 Thanh-Phuong Le
Thanh-Phuong Le To-Ha Thai
To-Ha Thai- Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Research on adult cancer immunotherapy is proceeding at a rapid pace resulting in an impressive success rate exemplified by a few high profile cases. However, this momentum is not readily extended to pediatric immunotherapy, and it is not for lack of trying. Though reasons for the slower advance are not apparent, some issues can be raised. Pediatric cancer patients represent a distinct demographic group whose immune system is inherently different from that of mature adults. Treating pediatric patients with immunotherapy designed for adults may not yield objective clinical responses. Here, we will present an update on adoptive T-cell and natural killer-cell therapies for neuroblastoma and other childhood solid tumors. Additionally, we will delineate key differences between human fetal/neonatal and adult immune systems. We hope this will generate interests leading to the discussion of potential future directions for improving adoptive cancer immunotherapy for children.
Introduction
In the past few decades, pivotal studies have yielded invaluable information on pediatric oncology leading to the formulation of standard therapies still being performed today (1–9). However, it is now evident that the majority of resistant, metastatic, recurrent/refractory tumors are non-responsive to conventional therapies (10–20). In addition, current approaches often rely on non-specific, cytotoxic chemotherapy and/or radiotherapy that result in long lasting, debilitating toxicities, and in some instance morbidity (21–26). Therefore, there is a need to explore new avenues to eradicate pediatric cancers.
Two seminal reports, published by the surgeon and cancer researcher William Bradley Coley in the late 19th century, show sarcoma tumor regression in patients repeatedly immunized with live or killed streptococcus bacteria (27, 28). His observations suggest an active function for the immune system to control tumor growth, thus laying the foundation for modern cancer immunotherapy. Today, harnessing the immune system to control cancer is proven effective and garnering momentum. Currently, immunotherapy is largely classified into two functional treatment groups: (1) those that amplify/reactivate host existing innate and adaptive tumor immunity including check point inhibitors, dendritic cell (DC) vaccines and cytokines; (2) those that involve the adoptive transfer of genetically manipulated immune cells to target tumor cells in vivo such as chimeric antigen receptor (CAR) T cells, genetically enhanced effector T cells and natural killer (NK) cells. We will focus primarily on T- and NK-cell adoptive immunotherapy in this perspective.
Adoptive T-Cell Therapy (ACT)
Adoptive T-cell therapy represents an attractive viable option for the control of solid tumor growth for the following reasons: T cells can systemically home to tumor sites through the entire body and can cross the blood–brain barrier (BBB). By contrast, antibodies such as checkpoint inhibitors cannot effectively cross the BBB and are not consistently or adequately distributed deep inside solid tumors. To date, ACT with CAR T cells is the prevalent type of immunotherapy to treat solid tumors.
Neuroblastoma (NB)
Neuroblastoma is the most common extracranial solid tumor of childhood and the third most common cause of pediatric cancer death (29–32). Despite conventional multimodal therapy, patients with high-risk NB have a poor prognosis due to high relapse rate (33). Since 1999, tremendous efforts and funds have been dedicated to discovering and testing various forms of immunotherapy to control refractory/recurrent NB (Table 1). To date the most studied NB-associated antigen (NBAA) identified is the ganglioside GD2 expressed on a subset of NB tumor cells (34). This discovery heralds in the era of GD2-based immunotherapy to treat human NB. The current standard of care for refractory/recurrent NB in human is anti-GD2 therapy, which recognizes and binds GD2 (34–39). However, because GD2 is also expressed on pain fibers, this therapy has side effects that include severe pain requiring continuous opiate infusions (34). Because of the paucity of identified NBAAs, all CAR clinical trials are GD2-based or variations thereof (38, 40–45) (Table 1). It is still early to know whether these GD2-dependent CAR trials will yield objective clinical responses.
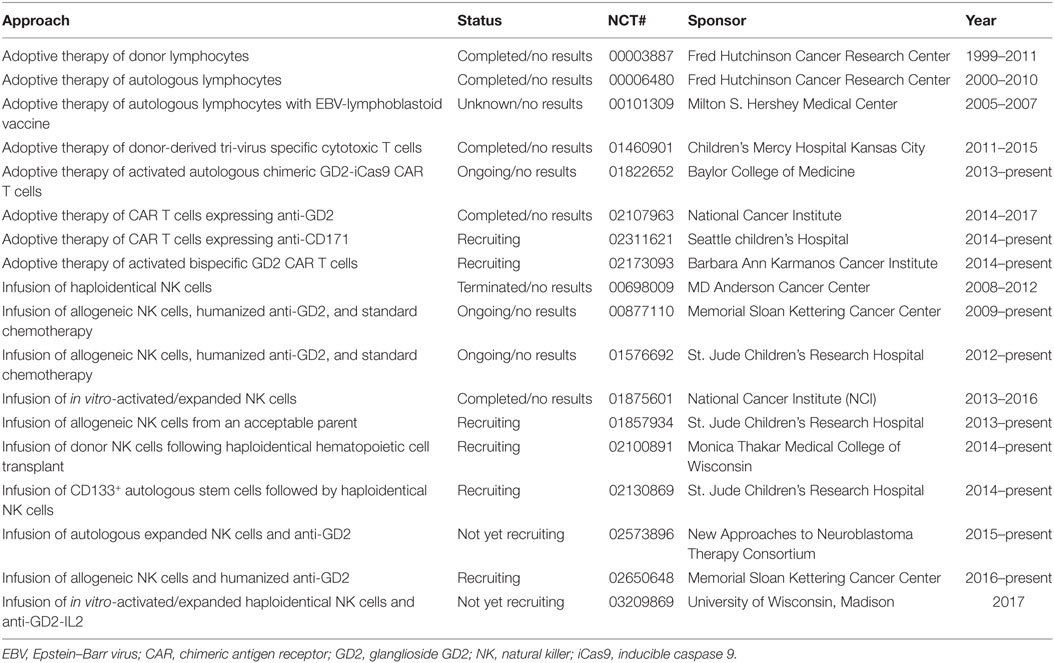
Table 1. Present and past adoptive immunotherapy trials for refractory/recurrent/relapsed neuroblastoma.
