 Meg L. Donovan
Meg L. Donovan Thomas E. Schultz
Thomas E. Schultz Antje Blumenthal
Antje Blumenthal- The University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, Brisbane, QLD, Australia
Tuberculosis (TB) remains a major global health threat. Urgent needs in the fight against TB include improved and innovative treatment options for drug-sensitive and -resistant TB as well as reliable biological indicators that discriminate active from latent disease and enable monitoring of treatment success or failure. Prominent interferon (IFN) inducible gene signatures in TB patients and animal models of Mycobacterium tuberculosis infection have drawn significant attention to the roles of type I IFNs in the host response to mycobacterial infections. Here, we review recent developments in the understanding of the innate immune pathways that drive type I IFN responses in mycobacteria-infected host cells and the functional consequences for the host defense against M. tuberculosis, with a view that such insights might be exploited for the development of targeted host-directed immunotherapies and development of reliable biomarkers.
Introduction
With an estimated 1.8 million TB-related annual deaths worldwide, approximately one-third of the global population harboring latent Mycobacterium tuberculosis infection, and significant numbers of drug-resistant cases, there is an urgent and compelling need for more sophisticated diagnostic and treatment options for tuberculosis (TB) (1). M. tuberculosis is an intracellular pathogen that mainly resides within macrophages. The multi-tiered immune response elicited upon M. tuberculosis infection is complex, and our understanding of the requirements for protective immunity remain incomplete (2). Observations in humans and mouse models have firmly established essential roles for interleukin-12 (IL-12) and interferon gamma (IFN-γ)-mediated T cell functions in the control of M. tuberculosis infection. Inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and IL-1 are critical contributors to the immune defense against M. tuberculosis (2). Recently described gene signatures in blood of patients with active TB disease (3–9) are being explored extensively for their utility as biomarkers for the reliable diagnosis of active TB, tracking of at-risk individuals, and monitoring of treatment outcome. Additionally, the discovery of IFN-related gene signatures in patients with active TB disease (3–9) has created significant momentum behind investigation of the innate immune pathways and pathophysiological consequences of type I IFN expression during M. tuberculosis infection. While IFN-γ is the sole type II IFN, type I IFNs in humans comprise several IFN-α subtypes, IFN-β, IFN-ω, IFN-ε, IFN-τ, and IFN-κ (10). All known type I IFNs signal through a common receptor, IFNAR, which consists of the low-affinity IFNAR1 and the high-affinity IFNAR2 (11, 12). It is increasingly appreciated that type I IFNs not only play a significant role in the antiviral response but are also a central aspect of the host response to bacterial infections (13, 14). In this context, type I IFNs appear to promote or impair pathogen control and disease pathology depending on the infectious agent, acute or chronic state of the infection, and possibly also the model system studied (15). Evidence from human case reports and animal models suggests complex contributions of type I IFNs to the host response during M. tuberculosis infection as both protective and detrimental roles for the host have been described. The impact of type I IFNs and related immune response networks on pathology and pathogen control during M. tuberculosis infection are also assessed with an eye to host-directed interventions in TB. In this review, we highlight recent advances in the study of TB patient blood gene expression signatures, and the definition of innate immune drivers and immunological consequences of type I IFN expression in the context of M. tuberculosis infection.
Blood Gene Expression Signatures in the Quest for TB Biomarkers
There are increasing numbers of cross-sectional and longitudinal studies exploring whole blood gene expression in TB patients as a potential diagnostic indicator of disease status, manifestation, and responsiveness to treatment [reviewed in more detail elsewhere, e.g. (16–18)]. While the individual genes identified across the various studies are remarkably discordant (18), overall signatures returned from many of these studies have implicated IFN signaling (5, 16). However, independent meta-analyses of publicly available datasets have expressed different views on the dominance and robustness of IFN signatures in active TB (6, 19). An overarching goal of current studies is the identification of minimal gene signatures in global gene expression profiles that can be utilized as reliable TB diagnostic markers in clinical settings. Potential applications of this approach include distinguishing active TB from latent M. tuberculosis infection and other infections; monitoring of treatment success/failure; and predicting risk of developing active disease (6–9, 20–22). Some, but not all, of these proposed minimal marker combinations contain IFN-regulated genes. The “omics” approaches currently being pursued clearly indicate innate and adaptive immune response signatures that significantly enhance our current understanding of peripheral host responses during active and latent TB. Consideration of whether or not these molecular signatures are unique to TB is important when designing and interpreting global host response profiles for the derivation of reliable diagnostic markers. Consideration of potential confounding factors, as well as inclusion of unrelated disease controls cohorts, is also critical for success in these endeavors.
Human immunodeficiency virus (HIV) remains a major risk factor for development of active TB. Approximately 1/3 of HIV-positive individuals worldwide are latently infected with M. tuberculosis (1). Currently, 55% of notified TB cases are HIV-positive and approximately 22% of TB-related deaths occur in HIV-positive individuals (1). Due to significant virus-associated immunological alterations in HIV-positive individuals, the utility of minimal gene signatures in distinguishing active from latent TB may be compromised in these cases (9, 21). Encouragingly, signatures that distinguish active TB from latent TB in HIV-positive and -negative individuals are emerging (8, 23). Additionally, type II diabetes is being increasingly recognized as a significant comorbidity that adversely affects TB severity, treatment responsiveness, and outcome in high TB burden countries (24). Thus, exploration of molecular signatures to better understand the nature of the TB/type II diabetes interaction is an important upcoming challenge. Notably, a recent study that compared blood transcriptomics in South-Indian TB patients with and without type II diabetes concluded that the utility of blood gene expression markers in the diagnosis of TB might not be confounded by type II diabetes (25). Independent confirmation of these findings in populations where high type II diabetes and high TB burden intersect will be of great significance for the development of new molecular marker-based TB diagnostics.
Interferon-associated genes are present not only in blood gene expression signatures reported for active TB, but also in patients with the granulomatous diseases meliodosis and sarcoidosis (7, 26–29). This suggests that peripheral blood gene expression signatures may be shaped by responses to tissue pathology rather than a specific underlying cause. Moreover, Blankley et al. highlight that severity of clinical symptoms in TB patients is associated with the magnitude of gene expression, regardless of the site of infection (7). Nevertheless, comparisons of global response patterns in active and latent TB with other infectious or inflammatory conditions have yielded encouraging results, as they suggest that molecular signatures that reliably discriminate active TB can be identified (3, 21, 28, 30). Patient cohort-based profiling in search of reliable TB diagnostics calls for careful study design, inclusion of clinical metadata, and clear definition of patient and control cohorts (17). Continuing developments in the analysis of “omics” data, including statistical frameworks, machine learning, multi-platform integration, and network analyses, will be important assets in defining molecular markers that perform robustly independent of age, gender, ethnicity, and comorbidities.
Molecular Pathways That Drive Type I IFN Production in Mycobacteria-Infected Host Cells
Mycobacterium tuberculosis infection induces type I IFN expression in human and mouse macrophages and myeloid dendritic cells (31–38). The serine/threonine kinase, TANK-binding kinase 1 (TBK1), is a central driver of type I IFN expression in M. tuberculosis-infected host cells (33). TBK1 facilitates phosphorylation of interferon regulatory factors (IRFs), and IRF3 and IRF5 have been reported to facilitate type I IFN expression in mycobacteria-infected host cells (39). IFN-β expression requires collaboration of IRFs with other transcription factors including NF-κB, ATF-2, and c-Jun (40), emphasizing the complexity of host signaling events that shape type I IFN expression. Pattern recognition receptors (PRRs) that have been linked to TBK1 phosphorylation and type I IFN expression in response to mycobacterial infection include the Nod-like receptor NOD2, stimulator of interferon genes (STING) either directly or via activation of cyclic GMP-AMP synthase (cGAS), as well as Toll-like receptor (TLR) 4 (Figure 1).
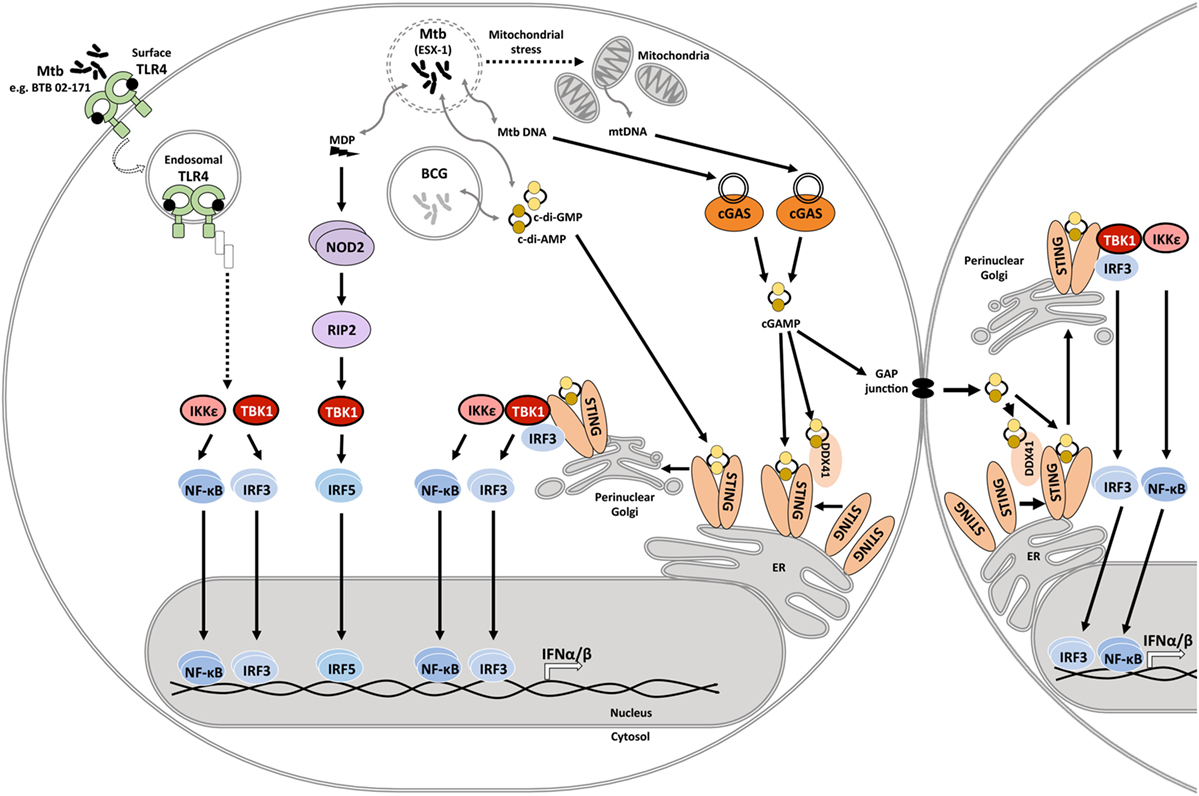
Figure 1. Innate immune signaling pathways that drive type I IFN responses in mycobacteria-infected host cells. During infection, the Mycobacterium tuberculosis ESX-1 secretion system facilitates disruption of the phagosomal membrane contributing to mitochondrial stress and leakage of mycobacterial products into the cytosol. These products include the cyclic dinucleotides cyclic diadenosine monophosphate (c-di-AMP) and cyclic diguanosine monophosphate (c-di-GMP), DNA, and N-glycolylated muramyl dipeptide (MDP). Recognition of cytosolic mitochondrial and mycobacterial DNA by cyclic GMP-AMP synthase (cGAS) initiates formation of the second messenger cyclic-GMP-AMP (cGAMP). cGAMP and bacterial cyclic dinucleotides interact with dimeric STING on the endoplasmic reticulum or the STING-accessory molecule DDX41. STING activation and relocation to the perinuclear Golgi initiates recruitment and activation of TANK-binding kinase 1 (TBK1), and possibly also IKKε. Subsequent activation and nuclear translocation of the functionally active dimeric transcription factors interferon regulatory factor (IRF)3 and NF-κB drives expression of type I IFNs. cGAMP may also access uninfected cells via gap junctions, triggering STING activation and type I IFN expression in bystander cells. MDP is recognized by NOD2, which leads to RIP2-mediated activation of TBK1, culminating in IRF5 dimerization and nuclear translocation. M. tuberculosis isolate BTB 02-171 induces type I IFN expression dependent on TLR4. It is inferred that the induction pathway is similar to the known toll-like receptor (TLR)4 endosomal signaling pathway. However, this awaits experimental confirmation. Mycobacterium bovis Bacillus Calmette–Guérin (BCG) lacks ESX-1, yet, can elicit type I IFN expression. Cyclic dinucleotide-mediated STING activation independent of ESX-1 has been described.
