 Fabrizio Antonangeli
Fabrizio Antonangeli Alessandra Soriani
Alessandra Soriani Cristina Cerboni
Cristina Cerboni Giuseppe Sciumè
Giuseppe Sciumè Angela Santoni
Angela Santoni- 1Department of Molecular Medicine, Sapienza University of Rome, Laboratory Affiliated to Istituto Pasteur Italia – Fondazione Cenci Bolognetti, Rome, Italy
- 2Neuromed I.R.C.C.S. – Istituto Neurologico Mediterraneo, Pozzilli, Italy
Mucosal epithelia encounter both physicochemical and biological stress during their life and have evolved several mechanisms to deal with them, including regulation of immune cell functions. Stressed and damaged cells need to be cleared to control local inflammation and trigger tissue healing. Engagement of the activating NKG2D receptor is one of the most direct mechanisms involved in the recognition of stressed cells by the immune system. Indeed, injured cells promptly express NKG2D ligands that in turn mediate the activation of lymphocytes of both innate and adaptive arms of the immune system. This review focuses on different conditions that are able to modulate NKG2D ligand expression on the epithelia. Special attention is given to the mechanisms of immunosurveillance mediated by natural killer cells, which are finely tuned by NKG2D. Different types of stress, including viral and bacterial infections, chronic inflammation, and cigarette smoke exposure, are discussed as paradigmatic conditions for NKG2D ligand modulation, and the implications for tissue homeostasis are discussed.
Introduction
Mucosal epithelia represent the frontline of multicellular organisms, and they are continuously exposed to several types of stress. Pathogens and environmental stress (thermal, oxidative, and chemical) can result in cell damage and loss of tissue function. To control inflammation and promote tissue repair, different mechanisms for the detection and elimination of stressed cells have evolved, including activation of the immune system.
NKG2D is a C-type lectin-like activating/co-stimulatory receptor expressed by innate and adaptive lymphocytes, such as natural killer (NK) cells, CD8+ αβ T cells, γδ T cells, and iNKT cells. Engagement of NKG2D triggers the cytolytic function of effector CD8+ T cells independently of TCR recognition in some circumstances (1–4) while directly activates the effector functions of NK cells, namely, cytolytic granule release and IFNγ secretion. Thus, expression of NKG2D ligands is strictly linked to the immunosurveillance of stressed cells by innate lymphoid cells, especially NK cells (5–7). In humans, NKG2D ligands are MICA and MICB (MHC class I chain-related proteins A and B), encoded by genes in the MHC region, and ULBP1-6 (UL16-binding proteins), with the encoding genes located on chromosome 6. Murine NKG2D ligands include five different isoforms of RAE-1 (retinoic acid early inducible-1), MULT-1 (murine ULBP-like transcript-1), and three different isoforms of H60 (histocompatibility 60) (8). These molecules are present at low or undetectable levels on normal cells (9, 10) but are rapidly induced upon cellular stress and are frequently upregulated in virus-infected and neoplastic cells (11–13). Even if it is known that the promoter region of the MICA gene contains a heat-shock element able to respond to cellular stress (14), only recently the molecular mechanisms driving NKG2D ligand expression during cellular stress have more deeply been investigated (15).
Expression of NKG2D ligands has been shown in tissues from patients with chronic inflammatory diseases, including rheumatoid arthritis (16, 17), type 1 diabetes (18), and atherosclerosis (19); much less is known regarding NKG2D ligand expression and relevance in mucosal epithelia during both normal and stress conditions.
This review focuses on different conditions that are able to modulate NKG2D ligands on epithelial cells, and, in particular, on the role of NKG2D/NKG2D ligands in controlling the homeostasis of the gut and lung epithelia during inflammation. Recent findings link toll-like receptor (TLR) signaling to NKG2D ligand expression, and we can now think of epithelial and immune cells as an integrated system able to deal promptly with environmental stress.
Sensing the Stress: Interplay Between TLRs and NKG2D Ligands
Among the pattern recognition receptors, TLRs play a key role in innate immunity, serving as first line sensors of structurally conserved bacterial and viral components, the so-called pathogen-associated molecular patterns. It is clear that TLRs can be triggered also by endogenous ligands, such as nucleic acids released by necrotic cells and matrix components generated during tissue injury, collectively called damage-associated molecular patterns. As TLR engagement leads to the production of inflammatory cytokines and chemokines, which contribute to local inflammation and leukocyte recruitment, the recent findings showing that TLR signaling is able to modulate the NKG2D axis and thus lymphocyte activation are of great interest (Table 1).
