 Richard E. Frye1,2*
Richard E. Frye1,2* Bistra Nankova3
Bistra Nankova3 Sudeepa Bhattacharyya1,2
Sudeepa Bhattacharyya1,2 Shannon Rose1,2
Shannon Rose1,2 Sirish C. Bennuri1,2
Sirish C. Bennuri1,2 Derrick F. MacFabe4
Derrick F. MacFabe4
- 1Department of Pediatrics, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 2Autism Research Program, Arkansas Children’s Research Institute, Little Rock, AR, United States
- 3New York Medical College, Valhalla, NY, United States
- 4Kilee Patchell-Evans Autism Research Group, Alberta Children’s Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
Propionic acid (PPA) is a ubiquitous short-chain fatty acid which is a fermentation product of the enteric microbiome and present or added to many foods. While PPA has beneficial effects, it is also associated with human disorders, including autism spectrum disorders (ASDs). We previously demonstrated that PPA modulates mitochondrial dysfunction differentially in subsets of lymphoblastoid cell lines (LCLs) derived from patients with ASD. Specifically, PPA significantly increases mitochondrial function in LCLs that have mitochondrial dysfunction at baseline [individuals with autistic disorder with atypical mitochondrial function (AD-A) LCLs] as compared to ASD LCLs with normal mitochondrial function [individuals with autistic disorder with normal mitochondrial function (AD-N) LCLs] and control (CNT) LCLs. PPA at 1 mM was found to have a minimal effect on expression of immune genes in CNT and AD-N LCLs. However, as hypothesized, Panther analysis demonstrated that 1 mM PPA exposure at 24 or 48 h resulted in significant activation of the immune system genes in AD-A LCLs. When the effect of PPA on ASD LCLs were compared to the CNT LCLs, both ASD groups demonstrated immune pathway activation, although the AD-A LCLs demonstrate a wider activation of immune genes. Ingenuity Pathway Analysis identified several immune-related pathways as key Canonical Pathways that were differentially regulated, specifically human leukocyte antigen expression and immunoglobulin production genes were upregulated. These data demonstrate that the enteric microbiome metabolite PPA can evoke atypical immune activation in LCLs with an underlying abnormal metabolic state. As PPA, as well as enteric bacteria which produce PPA, have been implicated in a wide variety of diseases which have components of immune dysfunction, including ASD, diabetes, obesity, and inflammatory diseases, insight into this metabolic modulator may have wide applications for both health and disease.
Introduction
The human microbiome represents a diverse ecosystem of microbes housed in the human body. Microbial cells outnumber the cells in the human body by a factor of 10 and microbial genes out number human genes by a factor of over 100 (1–3). There is a particular focus on the enteric (gut) microbiota since it represents about 99% of the human microbiome (4). The importance of the enteric microbiome in relation to human health and disease has been recognized since it appears to influence the immune system (5), metabolic processes (6), gene expression (7, 8), the nervous system (9, 10), and behavior (9, 10). Disruption of the enteric microbiome has been implicated in a wide range of human diseases including depression and anxiety (11), gastrointestinal disorders (12), inflammatory airway disease (13), diabetes (14–16), obesity (17, 18), atopic disease (5), and neurodegenerative conditions (19). The enteric microbiome may be particularly important early in life around the time of birth as it has been linked to early brain development and behavior (9, 10, 20) and disruption and/or treatments (i.e. early antibiotics) early in life can influence the development of childhood diseases, particularly atopic disease (9, 10).
The mechanism in which the enteric microbiome modulates particular effects on the host is not completely clear, although several mediators are potential vehicles for such influence. Such mediators include lipopolysaccharides, peptidoglycans, short-chain fatty acids (SCFAs), neurotransmitters and gaseous molecules (21–23). We are particularly interested in SCFAs because of their role as both mediators of physiology and mitochondrial fuels. SCFA are particularly intriguing as they are derived as a consequence of fermenting carbohydrates and some proteins, and also present naturally or as an additive in many foods, in particular wheat and dairy. Thus, dietary variations can have a larger influence on their production (19, 24, 25). Of the SCFAs, propionic acid (PPA) has been of key interest because it has several links to autism spectrum disorder (ASD), a disorder which affects as many as ~2% of children in the United States. What is intriguing about ASD is that the etiology is largely unknown but is strongly influenced by both genetic and environmental factors (26, 27).
The enteric microbiome is a major environmental factor that may contribute to the etiology of ASD (2, 9, 10, 28). First, several factors which may have a direct effect on health through disruption of the microbiome are associated with increased risk of developing ASD, including dietary alteration, environmental exposures that disrupt enteric microbiome bacteria content and diversity, being born by C-section delivery which reduces maternal transfer of enteric and vaginal bacteria, increased antibiotic use which can destroy key bacteria in the enteric microbiome, formula feeding and early hospitalization (2, 9, 28). Second, specific bacteria, such as Clostridia spp., a major SCFA producer, have been repeatedly reported to be overrepresented in the ASD microbiome (29, 30). Third, exposure to PPA has been demonstrated in several animal models to result in the development of ASD-like behaviors and physiological changes to the brain similar to those found in ASD are seen in adult rats acutely exposed to PPA (24, 25, 31) and in juvenile rats systematically exposed to PPA pre- and postnatally (32–34).
