate immune system by HIV-1 (97). HIV-1 suppresses TLR-induced maturation of DCs independent of SAMHD1 expression, although Vpx-mediated depletion of SAMHD1 enhanced the effect of HIV-1 infection on lipopolysaccharide-induced DC maturation (97). Vesicular stomatitis virus G-protein-pseudotyped HIV-1 suppressed maturation similar to strains containing HIV-1 envelope protein, suggesting that viral replication, not envelope-receptor interactions, is required for suppression of maturation (97). Removing the SAMHD1-mediated block of reverse transcription resulted in a stronger suppression of maturation. Although infection and subsequent innate immune sensing in DCs is blocked by SAMHD1, HIV-1 maintains an additional SAMHD1-independent mechanism of suppressing DC maturation through downregulation of TLRs (97).
Two additional models suggest that, in MDDCs, HIV-1 attempts to hide its genomic RNA and newly synthesized cDNA from cytosolic sensors by obstructing the nucleic acids using viral capsid. The models differ with respect to the effect of recruitment of cellular cyclophillins and cleavage and polyadenylation-specific factor 6 (CPSF6) by capsid. One model suggests increased cyclophillin A (CypA) binding to the capsid increases sensitivity to innate sensing (94), while another proposes CypA binding coordinates uncoating, reverse transcription, and nuclear import of the preintegration complex (98), all to minimize the exposure of viral nucleic acids to cytosolic sensors. Future work is needed to clarify the contribution of CypA and SAMHD1 to the negative regulation of the innate immune response in myeloid cells to provide insight into HIV-1 mechanisms of evasion.
Non-cycling CD4+ T-cells and macrophages are less permissive to HIV-1 because of SAMHD1. However, during HIV-1 infection in vivo, activated CD4+ T-cells and macrophages are infected due to phosphorylation of SAMHD1. Although cytosolic HIV-1 DNA is abundant in these permissive cells, a cell-autonomous IFN response is not triggered (99). This is due at least in part to host protein TREX1. As a single-stranded DNA exonuclease, TREX-1 digests cytoplasmic DNA from retroviral DNA intermediates, thereby preventing the activation of mislocalized DNA by an innate immune sensor (99). Cytosolic HIV-1 DNA is accumulated in HIV-1 infected TREX1-deficient CD4+ T-cells and macrophages, which leads to inhibition of TBK1-dependent IFN-I response (99). This suggests a competition between two DNA sensors: cGAS leading to antiviral effects, and TREX1 leading to enhanced viral replication (100).
Remaining Questions
Although it is clear that HIV-1 utilizes a variety of mechanisms to evade myeloid cell activation, controversial questions still exist. Conflicting reports could be due to the use of different cell types, and the differential use of clinical HIV-1 isolates, replication-competent lab strains, or pseudotyped virus. It is essential to confirm experimental findings with primary cells that accurately recapitulate in vivo mucosal infection sites. Further understanding of the strategies HIV-1 utilizes to evade the innate response will allow for better ideas on how to increase the innate immune response to HIV-1.
The existence of a yet-undiscovered mechanism of HIV-1 restriction that is dependent on phosphorylation cannot be overlooked (63). Pretreatment MDMs with Vpx enhances the rate of HIV-1 cDNA synthesis (101), suggesting that the decrease in reverse transcription kinetics conferred by SAMHD1-mediated modulation of dNTP levels negatively regulates the rate of proviral DNA synthesis in non-dividing cells. When transcription is silenced, integrated proviral DNA can lead to latency (102). Although SAMHD1 is highly expressed in cells purported to harbor latent provirus (19, 103) and the HIV-1 proviral promoter is activated by transcription factors (104), the effect of SAMHD1 expression on latency development or reversal has not been explored. It is possible that SAMHD1 utilizes its nucleic acid binding ability to restrict HIV-1 infection postintegration, although a recent study confirmed SAMHD1 exerts no effect on HIV-1 Gag synthesis, viral particle release, and virus infectivity in 293T cells transfected with a proviral DNA construct (55). SAMHD1 may exert a direct effect on proviral DNA through binding, as purified recombinant SAMHD1 was shown to bind in vitro transcribed fragments of gag and tat cDNA (72), or indirect effects may occur due to SAMHD1 modulation of inflammatory pathways. It is plausible that suppression of latency reactivation by SAMHD1 would further prevent activation of the innate antiviral response. Although viral nucleic acids can be sensed by IFI16 or cGAS in the absence of SAMHD1 (24, 105), whether other pro-inflammatory pathways are affected by SAMHD1 expression remains unknown.
