 Jo Mailleux1
Jo Mailleux1 Silke Timmermans1Katherine Nelissen1Jasmine Vanmol1Tim Vanmierlo1
Silke Timmermans1Katherine Nelissen1Jasmine Vanmol1Tim Vanmierlo1 Jack van Horssen2
Jack van Horssen2 Jeroen F. J. Bogie1
Jeroen F. J. Bogie1 Jerome J. A. Hendriks1*
Jerome J. A. Hendriks1*
- 1Biomedical Research Institute, Hasselt University, Diepenbeek, Belgium
- 2Molecular Cell Biology and Immunology, VU University Medical Center, Amsterdam, Netherlands
Objective: We aimed to determine the role of the low-density lipoprotein receptor (LDLr) in neuroinflammation by inducing experimental autoimmune encephalomyelitis (EAE) in ldlr knock out mice.
Methods: MOG35–55 induced EAE in male and female ldlr−/− mice was assessed clinically and histopathologically. Expression of inflammatory mediators and apolipoprotein E (apoE) was investigated by qPCR. Changes in protein levels of apoE and tumor necrosis factor alpha (TNFα) were validated by western blot and ELISA, respectively.
Results: Ldlr−/−-attenuated EAE disease severity in female, but not in male, EAE mice marked by a reduced proinflammatory cytokine production in the central nervous system of female ldlr−/− mice. Macrophages from female ldlr−/− mice showed a similar decrease in proinflammatory mediators, an impaired capacity to phagocytose myelin and enhanced secretion of the anti-inflammatory apoE. Interestingly, apoE/ldlr double knock out abrogated the beneficial effect of ldlr depletion in EAE.
Conclusion: Collectively, we show that ldlr−/− reduces EAE disease severity in female but not in male EAE mice, and that this can be explained by increased levels of apoE in female ldlr−/− mice. Although the reason for the observed sexual dimorphism remains unclear, our findings show that LDLr and associated apoE levels are involved in neuroinflammatory processes.
Introduction
Multiple sclerosis (MS) is an inflammatory autoimmune demyelinating disease of the central nervous system (CNS). Infiltrated macrophages and resident microglia are considered the dominant effector cells in MS and its animal model, experimental autoimmune encephalomyelitis (EAE) (1, 2). Effector mechanisms include phagocytosis of myelin and the secretion of inflammatory and toxic mediators (3–7). However, increasing evidence indicates that phagocytes can also acquire a disease-resolving phenotype in the CNS. For instance, we recently defined that ingestion of myelin by macrophages alters the inflammatory phenotype by activating the cholesterol- and fatty acid-sensing nuclear liver X receptors and peroxisome proliferator-activated receptors (8–10). These studies stress the pleiotropic role that phagocytes play in the pathophysiology of MS and indicate that lipid-signaling pathways drive the phenotype of phagocytes in MS lesions.
The impact of cholesterol and its metabolites on the pathophysiology of MS is a topic of interest in MS-research (11–16). In the MS brain, marked alterations in both myelin cholesterol and other lipid metabolites are found (13). Furthermore, plasma and cerebrospinal fluid (oxy)sterol levels are disturbed and closely correlate to neurodegenerative processes (14–17). In addition, we showed that lipoprotein levels and function are altered in relapsing-remitting MS patients (RRMS), which may advance disease progression in these patients (18). More specifically, we found smaller low-density lipoprotein (LDL) particles in RRMS patients compared to healthy controls. Interestingly, the average LDL particle size was smaller in male RRMS patients compared to female RRMS patients, which suggests gender differences in lipid metabolism in MS. LDL size is described to be important for LDL function indicating the lipoprotein may be involved in the pathophysiology of disease. Smaller particles have an increased susceptibility to oxidation and a decreased LDL receptor (LDLr) affinity, which may promote their proinflammatory properties (19–21).
The LDLr is a 160 kDa cell surface glycoprotein that plays a key role in plasma cholesterol homeostasis (22). It binds two physiologically important ligands, apolipoprotein B-100 (apoB) and apolipoprotein E (apoE). The only known ligand for the LDLr in the CNS is apoE since apoB is not synthesized in the CNS and, in contrast to apoE, cannot cross the blood–brain barrier (22). Malfunctioning of the receptor in humans results in familial hypercholesterolemia (FH) and is an important risk factor for cardiovascular disease (23). FH is caused by genetic mutations that directly or indirectly affect the function of the LDLr. It is characterized by defective catabolism of LDL, which results in increased plasma cholesterol concentrations and lipid accumulation in macrophages and other immune cells. This promotes proinflammatory responses, including enhanced NF-κB signaling, inflammasome activation, and increased production of neutrophils and monocytes in the bone marrow and spleen (24–28). The ability of lipoproteins to modulate neuroinflammation has been suggested in recent studies (29, 30). In addition, elevated LDL plasma levels were found to increase neurotoxicity in a mouse model for Alzheimer’s disease (30). The above findings indicate that lipoproteins modulate neuroinflammation and neurodegeneration. However, the direct contribution of LDL and LDLr to these processes remains unclear.
Here, we determined the role of the LDLr in neuroinflammation by using the EAE model and ldlr knock out (ldlr−/−) mice. We show that ldlr deficiency reduces disease severity in female, but not in male EAE mice. In line with this finding, the inflammatory burden was significantly lower in the CNS of female ldlr−/− mice compared to ldlr−/− male mice. Macrophages isolated from female ldlr−/− mice show an altered inflammatory profile and an impaired capacity to phagocytose myelin. Interestingly, apoE release was enhanced in macrophages derived from females and the beneficial effect of ldlr deficiency in EAE was absent in apoE/ldlr double knock out mice. This shows that elevated apoE levels likely attenuate EAE severity in female ldlr−/− mice. Collectively, our findings indicate that changes in ldlr expression contribute to the progression of neuroinflammatory diseases in a gender-specific manner.
Materials and Methods
Animals
Ldlr−/− and apoE−/− mice on a C57BL/6 background and C57BL/6 wild-type (WT) mice were purchased from Harlan Laboratories. Animals were fed a regular diet and housed in the animal facility of the Biomedical Research Institute of Hasselt University. All experiments were performed according to institutional guidelines and were approved by the ethical committee for animal experiments of Hasselt University.
Induction of EAE
Eleven-week-old mice were immunized subcutaneously at the base of the tail with 200 µg of recombinant human myelin oligodendrocyte glycoprotein MOG35–55 emulsified in 100 µl complete Freund’s adjuvant supplemented with 4 mg/ml of Mycobacterium tuberculosis (H37RA strain) according to manufacturer’s guidelines (Hooke Laboratories, Lawrence, USA). Within 2 h and after 22–26 h, mice were intraperitoneally injected with 0.1 ml pertussis toxin. Immunized mice were weighed and scored daily by following a five-point standardized rating of clinical symptoms: 0, no signs; 1, loss of tail tonus; 2, flaccid tail; 3, hind limb paresis; 4, hind limb paralysis; 5, death. Mice were sacrificed at day 18 (peak) and at day 27 post immunization.
Peritoneal Macrophage Cultures
Mice were injected intraperitoneally with 1.5 ml phosphate-buffered saline (PBS, Sigma-Aldrich) supplemented with 5 mM ethylenediamine teraacetic acid (EDTA, VWR, Leuven, Belgium). Peritoneal cells were cultured for 3 h in RPMI-1640 medium (Lonza, Verviers, Belgium), enriched with 10% fetal calf serum (FCS; Hyclone, Erembodegem, Belgium), 0.5% penicillin/streptomycin (P/S Invitrogen, Merelbeke, Belgium), in a 24-well plate (5 × 104 cells/well) at 37°C with 5% CO2. Cells were stimulated for 18 h with 100 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich) to assess their gene expression. Medium was stored for protein measurement.
