 Edward J. Steele
Edward J. Steele- CYO’Connor ERADE Village Foundation Inc., Piara Waters, WA, Australia
The evidence for the reverse transcriptase mechanism of somatic hypermutation is substantial and multifactorial. In this 60th anniversary year of the publication of Sir MacFarlane Burnet’s Clonal Selection Theory, the evidence is briefly reviewed and updated.
Overview
The molecular mechanism underlying somatic hypermutation (SHM) of rearranged immunoglobulin (Ig) genes (V[D]J) has been controversial for some time. Although the process of DNA deamination has dominated discussion in recent years, insufficient attention has been paid to a mechanism based on reverse transcription. One reason therefore for writing this Perspective is to counter balance a widely held view in the Ig SHM field that all relevant studies on the molecular mechanism deal only with the “DNA Deamination Model” which ended in complete consensus over 10 years ago sometime between 2004 and 2007 [Table 1 and Ref. (1) in particular]. The other is a personal tribute, in this anniversary year, to the founder of modern immunology, Sir Macfarlane Burnet. It is now 60 years since the publication of the first iteration of “The Clonal Selection Theory of Acquired Immunity” (2), the foundation stone of modern immunology. It was fully expounded in his 1959 book (3) where the main idea was clonal antigenic selection from a pre-existing diverse antibody repertoire from which somatic mutations might emerge as “forbidden” anti-self clones. Joshua Lederberg then gave the concept sharp molecular focus (4) as did Melvin Cohn and colleagues (5–7). Alastair Cunningham’s concept of “clonal variation around a theme” placed antigen-driven SHM firmly within the context of expanding B lymphocyte clones (8). Somatic mutation of Ig variable region genes has therefore been part and parcel of Burnet’s clonal selection concept since its inception and is central to a rational understanding of immunological diversification, self-tolerance and the emergence of cancer. We now have a very good idea of the molecular mechanism of SHM. I have chosen to fit this scientific progress within 60 key publications since the late 1950s (Table 1). The most plausible central molecular mechanism of Ig SHM, that fits with and explains all the evidence (9–11) is based on “Reverse Transcription” of the base-modified Ig pre-mRNA (Figure 1). That is, error-prone reverse transcription, by DNA Polymerase-η, of the Ig pre-mRNA template intermediate at rearranged V[D]J gene somatic loci. The Ig pre-mRNA encoding the V[D]J region is copied off the transcribed DNA strand carrying prior AID C-to-U deamination lesions (Uracils and Abasic sites), and it also accumulates ADAR-deaminase mediated RNA editing A-to-I modifications. This already base-modified pre-mRNA sequence is then copied back to the B lymphocyte genomic DNA and integrated at the rearranged V[D]J site (concurrent with antigen-mediated selection of Ig receptor bearing B lymphocytes, Centrocytes, in the Germinal Center). This is essentially the “Reverse Transcriptase Mechanism” which Jeff Pollard and I first published 30 years ago (12). The mechanistic steps, many logical, are clearly outlined in Figure 1, which shows that the characteristic A >> T and G >> C strand bias-generating mutagenic activity is firmly focused on the nascent RNA intermediate in the context of the Transcription Bubble (9–11, 13, 14). Recent publications should be consulted for further definitive ADAR A-to-I editing of both RNA and DNA moieties at RNA:DNA hybrids within Transcription Bubbles (11, 14, 15). Not only is it important to understand the correct molecular mechanism of SHM for cancer diagnosis and detection (16, 17) but also to the current efforts to better understand (18, 19) the origin of Ig diversity involving the mechanism of evolution of the sets germline V segments and the long IGHV and IGLV haplotypes in individual human beings (20, 21).