Discovering the mechanisms used by HIV-1 to avoid innate immune sensors is critical for the design of new therapies to eradicate HIV-1 infection. Therapeutic strategies aiming to inhibit host factors that promote HIV-1 replication and to stimulate the immune response could diminish viral infection and transmission. Current work aims to determine whether a role exists for drugs targeting SAMHD1. Expression of SAMHD1 can increase the susceptibility of HIV-1 to nucleoside reverse transcriptase inhibitors by reducing the levels of competitive dNTPs (106–109), suggesting modulation of SAMHD1 function may be a means to enhance drug effectiveness. Conversely, as SAMHD1 expression enables immune evasion by HIV-1 (13), it is tempting to hypothesize that SAMHD1 could be used as a drug target to enhance the innate immune response to viral infection. However, research is just beginning to uncover mechanisms to modify the dNTPase activity of SAMHD1 (110, 111). Importantly, as an ISG and a negative regulator of the innate immune system, SAMHD1 may be involved in an unknown negative feedback loop aimed at modulating the complex and delicate system of inflammatory pathways.
The effect of SAMHD1 on IFN-I induction during viral infection should be further studied in vivo. Although initial robust IFN-I responses can lead to an upregulation of antiviral genes and a block in infection, chronic immune hyperactivity could lead to desensitization of IFN-I and an eventual suppression of antiviral gene expression. This phenotype was observed when Sandler et al. manipulated the IFN-α2a response to SIV infection in rhesus macaques (112). The dismantling of the antiviral state after long-term IFN-α2a treatment led to an increase in SIV reservoir size and an accelerated CD4+ T-cell loss (112). Studies are necessary to determine whether stimulation of the IFN-I response through inhibition of SAMHD1 function leads to chronic inflammation and progression to AIDS in vivo.
Conclusion
The identification of SAMHD1 as a regulator of the innate immune response to viral infection has led to the development of an exciting field of research. The structural and functional studies of SAMHD1 connect the physiology of HIV-1 infection to the innate antiviral response and the dynamic regulatory mechanisms in cells. Further work will aid in the development of stratagems to enhance the antiviral effects of the intrinsic immune system.
Author Contributions
JA wrote the manuscript with input and edits from CG and LW.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Wu lab members for valuable discussions.
Funding
This work was supported by a grant (AI104483) from the National Institutes of Health (NIH) to LW. LW is also supported in part by NIH grants (AI120209 and GM128212). JA was supported by C. Glenn Barber funds from the College of Veterinary Medicine at The Ohio State University.