The Mycobacterial Type VII Secretion System ESX-1 Facilitates Activation of Cytosolic Surveillance Pathways and Type I IFN Expression
Early studies established that induction of type I IFN responses by M. tuberculosis was greatly diminished if the genomic region of difference-1 (RD-1), the RD-1 encoded type VII ESAT-6 secretion system 1 (ESX-1), or its key components were deleted (33, 34, 39). The ESX-1 secretion system is a major virulence determinant of M. tuberculosis that mediates secretion of mycobacterial products that shape host responses to infection (41). ESX-1 may also directly affect phagosome membrane integrity (42). Co-localization studies with galectin-3, a marker of damaged membranes, and ubiquitin suggest that within 24 h of infection approximately 10% of M. tuberculosis bacteria are in contact with the cytoplasm and that this is dependent on ESX-1 (43). Compromised phagosomal membranes facilitate interactions of M. tuberculosis and its products with the cytosol fostering recognition by cytosolic surveillance pathways (33, 35, 39, 44, 45).
NOD2 has been implicated as a cytosolic sensor of M. tuberculosis that drives IFN-α and IFN-β expression upon infection (39, 46). Polyubiquitination of the NOD2-effector RIP2 was significantly diminished upon infection with ESX-1-deficient M. tuberculosis when compared to wild-type bacteria, identifying RIP2 as a major downstream effector of NOD2 in the recognition of M. tuberculosis. The unusual mycobacterial N-glycolylated muramyl dipeptide is a driver of NOD2-mediated type I IFN responses via RIP2 and IRF5, but not IRF3 (39). Reports addressing the role of NOD2 in the host control of M. tuberculosis infection in the mouse model in vivo have returned varying results. Whereas NOD2-deficient (Card15−/−) mice were not impaired in their ability to control infection with M. tuberculosis in two independent studies (47, 48), one study reported impaired T helper 1 (Th1) responses associated with slightly impaired survival and a modest increase in bacterial burden late during infection (48). Similarly, reports on human polymorphisms in the NOD2 gene and susceptibility to, or protection from TB have returned varying results and will require further validation (49–53).
NOD2- and RIP2-deficiency only partially ablated M. tuberculosis-induced IFN-β and IFN-α expression (39), suggesting that additional molecular pathways of type I IFN induction exist. Several recent independent studies identified cGAS and STING as central drivers of IFN-β expression in human and murine macrophages during M. tuberculosis infection (35, 37, 38, 54). STING contains four transmembrane domains that anchor it into the endoplasmic reticulum membrane, as well as a large C-terminal domain involved in binding of TBK1 and IRF3. Binding of one molecule of cyclic di-nucleotide of either host or bacterial origin to STING dimers facilitates TBK1 and IRF3 activation (55). Endogenous 2′3′-cyclic GMP-AMP (cGAMP) is a second messenger generated by cGAS upon binding of cytoplasmic DNA. This cytoplasmic surveillance mechanism is a major driver of STING activation and type I IFN responses. Both cGAS- and STING-deficient human and mouse macrophages are impaired in their ability to express IFN-β in response to M. tuberculosis infection (35, 37, 38, 54). The underlying mechanisms that trigger this pathway are under intense investigation. It has been suggested that mycobacterial DNA gains access to the host cell cytoplasm and directly binds to cGAS, a process facilitated by ESX-1-dependent phagosome damage (37, 38). Others reported that the extent of macrophage IFN-β expression in response to different M. tuberculosis isolates correlated with mitochondrial stress, with no observable differences in the proportions of bacteria associated with damaged phagosomes (43). These observations suggest that the extent of mitochondrial DNA released during M. tuberculosis infection may contribute to cGAS-dependent activation of type I IFN expression by macrophages and determine the extent of the type I IFN response (43). Further amplification of STING-driven type I IFN expression may occur in uninfected bystander cells through shuttling of cGAMP through tight junction proteins (37, 56).
In addition, bacterial pathogens generate cyclic di-nucleotides that can activate STING independent of cGAS (55). The M. tuberculosis di-adenylate cyclase (disA or dacA) synthesizes c-di-AMP, which triggers IFN-β expression in macrophages and dendritic cells through STING activation, either through direct binding or via engagement of DEAD-box helicase 4 (DDX4) (35, 55, 57, 58). IFN-β expression in response to M. tuberculosis disA mutant bacteria was reduced compared to wild-type bacteria. Conversely, disA overexpression in M. tuberculosis CDC1551 and Erdman enhanced IFN-β expression (35). The discovery that M. tuberculosis expresses a phosphodiesterase, cdnP, that not only cleaves bacterial c-di-AMP and c-di-CMP but also the host-derived STING activator 2′3′-cGAMP (59, 60), is in keeping with the remarkable adaptation of this bacterium to its intracellular niche within the host. Accordingly, a cdnP M. tuberculosis transposon mutant induced elevated IFN-β, whereas cdnP overexpression impaired IFN-β expression by macrophages (60). Of note, the elevated IFN-β response elicited by cdnP deficient M. tuberculosis bacteria was independent of NOD2 (59), indicating that these cytoplasmic detection pathways are operating in parallel. Importantly, however, modulation of disA and cdnP expression did not only affect type I IFN expression but also pro-inflammatory cytokines like TNF, IL-6, and IL-1 (35, 60). These cytokines are important for the immune control of M. tuberculosis infection and can counter-regulate type I IFN expression (discussed below). This impact on the balance of key cytokines that are essential to control M. tuberculosis infection in vivo may account for the enhanced or attenuated virulence of the respective M. tuberculosis mutants in vivo (35, 59, 60). Moreover, the ability and extent of c-di-AMP production by M. tuberculosis, as well as cGAS and STING activation in M. tuberculosis-infected macrophages have been linked to autophagy (35, 38, 54). It will be valuable to further investigate molecular mechanisms of the engagement of pro-inflammatory cytokine responses and cell autonomous defense mechanisms by mycobacteria-derived c-di-AMP, which may involve other c-di-AMP-sensors such as DDX4 (35, 61) and RECON (62).
Despite the profound contributions of cGAS and STING to M. tuberculosis-induced type I IFN responses and autophagy activation reported by in vitro studies (35, 37, 38, 45, 54), bacterial burden in infected organs of mice deficient in cGAS or STING expression was comparable to wild-type controls (38, 54). One study reported impaired survival of cGAS- but not STING-deficient mice late during the chronic phase of infection (>100 days) (54). Whether this is linked to STING-independent functions of cGAS such as impaired autophagy (54, 63) remains to be established. Moreover, roles for DDX4 (61) and RECON (62) in regulating host responses and pathogen control during M. tuberculosis infection remain to be defined.
ESX-1-Independent Induction of Type I IFNs in Mycobacterial Infection
While the data discussed above support a central role for the ESX-1 secretion system in the induction of type I IFN expression in M. tuberculosis-infected host cells, there is compelling evidence for mycobacteria-induced type I IFN responses in the absence of ESX-1. Type I IFN expression in response to ESX-1-deficient M. tuberculosis strains is reduced but not ablated compared to WT mycobacteria (38, 39). Moreover, the attenuated Mycobacterium bovis strain Bacillus Calmette–Guérin (BCG) lacks the RD-1 region including the genes that encode the ESX-1 secretion system (64). Yet, BCG induces type I IFN expression in both human and mouse primary macrophages, albeit to a significantly lesser degree than virulent M. tuberculosis (34, 35). IFN-β expression in BCG-infected macrophages was significantly reduced in Sting−/− cells, indicating STING-driven type I IFN expression. Moreover, overexpression of disA in BCG enhanced IFN-β expression compared to wild-type BCG (35). These data suggest that mycobacteria-derived dinucleotides may gain access to the cytosol in the absence of ESX-1 and induce IFN-β expression via activation of STING. It seems plausible that bacteria-derived dinucleotides may also activate STING and type I IFN expression in uninfected bystander cells by passing the cell membrane or through gap-junction trans signaling as proposed for cGAMP (37, 56). The extent to which mitochondrial stress responses may contribute to this STING-dependent type I IFN response (43) remains to be established.