The antigen 4Ig-B7-H3 (CD276), a member of the B7 family of immune regulators such as CD80, CD86, PD-L1, PD-L2 and ICOSL, is expressed in a subset of NB tumors (46–49). In mouse and human, 4Ig-B7-H3 is expressed on many normal tissues such as spleen, lymph nodes, thymus and fetal liver as well as other tumors. However, its expression is induced on DCs and macrophages by inflammatory cytokines. 4Ig-B7-H3 binds to a yet to be identified cognate receptor induced on activated T and NK cells, and blockade of this interaction results in reduced interferon γ production and loss of cytotoxic activity of these cells. In mice, deficiency or blockade of B7-H3 leads to improved antitumor immunity suggesting that B7-H3 checkpoint may serve as a novel target for immunotherapy against cancers (50). Indeed, a mouse mAb anti-B7-H3 conjugated to iodine 131 (131I-burtomab) has been designed to bind and directly kill NB cells. 131I-burtomab is recently designated a breakthrough drug to treat metastatic NB by the Food and Drug Administration and a clinical trial has been filed (NCT03275402). The immune status of NB tumors treated with 131I-burtomab has not been published. It would be interesting to know whether the antitumor activity of burtomab results from direct killing of tumor cells or activated DCs thus reactivating existing anti-NB immunity. Although no published data are available to show the feasibility of B7-H3 CAR T cells for the treatment of human NB, two patents have been filed for such an invention (US Application No. 14/779,586; US Application No. 61/805,001; PCT Application No. PCT/US2014/031543 and PCT Application No. PCT/US2016/050887; US Application No. 62/216,447). It remains to be determined whether B7-H3 CAR T cells can inhibit NB tumor growth and whether the potential antitumor activity of B7-H3 CAR T cells is due to killing of intratumoral suppressive DCs/macrophages or tumor cells or both.
The anaplastic lymphoma kinase (ALK or CD246) is a receptor protein tyrosine kinase predominantly expressed in the central nervous system (CNS) and peripheral nervous system in mouse and human suggesting its role in normal brain development and function (51). A series of studies show that Alk is frequently mutated (mainly ALKR1275Q and ALKF1174L) and duplicated in high-risk NB tumors (52–58). The ALKR1275Q mutation results in a constitutively active kinase suggesting a role for ALK in NB development. However, mice harboring human ALKR1275Q or ALKF1174L alone do not develop aggressive NB irrespective of genetic background (53, 57). In the contrary, animals having both MYCN amplification and ALKR1275Q or ALKF1174L mutation succumb to NB at a higher rate (53, 57). These findings suggest that mutations in Alk are necessary but not sufficient to drive aggressive NB development. Because ALK is a cell surface kinase, developing CAR T cells targeting ALK has been suggested. Indeed, in a xenogeneic NSG mouse model for NB, human ALK CAR T cells can eradicate ALK-positive tumors; both tumor antigen and receptor density governs the efficacy of these CAR T cells (59). Clinical trials have not been initiated.
Although CAR T-cell therapy is being propelled to the forefront, problems exist that need further investigation. Production of CAR T cells requires the identification of tumor-associated antigen (TAA), generation of an antibody or T-cell receptor (TCR) capable of recognizing the TAA, cloning of genes encoding the antibody or TCR to be introduced into isolated tumor-infiltrating lymphocytes or haploidentical T cells. For most pediatric solid tumors, the identity of TAAs is still unknown and neoantigen load is low thus limiting the use of CAR T cells for this group. In patients with solid tumors for which TAAs have been identified, the use of CAR T cells has proven less effective than in patients with fluid tumors. Recent data are showing a previously unpredicted phenomenon observed in patients treated with CAR T cells: the emergence of tumor cells that have lost expression of the TAA targeted by CAR T cells, undoubtedly due to negative selection imposed by CAR therapy (60–62). New evidence demonstrates that CAR T cells once in the tumor microenvironment (TME) may suffer from exhaustion caused by suppressor cells including myeloid-derived suppressor cells or CD4+ regulatory T (Treg) cells present in the TME (63–65). Perhaps a more dangerous issue arisen is the development of cytokine release syndrome (CRS) (66, 67) and neurologic toxicity observed in patients undergoing CAR therapy (68).