References
1. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
2. Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol (2016) 14:360–73. doi:10.1038/nrmicro.2016.45
3. Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science (2013) 341:903–6. doi:10.1126/science.1240933
4. Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A (2013) 110:E4571–80. doi:10.1073/pnas.1311669110
5. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
6. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol (2016) 17:1142–9. doi:10.1038/ni.3558
7. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi:10.1084/jem.20101664
8. Doyle T, Goujon C, Malim MH. HIV-1 and interferons: who’s interfering with whom? Nat Rev Microbiol (2015) 13:403–13. doi:10.1038/nrmicro3449
9. Zaunders J, van Bockel D. Innate and adaptive immunity in long-term non-progression in HIV disease. Front Immunol (2013) 4:95. doi:10.3389/fimmu.2013.00095
10. Arcia D, Acevedo-Sáenz L, Rugeles MT, Velilla PA. Role of CD8(+) T cells in the selection of HIV-1 immune escape mutations. Viral Immunol (2017) 30:3–12. doi:10.1089/vim.2016.0095
12. Douville RN, Hiscott J. The interface between the innate interferon response and expression of host retroviral restriction factors. Cytokine (2010) 52:108–15. doi:10.1016/j.cyto.2010.04.010
13. Schott K, Riess M, König R. Role of innate genes in HIV replication. In: Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer. (2017). p. 1–43. doi:10.1007/82_2017_29
14. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature (2011) 474:654–7. doi:10.1038/nature10117
15. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature (2011) 474:658–61. doi:10.1038/nature10195
16. Gramberg T, Kahle T, Bloch N, Wittmann S, Müllers E, Daddacha W, et al. Restriction of diverse retroviruses by SAMHD1. Retrovirology (2013) 10:26. doi:10.1186/1742-4690-10-26
17. Hollenbaugh JA, Gee P, Baker J, Daly MB, Amie SM, Tate J, et al. Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog (2013) 9:e1003481. doi:10.1371/journal.ppat.1003481
18. Chen Z, Zhu M, Pan X, Zhu Y, Yan H, Jiang T, et al. Inhibition of Hepatitis B virus replication by SAMHD1. Biochem Biophys Res Commun (2014) 450:1462–8. doi:10.1016/j.bbrc.2014.07.023
19. Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, et al. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology (2012) 9:87. doi:10.1186/1742-4690-9-87
20. Berger A, Sommer AF, Zwarg J, Hamdorf M, Welzel K, Esly N, et al. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutières syndrome are highly susceptible to HIV-1 infection. PLoS Pathog (2011) 7:e1002425. doi:10.1371/journal.ppat.1002425
21. St Gelais C, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, et al. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology (2012) 9:105. doi:10.1186/1742-4690-9-105
22. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet (2009) 41:829–32. doi:10.1038/ng.373
23. Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D, et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep (2013) 4:689–96. doi:10.1016/j.celrep.2013.07.037
24. Maelfait J, Bridgeman A, Benlahrech A, Cursi C, Rehwinkel J. Restriction by SAMHD1 limits cGAS/STING-dependent innate and adaptive immune responses to HIV-1. Cell Rep (2016) 16:1492–501. doi:10.1016/j.celrep.2016.07.002
25. Rustagi A, Gale M. Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J Mol Biol (2014) 426:1161–77. doi:10.1016/j.jmb.2013.12.003
26. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498:380–4. doi:10.1038/nature12306
27. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi:10.1126/science.1232458
28. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi:10.1126/science.1229963