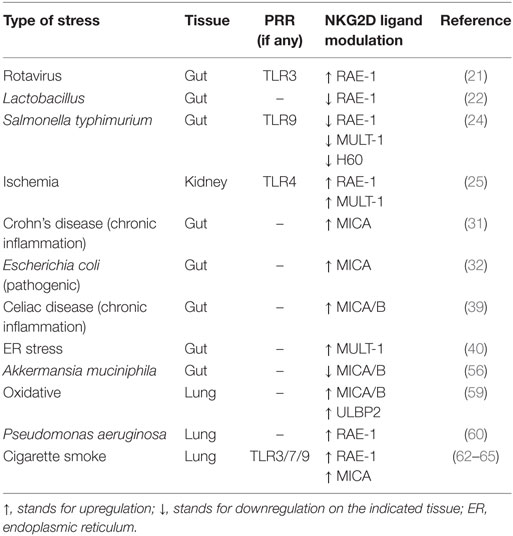
Table 1. Modulation of NKG2D ligand expression following different types of stress and pattern recognition receptor (PRR) involvement.
Rotaviruses are causative of intestinal alterations leading to diarrhea, and it has been reported that in mice viral dsRNA from rotavirus genome induces severe intestinal injury via TLR3 activation (20). Indeed, upon stimulation, TLR3 forces intestinal epithelial cells (IECs) to express both IL-15 and RAE-1, which promote mucosal damage by activating intraepithelial lymphocytes (IELs), in particular CD8αα+ T cells, engaging their NKG2D receptor (21). In this scenario, Tada and colleagues have shown that probiotics belonging to Lactobacillus strains are able to reduce the levels of IL-15 and RAE-1, and at the same time to increase the level of IL-10 in the intestine, thus performing immunomodulatory activities (22) (Figure 1). Interestingly, the NKG2D axis is modulated in an opposite way by TLR9 during Salmonella typhimurium infection. S. typhimurium is an important food-derived pathogen and unmethylated CpG-containing DNA from S. typhimurium is recognized by TLR9 (23). TLR9 triggering promotes the accumulation of IkBα, resulting in strong inhibition of the NF-kB pathway and thus controlling intestinal tissue inflammation. On the contrary, in TLR9-deficient mice, TLR9 signal deficiency releases its inhibition on NF-kB and leads to pro-IL-1β expression in IECs. In addition, lack of TLR9 signal causes activation of NLRP3 inflammasomes, resulting in pro-IL-1β processing and IL-1β secretion. Secreted IL-1β acts in an autocrine way stimulating IECs to expose NKG2D ligands (namely, RAE-1, MULT-1, and H60) and with a paracrine mechanism induces the expression of NKG2D on IELs (24). This positive loop augments the susceptibility of IECs to the cytotoxicity of IELs leading to the breaking of the epithelial barrier and to the spread of S. typhimurium infection (Figure 1).
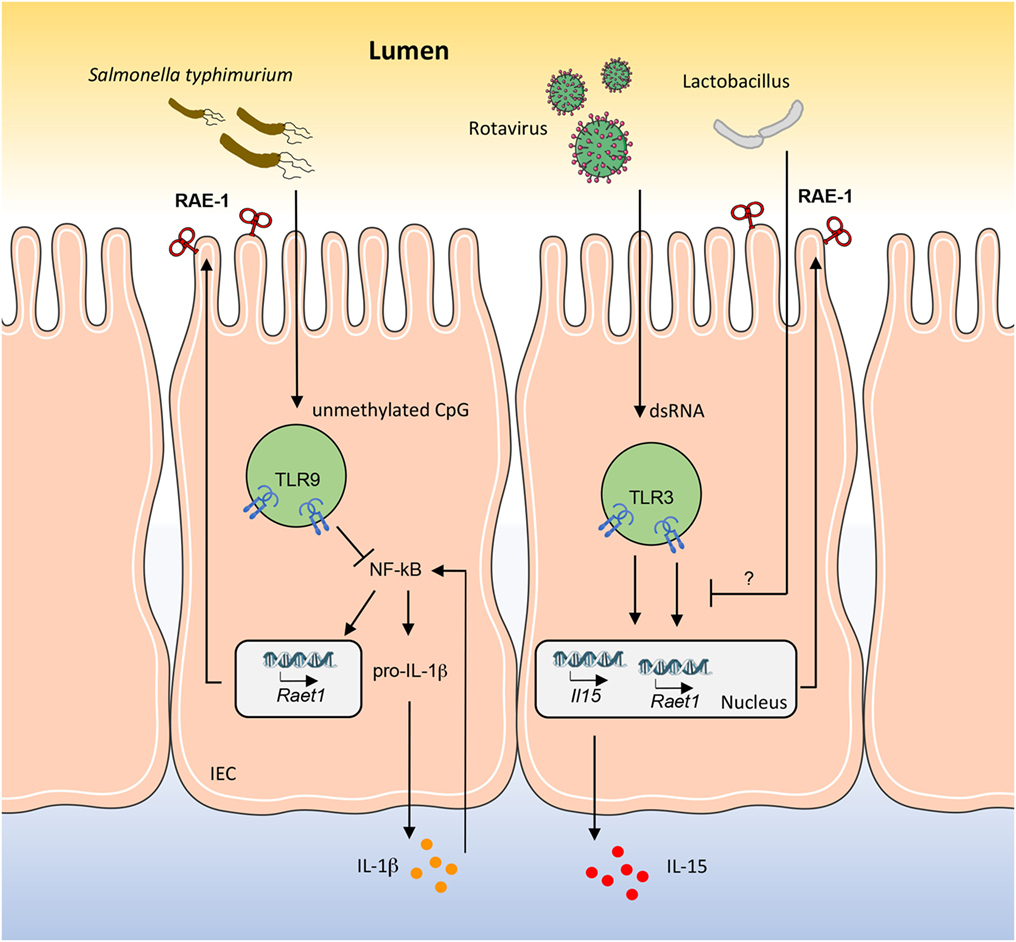
Figure 1. RAE-1 modulation in mouse intestinal epithelial cell (IEC) by gut pathogens and commensal bacteria. How RAE-1 (encoded by Raet1) is regulated after toll-like receptor (TLR) engagement is schematically depicted. The apical localization of RAE-1 is inferred based on its similarity to human ULBP1–3 structure. The mechanisms through which Lactobacillus strains are able to reduce the levels of RAE-1 and IL-15 are only partially known.