To circumvent problems posed by CAR T cells, we propose that perhaps the most effective strategy to control solid tumor growth is one that does not require identifying TAAs and corresponding tumor-reactive CD8+ T cells, can enhance effector activity of CD8+ T cells, and can simultaneously eliminate immune suppression within the TME. Our recent studies suggest that such an approach is attainable. We show that by targeting the histone reader Cbx3/HP1γ, we can enhance the tumor killing capacity of effector CD8+ T cells (69, 70). As a result, adoptive transfer of Cbx3/HP1γ-deficient CD8+ effector T cells alone into wild type (wt) tumor-bearing mice greatly reduces NB growth. Within the NB TME of Cbx3/HP1γ-deficient mice or wt mice treated with Cbx3/HP1γ-deficient CD8+ T cells, we detect an increase of Klrk1/NKG2D+ infiltrating CD8+ effector T cells and a decrease in CD4+ Treg cells. PD-1 and Pdl1 expression is not altered in CD8+ T cells or NB tumors, respectively. Moreover, Cbx3/HP1γ-deficient CD8+ T cells appear to have overcome exhaustion. These findings suggest that targeting Cbx3/HP1γ can represent an alternative and rational therapeutic approach to control NB as well as other solid tumors.
Other Pediatric Solid Tumors (CNS Tumors, Sarcomas, and Nasopharyngeal Sarcomas)
As for NB, there is a dearth of identified TAAs available for the formulation of CAR therapy to treat most pediatric solid tumors (71–73). Tumor immunity against pediatric solid tumors is not completely understood. The human epidermal growth factor receptor 2 (HER2) is expressed on pediatric as well as adult glioblastoma, glioma, and medulloblastoma tumors, and overexpression of HER2 has been associated with poorer prognosis. In animal models, HER2 CAR T cells efficiently cause the regression of CNS tumors. These preclinical studies have paved the way for a few HER2-based CAR clinical trials (74–77) (Table 2); results of these trials are not yet available. It would be crucial to determine whether HER2-targeted CAR therapy will induce the emergence of HER2-negative tumors as has been shown in animal models and in patients receiving CD19 targeted CAR therapy (60, 61) or will cause CRS as in adults (67).
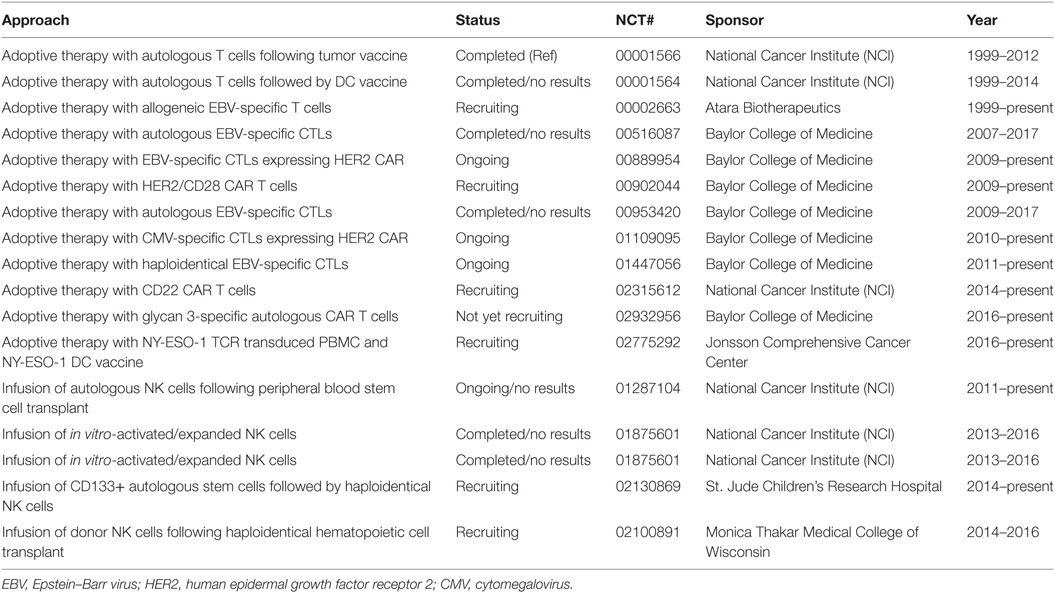
Table 2. Present and past adoptive immunotherapy for pediatric central nervous system tumors, sarcomas, and nasopharyngeal carcinomas.