29. Ablasser A, Gulen MF. The role of cGAS in innate immunity and beyond. J Mol Med (Berl) (2016) 94:1085–93. doi:10.1007/s00109-016-1423-2
30. Ma Z, Damania B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe (2016) 19:150–8. doi:10.1016/j.chom.2016.01.010
31. Campbell EM, Hope TJ. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol (2015) 13:471–83. doi:10.1038/nrmicro3503
32. Yamashita M, Engelman AN. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol (2017) 25:741–55. doi:10.1016/j.tim.2017.04.004
33. Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature (2011) 480:379–82. doi:10.1038/nature10623
34. Powell RD, Holland PJ, Hollis T, Perrino FW. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem (2011) 286:43596–600. doi:10.1074/jbc.C111.317628
35. Amie SM, Bambara RA, Kim B. GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J Biol Chem (2013) 288:25001–6. doi:10.1074/jbc.C113.493619
36. Aird KM, Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett (2015) 356:204–10. doi:10.1016/j.canlet.2014.01.017
37. Ji X, Wu Y, Yan J, Mehrens J, Yang H, DeLucia M, et al. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat Struct Mol Biol (2013) 20:1304–9. doi:10.1038/nsmb.2692
38. Rampazzo C, Miazzi C, Franzolin E, Pontarin G, Ferraro P, Frangini M, et al. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat Res (2010) 703:2–10. doi:10.1016/j.mrgentox.2010.06.002
39. Kohnken R, Kodigepalli KM, Wu L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer (2015) 14:176. doi:10.1186/s12943-015-0446-6
40. Kretschmer S, Wolf C, König N, Staroske W, Guck J, Häusler M, et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis (2015) 74:e17. doi:10.1136/annrheumdis-2013-204845
41. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell (2011) 145:435–46. doi:10.1016/j.cell.2011.03.044
42. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A (2015) 167A:296–312. doi:10.1002/ajmg.a.36887
43. Crow YJ. Aicardi-Goutières syndrome. Handb Clin Neurol (2013) 113:1629–35. doi:10.1016/B978-0-444-59565-2.00031-9
44. Kaliyadan F. Hiv and lupus erythematosus: a diagnostic dilemma. Indian J Dermatol (2008) 53:80–2. doi:10.4103/0019-5154.41652
45. Feng Q, Moran JV, Kazazian HH, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell (1996) 87:905–16. doi:10.1016/S0092-8674(00)81997-2
46. Mathias SL, Scott AF, Kazazian HH, Boeke JD, Gabriel A. Reverse transcriptase encoded by a human transposable element. Science (1991) 254:1808–10. doi:10.1126/science.1722352
47. Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X, et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutières syndrome-related SAMHD1. Cell Rep (2013) 4:1108–15. doi:10.1016/j.celrep.2013.08.019
48. Hu S, Li J, Xu F, Mei S, Le Duff Y, Yin L, et al. SAMHD1 inhibits LINE-1 retrotransposition by promoting stress granule formation. PLoS Genet (2015) 11:e1005367. doi:10.1371/journal.pgen.1005367
49. Crow MK. Long interspersed nuclear elements (LINE-1): potential triggers of systemic autoimmune disease. Autoimmunity (2010) 43:7–16. doi:10.3109/08916930903374865
50. Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog (2008) 4:e1000059. doi:10.1371/journal.ppat.1000059
51. Goujon C, Arfi V, Pertel T, Luban J, Lienard J, Rigal D, et al. Characterization of simian immunodeficiency virus SIVSM/human immunodeficiency virus type 2 Vpx function in human myeloid cells. J Virol (2008) 82:12335–45. doi:10.1128/JVI.01181-08
52. Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, et al. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem (2012) 287:12550–8. doi:10.1074/jbc.M112.340711
53. Wei W, Guo H, Han X, Liu X, Zhou X, Zhang W, et al. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell Microbiol (2012) 14:1745–56. doi:10.1111/j.1462-5822.2012.01835.x
54. Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol (2012) 13:223–8. doi:10.1038/ni.2236
55. Antonucci JM, St Gelais C, de Silva S, Yount JS, Tang C, Ji X, et al. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat Med (2016) 22:1072–4. doi:10.1038/nm.4163
56. Miazzi C, Ferraro P, Pontarin G, Rampazzo C, Reichard P, Bianchi V. Allosteric regulation of the human and mouse deoxyribonucleotide triphosphohydrolase sterile α-motif/histidine-aspartate domain-containing protein 1 (SAMHD1). J Biol Chem (2014) 289:18339–46. doi:10.1074/jbc.M114.571091
57. Ji X, Tang C, Zhao Q, Wang W, Xiong Y. Structural basis of cellular dNTP regulation by SAMHD1. Proc Natl Acad Sci U S A (2014) 111:E4305–14. doi:10.1073/pnas.1412289111
58. Li Y, Kong J, Peng X, Hou W, Qin X, Yu XF. Structural insights into the high-efficiency catalytic mechanism of the sterile α-Motif/Histidine-aspartate domain-containing protein. J Biol Chem (2015) 290:29428–37. doi:10.1074/jbc.M115.663658
59. Hansen EC, Seamon KJ, Cravens SL, Stivers JT. GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc Natl Acad Sci U S A (2014) 111:E1843–51. doi:10.1073/pnas.1401706111
60. Seamon KJ, Bumpus NN, Stivers JT. Single-stranded nucleic acids bind to the tetramer interface of SAMHD1 and prevent formation of the catalytic homotetramer. Biochemistry (2016) 55:6087–99. doi:10.1021/acs.biochem.6b00986
61. Cribier A, Descours B, Valadão AL, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep (2013) 3:1036–43. doi:10.1016/j.celrep.2013.03.017
62. St Gelais C, de Silva S, Hach JC, White TE, Diaz-Griffero F, Yount JS, et al. Identification of cellular proteins interacting with the retroviral restriction factor SAMHD1. J Virol (2014) 88:5834–44. doi:10.1128/JVI.00155-14
63. Welbourn S, Dutta SM, Semmes OJ, Strebel K. Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J Virol (2013) 87:11516–24. doi:10.1128/JVI.01642-13
64. Pauls E, Ruiz A, Badia R, Permanyer M, Gubern A, Riveira-Muñoz E, et al. Cell cycle control and HIV-1 susceptibility are linked by CDK6-dependent CDK2 phosphorylation of SAMHD1 in myeloid and lymphoid cells. J Immunol (2014) 193:1988–97. doi:10.4049/jimmunol.1400873
65. White TE, Brandariz-Nuñez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, et al. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe (2013) 13:441–51. doi:10.1016/j.chom.2013.03.005
66. Arnold LH, Groom HC, Kunzelmann S, Schwefel D, Caswell SJ, Ordonez P, et al. Phospho-dependent regulation of SAMHD1 oligomerisation couples catalysis and restriction. PLoS Pathog (2015) 11:e1005194. doi:10.1371/journal.ppat.1005194
67. Tang C, Ji X, Wu L, Xiong Y. Impaired dNTPase activity of SAMHD1 by phosphomimetic mutation of Thr-592. J Biol Chem (2015) 290:26352–9. doi:10.1074/jbc.M115.677435
68. Franzolin E, Pontarin G, Rampazzo C, Miazzi C, Ferraro P, Palumbo E, et al. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci U S A (2013) 110:14272–7. doi:10.1073/pnas.1312033110
69. Yan J, Hao C, DeLucia M, Swanson S, Florens L, Washburn MP, et al. CyclinA2-cyclin-dependent kinase regulates SAMHD1 protein phosphohydrolase domain. J Biol Chem (2015) 290:13279–92. doi:10.1074/jbc.M115.646588
70. Mauney CH, Rogers LC, Harris RS, Daniel LW, Devarie-Baez NO, Wu H, et al. The SAMHD1 dNTP triphosphohydrolase is controlled by a Redox switch. Antioxid Redox Signal (2017) 27:1317–31. doi:10.1089/ars.2016.6888
71. Welbourn S, Strebel K. Low dNTP levels are necessary but may not be sufficient for lentiviral restriction by SAMHD1. Virology (2016) 488:271–7. doi:10.1016/j.virol.2015.11.022
72. Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, et al. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J Biol Chem (2013) 288:8101–10. doi:10.1074/jbc.M112.431148