TLR-dependent NKG2D ligand expression has also been observed in mouse kidney during renal ischemia–reperfusion injury (25). HMGB1 is a DNA-binding protein showing inflammatory function after ischemia–reperfusion injury by binding TLR4 (26–28). Chen and colleagues have reported that HMGB1 can induce RAE-1 and MULT-1 upregulation on injured kidney cells through a TLR4/MyD88-dependent signaling (25), suggesting a contribution of the NKG2D axis during tissue damage after ischemia. Accordingly, NK cell depletion has been demonstrated to ameliorate kidney damage following ischemia, with NK cells having a direct role in killing tubular epithelial cells via perforin secretion (29).
Modulation of NKG2D Ligand Expression During Chronic Inflammation: The Gut Model
The gut system represents a peculiar challenge for the immune system, as it is continuously exposed to commensal bacteria and food antigens. IECs constitutively express MICA and MICB, even if at low levels (14, 30), while much less is known regarding ULBP proteins. In physiological conditions, this expression does not lead to an immune response, but during chronic inflammatory diseases, MICA/B-expressing cells become target of IELs, namely, intraepithelial NK cells, γδ T cells, and CD8+ αβ T cells. Although with different etiology, both Crohn’s disease and celiac disease are characterized by chronic inflammation. This type of stress strongly relies on the relationship between the environment and the immune system.
Crohn’s Disease
MICA expression has been found significantly increased on IECs isolated from patients with Crohn’s disease, with higher levels in the macroscopically affected areas (31). Whether this upregulation is causative of the inflammation or a consequence of tissue damage is still unclear. Nevertheless, the upregulation of MICA has been associated with the expansion of a mucosal NKG2D+CD4+ T cell population able to promote a Th1 response, thus contributing to tissue inflammation and damage (31). There is evidence of a direct link between persistent MICA expression, innate lymphocyte activation and Th1 cytokine production in a model of pathogenic Escherichia coli infection (32), suggesting that this host–bacteria interaction may be relevant to the pathogenesis of Crohn’s disease, as adherent E. coli strains have been isolated from inflammatory bowel disease (IBD) patients (33, 34). The presence of adherent pathogenic E. coli triggers a rapid increase of MICA expression on the surface of intestinal cells after the interaction of the microbial adhesin AfaE with the cellular protein CD55, also known as the decay-accelerating factor (32). This event leads to NK cell activation with production of high levels of IFN-γ. As other human enteric pathogens, such as enteroviruses, use CD55 for cell entry (35, 36), it is possible to speculate that the in loco MICA overexpression occurring during chronic inflammation in unresolved infections contributes to lymphocyte activation associated with Crohn’s disease. Based on these findings, targeting the interaction between NKG2D and MICA may be seen as a promising strategy to reduce inflammation. Results of a randomized controlled trial for the use of an anti-NKG2D monoclonal antibody (NNC0142-0002) in active Crohn’s disease have reported no major improvement, but further investigations regarding dose ranging and dose regimen are needed (37). Furthermore, NKG2D ligands may have a wider role than stress sensors, contributing to the homeostatic control of the immune system, since a study by La Scaleia and colleagues has shown that NKG2D ligands, namely MICA/B and ULBP1–2, are upregulated not only on the epithelium of gut but also on the immune infiltrate in IBD lesions (38).
Celiac Disease
Celiac disease is an immune-mediated disease characterized by damage to the small intestine in response to gluten exposure. Interestingly, there is evidence for a direct link between the expression of NKG2D ligands in the inflamed mucosa and cellular stress. Indeed, a similar MICA/B pattern of expression in the gut of celiac patients and in different in vitro models of cellular stress has been observed. In both cases, MICA/B were located in stress granules commonly observed after oxidative and endoplasmic reticulum (ER) stress (39). A recent study by Hosomi and colleagues has disclosed a molecular mechanism through which ER stress is associated with MULT-1 upregulation in IECs. In a mouse model of ER stress, the ER stress-related transcription factor C/EBP homology protein, a major component of the unfolded protein response, induces the transcription of the Ulbp1 gene leading to MULT-1 cellular surface expression. MULT-1 upregulation has been linked to the activation of intraepithelial group 1 innate lymphoid cells (NK cells and ILC1) and innate-like T cells (such as CD8αα+ T cells), which contribute to mucosal inflammation in an NKG2D-mediated manner (40). Expression of NKG2D ligands in celiac disease can also be induced by cytokines, among which IL-15 seems to play a major role (41–45). Indeed, IL-15 has been shown to rapidly induce the expression of MICA and to relocate it from the cytosol to the surface membrane of enterocytes, where can be engaged by NKG2D-expressing IELs (3, 46, 47). On the other hand, MICA/B expression has been found also in the cytoplasm of intraepithelial and lamina propria lymphocytes (39). This intracellular localization of MICA/B in T cells during active celiac disease has been postulated to avoid overactivated T cell homeostatic regulation, thus contributing to tissue inflammation and damage.