Although the mechanism by which PPA influences host function is still unclear, data from the animal model of PPA induced ASD demonstrates neuroinflammation and electrophysiological disturbances as well as disruptions in lipid, mitochondrial and redox metabolism (24, 25, 31). We have performed a series of studies to demonstrate that changes in mitochondrial metabolism similar to those found in the animal model exposed to PPA are also found in humans. For example, we found that the unique pattern of biomarkers of mitochondrial dysfunction found in the PPA rodent model was also found in a subset of children with ASD (28, 35, 36). We also demonstrated that PPA modulates mitochondrial respiration in lymphoblastoid cell lines (LCLs) derived from children with ASD differently than LCLs derived from age and gender matched typically developing control LCLs (37).
PPA also could induce changes in host physiology through modulation of the immune system. The animal models of PPA induced ASD behavior demonstrates neuroinflammation but inflammatory mediators induced by PPA in human ASD cells has not been investigated. In this study, we investigate whether PPA can differentially regulate immune genes using our LCL model of ASD. We have developed a cell line model of ASD in which LCLs derived from individuals with autistic disorder (AD) are classified into two groups: those with normal mitochondrial function (AD-N) and those with atypical mitochondrial function (AD-A) (38–40). The AD-A LCLs have respiratory rates approximately twice that of control and AD-N LCLs and are very sensitive to in vitro increases in reactive oxygen species (ROS) (38–40). We recently demonstrated that this atypical increase in mitochondrial function characteristic of AD-A LCLs was associated with more severe repetitive behaviors in the children from which these LCLs were derived (40). In this way, we believe that the AD-A LCLs may represent a more severe ASD phenotype. Given the connection between metabolism and immune system (41), we hypothesize that the AD-A LCLs will demonstrate a greater activation of immune genes with PPA exposure as compared to the control and AD-N LCLs.
Materials and Methods
LCLs and Culture Conditions
Lymphoblastoid cell lines were derived from white males diagnosed with AD chosen from pedigrees with at least other 1 affected male sibling (i.e., multiplex family) [mean (SD) age 7.3 (3.5) years]. These LCLs were obtained from the Autism Genetic Resource Exchange (Los Angeles, CA, USA) or the National Institutes of Mental Health (Bethesda, MD, USA) center for collaborative genomic studies on mental disorders. In our previous studies (37, 39, 40, 42–44), these LCLs where categorized into two different types of AD LCLs; ones with atypical mitochondrial respiration (AD-A) and those with normal respiration (AD-N). These metabolic groupings have been shown to be consistent and repeatable in our previous studies (37, 39, 40, 42–44). Eight pairs of AD-N and AD-A LCLs were age and gender matched to control LCLs. The sample size chosen was based on our previous studies. Control (CNT) LCLs were derived from healthy white male donors with no documented behavioral or neurological disorder and with no first degree relative suffering from any medical disorder that might involve mitochondrial dysfunction [mean (SD) age 7.5 (3.3) years]. CNT LCLs were obtained from Coriell Cell Repository (Camden, NJ, USA). Due to low availability of CNT LCLs which fit our criteria, a single CNT LCL line was paired with two AD LCL lines in one case (see Table 1). Also two AD-A LCLs were paired twice with AD-N LCLs. On average, cells were studied at passage 12, with a maximum passage of 15. Genomic stability is very high at this low passage number (45, 46). Cells were maintained in RPMI 1640 culture medium with 15% FBS and 1% penicillin/streptomycin (Invitrogen, Grand Island, NY, USA) in a humidified incubator at 37°C with 5% CO2.
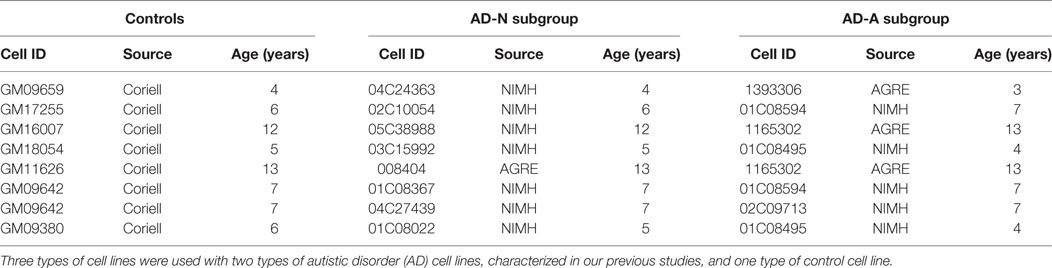
Table 1. Lymphoblastoid cell lines used in this study.