Pediatric nasopharyngeal carcinoma patients with local-regional bulky and metastatic disease have a poor prognosis (78). It is a rare tumor that is almost always associated with Epstein–Barr virus (EBV) (78, 79), and EBV-specific cytotoxic T lymphocytes (CTLs) can be found in individuals infected with this ubiquitous virus. These findings lead to the design of EBV-based T-cell therapy. In adults, ACT with EBV-specific CTLs is more effective in patients with low disease burden while results for pediatric trials are not available (80–83) (Table 2).
Prognosis for pediatric patients with recurrent/refractory sarcomas is poor, the survival rate ranges from 10 to 30% (73). For this group of children, few immunotherapy clinical trials are being tested, and past trials using autologous T cells (NCT00001566 and NCT 00001564) have not yielded much information to advance the field (Table 2).
Some studies have shown that the cancer-testes antigen NY-ESO-1 is expressed on a subset of pediatric tumors, which lead to a trial using T cells engineered to express NY-ESO-1-specific TCR (NCT02775292) (84) (Table 2). HER2 CAR therapy has also been proposed to treat sarcomas (85). CAR T cells targeting glypican-3, a proteoglycan expressed on a small number of solid tumors (86), are being tested in clinical trials to treat pediatric solid tumors (87) (NCT02932956). Data are not yet available for these trials.
The lack of available immunotherapy for pediatric solid tumors may be due to the paucity of identified TAAs, and few basic studies designed to understand tumor immunity during development from infancy to young adulthood in either mouse or human. As a result, most of these trials are based on those that have been designed to treat adult solid tumors.
Adoptive NK- and Natural Killer T (NKT)-Cell Therapy
Natural killer cells and NKT cells have been shown to play crucial roles in antitumor immunity by directly killing tumor cells or indirectly through antibody-dependent cellular cytotoxicity. Based on results from preclinical studies (88–90), several clinical trials have been initiated to test the ability of in vitro-activated/expanded or engineered NK cells and NKT cells to control pediatric solid tumors including NB (91, 92) (Tables 1 and 2). Results of these trials are not yet available to indicate whether NK- or NKT-cell therapy would be a viable option.
Nonetheless, clinical data have demonstrated that despite the large number of NK cells infused, the antitumor effects of these cells have been modest in adults. NK and NKT cells express a number of inhibitory receptors that bind to MHC class I and other molecules. Additionally, NK and NKT cells are sensitive to various inhibitory molecules within the TME (93). Moreover, we show that the frequency of NK and NKT cells in NB tumors is low, and no differences are detected in tumors from wt or Cbx3/HP1γ-deficient mice yet NB tumor growth is greatly abrogated in Cbx3/HP1γ-deficient mice (70). Our findings imply that NK or NKT cells may not play an important role in controlling NB tumor growth.
Discussion and Future Directions
In the past 40–50 years, pediatric oncologists have made significant, basic advances toward our understanding of molecular pathways driving the development of tumors in children. This achievement hinges on the belief that cancer pediatric patients represent a distinct demographic group, and the biology of their tumors is fundamentally different from that of adults.
Adult cancer immunotherapy is experiencing a renaissance while that of children is still at its infancy. The momentum that drives adult cancer immunotherapy is built upon decades of basic research designed to understand how an adult immune system responses to tumors developing within an adult host. Thus, there is a need to recognize that pediatric cancer patients represent a distinct demographic group whose immune system is fundamentally different than that of mature adults.
Fetal and adult T cells are distinct populations that arise from different hematopoietic stem cell populations present at different developmental stages (94), and human NK cells follow similar developmental evolution (95, 96). Notably, fetal CD4+ T cells are poised to differentiate into CD4+ Treg cells upon allogeneic stimulation. Indeed, human fetus and cord blood (CB) contains an abundance of phenotypically naïve CD25+CD4+ Treg cells, but functionally mature, capable of suppressing T- and NK-cell proliferation and function (97–101). Similar suppressive mechanism is observed in the mouse fetus (102). Thus, T- and NK-cell lineages in the developing human or mouse are biased toward immune tolerance mediated by active suppression of early immunity; in some instances, this suppression persists at least until early adulthood (101). The tolerogenic tendency of fetal/neonatal immune system can be attributed to marked differences in response to alloantigens between human fetal and adult DCs. Fetal DCs strongly promote Treg-cell induction and inhibit T-cell tumor-necrosis factor-α production when cultured with alloantigens (103). This may explain why CAR T cells once in the NB TME often suffer from exhaustion. In addition to functional disparities, neonatal and adult immune systems differ quantitatively. Overall, there is a greater number of circulating CD4+ T cells and a lower number of CD8+ T cells in neonates compared to adults (104). Consequently, the ratio of CD4:CD8 is higher in neonates than adults. Together these results suggest that for some children, the persistence of fetal immune suppression, mediated by fetal CD4+ Treg cells, and the lower number of CD8+ T cells may render their immune system incapable of surveilling and eradicating tumor. Therefore in the future, it might be essential to study the effects of persistent fetal immune suppression on tumor development and growth. If the persistence of fetal immune suppression does influence antitumor immunity in pediatric patients, it would be crucial to determine the developmental age at which intervention can be mounted to prevent tumorigenesis and growth without breaking tolerance. Clinically, it might be important to collect data on the immune status of children bearing solid tumors in addition to dissecting their tumor immune environment. Results from these studies might help direct the design of T- and NK-cell therapies that can circumvent suppression and prevent exhaustion induction.