An interesting issue to be considered for both Crohn’s disease and celiac disease regards NKG2D ligand polymorphism. MICA gene is highly polymorphic with more than one hundred alleles, which affect both RNA and protein expression levels (48). Some studies have assessed the prevalence of specific MICA alleles in IBD patients, with contrasting or not conclusive results (49). MICA allele*007 was associated with ulcerative colitis but not with Crohn’s disease in a British population (50), but this finding was not confirmed in a German cohort (51). MICA*008, instead, has been found overrepresented in celiac disease patients, but this could be ascribed to the linkage disequilibrium between HLA-B*08 and MICA*008 (52). Notably, MICA isoforms containing a methionine at position 129 bind NKG2D with high affinity, whereas those with a valine bind NKG2D with low affinity (53). Whether this diversity influences IBD is still unclear. A higher frequency of MICA-129met/met and a lower frequency of MICA-129val/met genotypes was observed in Spanish IBD patients compared with healthy controls (54). On the other hand, a study conducted in Chinese patients showed a higher frequency of the MICA-129val/val genotype in patients with ulcerative colitis (55).
The broad expression of NKG2D ligands on both epithelial cells and lymphocytes of the intestinal tract not only in pathological but also physiological conditions is intriguing but still unexplained. It may represent a state of alert ready to become active in response to stress, or, in more general terms, be part of tissue homeostasis regulation. On the other hand, it may be accountable for the state of permanent mild inflammation that characterizes the gut due to microbiota and food antigens. In support of the idea that the microbiota can be a main force driving the expression of NKG2D ligands on IECs is the observation that antibiotic administration and feeding are able to strongly modify NKG2D ligand expression (56). Germ-reducing conditions, such as ampicillin treatment, induce higher levels of NKG2D ligands, while food intake (i.e., xylooligosaccharides) or drug treatment supporting gut colonization by Akkermansia muciniphila decrease NKG2D ligand expression. An interesting association between the above conditions and the intestinal levels of IL-15 has been postulated, once again linking the inflammatory milieu to NKG2D ligands (56). Dietary contribution in the protection of gut inflammation has been investigated also in mice fed with a gluten-free diet. This food regimen leads to a reduced expression of NKG2D on DX5+ NK cells as well as to an increased number of CD8+ γδ T cells expressing TGF-β and the inhibitory receptor NKG2A, thus performing immunomodulatory functions (57, 58). Altogether, these findings reveal the importance of the NKG2D system in the homeostatic regulation of the intestinal mucosa.
Role of NKG2D in the Airway Epithelium Following Stress
The respiratory tract is often under attack of environmental pathogens and is equipped with different populations of NKG2D-expressing lymphocytes. Bronchial airway epithelial cells constitutively express MICA, MICB, and ULBP1–4 transcripts, while cell surface expression is largely absent in normal conditions. However, upon oxidative stress, NKG2D ligands become visible at the protein level on the cellular membrane, with a molecular mechanism based on the ERK pathway (59). The capability of stressed airway epithelial cells to rapidly express NKG2D ligands suggests that they are able to directly engage and activate the immune system. This finding has been confirmed in mice with lung Pseudomonas aeruginosa infection. Acute infection stimulates the alveolar epithelium to express RAE-1, and epithelial cell death (likely due to the activation of cytotoxic NKG2D-bearing lymphocytes) is associated with increased bacterial clearance (60). Pulmonary P. aeruginosa infection is also associated with increased levels of IL-1β, TNF-α, and IFN-γ, which may be another effect of NKG2D engagement. Thus, NKG2D ligand expression leads to a competent host response that can be blocked with NKG2D-specific antibody (60).
The NKG2D system plays an important role in lung homeostasis also in the event of physicochemical stress, as demonstrated in chronic obstructive pulmonary disease (COPD). COPD is a severe form of airway epithelium inflammation largely due to cigarette smoke exposure. Cigarette smoke is known to induce necrotic and apoptotic cell death with concomitant nucleic acid release (61). This leads to TLR3/7/9 activation and, with an unknown mechanism, to RAE-1 expression on mucosal airway epithelial cells. The consequent inflammation and NKG2D-driven cytotoxicity is responsible for further tissue damage, worsening the initial cigarette smoke exposure. NK cells become hyperresponsive breaking the balance between injury and repair (62–64). Notably, NKG2D ligand overexpression in transgenic mice is sufficient to induce pulmonary emphysema, strongly suggesting that NKG2D/NKG2D ligand axis via cytotoxic lymphocyte activation plays a major role in alveolar epithelial injury (65). The strong relationship between NKG2D ligand expression and COPD has been further confirmed by the finding of enhanced MICA expression in the airway epithelium specimens from smokers (65).