PPA Exposure
Each group of LCLs were cultured with PPA 1 mM for 24 or 48 h or left untreated (0 mM). This concentration was selected because it provided optimal metabolic activation in our previous studies (37). The sodium propionate was buffered with sodium bicarbonate in the culture medium to prevent changes in pH which could cause changes in influx of PPA (47). As PPA is mostly disassociated at physiological pH, the effects of the PPA treatment are most likely a combination of both PPA and propionate.
Expression Studies
Total RNA samples from each LCL group were pooled together and after DNase treatment and purified using RNeasy Mini Kit (Qiagen Sciences, MD, USA) as described in our previous studies (48). The cDNA synthesis and microarray analyses were performed at Keck Affymetrix GeneChip Resource at Yale, New Haven, CT, USA (NIH Neuroscience Microarray Consortium) as previously described (48).
Analytic Approach
Analysis of variance was conducted between the exposure conditions and different cell types. Genes showing expression of at least ≥2.0-fold were exported for functional annotation to several pathway analysis packages including Ingenuity Pathway Analysis (IPA) and Panther software. For the initial comparison of the effect of PPA for each exposure time on a particular LCL type, the statistical significance of the comparison was not considered as there was only an N of 1 for each example. When the ASD LCL types were compared to controls, the two PPA exposure times were combined and the genes selected not only showed a difference in expression of at least ≥2.0-fold but also a p < 0.05.
Results
The Effect of PPA on Gene Expression for Each LCL Type
The change in gene expression resulting from 1 mM exposure to PPA for 24 and 48 h was determined for each LCL type separately. Table S1 in Supplementary Material demonstrates the number of genes up- and downregulated more than 2.0-fold for each LCL type.
The CNT LCLs demonstrated no upregulation or downregulation of known genes with 24 h PPA exposure and only one gene upregulated and downregulated with 48 h PPA exposure. Only the downregulated gene was associated with immune function. Panther analysis demonstrated no overrepresentation of immune genes associated with PPA exposure in CNT LCLs.
Exposure of AD-N LCLs to PPA for 24 h demonstrated no upregulated genes and downregulation of several immune genes including two major histocompatibility complex genes. Exposure of AD-N LCLs to PPA for 48 h demonstrated upregulation of two microRNA genes not known to be involved in immune function and downregulation of the gene for complement C4B. Panther analysis demonstrated overrepresentation of genes associated with major histocompatibility complex antigen with 24 h PPA exposure in AD-N LCLs (see Table 2).
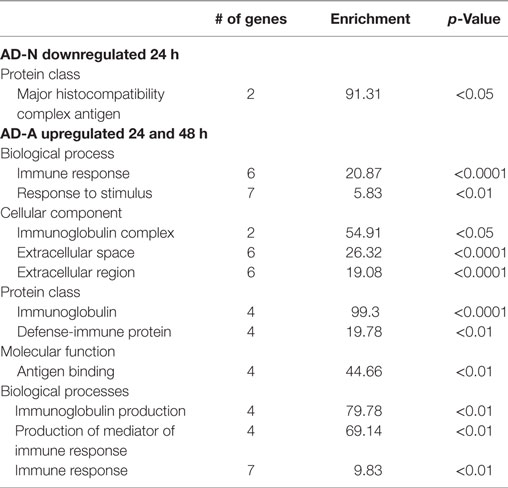
Table 2. Panther overrepresentation analysis of genes significantly upregulated and downregulated with 24 and 48 h PPA exposure.
Exposure of AD-A LCLs to PPA for 24 or 48 h demonstrated upregulation of several genes related to immune function, particularly several genes associated with immunoglobulin production and one gene related to activation of proinflammatory caspases. Downregulation of the gene for complement C4B was found for 24 h exposure and no genes were downregulated for 48 h exposure. Panther analysis demonstrated overrepresentation of many immune processes and proteins as result of PPA exposure to AD-A LCLs for 24 and 48 h, demonstrating that PPA did significantly activate immune processes for AD-A LCLs (Table 2).
Comparison of PPA Effect on ASD LCLs as Compared to Control LCLs
To better understand how PPA exposure affects ASD LCLs differently than control LCLs, gene expression was compared between CNT LCLs and each ASD LCL group independently. Both the 24- and 48-h PPA exposure data was combined since the previous analysis demonstrated little difference between the changes in gene expression with these two different exposure durations. Table S2 in Supplementary Material outlines the genes that were upregulated or downregulated with PPA exposure for each ASD LCL group as compared to CNT LCLs. Table 3 demonstrates the biological processes identified by the differential gene expression for AD-N and AD-A LCLs as compared to CNT LCLs. The major processes identified are also represented in Figure 1. Biological process was the only Panther analysis used as it was the most robust for representing the difference in pathway activation.