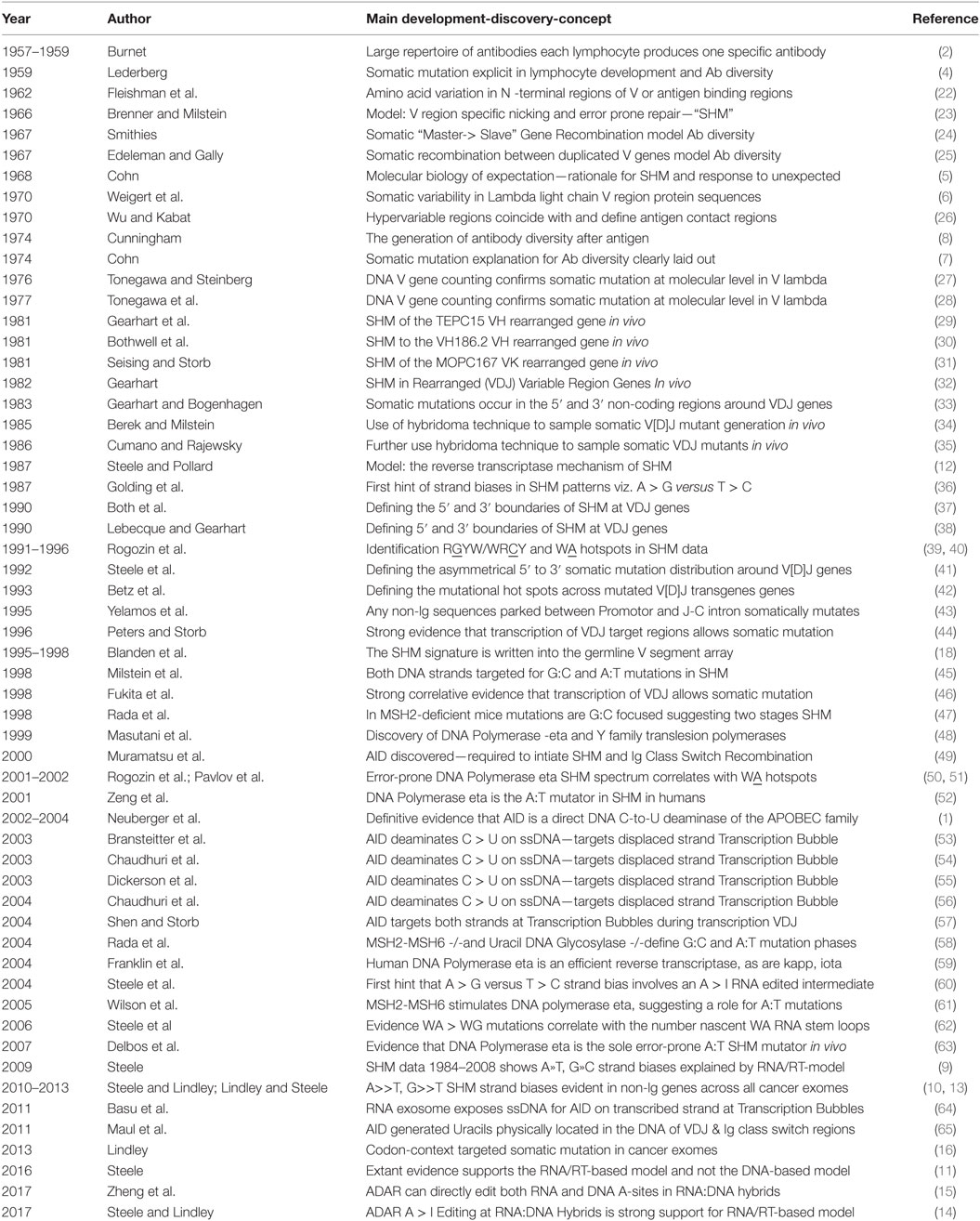
Table 1. History of somatic hypermutation (SHM): developments relevant to the reverse transcriptase mechanism.