Concluding Remarks
Expression of NKG2D ligands can be achieved in several circumstances, ranging from viral and bacterial infections to physiochemical stress (Table 1). This versatility is needed to face a plethora of stressful stimuli with common pathways leading to the involvement of the immune system. As a result, intracellular insults are associated with intercellular responses, with great advantage for the whole organism. However, only recently the molecular mechanisms underlining these processes have been investigated, and many questions remain unanswered:
I. Which ligand is displayed? Is this choice stress type dependent?
II. Do different ligands engage the receptor in different ways?
III. What is the contribution of NKG2D ligands to the pathogenesis of gut and lung inflammatory diseases? Does NKG2D ligand polymorphism play a role in IBD?
Indeed, both NKG2D and NKG2D ligand genes are highly polymorphic. This polymorphism has been associated with host–pathogen coevolution, with great relevance for viral infections, but functional consequences have also been described during hematopoietic transplantation and cancer immunosurveillance (48, 66, 67). The impact of this variability in the context of stress response and inflammation still needs to be fully elucidated.
Another important point to be considered is the strict polarity of epithelial tissues. In the gut, the apical side of the epithelial layer displays different functions from the basolateral side, where IELs are located. Human ULBP1–3 and murine RAE-1 are anchored to the cell membrane via a glycosylphosphatidylinositol molecule and thus are supposed to be trapped in the lipid raft-enriched apical side of epithelial cells (Figure 1), while MICA presents a basolateral-targeting motif in his structure (68). Hence, MICA expression is prone to a rapid recognition by NKG2D-bearing IELs, while ULBP1–3 and murine RAE-1 become available only in the event of epithelial polarity breaking, as result of infection or autoimmunity (69). Remarkably, MICA allele*008, due to its truncated cytoplasmic tail, lacks the basolateral-targeting motif and changes its localization, leading to hypothesize a different NKG2D engagement (68, 69). Furthermore, it is now clear that NKG2D ligand transcript and even protein expression does not always mean cell surface exposure, with obvious functional implications (70, 71).
Great interest is growing regarding the role of the microbiome in human pathophysiology, as commensal bacteria can establish an immune regulatory milieu in the intestine (72, 73). The articles here reported suggest that, beside the production of TGF-β and IL-10, modulation of the NKG2D axis is a key function in this context. Indeed, it is now emerging that the NKG2D receptor and NKG2D ligands not only function as stress sensor molecules but also play a pivotal role in shaping innate and adaptive lymphocyte populations, thus contributing to the homeostatic regulation of the mucosal immune system.
Author Contributions
FA searched for literature articles; conceived and wrote the manuscript. ANS conceived and wrote the manuscript. ALS, CC, and GS revised and critically contributed to the manuscript drafting. FA, ALS, CC, GS, and ANS approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Funding
The work of the authors was supported by grants from the Italian Association for Cancer Research (AIRC: project #16014 and AIRC 5xmille: project #9962), Istituto Pasteur Italia—Fondazione Cenci Bolognetti, the Italian Ministry of University and Research (PRIN 2015-W729WH to CC), and from Sapienza University of Rome (C26H15HBMT).
References
1. Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise EL, et al. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J Immunol (2005) 175(5):2825–33. doi:10.4049/jimmunol.175.5.2825
2. Markiewicz MA, Wise EL, Buchwald ZS, Pinto AK, Zafirova B, Polic B, et al. RAE1epsilon ligand expressed on pancreatic islets recruits NKG2D receptor-expressing cytotoxic T cells independent of T cell receptor recognition. Immunity (2012) 36(1):132–41. doi:10.1016/j.immuni.2011.11.014
3. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity (2004) 21(3):357–66. doi:10.1016/j.immuni.2004.06.020
4. Verneris MR, Karimi M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood (2004) 103(8):3065–72. doi:10.1182/blood-2003-06-2125
5. Antonangeli F, Soriani A, Ricci B, Ponzetta A, Benigni G, Morrone S, et al. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. Oncoimmunology (2016) 5(10):e1218105. doi:10.1080/2162402X.2016.1218105