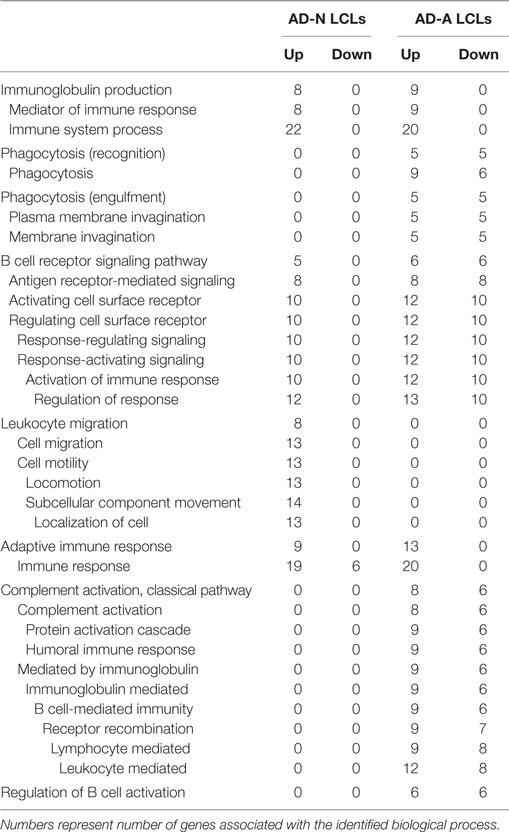
Table 3. Biological processes panther overrepresentation analysis of genes differentially expressed in autism cell lines as compared to control cell lines.
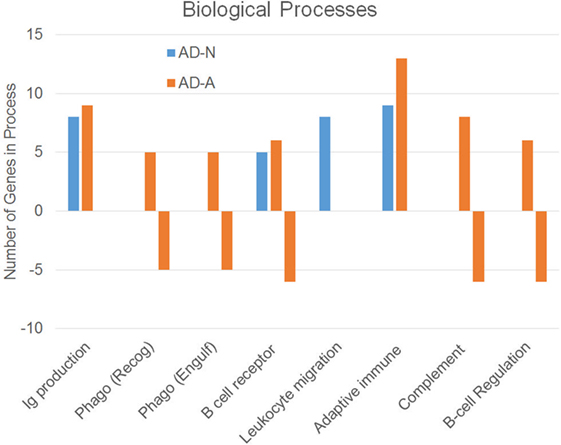
Figure 1. Biological processes associated with an increase or decrease in gene expression resulting from propionic acid exposure to autism cell lines as compared to control cell lines.
This analysis suggests that both the AD-N and AD-A LCLs demonstrate change in immune genes as compared to CNT LCLs. Both AD-A and AD-N LCLs demonstrate an upregulation in genes associated with immunoglobulin production and adaptive immune responses without any downregulation in genes involved in these processes. AD-A LCLs demonstrate both upregulation and downregulation of genes involved in a wider variety of immune responses as compared to AD-N LCLs, including phagocytosis, complement system activation, B cell regulation, and B cell receptors. This suggests that AD-A LCLs may have a wider network of immune genes activated as compared to AD-N LCLs as well as CNT LCLs.
Table 4 represents the top canonical pathways (p < 0.01) identified by IPA for the comparison between the AD-A and CNT LCLs. As we see, many of these processes are involved in immune activation and immune disorders. IPA also identified the top upstream regulators as RUNX3, ONECUT1, SNAI2, STAT5A, and TCF7. Interestingly, as will be discussed below, these genes are regulatory of both developmental and immune processes.

Table 4. Top canonical pathways identified using Ingenuity Pathway Analysis (IPA).
Discussion
In this study, we examined the effect of PPA, a SCFA produced by enteric bacteria that are overrepresented in the ASD gut, on transformed B cells (LCLs) derived from children with ASD as well as controls. We examined two types of LCLs derived from children with ASD, those with mitochondrial dysfunction (AD-A) and those found to have mitochondrial function similar to controls (AD-N). We hypothesized that PPA would activate immune pathways in ASD LCLs since the PPA animal model of ASD demonstrates neuroinflammation and immune activation, including increased GFAP immunoreactivity in the hippocampus, increased activation of microglia, and increased interleukin (IL)-6 (24, 25, 31). We further hypothesized that the AD-A LCLs would have a greater enhancement of immune pathways since this is a more severe ASD phenotype and since optimal mitochondrial function is required for appropriate immune function and response (41).
Exposure to PPA for either 24 or 48 h resulted in upregulation in genes associated with immune system activation in AD-A LCLs, particularly genes involved in immunoglobulin production. This effect was not seen in CNT or AD-N LCLs. In fact, there was a decrease in major histocompatibility complex antigen genes in AD-N LCLs exposed to PPA for 24 h. We then compared the effect of PPA on ASD LCLs as compared to the effect of PPA on CNT LCLs. We found that both the AD-N and AD-A LCLs demonstrated changes in gene expression as compared to the control LCLs with a significant change in genes related to immune pathways almost exclusively. Although the AD-N LCLs demonstrated activation of immune pathways, the AD-A LCLs demonstrated a wider range of genes and processes involved in immune pathways. In addition, IPA analysis of AD-A LCL gene expression changes identified canonical pathways almost exclusively related to immune function.