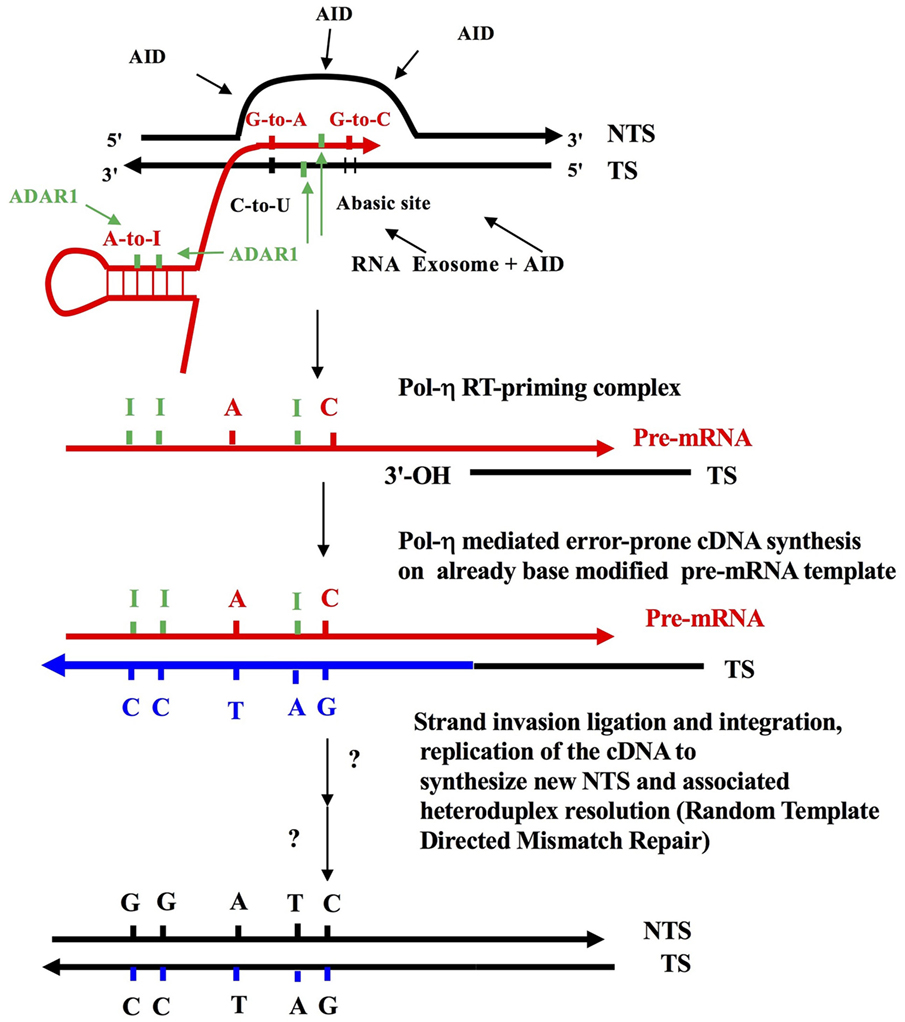
Figure 1. The reverse transcriptase mechanism of somatic hypermutation (SHM). Some elements of this figure have appeared before, and this figure in toto is a modified combination of parts from Figure 1 in Lindley and Steele (10), as well as from figures in Steele (9, 11) and Steele and Lindley (14). This is also an adaptation of the target site reverse transcription process of Luan et al. (66). Shown is an RNA Polymerase II-generated Transcription Bubble with C-site and A-site substrate deamination events by AID and ADAR proteins, which generates the strand-biased mutation signatures—A-to-G, G-to-A, G-to-T, and G-to-C (9, 11, 14). DNA strands shown by black lines; pre-mRNA as red lines; cDNA strands as thick blue lines due to DNA polymerase η (59). Green bars are Inosines. Shown also is the action of the RNA exosome (64) allowing access of AID deaminase to cytosines on the transcribed strand (TS). The ssDNA regions on the displaced non-transcribed strand (NTS) are established targets of AID action (53–56). Note that DNA mutations are first introduced as AID-mediated C-to-U, followed by excision of uracils by DNA glycosylase (UNG), which creates Abasic sites in the TS (these can mature into single strand nicks with 3′-OH ends via the action of AP endonuclease). These template Uracil and Abasic sites can be copied into pre-mRNA by RNA Pol II generating G-to-A and G-to-C modifications as shown (67). Following target site reverse transcription (66), this results in G-to-A and G-to-C mutations in the NTS, in a strand biased manner (9–11, 14). Separately at WA targets in nascent dsRNA substrates, adenosine-to-inosine (A-to-I) RNA editing events, mediated by ADAR1 deaminase, are copied back into DNA by reverse transcription via Pol-η (59). In theory, ADARs can also deaminate the RNA and DNA moieties in the RNA: DNA hybrid (14, 15). The strand invasion and integration of newly synthesized cDNA TS, as well as random-template mismatch repair (68) are hypothesized additional steps (not shown here). In short, RNA Pol II introduces modifications in the Ig pre-mRNA as it copies TS DNA with AID lesions and this is coupled to A-to-I in dsRNA stem-loops near the transcription bubble (62) as well as in RNA:DNA hybrids within the bubble (14, 15). Next, a RT-priming substrate is formed when the nicked TS strand with an exposed 3′-OH end anneals with the base modified pre-mRNA copying template allowing cDNA synthesis by Y Family translesion DNA polymerase-η (48), now acting in its reverse transcriptase mode (59). These 3′-OH annealed priming sites could arise due to excisions at previous AID-mediated Abasic sites. Alternatively, they could arise due to an endonuclease excision associated with the MSH2-MSH6 heterodimer engaging a U:G mispaired lesion (61). Shown is an A-to-T transversion generated at the RT step at a template Inosine. ADAR, Adenosine Deaminase that acts on RNA; AP, an Abasic, or apurinic/apyrimidinic, site; APOBEC family, generic abbreviation for the dC-to-dU deaminase family of which AID is a member (e.g., APOBEC1; APOBEC3 A, B, C, D, F, G, H); AID, activation induced cytidine deaminase causing C-to-U lesions at WRCY/RGYW C-site motifs in ssDNA; W, A, or U/T; WA-site, target motif for ADAR deaminase including DNA polymerase-η error prone incorporation in vitro (50, 51); Y, pyrimidines T/U or C.; R, purine A or G.