Interactions of bacterial or viral components with specific TLRs can drive type I IFN expression. TLRs are type I transmembrane proteins that localize to the cell membrane or endosomal membranes. They sense microbial components in the extracellular environment or internalized materials in the “extracellular” space of intracellular trafficking compartments, but not in the cytoplasm (65). Dimerization of TLRs upon ligand binding facilitates recruitment of adaptor proteins and the initiation of intracellular signaling (66, 67). MyD88-dependent signaling from endosomal TLR7, TLR8, and TLR9 activates IRF-7 and results in the expression of type I IFNs (68). TLR4 internalization into endosomes upon stimulation with lipopolysaccharide engages TRIF and TRAM, facilitating IKKε and TBK-1 activation. Subsequent phosphorylation of IRF-3 leads to the expression of type I IFNs (69, 70). Recent data suggest that TLR2 may similarly induce TRIF- and TRAM-dependent signaling from endosomes (71). Initial studies concluded that TLRs are not a major driver of type I IFN responses during M. tuberculosis infection. Ifnb1 expression by mouse macrophages infected with M. tuberculosis H37Rv was independent of TLR2, exhibited a minor defect in Trif−/− cells, and appeared to be negatively regulated by TLR4 (comparison of C3H/HeN versus C3H/HeJ) and MyD88 (33). A separate study showed that macrophage Ifnb1 expression upon infection with the M. tuberculosis clinical isolate 1254 was equivalent between wild-type cells and macrophages deficient in Tlr4, Tlr2, Tirap, Myd88, or Trif (32). In contrast, TLR4-driven Ifnb1 expression induced by the lineage 2 M. tuberculosis isolate BTB 02-171 was associated with TLR4-dependent enhanced virulence in mice (72, 73). While the TLR4 ligand in strain BTB 02-171 remains to be identified, these observations invite speculation that TLR4 signaling in this context may occur from maturing mycobacteria-containing phagosomes or endosomal compartments during infection. Whether such signaling engages TRIF, and/or also occurs downstream of other TLRs implicated in human susceptibility to M. tuberculosis infection, remains to be established. These findings encourage investigations into the TLR-activating properties of M. tuberculosis isolates dominant in populations or ethnic groups that show associations of TB susceptibility or severity with polymorphisms in TLR4 (74, 75), TLR2 and its co-receptors (76–81), as well as TLR8 (80, 82) and TLR9 (83). Comparisons with populations where such associations cannot be established (63, 84–87) will be of great value.
Consequences of Type I IFN Signaling During M. tuberculosis Infection
The IFN gene signatures reported by the landmark study by Berry et al. (3) and subsequently several other studies firmly establish that IFNs flavor the peripheral immune response in patients with active TB. It is important to note that both type I and type II IFNs are implicated in driving these IFN signatures, especially those attributable to STAT1 homodimerization-stimulated gamma-IFN activation sequence (GAS)-regulated gene expression, which occurs in response to both type I and type II IFNs (88).
Impact of Type I IFN Signaling in Mice and Humans on Pathogen Control and Disease Outcome
Mice
The functional consequences of type I IFN signaling in the context of M. tuberculosis infection are incompletely understood. The evidence so far suggests that the impact of type I IFN signaling on host resistance and disease severity are determined by the immune competence of the host, with contributions by the bacterial strain. The degree of M. tuberculosis-induced IFNα/β expression in mice has been correlated with the virulence of M. tuberculosis strains (43, 89). Ifnar1-deficiency in mice with a M. tuberculosis-susceptible genetic background (A129, 129S2) enhanced host survival upon infection with the high type I IFN-inducing hypervirulent HN878 strain but also the low IFN-inducing strain CDC1551 (90, 91). Ifnar1-deficiency also partially rescued accelerated mortality in the highly susceptible Il1r−/− mice infected with the moderate type I IFN-inducer, H37Rv (92). These data suggest that endogenous type I IFN signaling impairs host resistance to M. tuberculosis infection in susceptible hosts. A study in C57BL/6 mice, a mouse strain more resistant to M. tuberculosis infection, reported equivalent survival rates between Ifnar1−/− and C57BL/6 mice infected with the hypervirulent HN878 and other M. tuberculosis strains, despite diminished bacterial burden in the lungs of infected Ifnar1−/− mice (93). Other studies compared C57BL/6 and Ifnar1−/− mice during infection with H37Rv and the hypervirulent, high type I IFN-inducing BTB 02-171 strain for 70 and >200 days of infection. As neither C57BL/6 wild-type nor Ifnar1−/− mice succumbed to the infection during these time frames, the impact of Ifnar1 single deficiency on survival upon infection with these M. tuberculosis strains remained unclear (73, 94). Of note, Ifnar1−/− mice on C57BL/6 background showed diminished bacterial burden and pathology as well as a minor survival advantage during long-term infection (>200 days) with Mycobacterium africanum (95). While M. africanum is a poor inducer of type I IFN responses (43), these data suggest that type I IFN signaling may subvert effective long-term host control of this infection in a relatively resistant host.
Administration of neutralizing anti-IFNα/β antibodies to resistant B6D2/F1 mice prior to and upon infection with M. tuberculosis HN878 provided a long-term survival benefit, associated with lower IFN-α expression and STAT1 activation in lung tissue, albeit without significant effects on bacterial burden (90). This suggests that pre-existing type I IFN responses in the host at the time of exposure may influence the outcome of M. tuberculosis infection. This is supported by the observation that pre-infection of mice with influenza A virus accelerated host death (>160 days of M. tuberculosis infection). This was associated with exacerbated inflammation and some transient early and late elevation of bacterial burden, which was abrogated in Ifnar1−/− mice (96). Moreover, artificial exacerbation of type I IFN responses in M. tuberculosis-infected resistant mice, e.g., by intranasal administration of IFNα/β or the stabilized synthetic TLR3 agonist pICLC, significantly impaired host survival, exacerbated lung inflammation, and impaired the host’s ability to restrict M. tuberculosis (89, 92, 97).
While the reasons for variable effects on bacterial burden and host survival in the abovementioned studies remain to be elucidated, the evidence gathered through genetic and exogenous modulation indicate detrimental effects of type I IFN signaling in mouse models of M. tuberculosis infection across a spectrum of host genetic backgrounds and mycobacterial strains (Table 1). However, there is evidence that type I IFN signaling exerts some protective effects in the absence of IFN-γ. Mice that lack the IFN-γ receptor are highly susceptible to M. tuberculosis infection, as reflected by poor host control of the bacteria, extensive pathology, and accelerated death. Additional Ifnar1 deficiency in Ifngr−/− mice further impaired survival suggesting that type I IFN signaling played a non-redundant protective role relatively early during infection with M. tuberculosis H37Rv and BTB 02-171 that was only apparent in the absence of IFN-γ signaling (73, 94). These observations suggest that the relative balance of type I and type II IFN functions defines host control of M. tuberculosis and pathology during the infection. It is worth noting that in mice infected with M. bovis BCG, Mycobacterium avium, and Mycobacterium smegmatis, type I IFNs appear to have protective roles (98–100), encouraging further investigations into the functional contributions of type I IFNs to host protection across a wider spectrum of mycobacteria.
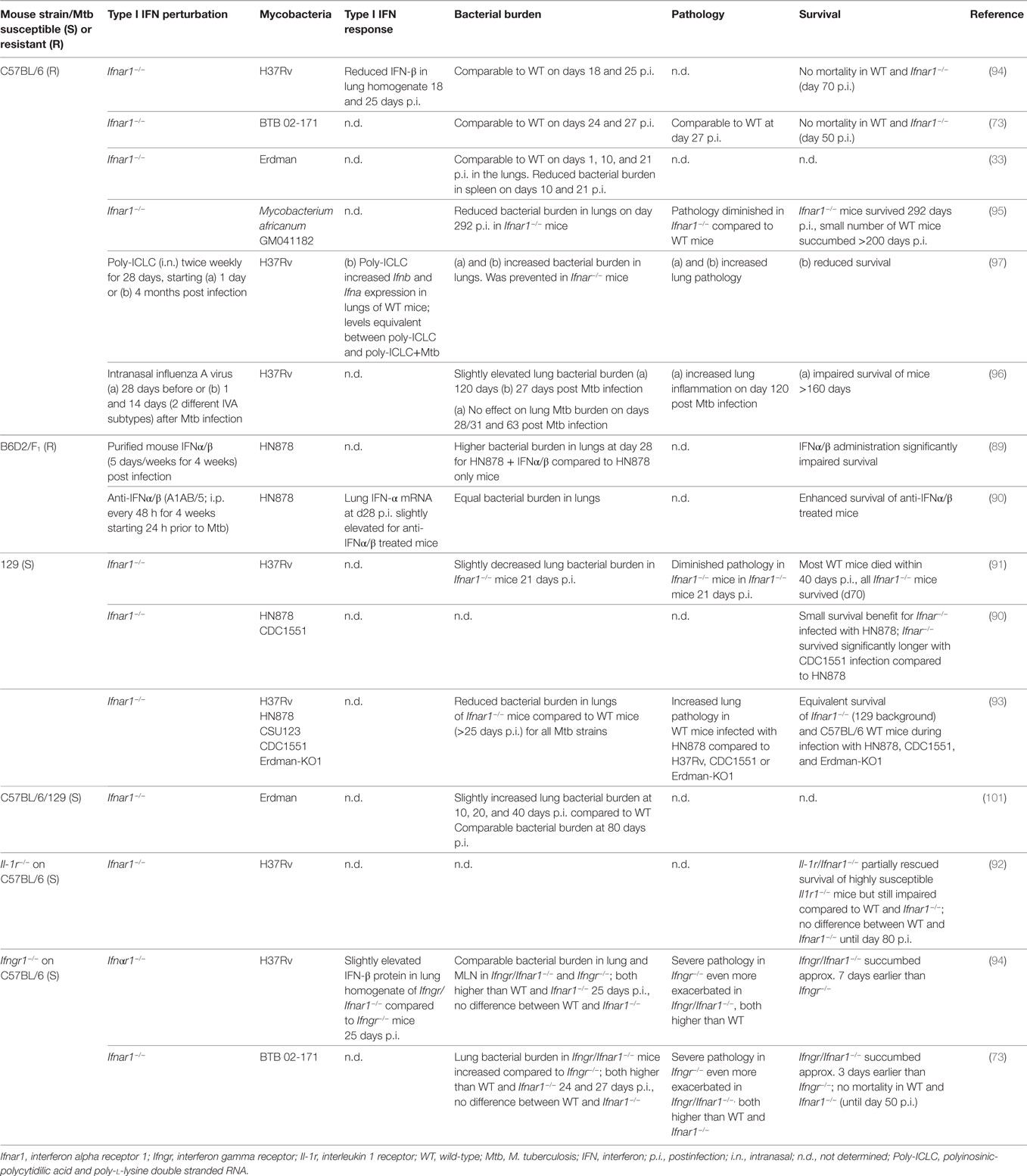
Table 1. Perturbations of type I IFN signaling in mouse models and consequences for infection with bacteria of the Mycobacterium tuberculosis complex.