T Cell Proliferation
Spleen and inguinal lymph nodes (LN) were isolated from mice 9 days after EAE induction. T cells were isolated from LN and spleen and cultured in RPMI medium containing 20 µM β-mercaptoethanol, 2% mouse serum, 1% non-essential amino acids, 1% sodium pyruvate, 0.5% P/S. Cells were plated in a 96-well plate at a density of 3 × 105 cells/well and subsequently stimulated with 10 µg/ml MOG for 48 h. Next, 1μCi [3H] thymidine (Amersham biosciences, UK) was added for 18 h after which cells were collected using an automatic cell harvester (Pharmacia, Uppsala, Sweden). A β plate liquid scintillation counter (Perkin Elmer, lifesciences, Wellesly, MA, USA) was used to quantify radioactivity and results are expressed as stimulation index (SI). The SI shows the relative proliferation of MOG-stimulated T cells compared to non-stimulated T cells (SI = 1).
Quantitative PCR (qPCR)
RNA was extracted from tissue or cells using the RNeasy mini kit (Qiagen, Venlo, The Netherlands). In short, lysis was performed with Qiazol Lysis reagent (Qiagen) supplemented with 1% β-mercaptoethanol (Sigma-Aldrich). RNA concentration and quality were determined with a Nanodrop spectrophotometer (Isogen Life Science, The Netherlands). cDNA synthesis was conducted using the Quanta qScript cDNA synthesis kit (Quanta Biosciences, Boston, MA, USA) per manufacturer’s instructions. qPCR was performed on a StepOnePlus™ Real-Time PCR system (Applied biosystems, Ghent, Belgium) using the SYBR green method (Applied Biosystems). The master mix contained 1× SYBR green, 10 µM primers, 12.5 ng cDNA, and nuclease free water. Data were normalized to the most stable reference genes and the ΔΔCt method was used to determine relative quantification of gene expression. Primers were designed using NCBI’s Primer-blast (details are shown in Table 1).
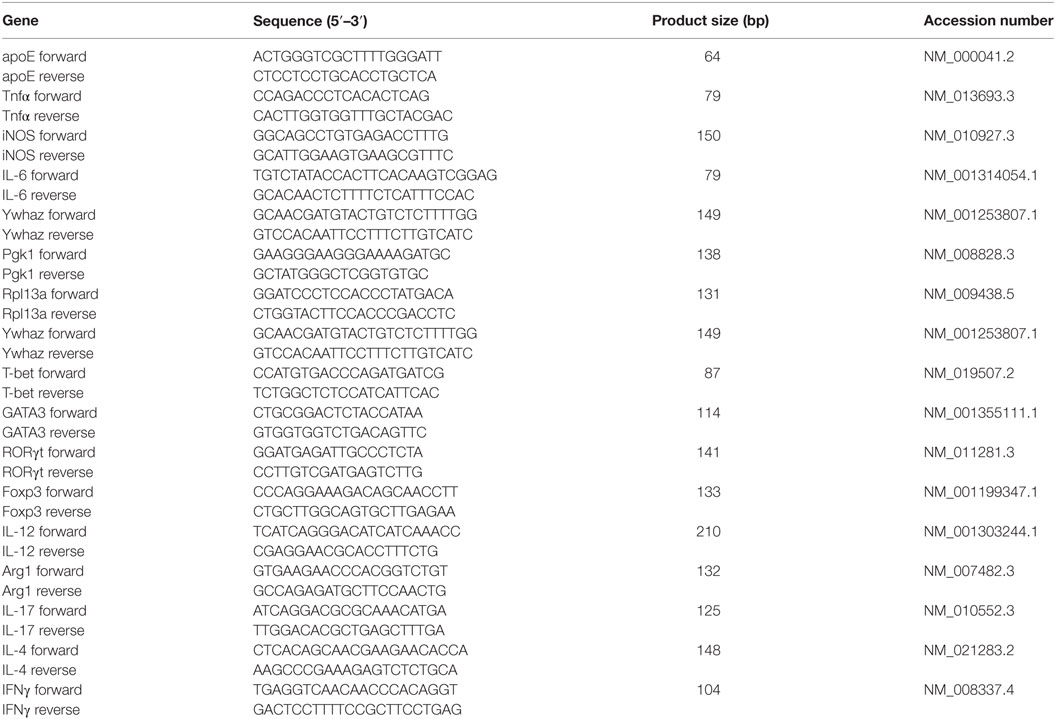
Table 1. Mouse primers for RT-qPCR.
NO Production
Peritoneal macrophages in culture were stimulated with 100 ng/ml LPS (Sigma-Aldrich) prior to the assay. NO production was determined in the supernatant of peritoneal macrophages using a Griess-reagent assay (Promega, Leuven, Belgium) according to the manufacturer’s instructions.
Immunohistochemistry
Animals were perfused transcardially through the ventricular catheter with ringer (containing heparin). Spinal cord tissue was isolated, snap-frozen, and sectioned with a Leica CM1900UV cryostat (Leica Microsystems, Wetzlar, Germany) to obtain 8 µm slices. Dried cryosections were fixed in acetone for 10 min and subsequently blocked for 0.5 h with 10% normal serum. Cryosections were incubated overnight at 4°C with primary antibodies. Next, secondary antibodies were added for 1 h. Primary antibodies used were rat anti-mouse CD3 (1:150; AbD Serotec) and rat anti-mouse F4/80 (1:100; AbD Serotec). Goat anti-rat Alexa Fluor®555 (1:400; Invitrogen) was used as a secondary antibody. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 1:2,500; Sigma-Aldrich). Stained sections were visualized under a Nikon Eclipse 80i fluorescence microscope (Nikon, Kingston, UK). Sections were analyzed using NIS Elements viewer software 4.0 (Nikon).
Western Blot
Apolipoprotein E secretion by peritoneal macrophages was determined using western blot. Culture medium was collected and separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Gels were transferred to a PVDF-membrane (VWR, Leuven, Belgium) and blots were blocked for 1 h in TBS-Tween 5% non-fat dry milk. Membranes were probed with mouse anti-apoE (Abbiotec, Antwerpen, Belgium). After washing steps with TBS-Tween, blots were incubated with horseradish peroxidase-labeled anti-mouse antibody (Dako, Heverlee, Belgium). Immunoreactive signals were detected with Enhanced Chemiluminescence (ECL Plus, GE Healthcare, Diegem, Belgium).
Phagocytosis Assay
Myelin was isolated from brain tissue of healthy adult WT C57BL/6 OlaHSD mice (Harlan) by means of sucrose density gradient centrifugation, as previously described (31). Next, myelin was labeled with the lypophilic dye 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (DiI) (Thermofisher Scientific, Erembodegem, Belgium). Peritoneal macrophages were incubated with DiI labeled myelin (25 µg/ml) for 90 min at 37°C and 5% CO2. Next, cells were rinsed with PBS (Sigma-Aldrich), detached with PBS/EDTA and resuspended in FACS buffer containing 1× PBS, 2% FCS (Hyclone) and sodium Azide. The fluorescent internalized myelin was measured using the FACS Calibur flow cytometer (BD biosciences, Erembodegem, Belgium). Results are expressed as mean fluorescence.
ELISA
Peritoneal mouse macrophages were stimulated with 100 ng/ml LPS for 18 h prior to the assay. TNFα concentration in peritoneal macrophage culture supernatant was determined using the TNFα Mouse Uncoated ELISA Kit (Thermofisher), following the manufacturer’s instructions. Absorption was measured at 450 nm using a microtiterplate reader (Biorad, Temse, Belgium).