6. Chan CJ, Smyth MJ, Martinet L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death Differ (2014) 21(1):5–14. doi:10.1038/cdd.2013.26
7. Zingoni A, Fionda C, Borrelli C, Cippitelli M, Santoni A, Soriani A. Natural killer cell response to chemotherapy-stressed cancer cells: role in tumor immunosurveillance. Front Immunol (2017) 8:1194. doi:10.3389/fimmu.2017.01194
8. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
9. Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood (2007) 110(2):606–15. doi:10.1182/blood-2006-10-052720
10. Eagle RA, Jafferji I, Barrow AD. Beyond stressed self: evidence for NKG2D ligand expression on healthy cells. Curr Immunol Rev (2009) 5(1):22–34. doi:10.2174/157339509787314369
11. Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev (2010) 235(1):267–85. doi:10.1111/j.0105-2896.2010.00893.x
12. Gleimer M, Parham P. Stress management: MHC class I and class I-like molecules as reporters of cellular stress. Immunity (2003) 19(4):469–77. doi:10.1016/S1074-7613(03)00272-3
13. Pignoloni B, Fionda C, Dell’Oste V, Luganini A, Cippitelli M, Zingoni A, et al. Distinct roles for human cytomegalovirus immediate early proteins IE1 and IE2 in the transcriptional regulation of MICA and PVR/CD155 expression. J Immunol (2016) 197(10):4066–78. doi:10.4049/jimmunol.1502527
14. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A (1996) 93(22):12445–50. doi:10.1073/pnas.93.22.12445
15. Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, et al. The DNA damage response: a common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol (2014) 4:508. doi:10.3389/fimmu.2013.00508
16. Andersson AK, Sumariwalla PF, McCann FE, Amjadi P, Chang C, McNamee K, et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: a potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum (2011) 63(9):2617–29. doi:10.1002/art.30460
17. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A (2003) 100(16):9452–7. doi:10.1073/pnas.1632807100
18. Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity (2004) 20(6):757–67. doi:10.1016/j.immuni.2004.05.008
19. Xia M, Guerra N, Sukhova GK, Yang K, Miller CK, Shi GP, et al. Immune activation resulting from NKG2D/ligand interaction promotes atherosclerosis. Circulation (2011) 124(25):2933–43. doi:10.1161/CIRCULATIONAHA.111.034850
20. Zhou R, Wei H, Sun R, Tian Z. Recognition of double-stranded RNA by TLR3 induces severe small intestinal injury in mice. J Immunol (2007) 178(7):4548–56. doi:10.4049/jimmunol.178.7.4548
21. Zhou R, Wei H, Sun R, Zhang J, Tian Z. NKG2D recognition mediates toll-like receptor 3 signaling-induced breakdown of epithelial homeostasis in the small intestines of mice. Proc Natl Acad Sci U S A (2007) 104(18):7512–5. doi:10.1073/pnas.0700822104
22. Tada A, Zelaya H, Clua P, Salva S, Alvarez S, Kitazawa H, et al. Immunobiotic Lactobacillus strains reduce small intestinal injury induced by intraepithelial lymphocytes after toll-like receptor 3 activation. Inflamm Res (2016) 65(10):771–83. doi:10.1007/s00011-016-0957-7
23. Zhan R, Han Q, Zhang C, Tian Z, Zhang J. Toll-like receptor 2 (TLR2) and TLR9 play opposing roles in host innate immunity against Salmonella enterica serovar Typhimurium infection. Infect Immun (2015) 83(4):1641–9. doi:10.1128/IAI.02870-14
24. Li Y, Liu M, Zuo Z, Liu J, Yu X, Guan Y, et al. TLR9 regulates the NF-kappaB-NLRP3-IL-1beta pathway negatively in Salmonella-induced NKG2D-mediated intestinal inflammation. J Immunol (2017) 199(2):761–73. doi:10.4049/jimmunol.1601416
25. Chen GE, Wu H, Ma J, Chadban SJ, Sharland A. Toll-like receptor 4 engagement contributes to expression of NKG2D ligands by renal tubular epithelial cells. Nephrol Dial Transplant (2011) 26(12):3873–81. doi:10.1093/ndt/gfr234
26. Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol (2005) 175(11):7661–8. doi:10.4049/jimmunol.175.11.7661
27. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med (2005) 201(7):1135–43. doi:10.1084/jem.20042614
28. Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol (2010) 21(11):1878–90. doi:10.1681/ASN.2009101048
29. Zhang ZX, Wang S, Huang X, Min WP, Sun H, Liu W, et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol (2008) 181(11):7489–98. doi:10.4049/jimmunol.181.11.7489
30. Perera L, Shao L, Patel A, Evans K, Meresse B, Blumberg R, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis (2007) 13(3):298–307. doi:10.1002/ibd.20026
31. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology (2007) 132(7):2346–58. doi:10.1053/j.gastro.2007.03.025