Humans
The evidence obtained from mouse models of M. tuberculosis infection suggests that in the immune competent host, type I IFN signaling impedes the host’s ability to limit lung pathology and/or bacterial replication, to the benefit of the pathogen. However, there are several clinical case reports that inhaled or subcutaneous administration of IFN-α to tuberculosis patients, in conjunction with co-administration of antimycobacterial antibiotics, improved clinical symptoms (Table 2). It is important to note that these patients were selected due to failure of conventional treatment options and/or recurrent disease, and often received IFN-α late in their infection. Underlying unidentified immune deficiencies may thus have been a confounding factor. Moreover, reports of therapeutic effects of IFN-α administration to patients that presented with non-tuberculous mycobacterial infections suggest contributions of type I IFNs to the antimycobacterial defense in humans in the context of genetically manifested IFN-γ signaling deficiencies (102, 103). The clinical observations in IFN-γ signaling deficient patients are somewhat reminiscent of the studies in Ifngr−/−Ifnar1−/− mice (73, 94). This encourages studies to enhance our understanding of the complex interplay between type I and type II IFN during mycobacterial infection and how this may be harnessed therapeutically in the context of compromised IFN-γ/IFNGR signaling. Recombinant IFN-α is commonly used in the treatment of chronic viral hepatitis. While some studies suggest that long-term administration of recombinant IFN-α does not bear a major risk for TB reactivation (104), case reports of TB reactivation associated with IFN-α therapy demand caution in the clinical management of co-infected individuals (105–109). Thus, detailed assessment of the direct and indirect roles of endogenous and therapeutically administered type I IFNs in the control of latent M. tuberculosis infection is an important area of future pursuit.
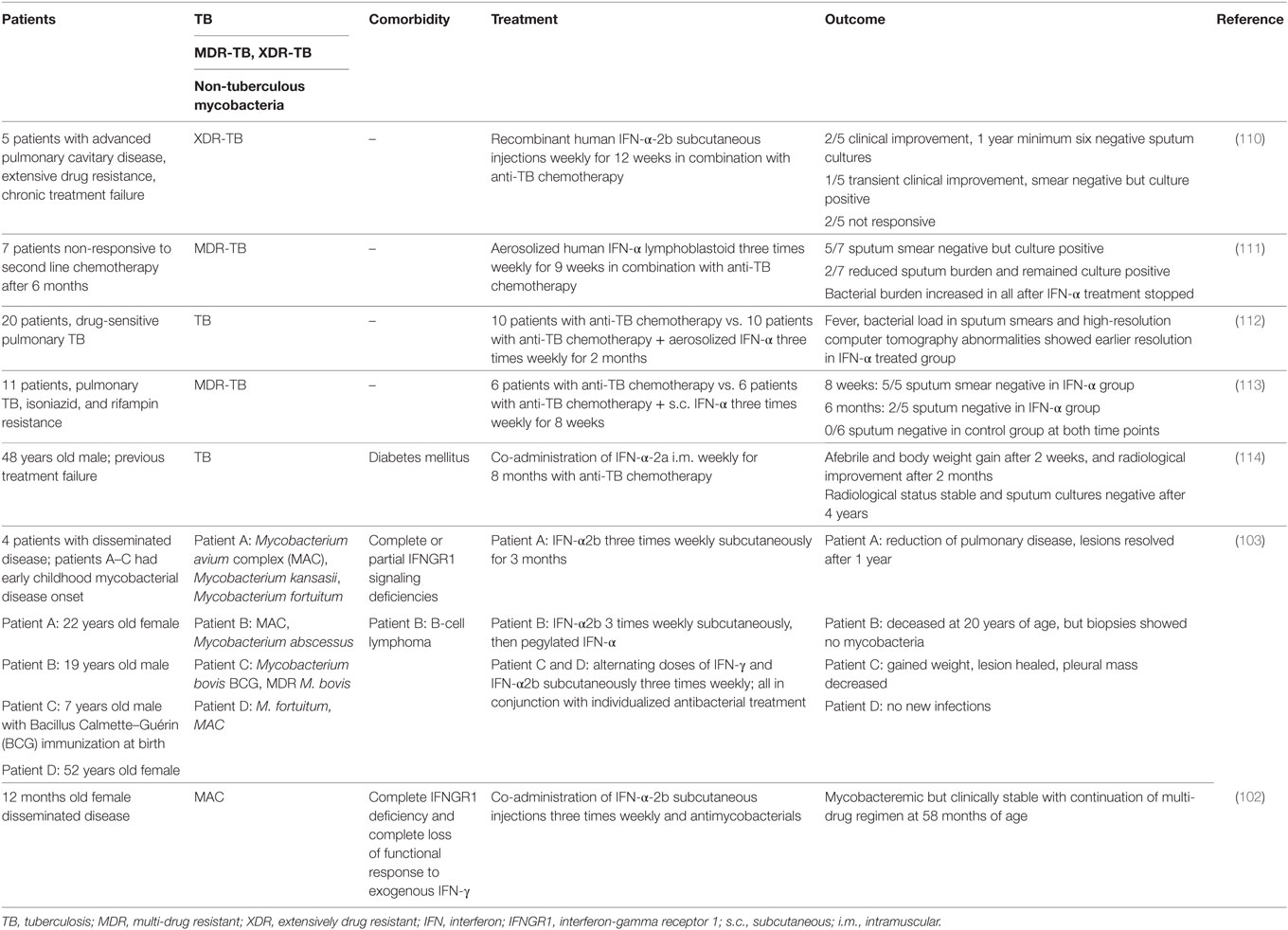
Table 2. Impact of type I IFNs in patients with mycobacterial infections.
It is obvious that our understanding of the contributions of type I IFN signaling to host control of mycobacterial infections is incomplete. Parallels between mouse models and human patients are emerging, but remain limited. In-depth knowledge of type I IFN responses and their contributions to host control in other model systems may be required to bridge important gaps between human and mouse immune pathogenesis during M. tuberculosis infection. The non-human primate model where IFN signaling transcriptional signatures are also highly associated with active TB disease may offer valuable opportunities (115). Confounding factors that require careful consideration in future studies include the impact of host genetic susceptibility, the host’s type I IFN status at the time of infection, the temporal contributions of type I IFNs in active and latent infection, the site of infection, and the infecting mycobacterial species. Detailed understanding of the timing and nature of the interactions between type I and type II IFNs may help to more clearly define intervention strategies and risk factors in the management of mycobacterial infections in patients with (known or unknown) underlying immune deficiencies and co-infections.
Mechanistic Insights into How Type I IFNs Regulate Immune Responses during M. tuberculosis Infection
In light of the potential implications for host-directed therapies in TB, the molecular and cellular mechanisms by which type I IFNs suppress, or in some contexts promote, effective immune control of M. tuberculosis are the subject of intense investigation and discussion (2, 116, 117) (Figure 2).
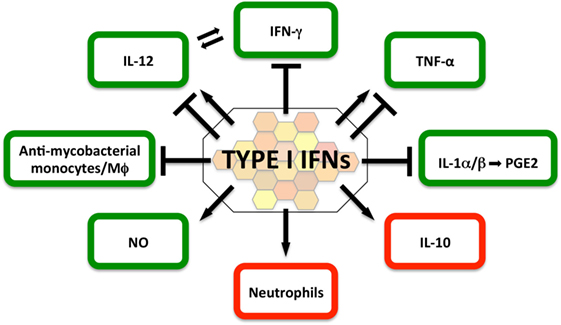
Figure 2. Impact of type I IFNs on protective (green) and detrimental (red) host responses in the immune-competent Mycobacterium tuberculosis-infected host. The current literature suggests that in the immune-competent host, type I IFNs suppress host-protective IFN-γ and IL-1α/β responses, as well as the recruitment of mycobacteria-restricting monocytes/macrophages. In contrast, type I IFN signaling can directly induce NO production, a contributor to effective antimycobacterial host defense. The evidence for the contributions of type I IFNs to host-protective interleukin-12 and TNF responses is currently ambiguous and may be determined by timing, dose, and source of the type I IFN response. Type I IFNs are a driver of IL-10, a cytokine that impairs antimycobacterial immune responses during all stages of infection. Type I IFN signatures in patients have been associated with neutrophils and studies in mouse models of heightened M. tuberculosis susceptibility suggest that type I IFN signaling facilitates infection of permissive neutrophils, which have been associated with damaging tissue pathology.
Myeloid Antimicrobial Defense
Independent studies in different mouse models suggest that type I IFN signaling during M. tuberculosis infection defines the nature of the early myeloid cell infiltrate into the lung, which may favor bacterial replication due to a diminished ability to restrict intracellular mycobacteria (91, 97, 118). Type I IFNs impair the IFN-γ-induced human and mouse macrophage control of M. tuberculosis and other mycobacteria (119–121). At least in human macrophages, this impaired ability to control intracellular mycobacteria is likely facilitated through the IL-10-mediated inhibition of the IFN-γ-driven vitamin D3/cathelicidin/defensin beta 4A antimicrobial defense pathway (120). The interferon-stimulated gene (ISG) ISG-15 has recently been implicated in promotion of early intracellular M. tuberculosis replication in mice, but seems to have protective roles later during infection (122). This later role supports findings in ISG15-deficient patients with mycobacterial disease that suggest that secreted ISG15 promotes IFN-γ secretion (123). Of note, ISG15 expression was detectable in several human leukocyte subpopulations including T cells, NK cells, myeloid and plasmacytoid dendritic cells, monocytes, with the highest levels detected in granulocytes. Exogenous IFN-a enhanced ISG15 expression most dramatically in granulocytes (123), which may provide a link for the functional importance of neutrophil-associated IFN-inducible gene signatures in humans with active disease (3). In contrast to the impaired IFN-γ-mediated macrophage control of mycobacteria, in the absence of IFN-γ signaling type I IFNs may facilitate the recruitment, differentiation and/or survival of myeloid cells that control M. tuberculosis to some extent. This may be due to type I IFN-mediated prevention of skewing of macrophages toward an alternatively activated phenotype (73, 94).
IL-12/IFN-γ and Th1 Response
High type I IFN responses in mice infected with hypervirulent HN878 are associated with diminished Th1 responses including lower IFN-γ production and proliferation by antigen-specific T cells (89, 90, 93). There is in vitro and in vivo evidence that type I IFNs exert these suppressive effects through impairment of myeloid-derived IL-12 production. In myeloid cells derived from M. tuberculosis-infected bone marrow chimeric mice, IFNAR-deficiency enhanced myeloid cell-derived IL-12p40 expression in response to re-stimulation with M. tuberculosis compared to wild-type cells (116). In murine M. tuberculosis-infected macrophages, exogenous addition of high concentrations of IFN-β suppressed IL-12 expression as well as IFN-γ-induced macrophage activation (121). Exogenously added type I IFNs also suppressed IFN-γ-induced cytokine responses in human monocytes (120, 124). Molecular mechanisms underlying type I IFN-mediated suppression of IL-12 responses include the induction of IL-10, downregulation of the IFN-γ receptor, and induction of protein arginine methyl transferase 1, a negative regulator of IFN signaling (120, 121, 124). However, diminished IL-12p40 release by M. tuberculosis-infected IFNAR−/− macrophages has been reported (121), suggesting that in isolated macrophage cultures, endogenous type I IFN signaling facilitates IL-12p40 responses. Future studies may need to address similarities and potential differences in the effects of endogenously produced versus exogenously added type I IFNs both in cell culture as well as in vivo.