Statistical Analysis
Data were statistically analyzed using GraphPad Prism for windows (version 5.0) and are reported as mean ± SEM. D’Agostino and Pearson omnibus normality test was used to test Gaussian distribution. A two-tailed unpaired student t-test was used for normally distributed data sets [t(df); P]. The Mann–Whitney analysis (MWU, n1, n2; P) was used for data sets that did not pass normality. EAE scores were analyzed using two-way ANOVA (Bonferroni’s post hoc multiple comparison test). *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Ldlr Deficiency Reduces EAE Severity in Female But Not in Male Mice
To elucidate whether the LDLr contributes to neuroinflammation, we induced EAE in male and female ldlr−/− mice. We show that ldlr deficiency has a sex-specific effect on the EAE course. Female ldlr−/− mice exhibited a decreased EAE disease severity compared to WT mice, whereas in male mice ldlr deficiency did not affect EAE disease severity (Figures 1A,B). In female animals, the mean peak of disease symptoms was reached around day 17. The mean disease score was attenuated in ldlr−/− (1.504 ± 0.1995) compared to WT (2.318 ± 0.1782) mice (Figure 1C). Accordingly, the maximum disease score was lower in LDLr−/− (1.70 ± 0.30) compared to WT (2.68 ± 0.29) mice (Figure 1D). The ldlr−/− and WT groups displayed a similar day of onset (14.29 ± 1.04 and 12.41 ± 0.34, respectively Figure 1E).
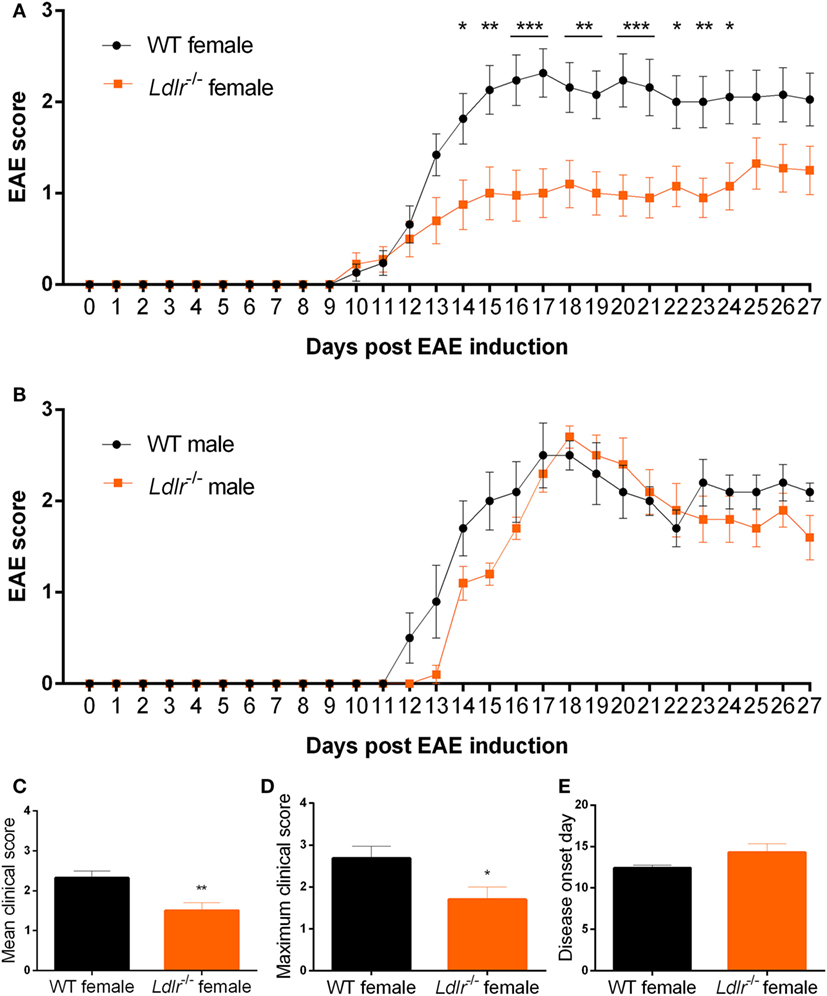
Figure 1. Ldlr−/− reduces experimental autoimmune encephalomyelitis (EAE) severity in female but not in male mice. Daily EAE score of female [p < 0.0079; wild-type (WT) vs ldlr−/− by two-way ANOVA] (n = 19–20, pooled from three independent experiments) (A) and male (p < 0.2221; WT vs ldlr−/− by two-way ANOVA) (n = 5) (B) mice. The mean score [t(31) = 3.050; p < 0.0048] (C), the maximum score [t(37) = 2.349; p < 0.0244] (D), and the day of onset of the disease [t(29) = 1.848; p < 0.0748] (E).
Ldlr Deficiency Has No Significant Influence on Immune Cell Infiltration into the CNS
Experimental autoimmune encephalomyelitis is characterized by the infiltration of peripheral immune cells into the CNS leading to a local inflammatory response. To determine whether this process is altered by ldlr deficiency in female mice, the accumulation of T cells (CD3) and macrophages (F4/80) in the CNS of WT and ldlr deficient female EAE mice was assessed by immunohistochemistry at day 18 and day 33 post immunization (Figure 2; Figures S1 and S2 in Supplementary Material). Despite a reduced disease severity, no significant differences in the number of infiltrated macrophages and T cells into the spinal cord tissue were observed comparing female WT EAE mice and ldlr-deficient EAE mice, although a trend on day 18 is visible.
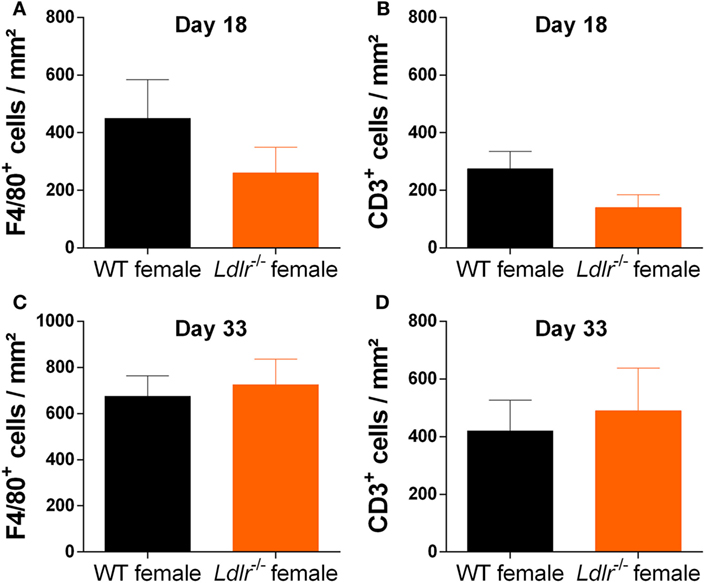
Figure 2. Ldlr deficiency has no significant influence on macrophage and T cell infiltration into the central nervous system. Immunohistological sections of spinal cord tissue from wild-type (WT) and ldlr−/− mice at day 18 (A,B) and day 33 (C,D) post immunization. Sections were stained against F4/80 for macrophages (A,C) and CD3 for T cells (B,D). At least six sections per mouse were quantified (n = 5). Representative images can be found in the supplementary material section S1/2.