In adult tumors, mutation load appears to correlate with tumor immunity. However, the number of somatic mutations in pediatric solid tumors is low. In the future, it would be necessary to determine mechanisms controlling tumor immunity independent of somatic mutation loads in pediatric patients.
For this demographic group, perhaps the most effective strategy to control solid tumor growth is one that does not require identifying TAAs or neoantigens and their corresponding reactive CD8+ T cells, can enhance effector activity of CD8+ T cells, and can simultaneously eliminate immune suppression within the TME.
We believe these are crucial issues that need to be addressed in order to move the field of pediatric adoptive cancer immunotherapy to the fore. Until there is a will to allow for the funding of such studies and those that are outside traditional belief, children with cancers will continue to be treated as adults, and may not benefit from the cancer immunotherapy renaissance.
Author Contributions
TPL and THT conceived and cowrote this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all those who have laid significant and important groundwork upon which we build ours. We apologize to those whose studies cannot be cited due to space constraint.
Funding
This work was funded by NIH/NCI grant CA198263, NIH/NIAID AI099012, The Mayer Family Fund, The Friends for Life Neuroblastoma Research Program.
References
1. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature (1992) 359(6391):162–5. doi:10.1038/359162a0
2. Grady-Leopardi EF, Schwab M, Ablin AR, Rosenau W. Detection of N-myc oncogene expression in human neuroblastoma by in situ hybridization and blot analysis: relationship to clinical outcome. Cancer Res (1986) 46(6):3196–9.
3. Jha P, Suri V, Singh G, Jha P, Purkait S, Pathak P, et al. Characterization of molecular genetic alterations in GBMs highlights a distinctive molecular profile in young adults. Diagn Mol Pathol (2011) 20(4):225–32. doi:10.1097/PDM.0b013e31821c30bc
4. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science (2011) 331(6016):435–9. doi:10.1126/science.1198056
5. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med (2014) 371(11):1005–15. doi:10.1056/NEJMoa1403088
6. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell (2012) 22(2):153–66. doi:10.1016/j.ccr.2012.06.005
7. Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature (1983) 305(5931):245–8. doi:10.1038/305245a0
8. Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ III, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet (1993) 5(3):230–5. doi:10.1038/ng1193-230
9. Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol (2010) 28(15):2625–34. doi:10.1200/JCO.2009.27.0421
10. Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol (2011) 13(3):317–23. doi:10.1093/neuonc/noq191
11. MacDonald TJ, Aguilera D, Kramm CM. Treatment of high-grade glioma in children and adolescents. Neuro Oncol (2011) 13(10):1049–58. doi:10.1093/neuonc/nor092
12. Feun LG, Savaraj N, Landy HJ. Drug resistance in brain tumors. J Neurooncol (1994) 20(2):165–76. doi:10.1007/bf01052726
14. McNeil DE, Cote TR, Clegg L, Rorke LB. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol (2002) 39(3):190–4. doi:10.1002/mpo.10121
15. Moreno L, Marshall LV, Pearson AD. At the frontier of progress for paediatric oncology: the neuroblastoma paradigm. Br Med Bull (2013) 108:173–88. doi:10.1093/bmb/ldt033
16. Lagmay JP, Krailo MD, Dang H, Kim A, Hawkins DS, Beaty O III, et al. Outcome of patients with recurrent osteosarcoma enrolled in seven phase II trials through children’s cancer group, pediatric oncology group, and children’s oncology group: learning from the past to move forward. J Clin Oncol (2016) 34(25):3031–8. doi:10.1200/JCO.2015.65.5381
17. Shankar AG, Ashley S, Craft AW, Pinkerton CR. Outcome after relapse in an unselected cohort of children and adolescents with Ewing sarcoma. Med Pediatr Oncol (2003) 40(3):141–7. doi:10.1002/mpo.10248
18. Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer (2011) 11(8):541–57. doi:10.1038/nrc3087
19. Duffner PK, Armstrong FD, Chen L, Helton KJ, Brecher ML, Bell B, et al. Neurocognitive and neuroradiologic central nervous system late effects in children treated on Pediatric Oncology Group (POG) P9605 (standard risk) and P9201 (lesser risk) acute lymphoblastic leukemia protocols (ACCL0131): a methotrexate consequence? A report from the Children’s Oncology Group. J Pediatr Hematol Oncol (2014) 36(1):8–15. doi:10.1097/MPH.0000000000000000
20. Galloway TJ, Indelicato DJ, Amdur RJ, Swanson EL, Smith AA, Marcus RB Jr. Second tumors in pediatric patients treated with radiotherapy to the central nervous system. Am J Clin Oncol (2012) 35(3):279–83. doi:10.1097/COC.0b013e318210f533