Exogenous addition of recombinant IFN-α to in vitro cultured T cell clones from TB patients enhanced the number of clones that produced IFN-γ (125) in line with observations that IFN-α can promote IFN-γ production by human CD4+ T cells (126). Thus, the overall impact of type I IFN signaling during mycobacterial infection may be a composite of differential effects on myeloid and lymphocyte cell functions. Mouse models that allow assessment of the impact of type I IFN signaling in specific cell subsets such as T cells (127) might prove valuable in dissecting the direct impact of type I IFNs on T cell functions during M. tuberculosis infection.
IL-10
Early studies associated higher virulence of M. tuberculosis isolates (defined by elevated bacterial burden and impaired survival of infected mice) with the capacity to elicit high type I IFN responses in vivo. This phenotype was accompanied by relatively lower pro-inflammatory cytokine responses (89, 90, 93). More recent data indicate that elevated virulence in the mouse model can be associated with induction of both high IFN-β and high pro-inflammatory cytokine responses (72). Association of elevated virulence with high type I IFN paired with high IL-10 levels may provide a more direct link between high type I IFN responses and impaired host control of the infection. Type I IFN signaling drives IL-10 expression by both M. tuberculosis-infected macrophages and CD4+ T cells in infected mice (121, 128). While there is great variability between observations made in different mouse strains and between different labs, several studies in mice suggest that IL-10 significantly impairs host control of M. tuberculosis infection during active, chronic, and latent infection (2). Overexpression of IL-10 in macrophages driven by the CD86 promoter led to increased bacterial burden and impaired survival of M. tuberculosis-infected mice without noticeable effects on T cell functions (129). In contrast, a recent study that utilized cell-specific deletion of Il10 expression in vivo suggests that a reduction of bacterial burden in lungs of IL-10-deficient mice during the chronic phase of infection (day 60) is attributable to IL-10 produced by CD4+ T cells rather than myeloid or B cells. However, while lung CD4+ T cells from M. tuberculosis-infected Ifnar1−/− showed diminished Il10 expression, lung bacterial burden in Ifnar1−/− mice was not decreased and did hence not mirror the reduction of lung bacterial load in Il10−/− and Il10fl/fl CD4Cre+ mice (128). Instead, IL-27 receptor signaling was implicated to drive IL-10 expression by CD4+ T cells, in parallel to type I IFNs, and IL27ra−/− mice exhibited diminished lung bacterial burden similar to Il10−/− and Il10fl/fl CD4Cre+ mice (128). Thus, cell-specific targeting of the cellular source of IL-10 that impairs host control of M. tuberculosis replication may be of interest for host-directed intervention but requires detailed characterization of the functionalities and impact of IL-10-producing host cell populations.
TNF-α
The increased risk of TB reactivation in individuals undergoing TNF neutralizing therapies for chronic inflammatory diseases underpins the central role for this cytokine in the host control of M. tuberculosis infection, which is further supported by mouse studies (2, 130). Macrophages deficient for IFNAR signaling showed diminished TNF expression in response to M. tuberculosis infection, which was interpreted as a promoting role for endogenous type I IFN responses in the M. tuberculosis-induced TNF response (121). However, an independent study reported no differences between IFNAR-deficient and WT macrophages (131). In contrast, exogenously added IFN-β suppressed TNF release by M. tuberculosis-infected macrophages (121). The impact of type I IFN signaling on the TNF response in vivo remains to be clearly defined. WT and IFNAR-deficient mice infected with the high type I IFN-inducing M. tuberculosis strain BTB 02-171 displayed equivalent lung Tnf mRNA expression (day 20 p.i.) (73). In contrast, decreased TNF concentrations were observed in lung homogenates of M. tuberculosis-infected IFNAR-deficient 129 mice (day 21 p.i.) (91) and a small decrease in lung TNF concentrations was also reported in M. africanum-infected IFNAR-deficient C57BL/6 mice (day 292 p.i.) (95). The reasons for the apparent differences between individual studies are not immediately obvious, but may be attributable to the different time points analyzed postinfection and the different models systems studied. These observations may indicate, however, that the concentration, source, and timing of the type I IFN response direct TNF responses in the context of M. tuberculosis infection.
IL-1, Eicosanoids, Nitric Oxide (NO), and Neutrophils
IL-1 signaling is an important driver of host resistance against M. tuberculosis infection in humans and mice (132–135). The underlying molecular mechanisms are emerging and TNF-driven antimicrobial activity, autophagy, as well as eicosanoid signaling have been implicated (92, 134, 136). Type I IFN signaling suppressed IL-1α and β production by macrophages in vitro and in vivo mouse infection (34, 121, 137). The type I IFN-mediated regulation of IL-1 expression by human and mouse macrophages was shown to be dependent on NO synthase 2 and IL-10, and was associated with enhanced IL-1Ra expression (121, 137).
Thus, type I IFN signaling regulates IL-1 responses through inhibition of ligand expression as well as receptor signaling. A third potential mechanism may be inferred from the observation that NO interference with NLRP3 inflammasome assembly impairs IL-1β processing by M. tuberculosis-infected macrophages (138). As endogenous and exogenously added type I IFNs drive NO production by M. tuberculosis-infected macrophages (32, 73), type I IFN signaling may also interfere with IL-1 processing. It is interesting to note that NO-mediated thiol nitrosylation inhibited assembly and activation of the NLRP3 but not AIM2 inflammasome (138). AIM2 has been reported to be dispensable for IL-1β production by murine dendritic cells infected with virulent M. tuberculosis in vitro. In contrast, AIM2 was required for IL-1β release induced by Mycobacterium smegmantis, Mycobacterium fortuitum, Mycobacterium kansasii and, to a lesser extent, the attenuated M. tuberculosis H37Ra (139). Intriguingly, infection of DCs with virulent M. tuberculosis prior to infection with M. smegmatis reduced the IL-1β release, which was associated with an impaired IFN-β response. The authors concluded that virulent M. tuberculosis actively suppressed AIM2 activation by a mechanism that required M. tuberculosis ESX-1 (139). Yet, AIM2-deficient mice infected intratracheally with a high dose of M. tuberculosis H37Rv displayed elevated bacterial loads in the lung and liver 4 weeks postinfection accompanied by exacerbated pathology, impaired IL-18 and IL-10 production, as well as an impaired antigen-specific CD4+ T cell-derived IFN-g response. The Aim2−/− mice succumbed to infection between 5 and 7 weeks postinoculation (140). It is important to note, however, that deficiency in caspase 1 and 11, inflammasome effector caspases that cleave pro-IL-1β, does not impair host control of M. tuberculosis infection in vivo (133, 141). This suggests that alternative mechanisms of pro-IL-1 processing during M. tuberculosis infection in vivo exist, and that AIM2 contributions to the host control of M. tuberculosis infection may exceed processing of pro-IL-1β.
An emerging pathway of type I IFN-mediated suppression of effective immune control of M. tuberculosis occurs via dysregulation of IL-1-controlled eicosanoid lipid mediators (116). In mice, IL-1 receptor signaling enhanced the ratio of host-protective prostaglandin E2 over host-detrimental 5-lipoxygenase products such as lipoxin A4 (116). Type I IFN signaling counter-regulates this IL-1-driven process, and conversely, IL-1 signaling impairs the type I IFN response (92). Poor control of M. tuberculosis infection in various mouse models is associated with an extensive neutrophil response in the lung. Pathogen control and host survival can be improved by neutrophil depletion and Ifnar1-deficiency in a susceptible mouse strain, which is associated with a shift of intracellular M. tuberculosis burden from permissive neutrophils toward monocytes (91, 142, 143). Recent findings suggest that in a permissive environment, 12/15-lipoxygenase products drive neutrophil infiltration into infected lungs, which is associated with poor host restriction of mycobacterial replication in a nutrient-rich environment (143). In the absence of NO production (Nos2−/− mice), this process was driven by inflammasome-dependent IL-1 production (143) linking back to the inhibitory effects of NO on inflammasome assembly (138).
Associations of IL-1, type I IFN, eicosanoids, neutrophil infiltration, and SNPs in associated genes with disease severity and treatment responsiveness in human TB patients (92, 135, 143) underpin a dynamic cytokine network that balances the protective or detrimental roles of eicosanoid lipid mediators and neutrophil functions in M. tuberculosis infection. Pharmacologic targeting of eicosanoids has been proposed as a possible avenue for host-directed therapeutic intervention in TB (92).
Concluding Remarks
As we continue to gain mechanistic insight into the interactions between M. tuberculosis and innate immune sensors, it is evident that multiple independent extracellular and intracellular surveillance pathways converge to drive type I IFN expression in infected host cells. One of the main questions remaining is which cells are the key producers of type I IFNs during M. tuberculosis infection in vivo. In vitro evidence strongly suggests that host cells that harbor M. tuberculosis bacteria such as macrophages and myeloid DCs are sources of type I IFN during infection. In the context of the close interactions between infected and uninfected cells within granulomas, the concept of dinucleotide-mediated STING-activation in bystander cells warrants further exploration as it may allow for type I IFN expression not only in myeloid cells but also in T and B cells. Moreover, mice infected with hypervirulent, high type I IFN-inducing HN878 harbored elevated numbers of plasmacytoid DCs (pDCs) in their lungs (93). Human peripheral blood pDCs responded with high IFN-α production to stimulation with high concentrations of mycobacterial DNA (144). However, in vitro cultured pDCs expressed little to no type I IFNs when co-cultured with M. tuberculosis- or M. bovis-BCG-infected myeloid DCs (144, 145). Advanced single cell functional profiling in TB granulomas and the employment of sensitive IFN reporter animal models are just two approaches that may provide a better understanding of the dynamics and cellular sources of type I IFN expression during M. tuberculosis infection both at the site of infection and in the periphery. Individual type I IFNs exhibit distinct functions in various homeostatic and pathological contexts, which has at least in part been attributed to varying ligand affinity, stability of the receptor complex, and unique signaling capacity of IFN-β through IFNAR1 (146–148). Thus, insights into the spatial and temporal expression of individual members of the type I IFN family may hold the key to providing context for some of the apparent opposing roles of type I IFN signaling deduced from model systems and patient observations. As the discoveries around blood gene expression signatures in patients with active TB disease are being explored for suitable biomarkers, they also raise questions related to TB pathophysiology. For example: are IFN signatures related to neutrophils in the periphery (3, 149) a natural consequence of M. tuberculosis infection in humans, or an indication of disease susceptibility of the individual due to a propensity to mount a neutrophil response upon encounter with M. tuberculosis? How reflective is the peripheral gene expression profile of the response mounted locally at the site of infection or within the relevant secondary lymphoid organs? Longitudinal analyses and immune profiling in at-risk individuals (8) will provide valuable insights, and suitable animal models are key for functional context. While type I IFN signaling may contribute to some aspects of the host defense against M. tuberculosis infection, it is clear that extensive type I IFN responses, e.g. in the context of viral infections (150), promote pathology to the detriment of the host. Nevertheless, the reported therapeutic benefits of type I IFN administration in conjunction with antimycobacterial antibiotics warrants detailed exploration to establish whether this is reflective of a patient immune status permissive to potential antimycobacterial activities of type I IFN or whether IFNs facilitate mycobacterial replication rendering them more susceptible to antibiotics. Understanding whether and when type I IFNs are “friend or foe” in the context of mycobacterial infections will define their place in the quest for tailored host-directed therapeutic interventions in TB.