Ldlr Deficiency in Female Mice Has No Influence on T Cell Proliferation
T cell proliferation is an important hallmark of EAE and is essential for the initiation of EAE pathogenesis. Since T cells are dependent on cholesterol in order to proliferate (32), we investigated the influence of ldlr on T cell proliferation during EAE. T cells from both ldlr−/− and WT mice were isolated on day 9 post immunization and a thymidine assay was used to determine their proliferation capacity. No significant differences in T cell proliferation between ldlr−/− and WT mice induced with EAE (Figure 3A). In addition, we determined mRNA expression of transcription factors such as T-bet, GATA3, RORγt, and Foxp3, which are involved in T cell differentiation into Th1, Th2, Th17, and Treg subsets, in LN and spleen but found no difference between ldlr−/− and WT female mice (Figures 3B,C). Lastly, we analyzed the mRNA expression levels of cytokines important in T cell differentiation (IFNγ, IL-4, IL-17, and IL-10) in T cells isolated from both spleen and LN. No differences in the gene expression of these cytokines were observed. The mRNA levels of IL-4 and IL-17 were not detectable. Data are shown in the supplementary section (Figure S3 in Supplementary Material). Together, these results indicate that altered T cell proliferation and differentiation do not account for the reduced EAE in ldlr deficient mice.
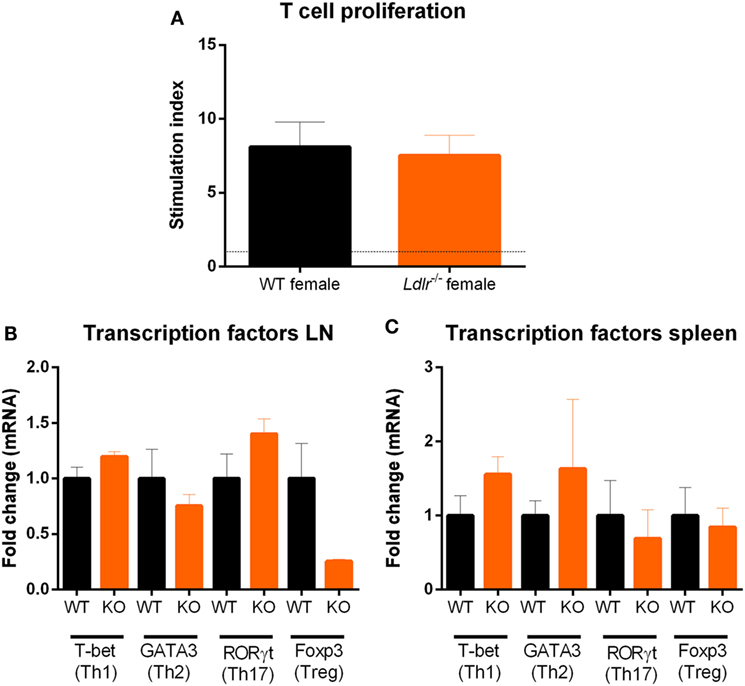
Figure 3. Ldlr deficiency has no influence on T cell proliferation. Stimulation index (SI) of T cells isolated from lymph nodes (LN) of wild-type (WT) and LDLR deficient mice (n = 3). Cells were collected on day 9 post immunization and proliferation was analyzed by means of a thymidine assay. Unstimulated cells an SI of 1 (A). Gene expression of transcription factors (T-bet, GATA3, RORγt, and Foxp3) in T cells isolated from LN (B) and spleen (C) from WT and ldlr−/− female mice on day 9 post immunization (n = 4).
Ldlr Deficiency Reduces Inflammation in the Spinal Cord of Female Mice Compared to Male Mice
Our data indicate that ldlr deficiency in female mice attenuates EAE severity without affecting T cell proliferation and the level of immune cell infiltration into the CNS. To further define the inflammatory burden in the CNS, whole spinal cord tissue of male and female ldlr−/− mice was analyzed for mRNA expression of proinflammatory genes. Female ldlr−/− mice exhibited a significantly lower expression of tumor necrosis factor alpha (Tnfα), Interleukin-6 (IL-6), and nitric oxide synthase (iNOS) compared to male ldlr−/− mice and female WT mice (Figures 4A,B), whereas the expression of these inflammatory genes is not changed in male ldlr−/− mice (Figure 4C). These data indicate that female ldlr−/− mice have decreased inflammation in the spinal cord compared to male ldlr−/− and female WT mice.
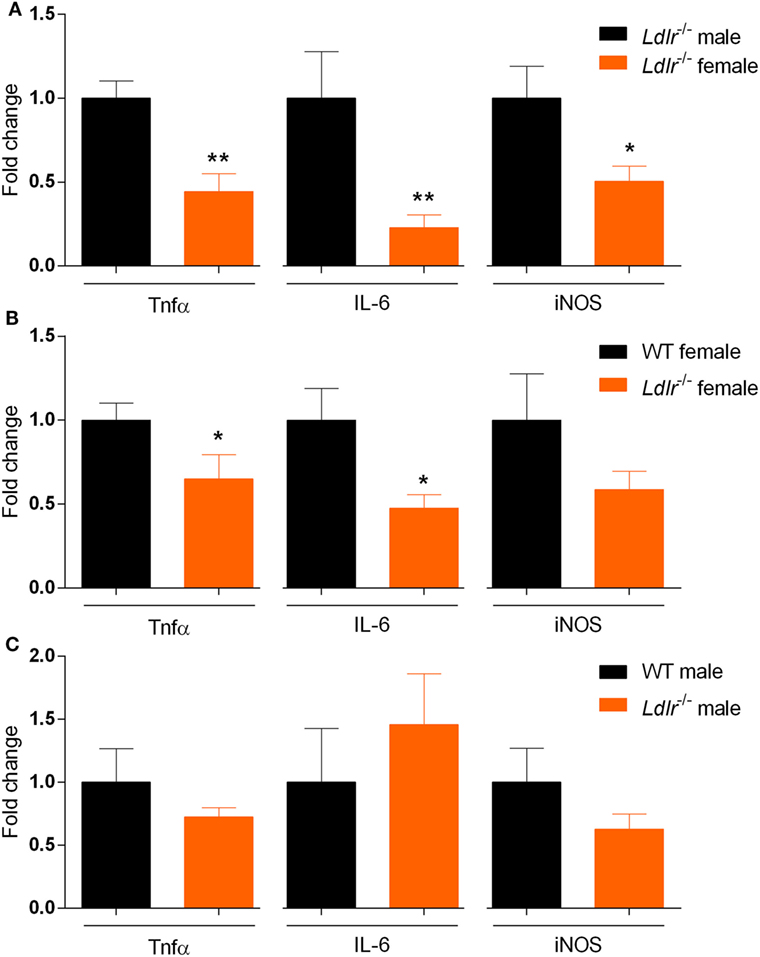
Figure 4. Relative expression of proinflammatory genes in the spinal cord of ldlr−/− mice. Expression of Tnfα [t(10) = 3.668; p < 0.0044], IL-6 (MWU = 0; 5; 7; p < 0.0026) and iNOS [t(10) = 2.588; p < 0.0271] for male and female ldlr−/− mice at experimental autoimmune encephalomyelitis (EAE) peak (n = 5–7) (A). Expression of Tnfα [t(13) = 2.467; p < 0.0284], IL-6 [t(12) = 2.553; p < 0.0254], and iNOS [t(13) = 1.313; p < 0.2118] for wild-type (WT) and ldlr−/− female mice at EAE peak (n = 5–7) (B). Expression of Tnfα [t(7) = 1.106; p < 0.3054], IL-6 [t(7) = 0.7745; p < 0.4641], and iNOS [t(7) = 1.370; p < 0.2129] for WT and ldlr−/− male mice at EAE peak (n = 5–7) (C). Data were normalized to the most stable reference genes, determined by Genorm (Ywhaz and Pgk1).