Critical Focus on the RNA/RT-Mechanism
The author has comprehensively reviewed the detailed evidence for the reverse transcription-based mechanism of SHM in previous and current studies (9–11). However, as flagged at the start of this article, many immunology researchers describe the mechanism of Ig SHM as being via DNA Polymerase-η-mediated DNA lesion repair independent of pre-mRNA in the context of the AID-initiated “DNA Deamination Model.” It will be informative then to not only refer to these literatures but also summarize the evidence directly supporting an Ig pre-mRNA intermediate and reverse transcription, as summarized in Figure 1.
The alternative to the RNA/RT-based mechanism is the “DNA Deamination Model,” which is assumed to be coupled to direct DNA-based error-prone repair via translesion DNA polymerase-η acting solely by error-prone copying of DNA templates (50, 51) during gap-repair surrounding AID-generated lesions (Uracils, Abasic sites, ssDNA nicks), as outlined in detail by Neuberger and associates (1, 58), Gearhart and associates (61, 65), and many other laboratories (53–57, 63) published mainly in the period 2002–2011. Quite apart from all the data at odds or inconsistent with this alternative theory, there have been three direct published tests of the Reverse Transcriptase Mechanism since 2001, one study was inconclusive and two studies reported positive data directly consistent with the RNA/RT-based mechanism.
In the first direct test of the RT model, Sack et al. (69) treated immunized mice with retroviral RT inhibitors, AZT, ddC and determined mutation frequencies in the anti-NP response of the rearranged VH186.2 sequence from control and test mice and showed a systematic lowering of the somatic mutation frequency by about 33–35% in both test groups compared to the control [see Table 2 in Ref. (69)]. The authors however concluded that these retroviral RT inhibitors had no statistically significant effect (the P values were P = 0.056 and P = 0.069, respectively), thus claiming that “standard reverse transcription is not required for antibody V region hypermutation in the mouse” (69). This study and the conclusions drawn have been critically evaluated, and the present author considers that the data published in Sack et al. (69) have been misinterpreted (9, 11, 70).
In the next test, Franklin et al. [(59), Figure 1 and legend] showed that the sole known error-prone DNA polymerase involved in Ig SHM, DNA Polymerase-η (52, 63) is a very efficient reverse transcriptase: as indeed are human DNA Polymerases iota (-ι) and kappa (-κ) although less active than eta (-η).
Lastly Steele et al. (62) tested directly if a quantitative relationship exists between the number of appropriate Ig VκOxJκ5 mRNA secondary structures bearing WA target sites for the ADAR1 RNA editor (adenosine to inosine, A-to-I) and the recorded incidence, across the full length of the in vivo mutated VκOx1Jκ5 sequence, of A-to-G mutations (the standard proxy for A-to-I RNA editing, where W = A or T). We showed that a highly significant and specific correlation (P < 0.002) existed between the frequency (or number) of WA-to-WG mutations and the number of mRNA hairpins that could potentially form at such WA mutation sites. This is still the best direct data-driven evidence for an RNA intermediary in Ig SHM as it implies a direct role for both RNA editing and reverse transcription during SHM in vivo, occurring at the highest frequency in the nascent RNA stem-loops presenting WA-sites in dsRNA substrates just emergent from the Transcription Bubble. We now also know that both the RNA and DNA moieties in the RNA:DNA hybrid in the Transcription Bubble can potentially be A-to-I edited and contribute to A-to-G and T-to-C somatic mutations (14, 15).
These two sets of positive results consistent with the RNA/RT-based model are completely outside the ambit of the “DNA Deamination Model” neither explained by it nor predicted by it (9, 11). This fact was pointed out explicitly in 2008 (71).