32. Tieng V, Le Bouguenec C, du Merle L, Bertheau P, Desreumaux P, Janin A, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci U S A (2002) 99(5):2977–82. doi:10.1073/pnas.032668099
33. Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology (1998) 115(6):1405–13. doi:10.1016/S0016-5085(98)70019-8
34. Masseret E, Boudeau J, Colombel JF, Neut C, Desreumaux P, Joly B, et al. Genetically related Escherichia coli strains associated with Crohn’s disease. Gut (2001) 48(3):320–5. doi:10.1136/gut.48.3.320
35. Lea SM, Powell RM, McKee T, Evans DJ, Brown D, Stuart DI, et al. Determination of the affinity and kinetic constants for the interaction between the human virus echovirus 11 and its cellular receptor, CD55. J Biol Chem (1998) 273(46):30443–7. doi:10.1074/jbc.273.46.30443
36. Vuorinen T, Vainionpaa R, Heino J, Hyypia T. Enterovirus receptors and virus replication in human leukocytes. J Gen Virol (1999) 80(Pt 4):921–7. doi:10.1099/0022-1317-80-4-921
37. Allez M, Skolnick BE, Wisniewska-Jarosinska M, Petryka R, Overgaard RV. Anti-NKG2D monoclonal antibody (NNC0142-0002) in active Crohn’s disease: a randomised controlled trial. Gut (2017) 66(11):1918–25. doi:10.1136/gutjnl-2016-311824
38. La Scaleia R, Stoppacciaro A, Oliva S, Morrone S, Di Nardo G, Santoni A, et al. NKG2D/ligand dysregulation and functional alteration of innate immunity cell populations in pediatric IBD. Inflamm Bowel Dis (2012) 18(10):1910–22. doi:10.1002/ibd.22899
39. Allegretti YL, Bondar C, Guzman L, Cueto Rua E, Chopita N, Fuertes M, et al. Broad MICA/B expression in the small bowel mucosa: a link between cellular stress and celiac disease. PLoS One (2013) 8(9):e73658. doi:10.1371/journal.pone.0073658
40. Hosomi S, Grootjans J, Tschurtschenthaler M, Krupka N, Matute JD, Flak MB, et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J Exp Med (2017) 214(10):2985–97. doi:10.1084/jem.20162041
41. Abadie V, Discepolo V, Jabri B. Intraepithelial lymphocytes in celiac disease immunopathology. Semin Immunopathol (2012) 34(4):551–66. doi:10.1007/s00281-012-0316-x
42. Abadie V, Jabri B. IL-15: a central regulator of celiac disease immunopathology. Immunol Rev (2014) 260(1):221–34. doi:10.1111/imr.12191
43. Londei M, Ciacci C, Ricciardelli I, Vacca L, Quaratino S, Maiuri L. Gliadin as a stimulator of innate responses in celiac disease. Mol Immunol (2005) 42(8):913–8. doi:10.1016/j.molimm.2004.12.005
44. Maiuri L, Ciacci C, Auricchio S, Brown V, Quaratino S, Londei M. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology (2000) 119(4):996–1006. doi:10.1053/gast.2000.18149
45. Maiuri L, Ciacci C, Vacca L, Ricciardelli I, Auricchio S, Quaratino S, et al. IL-15 drives the specific migration of CD94+ and TCR-gammadelta+ intraepithelial lymphocytes in organ cultures of treated celiac patients. Am J Gastroenterol (2001) 96(1):150–6. doi:10.1111/j.1572-0241.2001.03437.x
46. Hue S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity (2004) 21(3):367–77. doi:10.1016/j.immuni.2004.06.018
47. Martin-Pagola A, Perez-Nanclares G, Ortiz L, Vitoria JC, Hualde I, Zaballa R, et al. MICA response to gliadin in intestinal mucosa from celiac patients. Immunogenetics (2004) 56(8):549–54. doi:10.1007/s00251-004-0724-8
48. Shafi S, Vantourout P, Wallace G, Antoun A, Vaughan R, Stanford M, et al. An NKG2D-mediated human lymphoid stress surveillance response with high interindividual variation. Sci Transl Med (2011) 3(113):113ra24. doi:10.1126/scitranslmed.3002922
49. Ferguson LR, Shelling AN, Browning BL, Huebner C, Petermann I. Genes, diet and inflammatory bowel disease. Mutat Res (2007) 622(1–2):70–83. doi:10.1016/j.mrfmmm.2007.05.011
50. Orchard TR, Dhar A, Simmons JD, Vaughan R, Welsh KI, Jewell DP. MHC class I chain-like gene A (MICA) and its associations with inflammatory bowel disease and peripheral arthropathy. Clin Exp Immunol (2001) 126(3):437–40. doi:10.1046/j.1365-2249.2001.01662.x
51. Glas J, Martin K, Brunnler G, Kopp R, Folwaczny C, Weiss EH, et al. MICA, MICB and C1_4_1 polymorphism in Crohn’s disease and ulcerative colitis. Tissue Antigens (2001) 58(4):243–9. doi:10.1034/j.1399-0039.2001.580404.x
52. Bratanic N, Smigoc Schweiger D, Mendez A, Bratina N, Battelino T, Vidan-Jeras B. An influence of HLA-A, B, DR, DQ, and MICA on the occurrence of celiac disease in patients with type 1 diabetes. Tissue Antigens (2010) 76(3):208–15. doi:10.1111/j.1399-0039.2010.01501.x
53. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics (2001) 53(4):279–87. doi:10.1007/s002510100325