Ldlr Deficiency Suppresses the Inflammatory Phenotype of Macrophages in Female Mice
As the ldlr is highly expressed on macrophages and we observed a reduced expression of cytokines typically released by macrophages in female ldlr−/− EAE mice, we investigated the influence of ldlr deficiency on the inflammatory phenotype of macrophages in vitro. We found that female LPS-stimulated ldlr−/− macrophages produce less TNFα and NO compared to macrophages isolated from WT females (Figures 5A,B). In addition, we show that macrophages isolated from ldlr−/− female mice less efficiently internalize myelin compared to macrophages isolated from WT females (Figure 5C). Interestingly, expression of lrp1, a major receptor involved in the phagocytosis of myelin (33), was reduced as well (Figure 5D). The expression of the proinflammatory genes Tnfα, IL-6 and Il-12 is reduced in ldlr−/− female mice, whereas the anti-inflammatory gene Arg1 is increased in macrophages isolated from ldlr−/− female mice compared to WT mice (Figures 5E–I). These data indicate that macrophages isolated from female ldlr−/− mice have an attenuated inflammatory response upon stimulation and have a reduced phagocytosis capacity compared to their WT counterparts.
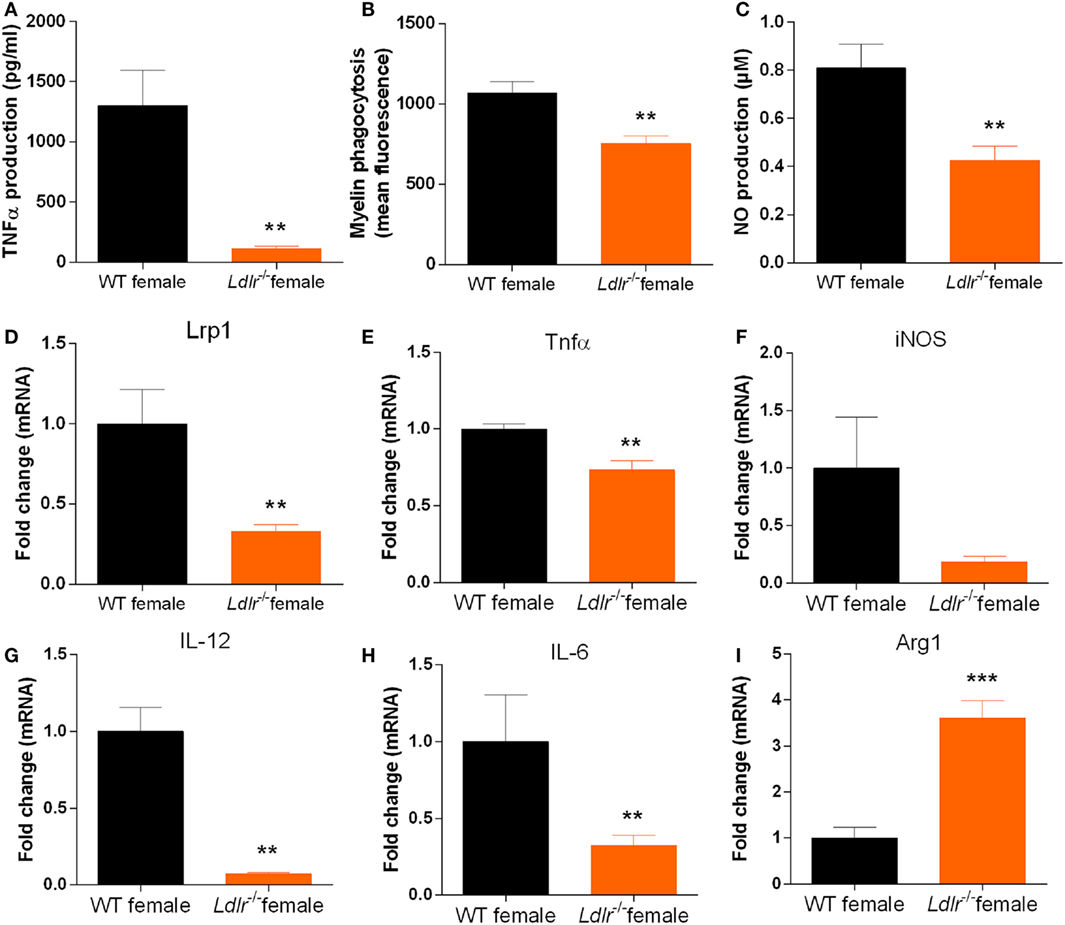
Figure 5. Ldlr deficiency influences the production of inflammatory mediators by macrophages. TNFα production [t(17) = 3.444; p < 0.0032] (n = 6) (A), NO production [t(22) = 3.368; p < 0.0029] (n = 6) (B), phagocytosis (mean fluorescence) [t(28) = 3.638; p < 0.0012] (n = 6) (C), lrp1 expression (MWU = 2; 5; 8; p < 0.0063) (n = 5–8) (D), Tnfα expression [t(9) = 3.630; p < 0.0055] (E), iNOS expression [t(9) = 2.020; p < 0.0742] (F), IL-12 expression (MWU = 0; 4; 6; p < 0.0095) (G), IL-6 expression [t(9) = 2.391; p < 0.0405] (H), and Arg1 expression [t(8) = 5.846; p < 0.0004] (I) in peritoneal macrophages isolated from wild-type (WT) and ldlr−/− female mice (n = 6).
ApoE Expression Is Increased in Activated Peritoneal Macrophages Isolated from ldlr−/− Female Mice
Apolipoprotein E is known to induce an anti-inflammatory phenotype in macrophages (34). Interestingly, we show that ApoE mRNA expression is significantly increased in activated peritoneal macrophages from ldlr−/− female mice compared to both male ldlr−/− mice and female WT mice (Figures 6A–C). Next, we used Western blot to determine the amount of apoE secreted into the culture medium. We found that only LPS-stimulated macrophages isolated from female ldlr−/− mice, and not those from WT and male ldlr−/− mice, secreted significant levels of apoE (Figure 6D). These data suggest that ldlr deficiency in female mice is responsible for the increase and subsequent secretion of apoE by macrophages.
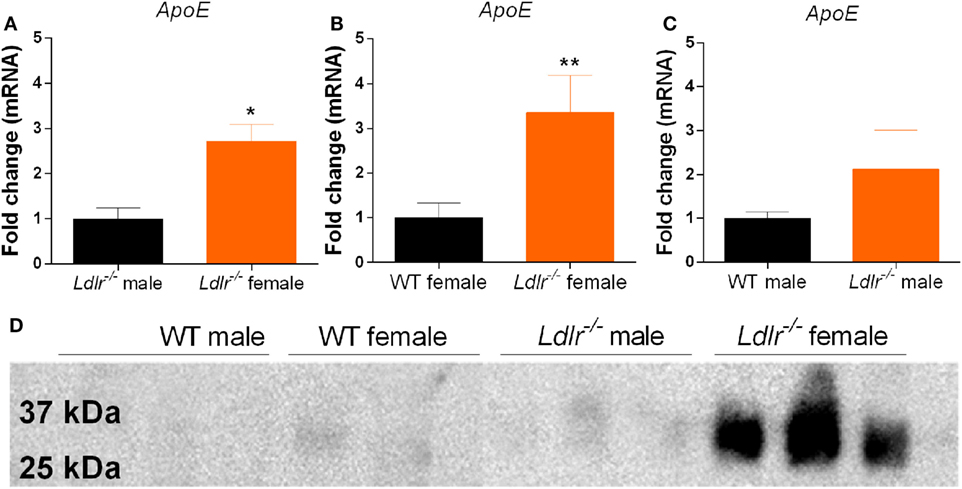
Figure 6. Apolipoprotein E (ApoE) expression by peritoneal macrophages. Comparison of apoE gene expression between male and female ldlr−/− mice (MWU = 1; 6; 6; p < 0.0476) (n = 6) (A), wild-type (WT) and ldlr−/− female mice (MWU = 1; 6; 6; p < 0.0044) (n = 6) (B), and WT and ldlr−/− male mice (n = 6) (C). Western blot analysis of cell culture medium from LPS-stimulated peritoneal macrophages, stained for apoE (n = 3) (D).