The reader is referred to the considerable detail reviewed in Steele (9, 11) and Lindley and Steele (10), but attention should also be drawn to an awkward fact that cannot be explained by the “DNA Deamination” model yet is readily explained and predicted by the RNA/RT-mechanism (Figure 1)—these are the clear strand biases of somatic mutations whereby mutations off A exceed mutations off T (A >> T, mainly A-to-G >> T-to-C) and yet paradoxically in the same data set or experiment, somatic mutations off G exceed mutations of C (G >> C, mainly G-to-A >> C-to-T). We have illustrated the contradictions of this paradox clearly in Lindley and Steele (10)—as these characteristic strand biases are noted not only in Ig SHM datasets but also in AID/APOBEC driven “Ig-SHM-like responses” in cancer genomes (10, 16).
The other foundation inspiration for our work is the series of discoveries, begun in the 1950s (72, 73), which led to the demonstration in 1970 of reverse transcription in RNA tumor viruses by Howard Temin and David Baltimore (74, 75).
In summary, the DNA-based model of Neuberger and Gearhart, or the “DNA Deamination Model,” is based on AID-induced C-to-U lesions and short-patch error-prone DNA repair by DNA Polymerase-η operating around such lesions (1, 61, 65). However, the RNA/RT-based mechanism (“Reverse Transcriptase Model”) actually subsumes this initiating AID-mediated step and then couples it in the production of the full spectrum of strand-biased mutations at both G:C and A:T base pairs: error-prone cDNA synthesis via an RNA-dependent DNA polymerase (Pol-η) copying the base-modified Ig pre-mRNA template and leading to this now error-filled cDNA copy being integrated back into the normal chromosomal site (Figure 1). The modern form of this mechanism thus depends both on initiating AID C-to-U lesions in DNA and then long-tract error-prone cDNA synthesis of the TS by DNA Polymerase-η acting in its reverse transcriptase mode (59). There are several possible tests. The first could involve measuring the outcome of ADAR A-to-I editing of the RNA and DNA moieties at RNA:DNA hybrids (15) during SHM in vivo. Thus on a DNA polymerase-η deficient background (52, 63) the lowered number of mutations at A:T base pairs may allow A-to-I editing of the RNA:DNA hybrid and nascent dsRNA stem loops (Figure 1), but the lack of a RNA-to-DNA copying step could show that T-to-C mutations now balance or exceed A-to-G mutations. Furthermore, a direct test of ADAR deamination in Ig SHM in vivo could be achieved in either ADAR1 deficient Aicardi-Goutières Syndrome (AGS) patients (76, 77) or catalytically inactive ADAR1 mouse strains, such as Adar1E861A/E861A Ifih1−/− (78). The caveat to both approaches is a statistically sufficient numbers of A/T mutations and a strategy to avoid or minimize strand bias blunting PCR recombinant artifacts (9).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author appreciates the critical comments of reviewers and editors in preparation of this MS for publication.
References
1. Noia JM, Neuberger MS. Molecular mechanisms of somatic hypermutation. Annu Rev Biochem (2007) 76:1–22. doi:10.1146/annurev.biochem.76.061705.090740
2. Burnet FM. A modification of Jerne’s theory of antibody production using the concept of clonal selection. Aust J Science (1957) 20:67–9.
3. Burnet FM. The Clonal Selection Theory of Acquired Immunity. Cambridge: Cambridge University Press (1959).
5. Cohn M. The molecular biology of expectation. In: Plescia OJ, Brain W, editors. Nucleic Acids in Immunology. New York: Springer-Verlag NY (1968). p. 671–715.
6. Weigert MG, Cesari IM, Yonkovich SJ, Cohn M. Variability in the Lambda light chain sequences. Nature (1970) 228:1045–7. doi:10.1038/2281045a0
7. Cohn M. A rationale for ordering the data on antibody diversity. In: Brent L, Holborow J, editors. Progress Immunol. II Biol Aspects I. Amsterdam-Oxford-New York: Nth Holl Publ Co (1974). p. 261–84.