73. Tüngler V, Staroske W, Kind B, Dobrick M, Kretschmer S, Schmidt F, et al. Single-stranded nucleic acids promote SAMHD1 complex formation. J Mol Med (Berl) (2013) 91:759–70. doi:10.1007/s00109-013-0995-3
74. Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, et al. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to Aicardi-Goutières syndrome-associated mutations. Hum Mutat (2012) 33:1116–22. doi:10.1002/humu.22087
75. Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim SY, et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med (2014) 20:936–41. doi:10.1038/nm.3626
76. Choi J, Ryoo J, Oh C, Hwang S, Ahn K. SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology (2015) 12:46. doi:10.1186/s12977-015-0174-4
77. Seamon KJ, Sun Z, Shlyakhtenko LS, Lyubchenko YL, Stivers JT. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res (2015) 43:6486–99. doi:10.1093/nar/gkv633
78. Bloch N, Gläsker S, Sitaram P, Hofmann H, Shepard CN, Schultz ML, et al. A highly active isoform of lentivirus restriction factor SAMHD1 in mouse. J Biol Chem (2017) 292:1068–80. doi:10.1074/jbc.M116.743740
79. Wittmann S, Behrendt R, Eissmann K, Volkmann B, Thomas D, Ebert T, et al. Phosphorylation of murine SAMHD1 regulates its antiretroviral activity. Retrovirology (2015) 12:103. doi:10.1186/s12977-015-0229-6
80. Brandariz-Nuñez A, Valle-Casuso JC, White TE, Laguette N, Benkirane M, Brojatsch J, et al. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology (2012) 9:49. doi:10.1186/1742-4690-9-49
81. Ryoo J, Hwang SY, Choi J, Oh C, Ahn K. Reply to SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat Med (2016) 22:1074–5. doi:10.1038/nm.4164
82. Ryoo J, Hwang SY, Choi J, Oh C, Ahn K. SAMHD1, the Aicardi-Goutières syndrome gene and retroviral restriction factor, is a phosphorolytic ribonuclease rather than a hydrolytic ribonuclease. Biochem Biophys Res Commun (2016) 477:977–81. doi:10.1016/j.bbrc.2016.07.013
83. Li N, Zhang W, Cao X. Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol Lett (2000) 74:221–4. doi:10.1016/S0165-2478(00)00276-5
84. Pauls E, Jimenez E, Ruiz A, Permanyer M, Ballana E, Costa H, et al. Restriction of HIV-1 replication in primary macrophages by IL-12 and IL-18 through the upregulation of SAMHD1. J Immunol (2013) 190:4736–41. doi:10.4049/jimmunol.1203226
85. Chen Z, Zhang L, Ying S. SAMHD1: a novel antiviral factor in intrinsic immunity. Future Microbiol (2012) 7:1117–26. doi:10.2217/fmb.12.81
86. Yang S, Zhan Y, Zhou Y, Jiang Y, Zheng X, Yu L, et al. Interferon regulatory factor 3 is a key regulation factor for inducing the expression of SAMHD1 in antiviral innate immunity. Sci Rep (2016) 6:29665. doi:10.1038/srep29665
87. Riess M, Fuchs NV, Idica A, Hamdorf M, Flory E, Pedersen IM, et al. Interferons induce expression of SAMHD1 in monocytes through down-regulation of miR-181a and miR-30a. J Biol Chem (2017) 292:264–77. doi:10.1074/jbc.M116.752584
88. Goujon C, Schaller T, Galão RP, Amie SM, Kim B, Olivieri K, et al. Evidence for IFNα-induced, SAMHD1-independent inhibitors of early HIV-1 infection. Retrovirology (2013) 10:23. doi:10.1186/1742-4690-10-23
89. Schmidt S, Schenkova K, Adam T, Erikson E, Lehmann-Koch J, Sertel S, et al. SAMHD1’s protein expression profile in humans. J Leukoc Biol (2015) 98:5–14. doi:10.1189/jlb.4HI0714-338RR
90. Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med (2012) 18:1682–7. doi:10.1038/nm.2964
91. Che KF, Sabado RL, Shankar EM, Tjomsland V, Messmer D, Bhardwaj N, et al. HIV-1 impairs in vitro priming of naïve T cells and gives rise to contact-dependent suppressor T cells. Eur J Immunol (2010) 40:2248–58. doi:10.1002/eji.201040377
92. Miller E, Bhardwaj N. Dendritic cell dysregulation during HIV-1 infection. Immunol Rev (2013) 254:170–89. doi:10.1111/imr.12082
93. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
94. Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature (2010) 467:214–7. doi:10.1038/nature09337
95. Zhang C, de Silva S, Wang JH, Wu L. Co-evolution of primate SAMHD1 and lentivirus Vpx leads to the loss of the vpx gene in HIV-1 ancestor. PLoS One (2012) 7:e37477. doi:10.1371/journal.pone.0037477
96. Su B, Biedma ME, Lederle A, Peressin M, Lambotin M, Proust A, et al. Dendritic cell-lymphocyte cross talk downregulates host restriction factor SAMHD1 and stimulates HIV-1 replication in dendritic cells. J Virol (2014) 88:5109–21. doi:10.1128/JVI.03057-13
97. Hertoghs N, van der Aar AM, Setiawan LC, Kootstra NA, Gringhuis SI, Geijtenbeek TB. SAMHD1 degradation enhances active suppression of dendritic cell maturation by HIV-1. J Immunol (2015) 194:4431–7. doi:10.4049/jimmunol.1403016
98. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature (2013) 503:402–5. doi:10.1038/nature12769
99. Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol (2010) 11:1005–13. doi:10.1038/ni.1941
100. Hasan M, Yan N. Safeguard against DNA sensing: the role of TREX1 in HIV-1 infection and autoimmune diseases. Front Microbiol (2014) 5:193. doi:10.3389/fmicb.2014.00193
101. Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem (2012) 287:21570–4. doi:10.1074/jbc.C112.374843
102. Ruelas DS, Greene WC. An integrated overview of HIV-1 latency. Cell (2013) 155:519–29. doi:10.1016/j.cell.2013.09.044
103. Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med (1999) 5:512–7. doi:10.1038/8394
104. Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J (2003) 22:1868–77. doi:10.1093/emboj/cdg188
105. Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun (2017) 8:14391. doi:10.1038/ncomms14391
106. Amie SM, Noble E, Kim B. Intracellular nucleotide levels and the control of retroviral infections. Virology (2013) 436:247–54. doi:10.1016/j.virol.2012.11.010
107. Huber AD, Michailidis E, Schultz ML, Ong YT, Bloch N, Puray-Chavez MN, et al. SAMHD1 has differential impact on the efficacies of HIV nucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother (2014) 58:4915–9. doi:10.1128/AAC.02745-14
108. Ballana E, Badia R, Terradas G, Torres-Torronteras J, Ruiz A, Pauls E, et al. SAMHD1 specifically affects the antiviral potency of thymidine analog HIV reverse transcriptase inhibitors. Antimicrob Agents Chemother (2014) 58:4804–13. doi:10.1128/AAC.03145-14
109. Ordonez P, Kunzelmann S, Groom HC, Yap MW, Weising S, Meier C, et al. SAMHD1 enhances nucleoside-analogue efficacy against HIV-1 in myeloid cells. Sci Rep (2017) 7:42824. doi:10.1038/srep42824
110. Hollenbaugh JA, Shelton J, Tao S, Amiralaei S, Liu P, Lu X, et al. Substrates and Inhibitors of SAMHD1. PLoS One (2017) 12:e0169052. doi:10.1371/journal.pone.0169052
111. Seamon KJ, Hansen EC, Kadina AP, Kashemirov BA, McKenna CE, Bumpus NN, et al. Small molecule inhibition of SAMHD1 dNTPase by tetramer destabilization. J Am Chem Soc (2014) 136:9822–5. doi:10.1021/ja5035717
Keywords: HIV-1, sterile alpha motif and HD-domain containing protein 1, infection, innate immunity, myeloid cells
Citation: Antonucci JM, St. Gelais C and Wu L (2017) The Dynamic Interplay between HIV-1, SAMHD1, and the Innate Antiviral Response. Front. Immunol. 8:1541. doi: 10.3389/fimmu.2017.01541
Received: 30 September 2017; Accepted: 30 October 2017;
Published: 10 November 2017
Edited by:
Persephone Borrow, University of Oxford, United KingdomReviewed by:
Jonathan Paul Stoye, Francis Crick Institute, United KingdomStephen Kent, University of Melbourne, Australia
Copyright: © 2017 Antonucci, St. Gelais and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Wu, d3UuODQwQG9zdS5lZHU=