Several of the genes identified by the IPA analysis are involved in regulation of the immune system and may be linked to ASD. Several genes are linked to regulation of T cells. TCF7 is a T lymphocyte-specific enhancer of the CD3-Epsilon T cell antigen receptor complex. Interestingly TCF7 expression may be regulated by beta-catenin (49). This is intriguing since beta-catenin has been shown to be dysregulated in an animal model of ASD (50). STAT5 is induced in response to T cell activation with cytokines, most notably IL-2, and is believed to be involved in the effect of IL-2 in the immune response and may be involved in the suppression of IL-3 production. This is interesting as IL-2 is produced by neurons and astrocytes, is important in brain development and normal brain physiology and has been implicated in neurodegenerative disease, cognitive dysfunction and has been linked to ASD (51). RUNX3 is also important in immune system function as well as neuronal development. RUNX3 is essential during thymopoiesis where it modulates the development of CD8 T cells, thus having an important role in immune system development through lineage specification (52). Interestingly, RUNX3 is involved in the TNF-beta signaling cascade (53), a cytokine whose dysregulation has been correlated with ASD severity (51). RUNX3 appears to have an important role in the development of proprioceptive afferent neurons in mice, resulting in ataxia (54), a neurological finding that is not uncommon in ASD. Other genes identified are related to B cell function. SNAI2 is an evolutionarily conserved zinc finger transcription factor which plays an important role in prenatal fetal development, most notably the development of neural crest-derived cells and adipocytes (55). SNAI2 is also involved in regulation of B cells and can promote the aberrant survival and malignant transformation of mammalian pro-B cells otherwise slated for apoptotic death (56) and has antiapoptotic effects (57).
In conclusion, ASD is being recognized as having a very strong immune component to its etiology (58). Several models of ASD demonstrate immune dysregulation, including prenatal exposure to immune challenges (59, 60). In fact two animal models have been developed to parallel prenatal exposure to autoantibodies (61), including fetal brain antibodies (62) and antibodies to the folate transporter (63, 64). The microbiome is being recognized as important in the etiology of neurodevelopmental disorders (9, 10), potentially through modulation of the immune system (65) through enteric metabolites (65) including SCFAs like PPA (24, 25, 31). It is important to note the effects of SCFA on gene expression and inflammation are complex, and include histone deacetylase activity, activation of free fatty acid G-coupled receptor and mitochondrial inflammatory signaling cascades, which may or may not be mutually reinforcing. Furthermore, we do not yet know if the effects found in our LCL model also occur in patients, as many effects of SCFA, in particular PPA and butyrate, are dose and tissue dependent, and have different effects at key developmental time periods (9, 10, 24, 31, 48, 66, 67). Nonetheless, this study provides insight into the mechanism in which the microbiome may influence the immune system to result in disease and demonstrates the predisposition of certain cells to be sensitive to microbiome metabolites. It also may lead to further reevaluation of the widespread use of PPA in agriculture and the food industry (24, 31). Certainly, further research is needed in this area to better define the role of the microbiome and microbial metabolites in immune modulation and disease.
Author Contributions
The conception and design of the work was agreed upon by all authors as was the drafting and final approval of the manuscript. BN, SR, and SB were involved in laboratory analysis. SB was involved in data analysis. RF, DM, SB, SR, and SB were involved in interpretation of data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the autism families that participated in the Autism Genetic Research Exchange and the studies at the National Institutes of Mental Health. We would also like to express our utmost thanks to David Patchell-Evans, for his tireless devotion to persons with autism, and his daughter, Kilee Patchell-Evans. Our heartfelt thanks go out to countless parents and caregivers of persons with autism who have shared their stories.
Funding
This research was supported by the Arkansas Biosciences Institute (Little Rock, AR, USA), The Jonty Foundation (St Paul, MN), The Autism Research Institute (San Diego, CA), the Gupta Family Foundation (Atherton, CA) and the Jager Family Foundation (Chicago, IL) to REF, and GoodLife Children’s Charities, Autism Canada and Autism Research Institute to DFM.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01670/full#supplementary-material.