8. Cunningham AJ. The generation of antibody diversity: its dependence on antigenic stimulation. Contemp Top Mol Immunol (1974) 3:1–26. doi:10.1007/978-1-4684-2838-4_1
9. Steele EJ. Mechanism of somatic hypermutation: critical analysis of strand biased mutation signatures at A:T and G:C base pairs. Mol Immunol (2009) 46:305–20. doi:10.1016/j.molimm.2008.10.021
10. Lindley RA, Steele EJ. Critical analysis of strand-biased somatic mutation signatures in TP53 versus Ig genes, in genome-wide data and the etiology of cancer. ISRN Genomics (2013) 2013:18. doi:10.1155/2013/921418
11. Steele EJ. Somatic hypermutation in immunity and cancer: critical analysis of strand-biased and codon-context mutation signatures. DNA Repair (2016) 45:1–24. doi:10.1016/j.dnarep.2016.07.001
12. Steele EJ, Pollard JW. Hypothesis: somatic hypermutation by gene conversion via the error-prone DNA->RNA->DNA information loop. Mol Immunol (1987) 24:667–73. doi:10.1016/0161-5890(87)90049-6
13. Steele EJ, Lindley RA. Somatic mutation patterns in non-lymphoid cancers resemble the strand biased somatic hypermutation spectra of antibody genes. DNA Repair (2010) 9:600–3. doi:10.1016/j.dnarep.2010.03.007
14. Steele EJ, Lindley RA. ADAR deaminase A-to-I editing of DNA and RNA moieties of RNA:DNA hybrids has implications for the mechanism of Ig somatic hypermutation. DNA Repair (2017) 55:1–6. doi:10.1016/j.dnarep.2017.04.004
15. Zheng YC, Lorenzo C, Beal PA. DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucleic Acids Res (2017) 45:3369–77. doi:10.1093/nar/gkx050
16. Lindley R. The importance of condon context for understanding the Ig-like somatic hypermutation starnd-biased patterns in TP53 mutations in breast cancer. Cancer Genet (2013) 206:222–6. doi:10.1016/j.cancergen.2013.05.016
17. Lindley RA, Humbert P, Larmer C, Akmeemana EH, Pendlebury CRR. Association between targeted somatic mutation (TSM) signatures and HGS-OvCa progression. Cancer Med (2016) 5:2629–40. doi:10.1002/cam4.825
18. Blanden RV, Rothenfluh HS, Zylstra P, Weiller GF, Steele EJ. The signature of somatic hypermutation appears to be written into the germline IgV segment repertoire. Immunol Rev (1998) 162:117–32. doi:10.1111/j.1600-065X.1998.tb01435.x
19. Steele EJ, Lloyd SS. Soma-to-germline feedback is implied by the extreme polymorphism at IGHV relative to MHC. Bioessays (2015) 37:557–69. doi:10.1002/bies.201400213
20. Kidd MJ, Chen Z, Wang Y, Jackson KJ, Zhang L, Boyd SD, et al. The inference of phased haplotypes for the immunoglobulin H chain V region gene loci by analysis of VDJ rearrangements. J Immunol (2012) 188:1333–40. doi:10.4049/jimmunol.1102097
21. Watson CT, Steinberg KM, Huddleston J, Warren RL, Malig M, Schein J, et al. Complete haplotype sequence of the human immunoglobulin heavy- chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am J Hum Genet (2013) 92:530–46. doi:10.1016/j.ajhg.2013.03.004
22. Fleischman JB, Pain RH, RR Porter RR. Reduction of gamma-globulins. Arch Biochem Biophys (1962) Suppl 1:174–80.
23. Brenner S, Milstein C. Origin of antibody variation. Nature (1966) 211:242–6. doi:10.1038/211242a0
24. Smithies O. Antibody variability. Somatic recombination between the elements if “antibody gene pairs” may explain antibody variability. Science (1967) 157:267–73. doi:10.1126/science.157.3786.267
25. Edelman GM, Gally JA. Somatic recombination of duplicated genes: an hypothesis on the origin Ab diversity. Proc Natl Acad Sci U S A (1967) 57:353–8. doi:10.1073/pnas.57.2.353
26. Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complimentarity. J Exp Med (1970) 132:211–50. doi:10.1084/jem.132.2.211
27. Tonegawa S, Steinberg C. Too many chains – too few genes. In: Cunningham AJ, editor. Generation of Antibody Diversity: A New Look. New York: Academic Press (1976). p. 175–82.