21. Applebaum MA, Vaksman Z, Lee SM, Hungate EA, Henderson TO, London WB, et al. Neuroblastoma survivors are at increased risk for second malignancies: a report from the International Neuroblastoma Risk Group Project. Eur J Cancer (2017) 72:177–85. doi:10.1016/j.ejca.2016.11.022
22. Lancashire ER, Frobisher C, Reulen RC, Winter DL, Glaser A, Hawkins MM. Educational attainment among adult survivors of childhood cancer in Great Britain: a population-based cohort study. J Natl Cancer Inst (2010) 102(4):254–70. doi:10.1093/jnci/djp498
23. Frobisher C, Gurung PM, Leiper A, Reulen RC, Winter DL, Taylor AJ, et al. Risk of bladder tumours after childhood cancer: the British Childhood Cancer Survivor Study. BJU Int (2010) 106(7):1060–9. doi:10.1111/j.1464-410X.2010.09224.x
24. Reulen RC, Winter DL, Frobisher C, Lancashire ER, Stiller CA, Jenney ME, et al. Long-term cause-specific mortality among survivors of childhood cancer. JAMA (2010) 304(2):172–9. doi:10.1001/jama.2010.923
25. Taylor AJ, Little MP, Winter DL, Sugden E, Ellison DW, Stiller CA, et al. Population-based risks of CNS tumors in survivors of childhood cancer: the British Childhood Cancer Survivor Study. J Clin Oncol (2010) 28(36):5287–93. doi:10.1200/JCO.2009.27.0090
26. Kuhlthau KA, Pulsifer MB, Yeap BY, Rivera Morales D, Delahaye J, Hill KS, et al. Prospective study of health-related quality of life for children with brain tumors treated with proton radiotherapy. J Clin Oncol (2012) 30(17):2079–86. doi:10.1200/JCO.2011.37.0577
27. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res (1991) 262:3–11.
28. Coley WB. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med (1910) 3(Surg Sect):1–48.
29. Seeger RC. Immunology and immunotherapy of neuroblastoma. Semin Cancer Biol (2011) 21(4):229–37. doi:10.1016/j.semcancer.2011.09.012
30. Speleman F, Park JR, Henderson TO. Neuroblastoma: a tough nut to crack. Am Soc Clin Oncol Educ Book (2016) 35:e548–57. doi:10.14694/EDBK_159169
31. Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer (2013) 13(6):397–411. doi:10.1038/nrc3526
32. Sharp SE, Gelfand MJ, Shulkin BL. Pediatrics: diagnosis of neuroblastoma. Semin Nucl Med (2011) 41(5):345–53. doi:10.1053/j.semnuclmed.2011.05.001
33. Li R, Polishchuk A, DuBois S, Hawkins R, Lee SW, Bagatell R, et al. Patterns of relapse in high-risk neuroblastoma patients treated with and without total body irradiation. Int J Radiat Oncol Biol Phys (2017) 97(2):270–7. doi:10.1016/j.ijrobp.2016.10.047
34. Suzuki M, Cheung NK. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert Opin Ther Targets (2015) 19(3):349–62. doi:10.1517/14728222.2014.986459
35. Yang RK, Sondel PM. Anti-GD2 strategy in the treatment of neuroblastoma. Drugs Future (2010) 35(8):665. doi:10.1358/dof.2010.035.08.1513490
36. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi:10.1056/NEJMoa0911123
37. Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol (2012) 30(26):3264–70. doi:10.1200/JCO.2011.41.3807
38. Hoseini SS, Dobrenkov K, Pankov D, Xu XL, Cheung N-KV. Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. Oncoimmunology (2017) 6(6):e1320625. doi:10.1080/2162402X.2017.1320625
39. Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, et al. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol (2014) 32(14):1445–52. doi:10.1200/JCO.2013.50.4423
40. Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using bispecific humanized antibody. Cancer Immunol Res (2015) 3(3):266–77. doi:10.1158/2326-6066.CIR-14-0230-T
41. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther (2016) 24(6):1135–49. doi:10.1038/mt.2016.63
42. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther (2017) 25(9):2214–24. doi:10.1016/j.ymthe.2017.05.012
43. Prapa M, Caldrer S, Spano C, Bestagno M, Golinelli G, Grisendi G, et al. A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget (2015) 6(28):24884–94. doi:10.18632/oncotarget.4670
44. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res (2014) 74(18):5195–205. doi:10.1158/0008-5472.CAN-14-0697
45. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14(11):1264–70. doi:10.1038/nm.1882
46. Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci U S A (2004) 101(34):12640–5. doi:10.1073/pnas.0405025101
47. Steinberger P, Majdic O, Derdak SV, Pfistershammer K, Kirchberger S, Klauser C, et al. Molecular characterization of human 4Ig-B7-H3, a member of the B7 family with four Ig-like domains. J Immunol (2004) 172(4):2352–9. doi:10.4049/jimmunol.172.4.2352