References
1. Rosenfeld CS. Microbiome disturbances and autism spectrum disorders. Drug Metab Dispos (2015) 43(10):1557–71. doi:10.1124/dmd.115.063826
2. Frye RE, Slattery J, MacFabe DF, Allen-Vercoe E, Parker W, Rodakis J, et al. Approaches to studying and manipulating the enteric microbiome to improve autism symptoms. Microb Ecol Health Dis (2015) 26:26878. doi:10.3402/mehd.v26.26878
3. Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol (2009) 587(Pt 17):4153–8. doi:10.1113/jphysiol.2009.174136
4. Naviaux RK. Metabolic features of the cell danger response. Mitochondrion (2014) 16:7–17. doi:10.1016/j.mito.2013.08.006
5. Johnson CC, Ownby DR. Allergies and asthma: do atopic disorders result from inadequate immune homeostasis arising from infant gut dysbiosis? Expert Rev Clin Immunol (2016) 12(4):379–88. doi:10.1586/1744666X.2016.1139452
6. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science (2012) 336(6086):1262–7. doi:10.1126/science.1223813
7. Cureau N, AlJahdali N, Vo N, Carbonero F. Epigenetic mechanisms in microbial members of the human microbiota: current knowledge and perspectives. Epigenomics (2016) 8(9):1259–73. doi:10.2217/epi-2016-0057
8. Woo V, Alenghat T. Host-microbiota interactions: epigenomic regulation. Curr Opin Immunol (2017) 44:52–60. doi:10.1016/j.coi.2016.12.001
9. Slattery J, MacFabe DF, Frye RE. The significance of the enteric microbiome on the development of childhood disease: a review of prebiotic and probiotic therapies in disorders of childhood. Clin Med Insights Pediatr (2016) 10:91–107. doi:10.4137/CMPed.S38338
10. Slattery J, MacFabe DF, Kahler SG, Frye RE. Enteric ecosystem disruption in autism spectrum disorder: can the microbiota and macrobiota be restored? Curr Pharm Des (2016) 22(40):6107–21. doi:10.2174/1381612822666160905123953
11. Borre YE, Moloney RD, Clarke G, Dinan TG, Cryan JF. The impact of microbiota on brain and behavior: mechanisms & therapeutic potential. Adv Exp Med Biol (2014) 817:373–403. doi:10.1007/978-1-4939-0897-4_17
12. Bull MJ, Plummer NT. Part 2: treatments for chronic gastrointestinal disease and gut dysbiosis. Integr Med (Encinitas) (2015) 14(1):25–33.
13. Gollwitzer ES, Marsland BJ. Microbiota abnormalities in inflammatory airway diseases – potential for therapy. Pharmacol Ther (2014) 141(1):32–9. doi:10.1016/j.pharmthera.2013.08.002
14. Palacios T, Vitetta L, Coulson S, Madigan CD, Denyer GS, Caterson ID. The effect of a novel probiotic on metabolic biomarkers in adults with prediabetes and recently diagnosed type 2 diabetes mellitus: study protocol for a randomized controlled trial. Trials (2017) 18(1):7. doi:10.1186/s13063-016-1762-x
15. Paun A, Yau C, Danska JS. The influence of the microbiome on type 1 diabetes. J Immunol (2017) 198(2):590–5. doi:10.4049/jimmunol.1601519
16. Wegielska I, Suliburska J. The role of intestinal microbiota in the pathogenesis of metabolic diseases. Acta Sci Pol Technol Aliment (2016) 15(2):201–11. doi:10.17306/J.AFS.2016.2.20
17. Bischoff SC, Boirie Y, Cederholm T, Chourdakis M, Cuerda C, Delzenne NM, et al. Towards a multidisciplinary approach to understand and manage obesity and related diseases. Clin Nutr (2017) 36(4):917–38. doi:10.1016/j.clnu.2016.11.007
18. Fandriks L. Roles of the gut in the metabolic syndrome: an overview. J Intern Med (2017) 281(4):319–36. doi:10.1111/joim.12584
19. Bourassa MW, Alim I, Bultman SJ, Ratan RR. Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci Lett (2016) 625:56–63. doi:10.1016/j.neulet.2016.02.009
20. Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A (2011) 108(7):3047–52. doi:10.1073/pnas.1010529108
21. Hersoug LG, Moller P, Loft S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: implications for inflammation and obesity. Obes Rev (2016) 17(4):297–312. doi:10.1111/obr.12370
22. Dworkin J. The medium is the message: interspecies and interkingdom signaling by peptidoglycan and related bacterial glycans. Annu Rev Microbiol (2014) 68:137–54. doi:10.1146/annurev-micro-091213-112844
23. Pimentel M, Mathur R, Chang C. Gas and the microbiome. Curr Gastroenterol Rep (2013) 15(12):356. doi:10.1007/s11894-013-0356-y
24. Macfabe D. Autism: metabolism, mitochondria, and the microbiome. Global advances in health and medicine: improving healthcare outcomes worldwide. Glob Adv Health Med (2013) 2(6):52–66. doi:10.7453/gahmj.2013.089
25. Macfabe DF. Short-chain fatty acid fermentation products of the gut microbiome: implications in autism spectrum disorders. Microb Ecol Health Dis (2012):23. doi:10.3402/mehd.v23i0.19260