Lack of apoE Abrogates the Reduced Neuroinflammatory Response in Female ldlr−/− Mice
To define to what extend an increase in apoE accounts for the observed reduction in EAE disease severity in ldlr−/− female mice and the observed less-inflammatory phenotype of macrophages isolated from these mice, we crossbred apoE−/− and ldlr−/− mice. In contrast to female ldlr−/− mice, female apoE−/− ldlr−/− mice did not exhibit an attenuated EAE disease course compared to WT mice (Figure 7A). In addition, the observed decreased capacity to phagocytose myelin and produce TNFα by macrophages from ldlr−/− female mice was counteracted by depletion of apoE (Figures 7B,C). Interestingly, the impairment in NO production observed in ldlr−/− macrophages was also present in apoE−/− ldlr−/− macrophages indicating apoE is not accountable for this effect (Figure 7D). ApoE−/− alone did not alter EAE severity in female mice compared to WT mice (Figure 7E). These data indicate that the lack of apoE largely restores the impaired inflammatory response induced by ldlr deficiency in female mice and that the effect of ldlr−/− on NO production is not responsible for the ameliorated disease course in these mice.
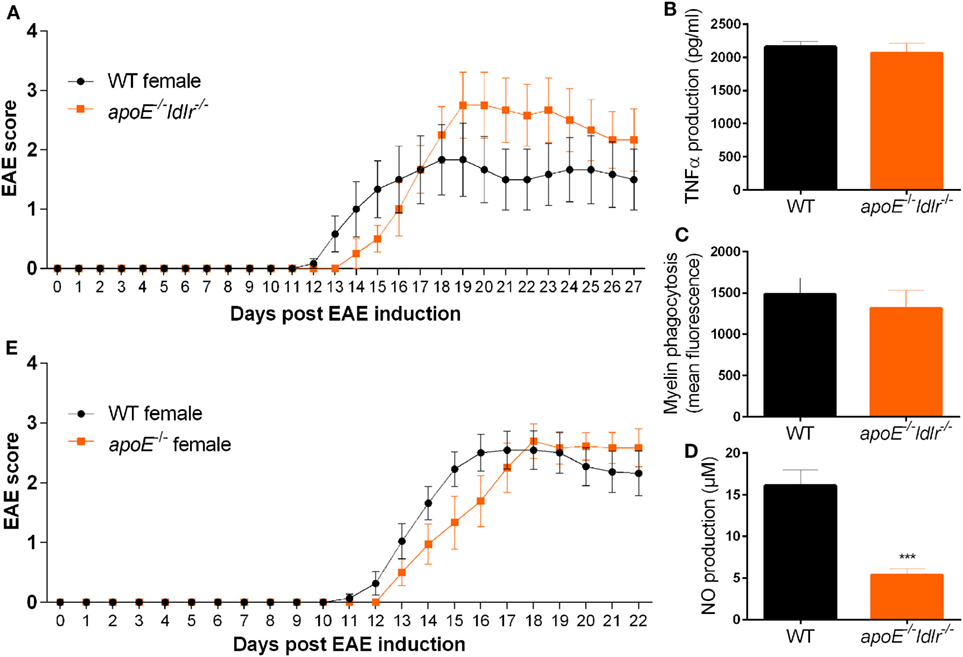
Figure 7. Lack of apolipoprotein E (apoE) abrogates the reduced neuroinflammatory response in female ldlr−/− mice. Daily clinical EAE score of female WT and apoE−/− and ldlr−/− mice (n = 6) (A). ELISA to measure TNFα production (n = 6) (B), myelin phagocytosis assay (n = 6) (C), Griess assay to measure NO production [t(15) = 5.091; p < 0.0001] (n = 6) (D), for WT and apoE−/− and ldlr−/− female mice. ApoE−/− female mice show a similar EAE disease score compared to WT female mice (n = 9–11) (E).
Discussion
In this study, we show that ldlr−/− attenuates neuroinflammation in female but not in male EAE mice, and that macrophages isolated from female mice have an attenuated inflammatory response and reduced phagocytic capacity. Interestingly, apoE deficiency counteracts the impact of ldlr deficiency in females on the course and pathology of EAE. These findings indicate that an increased apoE expression in macrophages apoE in female ldlr−/− mice is at least partially responsible for the observed reduction in EAE severity.
We show that ldlr deficiency in female mice decreases the inflammatory burden in the CNS but does not affect T cell proliferation and differentiation, and immune cell infiltration into the CNS. However, the levels of proinflammatory mediators in the CNS were reduced in these mice and isolated macrophages showed an impaired inflammatory response. These findings suggest that ldlr deficiency affects the inflammatory and phagocytic properties of macrophages, thereby altering EAE disease severity. In concordance, levels of the proinflammatory markers Tnfα, IL-6, and Il-12 were reduced while an increased gene expression of the anti-inflammatory marker Arg1 was observed in macrophages isolated from ldlr−/− female mice compared to WT macrophages. Moreover, ldlr deficiency decreases macrophage mediated myelin phagocytosis and expression of lrp1. LRP1 is a member of the LDLr family that also functions as a scavenger receptor and has been shown to be directly involved in myelin phagocytosis (33). Myelin degradation and phagocytosis by macrophages enhances demyelination which leads to neurological symptoms in MS (3). Although speculative, it is possible that lack of ldlr results in decreased Lrp1 expression, thereby indirectly impairing myelin phagocytosis. Together, an altered macrophage response at least partially accounts for the beneficial effects of ldlr deficiency on EAE severity.
In contrast to the reduction in EAE severity in ldlr−/− mice, the clinical outcome was not altered in apoE−/− ldlr−/− mice. Activated peritoneal macrophages isolated from ldlr−/− female mice significantly increased apoE expression compared to WT or their male counterparts and, the beneficial effect of ldlr deficiency on myelin phagocytosis and TNFα production was absent in apoE−/− ldlr−/− macrophages. Although the LDLr is described to directly regulate apoE levels in the CNS (35), we found no elevated apoE gene expression in the CNS of ldlr−/− EAE mice (data not shown). Studies have reported that increased expression of apoE leads to a less-inflammatory phenotype in macrophages by downregulating M1-like and upregulating M2-like markers. In addition, apoE suppresses microglial activation and the release of TNFα (34, 36, 37). Furthermore, treatment of EAE animals with apoE-derived peptides ameliorates the clinical course of EAE and apoE deficiency exacerbates EAE by increasing immune reactivity and hampering CNS repair mechanisms (38, 39). These findings suggest that the LDLr suppresses ApoE expression thereby exacerbating neuroinflammatory responses in females.
The reason for the sexual dimorphism in apoE expression remains elusive. It is reported that apoE exerts moderate sex-specific effects on neuroinflammation. Schrewe and coworkers found that the EAE disease course was slightly attenuated in male apoE−/− mice, whereas EAE was more severe in female apoE−/− mice compared to WT mice. In our hands apoE−/− did not alter EAE disease outcome in female mice compared to WT mice. Methodological differences (e.g., immunization protocol) may explain these discrepancies. Moreover, in Schrewe’s study differences between WT and apoE−/− female mice were only visible after 30–36 days post immunization, whereas our experiment lasted for up to 22 days post immunization. Nevertheless, the question remains why only macrophages from ldlr−/− female mice abundantly secrete apoE. Macrophages express receptors for sex hormones such as estrogen and testosterone and these hormones modulate macrophage activity (40, 41). Interestingly, administration of estrogens has been shown to directly regulate hepatic LDLr activity in human cells in vitro and 17β-estradiol increases the expression of apoE during the maturation stage of human monocytes to macrophages (42–44). As lack of LDLr has been shown to increase apoE levels also (35), both mechanisms may contribute to the increased apoE levels observed in female ldlr−/− mice.