48. Sun M, Richards S, Prasad DVR, Mai XM, Rudensky A, Dong C. Characterization of mouse and human B7-H3 genes. J Immunol (2002) 168(12):6294–7. doi:10.4049/jimmunol.168.12.6294
49. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-[gamma] production. Nat Immunol (2001) 2(3):269–74. doi:10.1038/85339
50. Lee Y-H, Martin-Orozco N, Zheng P, Li J, Zhang P, Tan H, et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res (2017) 27(8):1034–45. doi:10.1038/cr.2017.90
51. Morris S, Kirstein M, Valentine M, Dittmer K, Shapiro D, Saltman D, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science (1994) 263(5151):1281–4. doi:10.1126/science.8122112
52. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature (2008) 455(7215):971–4. doi:10.1038/nature07399
53. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell (2012) 22(1):117–30. doi:10.1016/j.ccr.2012.06.001
54. George RE, Sanda T, Hanna M, Frohling S, Ii WL, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature (2008) 455(7215):975–8. doi:10.1038/nature07397
55. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature (2008) 455(7215):930–5. doi:10.1038/nature07261
56. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45(3):279–84. doi:10.1038/ng.2529
57. Ueda T, Nakata Y, Yamasaki N, Oda H, Sentani K, Kanai A, et al. ALKR1275Q perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene (2016) 35(34):4447–58. doi:10.1038/onc.2015.519
58. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature (2008) 455(7215):967–70. doi:10.1038/nature07398
59. Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther (2017) 25(9):2189–201. doi:10.1016/j.ymthe.2017.06.008
60. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood (2013) 121(7):1165–74. doi:10.1182/blood-2012-06-438002
61. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3
62. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest (2016) 126(8):3036–52. doi:10.1172/JCI83416
63. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med (2015) 21(5):524–9. doi:10.1038/nm.3833
64. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi:10.1038/nm.3838
65. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res (2016) 4(10):869–80. doi:10.1158/2326-6066.cir-15-0230
66. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124(2):188–95. doi:10.1182/blood-2014-05-552729
67. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi:10.1038/mt.2010.24
68. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy [mdash] assessment and management of toxicities. Nat Rev Clin Oncol (2017). doi:10.1038/nrclinonc.2017.148
69. Ha N, Pham DH, Shahsafaei A, Naruse C, Asano M, Thai TH. HP-1gamma controls high-affinity antibody response to T-dependent antigens. Front Immunol (2014) 5:271. doi:10.3389/fimmu.2014.00271
70. Sun M, Ha N, Pham DH, Frederick M, Sharma B, Naruse C, et al. Cbx3/HP1gamma deficiency confers enhanced tumor-killing capacity on CD8+ T cells. Sci Rep (2017) 7:42888. doi:10.1038/srep42888
71. Bielamowicz K, Khawja S, Ahmed N. Adoptive cell therapies for glioblastoma. Front Oncol (2013) 3:275. doi:10.3389/fonc.2013.00275
72. Hegde M, Bielamowicz KJ, Ahmed N. Novel approaches and mechanisms of immunotherapy for glioblastoma. Discov Med (2014) 17(93):145–54.
73. Hegde M, Moll AJ, Byrd TT, Louis CU, Ahmed N. Cellular immunotherapy for pediatric solid tumors. Cytotherapy (2015) 17(1):3–17. doi:10.1016/j.jcyt.2014.05.019
74. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi:10.1200/JCO.2014.58.0225
75. Ahmed N, Ratnayake M, Savoldo B, Perlaky L, Dotti G, Wels WS, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res (2007) 67(12):5957–64. doi:10.1158/0008-5472.CAN-06-4309
76. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res (2010) 16(2):474–85. doi:10.1158/1078-0432.CCR-09-1322
77. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther (2009) 17(10):1779–87. doi:10.1038/mt.2009.133
78. Chan AT, Teo PM, Johnson PJ. Nasopharyngeal carcinoma. Ann Oncol (2002) 13(7):1007–15. doi:10.1093/annonc/mdf179
79. Raab-Traub N. Epstein-Barr virus in the pathogenesis of NPC. Semin Cancer Biol (2002) 12(6):431–41. doi:10.1016/S1044579X0200086X
80. Comoli P, Pedrazzoli P, Maccario R, Basso S, Carminati O, Labirio M, et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein-Barr virus-targeted cytotoxic T lymphocytes. J Clin Oncol (2005) 23(35):8942–9. doi:10.1200/JCO.2005.02.6195
81. Straathof KC, Leen AM, Buza EL, Taylor G, Huls MH, Heslop HE, et al. Characterization of latent membrane protein 2 specificity in CTL lines from patients with EBV-positive nasopharyngeal carcinoma and lymphoma. J Immunol (2005) 175(6):4137–47. doi:10.4049/jimmunol.175.6.4137
82. Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood (2005) 105(11):4247–54. doi:10.1182/blood-2004-11-4564