26. Frye RE, Rossignol DA. Identification and treatment of pathophysiological comorbidities of autism spectrum disorder to achieve optimal outcomes. Clin Med Insights Pediatr (2016) 10:43–56. doi:10.4137/CMPed.S38337
27. Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry (2012) 17(4):389–401. doi:10.1038/mp.2011.165
28. Frye RE, Rose S, Slattery J, MacFabe DF. Gastrointestinal dysfunction in autism spectrum disorder: the role of the mitochondria and the enteric microbiome. Microb Ecol Health Dis (2015) 26:27458. doi:10.3402/mehd.v26.27458
29. Williams BL, Hornig M, Buie T, Bauman ML, Cho Paik M, Wick I, et al. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One (2011) 6(9):e24585. doi:10.1371/journal.pone.0024585
30. Finegold SM, Molitoris D, Song Y, Liu C, Vaisanen ML, Bolte E, et al. Gastrointestinal microflora studies in late-onset autism. Clin Infect Dis (2002) 35(Suppl 1):S6–16. doi:10.1086/341914
31. MacFabe DF. Enteric short-chain fatty acids: microbial messengers of metabolism, mitochondria, and mind: implications in autism spectrum disorders. Microb Ecol Health Dis (2015) 26:28177. doi:10.3402/mehd.v26.28177
32. Foley KA, MacFabe DF, Kavaliers M, Ossenkopp KP. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: relevance to autism spectrum disorders. Behav Brain Res (2015) 278:244–56. doi:10.1016/j.bbr.2014.09.032
33. Foley KA, MacFabe DF, Vaz A, Ossenkopp KP, Kavaliers M. Sexually dimorphic effects of prenatal exposure to propionic acid and lipopolysaccharide on social behavior in neonatal, adolescent, and adult rats: implications for autism spectrum disorders. Int J Dev Neurosci (2014) 39:68–78. doi:10.1016/j.ijdevneu.2014.04.001
34. Foley KA, Ossenkopp KP, Kavaliers M, Macfabe DF. Pre- and neonatal exposure to lipopolysaccharide or the enteric metabolite, propionic acid, alters development and behavior in adolescent rats in a sexually dimorphic manner. PLoS One (2014) 9(1):e87072. doi:10.1371/journal.pone.0087072
35. Frye RE, Melnyk S, Macfabe DF. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl Psychiatry (2013) 3:e220. doi:10.1038/tp.2012.143
36. Frye RE. Biomarkers of abnormal energy metabolism in children with autism spectrum disorder. N A J Med Sci (2012) 5(3):141–7. doi:10.7156/v5i3p141
37. Frye RE, Rose S, Chacko J, Wynne R, Bennuri SC, Slattery JC, et al. Modulation of mitochondrial function by the microbiome metabolite propionic acid in autism and control cell lines. Transl Psychiatry (2016) 6(10):e927. doi:10.1038/tp.2016.189
38. Rose S, Melnyk S, Pavliv O, Bai S, Nick TG, Frye RE, et al. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl Psychiatry (2012) 2:e134. doi:10.1038/tp.2012.61
39. Rose S, Frye RE, Slattery J, Wynne R, Tippett M, Pavliv O, et al. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS One (2014) 9(1):e85436. doi:10.1371/journal.pone.0085436
40. Rose S, Bennuri SC, Wynne R, Melnyk S, James SJ, Frye RE. Mitochondrial and redox abnormalities in autism lymphoblastoid cells: a sibling control study. FASEB J (2017) 31(3):904–9. doi:10.1096/fj.201601004R
41. Mehta MM, Weinberg SE, Chandel NS. Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol (2017) 17(10):608–20. doi:10.1038/nri.2017.66
42. Rose S, Wynne R, Frye RE, Melnyk S, James SJ. Increased susceptibility to ethylmercury-induced mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines. J Toxicol (2015) 2015:573701. doi:10.1155/2015/573701
43. Rose S, Frye RE, Slattery J, Wynne R, Tippett M, Melnyk S, et al. Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines. Transl Psychiatry (2014) 4:e377. doi:10.1038/tp.2014.15
44. Frye RE, Rose S, Wynne R, Bennuri SC, Blossom S, Gilbert KM, et al. Oxidative stress challenge uncovers trichloroacetaldehyde hydrate-induced mitoplasticity in autistic and control lymphoblastoid cell lines. Sci Rep (2017) 7(1):4478. doi:10.1038/s41598-017-04821-3
45. Oh JH, Kim YJ, Moon S, Nam HY, Jeon JP, Lee JH, et al. Genotype instability during long-term subculture of lymphoblastoid cell lines. J Hum Genet (2013) 58(1):16–20. doi:10.1038/jhg.2012.123
46. Nickles D, Madireddy L, Yang S, Khankhanian P, Lincoln S, Hauser SL, et al. In depth comparison of an individual’s DNA and its lymphoblastoid cell line using whole genome sequencing. BMC Genomics (2012) 13:477. doi:10.1186/1471-2164-13-477
47. Karuri AR, Dobrowsky E, Tannock IF. Selective cellular acidification and toxicity of weak organic acids in an acidic microenvironment. Br J Cancer (1993) 68(6):1080–7. doi:10.1038/bjc.1993.485