However, it remains to be elucidated whether sex hormones are exclusively responsible for the observed effects. Studies investigating plasma lipid and lipoprotein concentrations across the menstrual cycle in women report virtually no variation (45–47), and those that do, report only small reductions in LDL during the luteal phase and no changes in plasma triglyceride or HDL concentrations (45, 46). Therefore, the sexual dimorphism in lipid metabolism and its impact on neuroinflammation appears to be the result of an intricate network of sex hormones in combination with other modulators of lipid metabolism. The key pathways and mediators of this network remain to be elucidated, but there are many other factors involved such as insulin and adipokines (48), and gene expression and imprinting (49), that need to be explored in the future.
In addition to apoE, differences in female and male mice may be the result of differences in LDLr levels and function. However, there are few studies about sex-related differences in LDLr behavior. Segatto and coworkers determined age- and sex-related differences in extra-hepatic LDLr expression and found that LDLr protein expression was decreased in 12-month-old (onset of menopause) female rat brains, which was completely restored by estrogen treatment. In skeletal muscle, LDLr levels were increased in both male and female aged rats, whereas in the heart no modifications were observed either in aged rats or rats of a specific gender (50). These data illustrate the complex age- and sex-related and tissue-specific regulation of LDLr expression, and warrant the need of more research to clarify the implications of these differences for neuroinflammatory diseases and other conditions involving the LDLr.
Collectively, we show that ldlr−/− attenuates EAE disease severity in female but not in male EAE mice. Although the nature of the observed sexual dimorphism remains unclear, our findings show that LDLr and associated apoE levels are involved in neuroinflammatory diseases such as multiple sclerosis, which may have implications for future treatment strategies.
Ethics Statement
Experiments were conducted in according to institutional guidelines and were approved by the ethical committee on animal experiments (ECAE) of Hasselt University.
Author Contributions
JM, ST, KN, JV, TV, and JB performed the experiments and analyzed the data. JM wrote the manuscript. JM, ST, KN, JV, TV, JB, JH, and JH participated in the design and coordination of the project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Joke Vanhoof and Katrien Wauterickx for excellent technical assistance. This work was supported by the Scientific Research-Flanders (FWO) (Grants 1506916N & 1512314N and 1264114N) and the Agentschap voor Innovatie van Wetenschap en Technologie (IWT) (Grant 131086).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fimmu.2017.01701/full#supplementary-material.
Figure S1. Fluorescent staining of macrophages (F4/80) in experimental autoimmune encephalomyelitis spinal cord. Staining of macrophages in wild-type female (A,C) and ldlr−/− female mice (B,D) on days 18 and 33, respectively.
Figure S2. Fluorescent staining of T cells (CD3) in experimental autoimmune encephalomyelitis spinal cord. Staining of T cells in wild-type female (A,C) and ldlr−/− female mice (B,D) on day 18 and 33, respectively.
Figure S3. Cytokine important in T cell differentiation. mRNA expression levels of cytokines important in T cell differentiation (IFNγ, IL-4, IL-17, and IL-10) in T cells isolated from both spleen and lymph nodes from WT and ldlr−/− female mice (n = 4). No differences in the gene expression of these cytokines were observed. The mRNA levels of IL-4 and IL-17 were not detectable.
References
1. Huitinga I, Ruuls SR, Jung S, Van Rooijen N, Hartung HP, Dijkstra CD. Macrophages in T cell line-mediated, demyelinating, and chronic relapsing experimental autoimmune encephalomyelitis in Lewis rats. Clin Exp Immunol (1995) 100(2):344–51. doi:10.1111/j.1365-2249.1995.tb03675.x
2. Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J Immunol (1981) 126(2):614–20.
3. Hendriks JJ, Teunissen CE, de Vries HE, Dijkstra CD. Macrophages and neurodegeneration. Brain Res Brain Res Rev (2005) 48(2):185–95. doi:10.1016/j.brainresrev.2004.12.008
4. Barnett MH, Henderson AP, Prineas JW. The macrophage in MS: just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult Scler (2006) 12(2):121–32. doi:10.1191/135248506ms1304rr
5. Williams K, Ulvestad E, Waage A, Antel JP, McLaurin J. Activation of adult human derived microglia by myelin phagocytosis in vitro. J Neurosci Res (1994) 38(4):433–43. doi:10.1002/jnr.490380409
6. Bogie JF, Mailleux J, Wouters E, Jorissen W, Grajchen E, Vanmol J, et al. Scavenger receptor collectin placenta 1 is a novel receptor involved in the uptake of myelin by phagocytes. Sci Rep (2017) 7:44794. doi:10.1038/srep44794
7. Bogie JF, Stinissen P, Hendriks JJ. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol (2014) 128(2):191–213. doi:10.1007/s00401-014-1310-2
8. Bogie JF, Timmermans S, Huynh-Thu VA, Irrthum A, Smeets HJ, Gustafsson JA, et al. Myelin-derived lipids modulate macrophage activity by liver X receptor activation. PLoS One (2012) 7(9):e44998. doi:10.1371/journal.pone.0044998
9. Mailleux J, Vanmierlo T, Bogie JF, Wouters E, Lutjohann D, Hendriks JJ, et al. Active liver X receptor signaling in phagocytes in multiple sclerosis lesions. Mult Scler (2017). doi:10.1177/1352458517696595
10. Bogie JF, Jorissen W, Mailleux J, Nijland PG, Zelcer N, Vanmierlo T, et al. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol Commun (2013) 1:43. doi:10.1186/2051-5960-1-43
11. Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, et al. High cholesterol level is essential for myelin membrane growth. Nat Neurosci (2005) 8(4):468–75. doi:10.1038/nn1426
12. Weinstock-Guttman B, Zivadinov R, Mahfooz N, Carl E, Drake A, Schneider J, et al. Serum lipid profiles are associated with disability and MRI outcomes in multiple sclerosis. J Neuroinflammation (2011) 8:127. doi:10.1186/1742-2094-8-127
13. Giubilei F, Antonini G, Di Legge S, Sormani MP, Pantano P, Antonini R, et al. Blood cholesterol and MRI activity in first clinical episode suggestive of multiple sclerosis. Acta Neurol Scand (2002) 106(2):109–12. doi:10.1034/j.1600-0404.2002.01334.x
14. Teunissen CE, Dijkstra CD, Polman CH, Hoogervorst EL, von Bergmann K, Lutjohann D. Decreased levels of the brain specific 24S-hydroxycholesterol and cholesterol precursors in serum of multiple sclerosis patients. Neurosci Lett (2003) 347(3):159–62. doi:10.1016/S0304-3940(03)00667-0
15. van de Kraats C, Killestein J, Popescu V, Rijkers E, Vrenken H, Lutjohann D, et al. Oxysterols and cholesterol precursors correlate to magnetic resonance imaging measures of neurodegeneration in multiple sclerosis. Mult Scler (2014) 20(4):412–7. doi:10.1177/1352458513499421
16. Leoni V, Masterman T, Diczfalusy U, De Luca G, Hillert J, Bjorkhem I. Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci Lett (2002) 331(3):163–6. doi:10.1016/S0304-3940(02)00887-X
17. Mandoj C, Renna R, Plantone D, Sperduti I, Cigliana G, Conti L, et al. Anti-annexin antibodies, cholesterol levels and disability in multiple sclerosis. Neurosci Lett (2015) 606:156–60. doi:10.1016/j.neulet.2015.08.054
18. Jorissen W, Wouters E, Bogie JF, Vanmierlo T, Noben JP, Sviridov D, et al. Relapsing-remitting multiple sclerosis patients display an altered lipoprotein profile with dysfunctional HDL. Sci Rep (2017) 7:43410. doi:10.1038/srep43410
19. de Graaf J, Hak-Lemmers HL, Hectors MP, Demacker PN, Hendriks JC, Stalenhoef AF. Enhanced susceptibility to in vitro oxidation of the dense low density lipoprotein subfraction in healthy subjects. Arterioscler Thromb (1991) 11(2):298–306. doi:10.1161/01.ATV.11.2.298
20. Chancharme L, Therond P, Nigon F, Lepage S, Couturier M, Chapman MJ. Cholesteryl ester hydroperoxide lability is a key feature of the oxidative susceptibility of small, dense LDL. Arterioscler Thromb Vasc Biol (1999) 19(3):810–20. doi:10.1161/01.ATV.19.3.810
21. Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res (2002) 43(9):1363–79. doi:10.1194/jlr.R200004-JLR200
22. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science (1986) 232(4746):34–47. doi:10.1126/science.3513311
23. Miyake Y, Tajima S, Funahashi T, Yamamoto A. Analysis of a recycling-impaired mutant of low density lipoprotein receptor in familial hypercholesterolemia. J Biol Chem (1989) 264(28):16584–90.
24. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol (2013) 14(8):812–20. doi:10.1038/ni.2639
25. Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci (2002) 115(Pt 12):2603–11.
26. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (2013) 339(6116):161–6. doi:10.1126/science.1230719
27. Tolani S, Pagler TA, Murphy AJ, Bochem AE, Abramowicz S, Welch C, et al. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis (2013) 229(1):79–85. doi:10.1016/j.atherosclerosis.2013.03.031
28. Feng Y, Schouteden S, Geenens R, Van Duppen V, Herijgers P, Holvoet P, et al. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS One (2012) 7(11):e47286. doi:10.1371/journal.pone.0047286
29. Jurukovska-Nospal M, Arsova V, Levchanska J, Sidovska-Ivanovska B. Effects of statins (atorvastatin) on serum lipoprotein levels in patients with primary hyperlipidemia and coronary heart disease. Prilozi (2007) 28(2):137–48.
30. de Oliveira J, Moreira EL, dos Santos DB, Piermartiri TC, Dutra RC, Pinton S, et al. Increased susceptibility to amyloid-beta-induced neurotoxicity in mice lacking the low-density lipoprotein receptor. J Alzheimers Dis (2014) 41(1):43–60. doi:10.3233/JAD-132228
31. Norton WT, Poduslo SE. Myelination in rat brain: changes in myelin composition during brain maturation. J Neurochem (1973) 21(4):759–73. doi:10.1111/j.1471-4159.1973.tb07520.x
32. Dimeloe S, Burgener AV, Grahlert J, Hess C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology (2017) 150(1):35–44. doi:10.1111/imm.12655
33. Gaultier A, Wu X, Le Moan N, Takimoto S, Mukandala G, Akassoglou K, et al. Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J Cell Sci (2009) 122(Pt 8):1155–62. doi:10.1242/jcs.040717
34. Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol (2011) 31(5):1160–8. doi:10.1161/ATVBAHA.111.222745
35. Fryer JD, Demattos RB, McCormick LM, O’Dell MA, Spinner ML, Bales KR, et al. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem (2005) 280(27):25754–9. doi:10.1074/jbc.M502143200
36. Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, et al. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol (2001) 167(1):74–85. doi:10.1006/exnr.2001.7541
37. Christensen DJ, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, et al. Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J Immunol (2011) 186(4):2535–42. doi:10.4049/jimmunol.1002847
38. Li FQ, Sempowski GD, McKenna SE, Laskowitz DT, Colton CA, Vitek MP. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J Pharmacol Exp Ther (2006) 318(3):956–65. doi:10.1124/jpet.106.103671
39. Karussis D, Michaelson DM, Grigoriadis N, Korezyn AD, Mizrachi-Koll R, Chapman S, et al. Lack of apolipoprotein-E exacerbates experimental allergic encephalomyelitis. Mult Scler (2003) 9(5):476–80. doi:10.1191/1352458503ms950oa
40. Trigunaite A, Dimo J, Jorgensen TN. Suppressive effects of androgens on the immune system. Cell Immunol (2015) 294(2):87–94. doi:10.1016/j.cellimm.2015.02.004
41. Garcia-Ovejero D, Veiga S, Garcia-Segura LM, Doncarlos LL. Glial expression of estrogen and androgen receptors after rat brain injury. J Comp Neurol (2002) 450(3):256–71. doi:10.1002/cne.10325
42. Semenkovich CF, Ostlund RE Jr. Estrogens induce low-density lipoprotein receptor activity and decrease intracellular cholesterol in human hepatoma cell line Hep G2. Biochemistry (1987) 26(16):4987–92. doi:10.1021/bi00390a016
43. Smith PM, Cowan A, White BA. The low-density lipoprotein receptor is regulated by estrogen and forms a functional complex with the estrogen-regulated protein ezrin in pituitary GH3 somatolactotropes. Endocrinology (2004) 145(7):3075–83. doi:10.1210/en.2004-0228
44. Napolitano M, Blotta I, Montali A, Bravo E. 17beta-estradiol enhances the flux of cholesterol through the cholesteryl ester cycle in human macrophages. Biosci Rep (2001) 21(5):637–52. doi:10.1023/A:1014721026280
45. Mumford SL, Schisterman EF, Siega-Riz AM, Browne RW, Gaskins AJ, Trevisan M, et al. A longitudinal study of serum lipoproteins in relation to endogenous reproductive hormones during the menstrual cycle: findings from the BioCycle study. J Clin Endocrinol Metab (2010) 95(9):E80–5. doi:10.1210/jc.2010-0109
46. Barnett JB, Woods MN, Lamon-Fava S, Schaefer EJ, McNamara JR, Spiegelman D, et al. Plasma lipid and lipoprotein levels during the follicular and luteal phases of the menstrual cycle. J Clin Endocrinol Metab (2004) 89(2):776–82. doi:10.1210/jc.2003-030506
47. Magkos F, Patterson BW, Mittendorfer B. No effect of menstrual cycle phase on basal very-low-density lipoprotein triglyceride and apolipoprotein B-100 kinetics. Am J Physiol Endocrinol Metab (2006) 291(6):E1243–9. doi:10.1152/ajpendo.00246.2006
48. Perez-Lopez FR, Larrad-Mur L, Kallen A, Chedraui P, Taylor HS. Gender differences in cardiovascular disease: hormonal and biochemical influences. Reprod Sci (2010) 17(6):511–31. doi:10.1177/1933719110367829
49. Kim AM, Tingen CM, Woodruff TK. Sex bias in trials and treatment must end. Nature (2010) 465(7299):688–9. doi:10.1038/465688a
Keywords: neuroinflammation, multiple sclerosis, experimental autoimmune encephalomyelitis, low-density lipoprotein receptor, apolipoprotein E
Citation: Mailleux J, Timmermans S, Nelissen K, Vanmol J, Vanmierlo T, van Horssen J, Bogie JFJ and Hendriks JJA (2017) Low-Density Lipoprotein Receptor Deficiency Attenuates Neuroinflammation through the Induction of Apolipoprotein E. Front. Immunol. 8:1701. doi: 10.3389/fimmu.2017.01701
Received: 11 September 2017; Accepted: 17 November 2017;
Published: 30 November 2017
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Iain Comerford, University of Adelaide, AustraliaRobert Adam Harris, Karolinska Institute (KI), Sweden
Copyright: © 2017 Mailleux, Timmermans, Nelissen, Vanmol, Vanmierlo, van Horssen, Bogie and Hendriks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome J. A. Hendriks, amVyb21lLmhlbmRyaWtzQHVoYXNzZWx0LmJl