54. Lopez-Hernandez R, Valdes M, Lucas D, Campillo JA, Martinez-Garcia P, Salama H, et al. Association analysis of MICA gene polymorphism and MICA-129 dimorphism with inflammatory bowel disease susceptibility in a Spanish population. Hum Immunol (2010) 71(5):512–4. doi:10.1016/j.humimm.2010.02.003
55. Zhao J, Jiang Y, Lei Y, Zou K, Wang C, Huang S, et al. Functional MICA-129 polymorphism and serum levels of soluble MICA are correlated with ulcerative colitis in Chinese patients. J Gastroenterol Hepatol (2011) 26(3):593–8. doi:10.1111/j.1440-1746.2010.06524.x
56. Hansen CH, Holm TL, Krych L, Andresen L, Nielsen DS, Rune I, et al. Gut microbiota regulates NKG2D ligand expression on intestinal epithelial cells. Eur J Immunol (2013) 43(2):447–57. doi:10.1002/eji.201242462
57. Adlercreutz EH, Weile C, Larsen J, Engkilde K, Agardh D, Buschard K, et al. A gluten-free diet lowers NKG2D and ligand expression in BALB/c and non-obese diabetic (NOD) mice. Clin Exp Immunol (2014) 177(2):391–403. doi:10.1111/cei.12340
58. Bhagat G, Naiyer AJ, Shah JG, Harper J, Jabri B, Wang TC, et al. Small intestinal CD8+TCRgammadelta+NKG2A+ intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease. J Clin Invest (2008) 118(1):281–93. doi:10.1172/JCI30989
59. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2006) 291(2):L222–31. doi:10.1152/ajplung.00327.2005
60. Borchers MT, Harris NL, Wesselkamper SC, Zhang S, Chen Y, Young L, et al. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun (2006) 74(5):2578–86. doi:10.1128/IAI.74.5.2578-2586.2006
61. Hodge S, Hodge G, Holmes M, Reynolds PN. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J (2005) 25(3):447–54. doi:10.1183/09031936.05.00077604
62. Motz GT, Eppert BL, Wortham BW, Amos-Kroohs RM, Flury JL, Wesselkamper SC, et al. Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease. J Immunol (2010) 184(8):4460–9. doi:10.4049/jimmunol.0903654
63. Wortham BW, Eppert BL, Flury JL, Morgado Garcia S, Borchers MT. TLR and NKG2D signaling pathways mediate CS-induced pulmonary pathologies. PLoS One (2013) 8(10):e78735. doi:10.1371/journal.pone.0078735
64. Wortham BW, Eppert BL, Motz GT, Flury JL, Orozco-Levi M, Hoebe K, et al. NKG2D mediates NK cell hyperresponsiveness and influenza-induced pathologies in a mouse model of chronic obstructive pulmonary disease. J Immunol (2012) 188(9):4468–75. doi:10.4049/jimmunol.1102643
65. Borchers MT, Wesselkamper SC, Curull V, Ramirez-Sarmiento A, Sanchez-Font A, Garcia-Aymerich J, et al. Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J Clin Invest (2009) 119(3):636–49. doi:10.1172/JCI34462
66. Isernhagen A, Malzahn D, Bickeboller H, Dressel R. Impact of the MICA-129Met/Val dimorphism on NKG2D-mediated biological functions and disease risks. Front Immunol (2016) 7:588. doi:10.3389/fimmu.2016.00588
67. Zingoni A, Cecere F, Vulpis E, Fionda C, Molfetta R, Soriani A, et al. Genotoxic stress induces senescence-associated ADAM10-dependent release of NKG2D MIC ligands in multiple myeloma cells. J Immunol (2015) 195(2):736–48. doi:10.4049/jimmunol.1402643
68. Suemizu H, Radosavljevic M, Kimura M, Sadahiro S, Yoshimura S, Bahram S, et al. A basolateral sorting motif in the MICA cytoplasmic tail. Proc Natl Acad Sci U S A (2002) 99(5):2971–6. doi:10.1073/pnas.052701099
69. Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol (2007) 7(9):737–44. doi:10.1038/nri2144
70. Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, et al. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J Immunol (2008) 180(7):4606–14. doi:10.4049/jimmunol.180.7.4606
71. Nice TJ, Coscoy L, Raulet DH. Posttranslational regulation of the NKG2D ligand Mult1 in response to cell stress. J Exp Med (2009) 206(2):287–98. doi:10.1084/jem.20081335
72. Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology (2008) 134(2):577–94. doi:10.1053/j.gastro.2007.11.059
Keywords: NKG2D, natural killer cells, toll-like receptor, inflammation, stress, gut, epithelia, innate lymphocytes
Citation: Antonangeli F, Soriani A, Cerboni C, Sciumè G and Santoni A (2017) How Mucosal Epithelia Deal with Stress: Role of NKG2D/NKG2D Ligands during Inflammation. Front. Immunol. 8:1583. doi: 10.3389/fimmu.2017.01583
Received: 14 September 2017; Accepted: 03 November 2017;
Published: 20 November 2017
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Mary A. Markiewicz, University of Kansas Medical Center, United StatesYasser El-Sherbiny, University of Leeds, United Kingdom
Copyright: © 2017 Antonangeli, Soriani, Cerboni, Sciumè and Santoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Santoni, YW5nZWxhLnNhbnRvbmlAdW5pcm9tYTEuaXQ=