28. Tonegawa S, Hozumai N, Matthyssens GH, Schuller R. Somatic changes in the context and content of immunoglobulin genes. Cold Spring Harb Symp Quant Biol (1977) 41(Pt 2):872–89. doi:10.1101/SQB.1977.041.01.097
29. Gearhart PJ, Johnson ND, Douglas R, Hood L. IgG Antibodies to phosphorylcholine exhibits more diversity than their IgM counterparts. Nature (1981) 291:29–34. doi:10.1038/291029a0
30. Bothwell ALM, Pashkind M, Reth M, Imanishi-Kari T, Rajewsky T, Baltimore D. Heavy chain variable region contribution to the NPb family of antibodies: somatic mutation evident in a IgG2a variable region. Cell (1981) 24:625–37. doi:10.1016/0092-8674(81)90089-1
31. Selsing E, Storb U. Somatic mutation of immunoglobulin light-chain variable region genes. Cell (1981) 25:47–58. doi:10.1016/0092-8674(81)90230-0
32. Gearhart PJ. Generation of immunoglobulin variable gene diversity. Immunol Today (1982) 3:107–12. doi:10.1016/S0167-5699(82)80026-1
33. Gearhart PJ, Bogenhagen DF. Clusters of point mutations are found exclusively around rearranged antibody variable gene. Proc Natl Acad Sci U S A (1983) 80:3439–43. doi:10.1073/pnas.80.11.3439
34. Berek C, Griffith GM, Milstein C. Molecular events during maturation of the immune response to oxazolone. Nature (1985) 316:412–8. doi:10.1038/316412a0
35. Cumano A, Rajewsky K. Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. EMBO J (1986) 5:2459–68.
36. Golding GB, Gearhart PJ, Glickman RW. Patterns of somatic mutations in immunoglobulin variable genes. Genetics (1987) 115:169–76.
37. Both GW, Taylor L, Pollard JW, Steele EJ. Distribution of mutations around rearranged heavy-chain antibody variable-region genes. Mol Cell Biol (1990) 10:5187–96. doi:10.1128/MCB.10.10.5187
38. Lebecque SG, Gearhart PJ. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5′ boundary is near the promoter, 3′ boundary is approximately 1 kb from V-D-J gene. J Exp Med (1990) 172:1717–27. doi:10.1084/jem.172.6.1717
39. Rogozin IB, Solovyov VV, Kolchanov NA. Somatic hypermutagenesis in immunoglobulin genes. I. Correlation between somatic mutations and repeats. Somatic mutation properties and clonal selection. Biochim Biophys Acta (1991) 1089:175–82. doi:10.1016/0167-4781(91)90005-7
40. Rogozin IB, Sredneva NE, Kolchanov NA. Somatic hypermutagenesis in immunoglobulin genes. III. Somatic mutations in the chicken light chain locus. Biochim Biophys Acta (1996) 1306:171–8. doi:10.1016/0167-4781(95)00241-3
41. Steele EJ, Rothenfluh HS, Both GW. Defining the nucleic acid substrate for somatic hypermutation. Immunol Cell Biol (1992) 70:129–44. doi:10.1038/icb.1992.18
42. Betz AG, Neuberger MS, Milstein C. Discriminating intrinsic and antigen-selected mutational hotspots in immunoglobulin V genes. Immunol Today (1993) 14:405–11. doi:10.1016/0167-5699(93)90144-A
43. Yelamos J, Klix N, Goyenechea B, Lozano F, Chui YL, Gonzalez Fernandez A, et al. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature (1995) 376:225–9. doi:10.1038/376225a0
44. Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity (1996) 4:57–65. doi:10.1016/S1074-7613(00)80298-8
45. Milstein C, Neuberger MS, Staden R. Both DNA strands of antibody genes are hypermutation targets. Proc Natl Acad Sci U S A (1998) 95:8791–4. doi:10.1073/pnas.95.15.8791
46. Fukita Y, Jacobs H, Rajewsky K. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity (1998) 9:105–14. doi:10.1016/S1074-7613(00)80592-0
47. Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity (1998) 9:135–41. doi:10.1016/S1074-7613(00)80595-6
48. Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem (2002) 71:17–50. doi:10.1146/annurev.biochem.71.083101.124707
49. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102:553–63. doi:10.1016/S0092-8674(00)00078-7
50. Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol (2001) 2:530–6. doi:10.1038/88732
51. Pavlov YI, Rogozin IB, Galkin AP, Aksenova AV, Hanaoka F, Rada C, et al. Correlation of somatic hypermutation specificity and A-T base pair substitution errors by DNA polymerase-eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci U S A (2002) 99:9954–9. doi:10.1073/pnas.152126799
52. Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase-eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol (2001) 2:537–41. doi:10.1038/88740
53. Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxcytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A (2003) 100:4102–7. doi:10.1073/pnas.0730835100
54. Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature (2003) 422:726–30. doi:10.1038/nature01574
55. Dickerson SK, Market E, Besmer E, Papvasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med (2003) 197:1291–6. doi:10.1084/jem.20030481
56. Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature (2004) 430:992–8. doi:10.1038/nature02821
57. Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci U S A (2004) 101:12997–3002. doi:10.1073/pnas.0404974101
58. Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell (2004) 16:163–71. doi:10.1016/j.molcel.2004.10.011
59. Franklin A, Milburn PJ, Blanden RV, Steele EJ. Human DNA polymerase-eta an A-T mutator in somatic hypermutation of rearranged immunoglobulin genes, is a reverse transcriptase. Immunol Cell Biol (2004) 82:219–25. doi:10.1046/j.0818-9641.2004.01221.x
60. Steele EJ, Franklin A, Blanden RV. Genesis of the strand biased signature in somatic hypermutation of rearranged immunoglobulin variable genes. Immunol Cell Biol (2004) 82:208–18. doi:10.1046/j.0818-9641.2004.01224.x
61. Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, et al. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J Exp Med (2005) 201:637–45. doi:10.1084/jem.20042066
62. Steele EJ, Lindley RA, Wen J, Weiler GF. Computational analyses show A-to-G mutations correlate with nascent mRNA hairpins at somatic hypermutation hotspots. DNA Repair (2006) 5:1346–63. doi:10.1016/j.dnarep.2006.06.002
63. Delbos F, Aoufouchi S, Faili A, Weill J-C, Reynaud C-A. DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J Exp Med (2007) 204:17–23. doi:10.1084/jem.20062131
64. Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell (2011) 144:353–63. doi:10.1016/j.cell.2011.01.001
65. Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol (2011) 12:70–6. doi:10.1038/ni.1970
66. Luan DD, Korman MH, Jakubczak JL, Eichbush TH. Reverse transcription of R2B mRNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell (1993) 72:595–605. doi:10.1016/0092-8674(93)90078-5
67. Kuraoka I, Endou M, Yamaguchi Y, Wada Y, Handa H, Tanaka K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. J Biol Chem (2003) 278:7294–9. doi:10.1074/jbc.M208102200
68. MacPhee DG. Mismatch repair, somatic mutations and the origins of cancer. Cancer Res (1995) 55:5489–92.
69. Sack SZ, Bardwell PD, Scharff MD. Testing the reverse transcriptase model of somatic mutation. Mol Immunol (2001) 38:303–11. doi:10.1016/S0161-5890(01)00058-X
70. Steele EJ. DNA polymerase-η as a reverse transcriptase: implications for mechanisms of hypermutation in innate anti-retroviral defences and antibody SHM systems. DNA Repair (2004) 3:687–92. doi:10.1016/j.dnarep.2004.03.040
71. Steele EJ. Reflections on the state of play in somatic hypermutation. Mol Immunol (2008) 45:2723–6. doi:10.1016/j.molimm.2008.02.002
72. Temin HM. Malignant transformation of cells by viruses. Perspect Biol Med (1970) 14:11–26. doi:10.1353/pbm.1970.0006
73. Temin HM. The protovitus hypothesis: speculations on the significance of RNA directed DNA synthesis for normal devlopment and for carcinogenesis. J Natl Cancer Inst (1971) 46:3–7.
74. Temin HM, Mizutani S. RNA-dependent DNA polyerase in virions of Rous Sarcoma Virus. Nature (1970) 226:1211–3. doi:10.1038/2261211a0
75. Baltimore D. RNA-dependent DNA polymerase in in virions of RNA tumor virus. Nature (1970) 226:1209–11.
76. O’Connell MA, Mannion NM, Keegan LP. The epitranscriptome and innate immunity. PLoS Genet (2015) 11(12):e1005687. doi:10.1371/journal.pgen.1005687
77. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet (2012) 44:1243–8. doi:10.1038/ng.2414
Keywords: somatic hypermutation, strand-biased mutations, DNA polymerase-η, A-to-I RNA and DNA editing, RNA exosome, AID-deaminase, reverse transcription
Citation: Steele EJ (2017) Reverse Transcriptase Mechanism of Somatic Hypermutation: 60 Years of Clonal Selection Theory. Front. Immunol. 8:1611. doi: 10.3389/fimmu.2017.01611
Received: 23 August 2017; Accepted: 07 November 2017;
Published: 23 November 2017
Edited by:
Thomas L. Rothstein, Western Michigan University Homer Stryker M.D. School of Medicine, United StatesReviewed by:
Paolo Casali, The University of Texas Health Science Center San Antonio, United StatesMasaki Hikida, Kyoto University, Japan
Copyright: © 2017 Steele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward J. Steele, ZWpzdGVlbGVAY3lvLmVkdS5hdQ==