Author Contributions
All authors contributed to the critical review of the literature and writing of the manuscript. MD and AB developed the figures and tables and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Katharina Ronacher and Carl Feng for critical review and feedback on the manuscript.
Funding
AB acknowledges funding by the National Health and Medical Research Council of Australia (GNT1120230) and The University of Queensland Diamantina Institute. MD and TS gratefully acknowledge Research Higher Degree Scholarships by The University of Queensland and The University of Queensland Diamantina Institute.
References
2. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi:10.1146/annurev-immunol-032712-095939
3. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466(7309):973–7. doi:10.1038/nature09247
4. Cliff JM, Lee JS, Constantinou N, Cho JE, Clark TG, Ronacher K, et al. Distinct phases of blood gene expression pattern through tuberculosis treatment reflect modulation of the humoral immune response. J Infect Dis (2013) 207(1):18–29. doi:10.1093/infdis/jis499
5. Blankley S, Berry MP, Graham CM, Bloom CI, Lipman M, O’Garra A. The application of transcriptional blood signatures to enhance our understanding of the host response to infection: the example of tuberculosis. Philos Trans R Soc Lond B Biol Sci (2014) 369(1645):20130427. doi:10.1098/rstb.2013.0427
6. Blankley S, Graham CM, Levin J, Turner J, Berry MP, Bloom CI, et al. A 380-gene meta-signature of active tuberculosis compared with healthy controls. Eur Respir J (2016) 47(6):1873–6. doi:10.1183/13993003.02121-2015
7. Blankley S, Graham CM, Turner J, Berry MP, Bloom CI, Xu Z, et al. The transcriptional signature of active tuberculosis reflects symptom status in extra-pulmonary and pulmonary tuberculosis. PLoS One (2016) 11(10):e0162220. doi:10.1371/journal.pone.0162220
8. Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet (2016) 387(10035):2312–22. doi:10.1016/s0140-6736(15)01316-1
9. Sambarey A, Devaprasad A, Mohan A, Ahmed A, Nayak S, Swaminathan S, et al. Unbiased identification of blood-based biomarkers for pulmonary tuberculosis by modeling and mining molecular interaction networks. EBioMedicine (2017) 15:112–26. doi:10.1016/j.ebiom.2016.12.009
10. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5(9):675–87. doi:10.1038/nri1684
11. de Weerd NA, Nguyen T. The interferons and their receptors – distribution and regulation. Immunol Cell Biol (2012) 90(5):483–91. doi:10.1038/icb.2012.9
12. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem (2017) 292(18):7285–94. doi:10.1074/jbc.R116.774562
13. Kovarik P, Castiglia V, Ivin M, Ebner F. Type I interferons in bacterial infections: a balancing act. Front Immunol (2016) 7:652. doi:10.3389/fimmu.2016.00652
14. Snyder DT, Hedges JF, Jutila MA. Getting “inside” type I IFNs: type I IFNs in intracellular bacterial infections. J Immunol Res (2017) 2017:9361802. doi:10.1155/2017/9361802
15. Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe (2016) 19(6):760–9. doi:10.1016/j.chom.2016.05.016
16. Cliff JM, Kaufmann SHE, McShane H, van Helden P, O’Garra A. The human immune response to tuberculosis and its treatment: a view from the blood. Immunol Rev (2015) 264(1):88–102. doi:10.1111/imr.12269
17. Deffur A, Wilkinson RJ, Coussens AK. Tricks to translating TB transcriptomics. Ann Transl Med (2015) 3(Suppl 1):S43. doi:10.3978/j.issn.2305-5839.2015.04.12
18. Haas CT, Roe JK, Pollara G, Mehta M, Noursadeghi M. Diagnostic ’omics’ for active tuberculosis. BMC Med (2016) 14:37. doi:10.1186/s12916-016-0583-9
19. Joosten SA, Fletcher HA, Ottenhoff TH. A helicopter perspective on TB biomarkers: pathway and process based analysis of gene expression data provides new insight into TB pathogenesis. PLoS One (2013) 8(9):e73230. doi:10.1371/journal.pone.0073230
20. Maertzdorf J, McEwen G, Weiner J III, Tian S, Lader E, Schriek U, et al. Concise gene signature for point-of-care classification of tuberculosis. EMBO Mol Med (2016) 8(2):86–95. doi:10.15252/emmm.201505790
21. Roe JK, Thomas N, Gil E, Best K, Tsaliki E, Morris-Jones S, et al. Blood transcriptomic diagnosis of pulmonary and extrapulmonary tuberculosis. JCI Insight (2016) 1(16):e87238. doi:10.1172/jci.insight.87238
22. Sweeney TE, Braviak L, Tato CM, Khatri P. Genome-wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respir Med (2016) 4(3):213–24. doi:10.1016/s2213-2600(16)00048-5
23. Cattamanchi A, Walter ND, Metcalfe JZ, Davis JL. A transcriptional signature for active TB: have we found the needle in the haystack? PLoS Med (2013) 10(10):e1001539. doi:10.1371/journal.pmed.1001539
24. Harries AD, Lin Y, Satyanarayana S, Lonnroth K, Li L, Wilson N, et al. The looming epidemic of diabetes-associated tuberculosis: learning lessons from HIV-associated tuberculosis. Int J Tuberc Lung Dis (2011) 15(11):1436–44. doi:10.5588/ijtld.11.0503
25. Prada-Medina CA, Fukutani KF, Pavan Kumar N, Gil-Santana L, Babu S, Lichtenstein F, et al. Systems immunology of diabetes-tuberculosis comorbidity reveals signatures of disease complications. Sci Rep (2017) 7(1):1999. doi:10.1038/s41598-017-01767-4
26. Koth LL, Solberg OD, Peng JC, Bhakta NR, Nguyen CP, Woodruff PG. Sarcoidosis blood transcriptome reflects lung inflammation and overlaps with tuberculosis. Am J Respir Crit Care Med (2011) 184(10):1153–63. doi:10.1164/rccm.201106-1143OC
27. Maertzdorf J, Weiner J III, Mollenkopf HJ, Bauer T, Prasse A, Muller-Quernheim J, et al. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci U S A (2012) 109(20):7853–8. doi:10.1073/pnas.1121072109
28. Bloom CI, Graham CM, Berry MP, Rozakeas F, Redford PS, Wang Y, et al. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS One (2013) 8(8):e70630. doi:10.1371/journal.pone.0070630
29. Koh GC, Schreiber MF, Bautista R, Maude RR, Dunachie S, Limmathurotsakul D, et al. Host responses to melioidosis and tuberculosis are both dominated by interferon-mediated signaling. PLoS One (2013) 8(1):e54961. doi:10.1371/journal.pone.0054961
30. Kaforou M, Wright VJ, Oni T, French N, Anderson ST, Bangani N, et al. Detection of tuberculosis in HIV-infected and -uninfected African adults using whole blood RNA expression signatures: a case-control study. PLoS Med (2013) 10(10):e1001538. doi:10.1371/journal.pmed.1001538
31. Remoli ME, Giacomini E, Lutfalla G, Dondi E, Orefici G, Battistini A, et al. Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J Immunol (2002) 169(1):366–74. doi:10.4049/jimmunol.169.1.366
32. Shi S, Blumenthal A, Hickey CM, Gandotra S, Levy D, Ehrt S. Expression of many immunologically important genes in Mycobacterium tuberculosis-infected macrophages is independent of both TLR2 and TLR4 but dependent on IFN-alphabeta receptor and STAT1. J Immunol (2005) 175(5):3318–28. doi:10.4049/jimmunol.175.5.3318
33. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol (2007) 178(5):3143–52. doi:10.4049/jimmunol.178.5.3143
34. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol (2011) 187(5):2540–7. doi:10.4049/jimmunol.1100926
35. Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med (2015) 21(4):401–6. doi:10.1038/nm.3813
36. Etna MP, Giacomini E, Pardini M, Severa M, Bottai D, Cruciani M, et al. Impact of Mycobacterium tuberculosis RD1-locus on human primary dendritic cell immune functions. Sci Rep (2015) 5:17078. doi:10.1038/srep17078
37. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe (2015) 17(6):799–810. doi:10.1016/j.chom.2015.05.003
38. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe (2015) 17(6):811–9. doi:10.1016/j.chom.2015.05.004
39. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog (2009) 5(7):e1000500. doi:10.1371/journal.ppat.1000500
40. Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell (1998) 1(4):507–18. doi:10.1016/S1097-2765(00)80051-9
41. Groschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol (2016) 14(11):677–91. doi:10.1038/nrmicro.2016.131
42. Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, et al. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci U S A (2017) 114(6):1371–6. doi:10.1073/pnas.1620133114
43. Wiens KE, Ernst JD. The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog (2016) 12(8):e1005809. doi:10.1371/journal.ppat.1005809
44. Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog (2012) 8(2):e1002507. doi:10.1371/journal.ppat.1002507
45. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150(4):803–15. doi:10.1016/j.cell.2012.06.040
46. Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog (2008) 4(1):e6. doi:10.1371/journal.ppat.0040006
47. Gandotra S, Jang S, Murray PJ, Salgame P, Ehrt S. Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infect Immun (2007) 75(11):5127–34. doi:10.1128/iai.00458-07
48. Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol (2008) 181(10):7157–65. doi:10.4049/jimmunol.181.10.7157
49. Austin CM, Ma X, Graviss EA. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect Dis (2008) 197(12):1713–6. doi:10.1086/588384