83. Louis CU, Straathof K, Bollard CM, Gerken C, Huls MH, Gresik MV, et al. Enhancing the in vivo expansion of adoptively transferred EBV-specific CTL with lymphodepleting CD45 monoclonal antibodies in NPC patients. Blood (2009) 113(11):2442–50. doi:10.1182/blood-2008-05-157222
84. Jungbluth AA, Antonescu CR, Busam KJ, Iversen K, Kolb D, Coplan K, et al. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int J Cancer (2001) 94(2):252–6. doi:10.1002/ijc.1451
85. Rainusso N, Brawley VS, Ghazi A, Hicks MJ, Gottschalk S, Rosen JM, et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther (2012) 19(3):212–7. doi:10.1038/cgt.2011.83
86. Kinoshita Y, Tanaka S, Souzaki R, Miyoshi K, Kohashi K, Oda Y, et al. Glypican 3 expression in pediatric malignant solid tumors. Eur J Pediatr Surg (2015) 25(1):138–44. doi:10.1055/s-0034-1393961
87. Bi Y, Jiang H, Wang P, Song B, Wang H, Kong X, et al. Treatment of hepatocellular carcinoma with a GPC3-targeted bispecific T cell engager. Oncotarget (2017) 8(32):52866–76. doi:10.18632/oncotarget.17905
88. Seidel D, Shibina A, Siebert N, Wels WS, Reynolds CP, Huebener N, et al. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol Immunother (2015) 64(5):621–34. doi:10.1007/s00262-015-1669-5
89. Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med (2012) 16(3):569–81. doi:10.1111/j.1582-4934.2011.01343.x
90. Liu Y, Wu H-W, Sheard MA, Sposto R, Somanchi SS, Cooper LJN, et al. Growth and activation of natural killer cells ex vivo from children with neuroblastoma for adoptive cell therapy. Clin Cancer Res (2013) 19(8):2132–43. doi:10.1158/1078-0432.ccr-12-1243
91. Perez-Martinez A, Leung W, Munoz E, Iyengar R, Ramirez M, Vicario JL, et al. KIR-HLA receptor-ligand mismatch associated with a graft-versus-tumor effect in haploidentical stem cell transplantation for pediatric metastatic solid tumors. Pediatr Blood Cancer (2009) 53(1):120–4. doi:10.1002/pbc.21955
92. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest (2009) 119(6):1524–36. doi:10.1172/JCI37869
93. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
94. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science (2010) 330(6011):1695–9. doi:10.1126/science.1196509
95. Uksila J, Lassila O, Hirvonen T, Toivanen P. Development of natural killer cell function in the human fetus. J Immunol (1983) 130(1):153–6.
96. Schönberg K, Fischer JC, Kögler G, Uhrberg M. Neonatal NK-cell repertoires are functionally, but not structurally, biased toward recognition of self HLA class I. Blood (2011) 117(19):5152–6. doi:10.1182/blood-2011-02-334441
97. Takahata Y, Nomura A, Takada H, Ohga S, Furuno K, Hikino S, et al. CD25+CD4+ T cells in human cord blood: an immunoregulatory subset with naive phenotype and specific expression of forkhead box p3 (Foxp3) gene. Exp Hematol (2004) 32(7):622–9. doi:10.1016/j.exphem.2004.03.012
98. Dalle J-H, Menezes J, Wagner E, Blagdon M, Champagne J, Champagne MA, et al. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr Res (2005) 57(5 Pt 1):649–55. doi:10.1203/01.PDR.0000156501.55431.20
99. Ivarsson MA, Loh L, Marquardt N, Kekalainen E, Berglin L, Bjorkstrom NK, et al. Differentiation and functional regulation of human fetal NK cells. J Clin Invest (2013) 123(9):3889–901. doi:10.1172/JCI68989
100. Michaëlsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol (2006) 176(10):5741–8. doi:10.4049/jimmunol.176.10.5741
101. Mold JE, Michaëlsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science (2008) 322(5907):1562–5. doi:10.1126/science.1164511
102. Marcoe JP, Lim JR, Schaubert KL, Fodil-Cornu N, Matka M, McCubbrey AL, et al. TGF-[beta] is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat Immunol (2012) 13(9):843–50. doi:10.1038/ni.2388
103. McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature (2017) 546(7660):662–6. doi:10.1038/nature22795
Keywords: pediatric solid tumors, recurrent/refractory/relapsed neuroblastoma, adoptive T-cell therapy, immune cell-based therapy, natural killer cells, Cbx3/HP1γ, CD4+ regulatory T cells, effector CD8+ T cells
Citation: Le TP and Thai TH (2017) The State of Cellular Adoptive Immunotherapy for Neuroblastoma and Other Pediatric Solid Tumors. Front. Immunol. 8:1640. doi: 10.3389/fimmu.2017.01640
Received: 27 September 2017; Accepted: 09 November 2017;
Published: 24 November 2017
Edited by:
Katy Rezvani, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Niels Halama, National Center for Tumor Diseases, GermanyKrithika Kodumudi, Moffitt Cancer Center, United States
Copyright: © 2017 Le and Thai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: To-Ha Thai, dHRoYWlAYmlkbWMuaGFydmFyZC5lZHU=