48. Nankova BB, Agarwal R, MacFabe DF, La Gamma EF. Enteric bacterial metabolites propionic and butyric acid modulate gene expression, including CREB-dependent catecholaminergic neurotransmission, in PC12 cells – possible relevance to autism spectrum disorders. PLoS One (2014) 9(8):e103740. doi:10.1371/journal.pone.0103740
49. Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, et al. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science (1999) 285(5435):1923–6. doi:10.1126/science.285.5435.1923
50. Mohn JL, Alexander J, Pirone A, Palka CD, Lee SY, Mebane L, et al. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry (2014) 19(10):1133–42. doi:10.1038/mp.2014.61
51. Xu N, Li X, Zhong Y. Inflammatory cytokines: potential biomarkers of immunologic dysfunction in autism spectrum disorders. Mediators Inflamm (2015) 2015:531518. doi:10.1155/2015/531518
52. Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell (2002) 111(5):621–33. doi:10.1016/S0092-8674(02)01111-X
53. Fainaru O, Woolf E, Lotem J, Yarmus M, Brenner O, Goldenberg D, et al. Runx3 regulates mouse TGF-beta-mediated dendritic cell function and its absence results in airway inflammation. EMBO J (2004) 23(4):969–79. doi:10.1038/sj.emboj.7600085
54. Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N, et al. Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nat Neurosci (2002) 5(10):946–54. doi:10.1038/nn925
55. Perez-Mancera PA, Bermejo-Rodriguez C, Gonzalez-Herrero I, Herranz M, Flores T, Jimenez R, et al. Adipose tissue mass is modulated by SLUG (SNAI2). Hum Mol Genet (2007) 16(23):2972–86. doi:10.1093/hmg/ddm278
56. Inukai T, Inoue A, Kurosawa H, Goi K, Shinjyo T, Ozawa K, et al. SLUG, a ces-1-related zinc finger transcription factor gene with antiapoptotic activity, is a downstream target of the E2A-HLF oncoprotein. Mol Cell (1999) 4(3):343–52. doi:10.1016/S1097-2765(00)80336-6
57. Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell (2005) 123(4):641–53. doi:10.1016/j.cell.2005.09.029
58. Masi A, Glozier N, Dale R, Guastella AJ. The immune system, cytokines, and biomarkers in autism spectrum disorder. Neurosci Bull (2017) 33(2):194–204. doi:10.1007/s12264-017-0103-8
59. Bilbo SD, Block CL, Bolton JL, Hanamsagar R, Tran PK. Beyond infection – maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp Neurol (2017). doi:10.1016/j.expneurol.2017.07.002
60. Varghese M, Keshav N, Jacot-Descombes S, Warda T, Wicinski B, Dickstein DL, et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol (2017) 134(4):537–66. doi:10.1007/s00401-017-1736-4
61. Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry (2017) 81(5):391–401. doi:10.1016/j.biopsych.2016.10.020
62. Matelski L, Van de Water J. Risk factors in autism: thinking outside the brain. J Autoimmun (2016) 67:1–7. doi:10.1016/j.jaut.2015.11.003
63. Desai A, Sequeira JM, Quadros EV. Prevention of behavioral deficits in rats exposed to folate receptor antibodies: implication in autism. Mol Psychiatry (2017) 22(9):1291–7. doi:10.1038/mp.2016.153
64. Sequeira JM, Desai A, Berrocal-Zaragoza MI, Murphy MM, Fernandez-Ballart JD, Quadros EV. Exposure to folate receptor alpha antibodies during gestation and weaning leads to severe behavioral deficits in rats: a Pilot study. PLoS One (2016) 11(3):e0152249. doi:10.1371/journal.pone.0152249
65. Morris G, Berk M, Carvalho A, Caso JR, Sanz Y, Walder K, et al. The role of the microbial metabolites including tryptophan catabolites and short chain fatty acids in the pathophysiology of immune-inflammatory and neuroimmune disease. Mol Neurobiol (2017) 54(6):4432–51. doi:10.1007/s12035-016-0004-2
66. Alvarez-Curto E, Milligan G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol (2016) 114:3–13. doi:10.1016/j.bcp.2016.03.017
Keywords: mitochondrial disease, autism, propionic acid, short-chain fatty acids, microbiome, inflammation, epigenetics, histone deacetylase inhibitor
Citation: Frye RE, Nankova B, Bhattacharyya S, Rose S, Bennuri SC and MacFabe DF (2017) Modulation of Immunological Pathways in Autistic and Neurotypical Lymphoblastoid Cell Lines by the Enteric Microbiome Metabolite Propionic Acid. Front. Immunol. 8:1670. doi: 10.3389/fimmu.2017.01670
Received: 31 July 2017; Accepted: 14 November 2017;
Published: 22 December 2017
Edited by:
Gayane Manukyan, Institute of Molecular Biology (NAS RA), ArmeniaReviewed by:
William Parker, Duke University, United StatesJoao Luiz Mendes Wanderley, Universidade Federal do Rio de Janeiro, Brazil
Copyright: © 2017 Frye, Nankova, Bhattacharyya, Rose, Bennuri and MacFabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard E. Frye, cmZyeWVAcGhvZW5peGNoaWxkcmVucy5jb20=