50. Pan H, Dai Y, Tang S, Wang J. Polymorphisms of NOD2 and the risk of tuberculosis: a validation study in the Chinese population. Int J Immunogenet (2012) 39(3):233–40. doi:10.1111/j.1744-313X.2011.01079.x
51. Singh V, Gaur R, Mittal M, Biswas SK, Das R, Girdhar BK, et al. Absence of nucleotide-binding oligomerization domain-containing protein 2 variants in patients with leprosy and tuberculosis. Int J Immunogenet (2012) 39(4):353–6. doi:10.1111/j.1744-313X.2012.01085.x
52. Zhao M, Jiang F, Zhang W, Li F, Wei L, Liu J, et al. A novel single nucleotide polymorphism within the NOD2 gene is associated with pulmonary tuberculosis in the Chinese Han, Uygur and Kazak populations. BMC Infect Dis (2012) 12:91. doi:10.1186/1471-2334-12-91
53. Wang C, Chen ZL, Pan ZF, Wei LL, Xu DD, Jiang TT, et al. NOD2 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. Int J Biol Sci (2013) 10(1):103–8. doi:10.7150/ijbs.7585
54. Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe (2015) 17(6):820–8. doi:10.1016/j.chom.2015.05.005
55. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol (2016) 17(10):1142–9. doi:10.1038/ni.3558
56. Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature (2013) 503(7477):530–4. doi:10.1038/nature12640
57. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature (2011) 478(7370):515–8. doi:10.1038/nature10429
58. Bai Y, Yang J, Zhou X, Ding X, Eisele LE, Bai G. Mycobacterium tuberculosis Rv3586 (DacA) is a diadenylate cyclase that converts ATP or ADP into c-di-AMP. PLoS One (2012) 7(4):e35206. doi:10.1371/journal.pone.0035206
59. Yang J, Bai Y, Zhang Y, Gabrielle VD, Jin L, Bai G. Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol Microbiol (2014) 93(1):65–79. doi:10.1111/mmi.12641
60. Dey RJ, Dey B, Zheng Y, Cheung LS, Zhou J, Sayre D, et al. Inhibition of innate immune cytosolic surveillance by an M. tuberculosis phosphodiesterase. Nat Chem Biol (2017) 13(2):210–7. doi:10.1038/nchembio.2254
61. Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol (2012) 13(12):1155–61. doi:10.1038/ni.2460
62. McFarland AP, Luo S, Ahmed-Qadri F, Zuck M, Thayer EF, Goo YA, et al. Sensing of bacterial cyclic dinucleotides by the oxidoreductase RECON promotes NF-kappaB activation and shapes a proinflammatory antibacterial state. Immunity (2017) 46(3):433–45. doi:10.1016/j.immuni.2017.02.014
63. Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe (2014) 15(2):228–38. doi:10.1016/j.chom.2014.01.009
64. Majlessi L, Prados-Rosales R, Casadevall A, Brosch R. Release of mycobacterial antigens. Immunol Rev (2015) 264(1):25–45. doi:10.1111/imr.12251
65. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34(5):637–50. doi:10.1016/j.immuni.2011.05.006
66. Botos I, Segal DM, Davies DR. The structural biology of toll-like receptors. Structure (2011) 19(4):447–59. doi:10.1016/j.str.2011.02.004
67. Kang JY, Lee JO. Structural biology of the toll-like receptor family. Annu Rev Biochem (2011) 80:917–41. doi:10.1146/annurev-biochem-052909-141507
68. O’Neill LA, Golenbock D, Bowie AG. The history of toll-like receptors – redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi:10.1038/nri3446
69. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of toll-like receptor 4 to the induction of interferon-beta. Nat Immunol (2008) 9(4):361–8. doi:10.1038/ni1569
70. Brown MG, McAlpine SM, Huang YY, Haidl ID, Al-Afif A, Marshall JS, et al. RNA sensors enable human mast cell anti-viral chemokine production and IFN-mediated protection in response to antibody-enhanced dengue virus infection. PLoS One (2012) 7(3):e34055. doi:10.1371/journal.pone.0034055
71. Nilsen NJ, Vladimer GI, Stenvik J, Orning MP, Zeid-Kilani MV, Bugge M, et al. A role for the adaptor proteins TRAM and TRIF in toll-like receptor 2 signaling. J Biol Chem (2015) 290(6):3209–22. doi:10.1074/jbc.M114.593426
72. Carmona J, Cruz A, Moreira-Teixeira L, Sousa C, Sousa J, Osorio NS, et al. Mycobacterium tuberculosis strains are differentially recognized by TLRs with an impact on the immune response. PLoS One (2013) 8(6):e67277. doi:10.1371/journal.pone.0067277
73. Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-gamma signaling. J Immunol (2016) 197(12):4714–26. doi:10.4049/jimmunol.1600584
74. Ferwerda G, Kullberg BJ, de Jong DJ, Girardin SE, Langenberg DM, van Crevel R, et al. Mycobacterium paratuberculosis is recognized by toll-like receptors and NOD2. J Leukoc Biol (2007) 82(4):1011–8. doi:10.1189/jlb.0307147
75. Pulido I, Leal M, Genebat M, Pacheco YM, Saez ME, Soriano-Sarabia N. The TLR4 ASP299GLY polymorphism is a risk factor for active tuberculosis in Caucasian HIV-infected patients. Curr HIV Res (2010) 8(3):253–8. doi:10.2174/157016210791111052
76. Ogus AC, Yoldas B, Ozdemir T, Uguz A, Olcen S, Keser I, et al. The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. Eur Respir J (2004) 23(2):219–23. doi:10.1183/09031936.03.00061703
77. Ma X, Liu Y, Gowen BB, Graviss EA, Clark AG, Musser JM. Full-exon resequencing reveals toll-like receptor variants contribute to human susceptibility to tuberculosis disease. PLoS One (2007) 2(12):e1318. doi:10.1371/journal.pone.0001318
78. Etokebe GE, Skjeldal F, Nilsen N, Rodionov D, Knezevic J, Bulat-Kardum L, et al. Toll-like receptor 2 (P631H) mutant impairs membrane internalization and is a dominant negative allele. Scand J Immunol (2010) 71(5):369–81. doi:10.1111/j.1365-3083.2010.02379.x
79. Xue Y, Jin L, Li AZ, Wang HJ, Li M, Zhang YX, et al. Microsatellite polymorphisms in intron 2 of the toll-like receptor 2 gene and their association with susceptibility to pulmonary tuberculosis in Han Chinese. Clin Chem Lab Med (2010) 48(6):785–9. doi:10.1515/cclm.2010.154
80. Dalgic N, Tekin D, Kayaalti Z, Soylemezoglu T, Cakir E, Kilic B, et al. Arg753Gln polymorphism of the human toll-like receptor 2 gene from infection to disease in pediatric tuberculosis. Hum Immunol (2011) 72(5):440–5. doi:10.1016/j.humimm.2011.02.001
81. Wang JJ, Xia X, Tang SD, Wang J, Deng XZ, Zhang Y, et al. Meta-analysis on the associations of TLR2 gene polymorphisms with pulmonary tuberculosis susceptibility among Asian populations. PLoS One (2013) 8(10):e75090. doi:10.1371/journal.pone.0075090
82. Davila S, Hibberd ML, Hari Dass R, Wong HE, Sahiratmadja E, Bonnard C, et al. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet (2008) 4(10):e1000218. doi:10.1371/journal.pgen.1000218
83. Velez DR, Wejse C, Stryjewski ME, Abbate E, Hulme WF, Myers JL, et al. Variants in toll-like receptors 2 and 9 influence susceptibility to pulmonary tuberculosis in Caucasians, African-Americans, and West Africans. Hum Genet (2010) 127(1):65–73. doi:10.1007/s00439-009-0741-7
84. Xue Y, Zhao ZQ, Wang HJ, Jin L, Liu CP, Wang Y, et al. Toll-like receptors 2 and 4 gene polymorphisms in a southeastern Chinese population with tuberculosis. Int J Immunogenet (2010) 37(2):135–8. doi:10.1111/j.1744-313X.2009.00892.x
85. Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol (2011) 2011:405310. doi:10.1155/2011/405310
86. Chen Z, Wang W, Liang J, Wang J, Feng S, Zhang G. Association between toll-like receptors 9 (TLR9) gene polymorphism and risk of pulmonary tuberculosis: meta-analysis. BMC Pulm Med (2015) 15:57. doi:10.1186/s12890-015-0049-4
87. Schurz H, Daya M, Moller M, Hoal EG, Salie M. TLR1, 2, 4, 6 and 9 variants associated with tuberculosis susceptibility: a systematic review and meta-analysis. PLoS One (2015) 10(10):e0139711. doi:10.1371/journal.pone.0139711
88. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem (2007) 282(28):20059–63. doi:10.1074/jbc.R700016200
89. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A (2001) 98(10):5752–7. doi:10.1073/pnas.091096998
90. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25(11):694–701. doi:10.1089/jir.2005.25.694
91. Dorhoi A, Yeremeev V, Nouailles G, Weiner J III, Jorg S, Heinemann E, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol (2014) 44(8):2380–93. doi:10.1002/eji.201344219
92. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511(7507):99–103. doi:10.1038/nature13489
93. Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol (2007) 179(1):522–31. doi:10.4049/jimmunol.179.1.522
94. Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol (2012) 188(12):6205–15. doi:10.4049/jimmunol.1200255
95. Wiens KE, Ernst JD. Type I interferon is pathogenic during chronic Mycobacterium africanum Infection. J Infect Dis (2016) 214(12):1893–6. doi:10.1093/infdis/jiw519
96. Redford PS, Mayer-Barber KD, McNab FW, Stavropoulos E, Wack A, Sher A, et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis (2014) 209(2):270–4. doi:10.1093/infdis/jit424
97. Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest (2010) 120(5):1674–82. doi:10.1172/jci40817
98. Denis M. Recombinant murine beta interferon enhances resistance of mice to systemic Mycobacterium avium infection. Infect Immun (1991) 59(5):1857–9.
99. Kuchtey J, Fulton SA, Reba SM, Harding CV, Boom WH. Interferon-alphabeta mediates partial control of early pulmonary Mycobacterium bovis bacillus Calmette-Guerin infection. Immunology (2006) 118(1):39–49. doi:10.1111/j.1365-2567.2006.02337.x
100. Ruangkiattikul N, Nerlich A, Abdissa K, Lienenklaus S, Suwandi A, Janze N, et al. cGAS-STING-TBK1-IRF3/7 induced interferon-beta contributes to the clearing of non tuberculous mycobacterial infection in mice. Virulence (2017) 19:1–13. doi:10.1080/21505594.2017.1321191
101. Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect Immun (2000) 68(12):6879–82. doi:10.1128/IAI.68.12.6879-6882.2000
102. Ward CM, Jyonouchi H, Kotenko SV, Smirnov SV, Patel R, Aguila H, et al. Adjunctive treatment of disseminated Mycobacterium avium complex infection with interferon alpha-2b in a patient with complete interferon-gamma receptor R1 deficiency. Eur J Pediatr (2007) 166(9):981–5. doi:10.1007/s00431-006-0339-1
103. Bax HI, Freeman AF, Ding L, Hsu AP, Marciano B, Kristosturyan E, et al. Interferon alpha treatment of patients with impaired interferon gamma signaling. J Clin Immunol (2013) 33(5):991–1001. doi:10.1007/s10875-013-9882-5
104. Lin SY, Chen TC, Lu PL, Lin CY, Lin WR, Yang YH, et al. Incidence rates of tuberculosis in chronic hepatitis C infected patients with or without interferon based therapy: a population-based cohort study in Taiwan. BMC Infect Dis (2014) 14:705. doi:10.1186/s12879-014-0705-y
105. Sabbatani S, Manfredi R, Marinacci G, Pavoni M, Cristoni L, Chiodo F. Reactivation of severe, acute pulmonary tuberculosis during treatment with pegylated interferon-alpha and ribavirin for chronic HCV hepatitis. Scand J Infect Dis (2006) 38(3):205–8. doi:10.1080/00365540500263268
106. Farah R, Awad J. The association of interferon with the development of pulmonary tuberculosis. Int J Clin Pharmacol Ther (2007) 45(11):598–600. doi:10.5414/CPP45598
107. Telesca C, Angelico M, Piccolo P, Nosotti L, Morrone A, Longhi C, et al. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J Infect (2007) 54(4):e223–6. doi:10.1016/j.jinf.2006.12.009
108. Belkahla N, Kchir H, Maamouri N, Ouerghi H, Hariz FB, Chouaib S, et al. [Reactivation of tuberculosis during dual therapy with pegylated interferon and ribavirin for chronic hepatitis C]. Rev Med Interne (2010) 31(11):e1–3. doi:10.1016/j.revmed.2009.11.017
109. Guardigni V, Fabbri G, Grilli A, Contini C. Successful antiviral treatment of chronic hepatitis C in patients with rare comorbidities. Two case-reports. Ann Hepatol (2012) 11(3):404–8.
110. Palmero D, Eiguchi K, Rendo P, Castro Zorrilla L, Abbate E, Gonzalez Montaner LJ. Phase II trial of recombinant interferon-alpha2b in patients with advanced intractable multidrug-resistant pulmonary tuberculosis: long-term follow-up. Int J Tuberc Lung Dis (1999) 3(3):214–8.
111. Giosue S, Casarini M, Ameglio F, Zangrilli P, Palla M, Altieri AM, et al. Aerosolized interferon-alpha treatment in patients with multi-drug-resistant pulmonary tuberculosis. Eur Cytokine Netw (2000) 11(1):99–104.
112. Giosue S, Casarini M, Alemanno L, Galluccio G, Mattia P, Pedicelli G, et al. Effects of aerosolized interferon-alpha in patients with pulmonary tuberculosis. Am J Respir Crit Care Med (1998) 158(4):1156–62. doi:10.1164/ajrccm.158.4.9803065
113. Mansoori D, Tavana S, Mirsaeidi M, Yazdanpanah M, Sohrabpour H. The efficacy of interferon-α in the treatment of multidrug resistant tuberculosis. Tanaffos (2002) 1(3):29–34.
114. Zarogoulidis P, Kioumis I, Papanas N, Manika K, Kontakiotis T, Papagianis A, et al. The effect of combination IFN-alpha-2a with usual antituberculosis chemotherapy in non-responding tuberculosis and diabetes mellitus: a case report and review of the literature. J Chemother (2012) 24(3):173–7. doi:10.1179/1973947812y.0000000005
115. Gideon HP, Skinner JA, Baldwin N, Flynn JL, Lin PL. Early whole blood transcriptional signatures are associated with severity of lung inflammation in cynomolgus macaques with Mycobacterium tuberculosis infection. J Immunol (2016) 197(12):4817–28. doi:10.4049/jimmunol.1601138
116. Mayer-Barber KD, Sher A. Cytokine and lipid mediator networks in tuberculosis. Immunol Rev (2015) 264(1):264–75. doi:10.1111/imr.12249
117. Chin KL, Anis FZ, Sarmiento ME, Norazmi MN, Acosta A. Role of interferons in the development of diagnostics, vaccines, and therapy for tuberculosis. J Immunol Res (2017) 2017:5212910. doi:10.1155/2017/5212910
118. Mariotti S, Teloni R, Iona E, Fattorini L, Romagnoli G, Gagliardi MC, et al. Mycobacterium tuberculosis diverts alpha interferon-induced monocyte differentiation from dendritic cells into immunoprivileged macrophage-like host cells. Infect Immun (2004) 72(8):4385–92. doi:10.1128/iai.72.8.4385-4392.2004
119. Bouchonnet F, Boechat N, Bonay M, Hance AJ. Alpha/beta interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect Immun (2002) 70(6):3020–5. doi:10.1128/IAI.70.6.3020-3025.2002
120. Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science (2013) 339(6126):1448–53. doi:10.1126/science.1233665
121. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol (2014) 193(7):3600–12. doi:10.4049/jimmunol.1401088
122. Kimmey JM, Campbell JA, Weiss LA, Monte KJ, Lenschow DJ, Stallings CL. The impact of ISGylation during Mycobacterium tuberculosis infection in mice. Microbes Infect (2017) 19(4–5):249–58. doi:10.1016/j.micinf.2016.12.006
123. Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science (2012) 337(6102):1684–8. doi:10.1126/science.1224026
124. de Paus RA, van Wengen A, Schmidt I, Visser M, Verdegaal EM, van Dissel JT, et al. Inhibition of the type I immune responses of human monocytes by IFN-alpha and IFN-beta. Cytokine (2013) 61(2):645–55. doi:10.1016/j.cyto.2012.12.005
125. Marchant A, Amedei A, Azzurri A, Vekemans J, Benagiano M, Tamburini C, et al. Polarization of PPD-specific T-cell response of patients with tuberculosis from Th0 to Th1 profile after successful antimycobacterial therapy or in vitro conditioning with interferon-alpha or interleukin-12. Am J Respir Cell Mol Biol (2001) 24(2):187–94. doi:10.1165/ajrcmb.24.2.4274
126. Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med (1993) 178(5):1655–63. doi:10.1084/jem.178.5.1655
127. Kavrochorianou N, Evangelidou M, Markogiannaki M, Tovey M, Thyphronitis G, Haralambous S. IFNAR signaling directly modulates T lymphocyte activity, resulting in milder experimental autoimmune encephalomyelitis development. J Leukoc Biol (2016) 99(1):175–88. doi:10.1189/jlb.3A1214-598R
128. Moreira-Teixeira L, Redford PS, Stavropoulos E, Ghilardi N, Maynard CL, Weaver CT, et al. T cell-derived IL-10 impairs host resistance to Mycobacterium tuberculosis infection. J Immunol (2017) 199(2):613–23. doi:10.4049/jimmunol.1601340
129. Schreiber T, Ehlers S, Heitmann L, Rausch A, Mages J, Murray PJ, et al. Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J Immunol (2009) 183(2):1301–12. doi:10.4049/jimmunol.0803567
130. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr (2016) 4(5). doi:10.1128/microbiolspec.TBTB2-0018-2016
131. Lienard J, Movert E, Valfridsson C, Sturegard E, Carlsson F. ESX-1 exploits type I IFN-signalling to promote a regulatory macrophage phenotype refractory to IFNgamma-mediated autophagy and growth restriction of intracellular mycobacteria. Cell Microbiol (2016) 18(10):1471–85. doi:10.1111/cmi.12594
132. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol (2007) 179(2):1178–89. doi:10.4049/jimmunol.179.2.1178
133. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol (2010) 184(7):3326–30. doi:10.4049/jimmunol.0904189
134. Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, et al. IL-1beta promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol (2013) 190(8):4196–204. doi:10.4049/jimmunol.1202688
135. Zhang G, Zhou B, Li S, Yue J, Yang H, Wen Y, et al. Allele-specific induction of IL-1beta expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog (2014) 10(10):e1004426. doi:10.1371/journal.ppat.1004426
136. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity (2012) 37(2):223–34. doi:10.1016/j.immuni.2012.04.015
137. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35(6):1023–34. doi:10.1016/j.immuni.2011.12.002
138. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol (2013) 14(1):52–60. doi:10.1038/ni.2474
139. Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VA, Vogel SN, et al. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-beta and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J Immunol (2013) 191(7):3514–8. doi:10.4049/jimmunol.1301331
140. Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol (2012) 24(10):637–44. doi:10.1093/intimm/dxs062
141. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One (2010) 5(8):e12320. doi:10.1371/journal.pone.0012320
142. Keller C, Lauber J, Blumenthal A, Buer J, Ehlers S. Resistance and susceptibility to tuberculosis analysed at the transcriptome level: lessons from mouse macrophages. Tuberculosis (Edinb) (2004) 84(3–4):144–58. doi:10.1016/j.tube.2003.12.003
143. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol (2017) 2:17072. doi:10.1038/nmicrobiol.2017.72
144. Lozza L, Farinacci M, Fae K, Bechtle M, Staber M, Dorhoi A, et al. Crosstalk between human DC subsets promotes antibacterial activity and CD8+ T-cell stimulation in response to bacille Calmette-Guerin. Eur J Immunol (2014) 44(1):80–92. doi:10.1002/eji.201343797
145. Lozza L, Farinacci M, Bechtle M, Staber M, Zedler U, Baiocchini A, et al. Communication between human dendritic cell subsets in tuberculosis: requirements for naive CD4(+) T cell stimulation. Front Immunol (2014) 5:324. doi:10.3389/fimmu.2014.00324
146. Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem (2008) 283(47):32925–36. doi:10.1074/jbc.M806019200
147. Strunk JJ, Gregor I, Becker Y, Li Z, Gavutis M, Jaks E, et al. Ligand binding induces a conformational change in ifnar1 that is propagated to its membrane-proximal domain. J Mol Biol (2008) 377(3):725–39. doi:10.1016/j.jmb.2008.01.017
148. de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat Immunol (2013) 14(9):901–7. doi:10.1038/ni.2667
149. Wu K, Wong KW, Deng WL, Zhang H, Li J, Lowrie DB, et al. The numerical predominance and large transcriptome differences of neutrophils in peripheral blood together inevitably account for a reported pulmonary tuberculosis signature. Int J Genomics (2017) 2017:5830971. doi:10.1155/2017/5830971
Keywords: Mycobacterium tuberculosis, type I interferon, innate immune signaling, pattern recognition receptors, immune responses, cytokines, patients, mouse models
Citation: Donovan ML, Schultz TE, Duke TJ and Blumenthal A (2017) Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front. Immunol. 8:1633. doi: 10.3389/fimmu.2017.01633
Received: 15 September 2017; Accepted: 09 November 2017;
Published: 27 November 2017
Edited by:
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
John R. Teijaro, The Scripps Research Institute, United StatesAnca Dorhoi, Friedrich Loeffler Institute Greifswald, Germany
Copyright: © 2017 Donovan, Schultz, Duke and Blumenthal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antje Blumenthal, YS5ibHVtZW50aGFsQHVxLmVkdS5hdQ==