 Nagisa Yoshida
Nagisa Yoshida Eva-Maria Frickel
Eva-Maria Frickel Serge Mostowy
Serge Mostowy- 1Host-Toxoplasma Interaction Laboratory, The Francis Crick Institute, London, United Kingdom
- 2Section of Microbiology, MRC Centre for Molecular Bacteriology and Infection, Imperial College London, London, United Kingdom
Macrophages provide front line defense against infections. The study of macrophage–microbe interplay is thus crucial for understanding pathogenesis and infection control. Zebrafish (Danio rerio) larvae provide a unique platform to study macrophage–microbe interactions in vivo, from the level of the single cell to the whole organism. Studies using zebrafish allow non-invasive, real-time visualization of macrophage recruitment and phagocytosis. Furthermore, the chemical and genetic tractability of zebrafish has been central to decipher the complex role of macrophages during infection. Here, we discuss the latest developments using zebrafish models of bacterial and fungal infection. We also review novel aspects of macrophage biology revealed by zebrafish, which can potentiate development of new therapeutic strategies for humans.
Introduction
Macrophages are a major component of the innate immune system, responding efficiently to tissue damage and infection (1, 2). During infection, macrophages have diverse roles including phagocytosis of foreign bodies, release of cytotoxic factors, and coordination of the inflammatory response via the secretion of chemokines and cytokines (3, 4). Phagocytosis can involve the recognition of pathogen- and damage-associated molecular patterns (PAMPs and DAMPs, respectively) through pattern recognition receptors (PRRs) on the macrophage surface (5, 6). Further, the complement system can mark pathogens for phagocytosis by opsonization (7). Once internalized, the pathogen resides inside a vacuole known as a phagosome (8). Subsequent phagosome maturation involves acidification of the lumen, which leads to lysosomal fusion and degradation of the internalized microbe (9). Pathogen restriction is enhanced by the nutrient-limiting ability of the phagolysosome and the input of antimicrobial agents into the lumen, such as reactive oxygen/nitrogen species (ROS/RNS) (10). Although the majority of microbes succumb to the microbicidal environment within the phagolysosome, some pathogens (including Mycobacterium tuberculosis and Salmonella Typhimurium) can survive and replicate within this harsh environment (11, 12). In contrast, some bacterial pathogens (including Listeria monocytogenes and Shigella flexneri) have mechanisms to escape from the phagosome and proliferate in the cytosol (13).
Mechanisms of cell-autonomous immunity are crucial for protection of the host cell cytosol (14). Autophagy is an evolutionarily conserved process of intracellular degradation, recognized as an important defense mechanism against intracellular pathogens (15). Targeting of bacterial pathogens by the autophagy machinery is often mediated by ubiquitination, a posttranslational modification (16, 17). In this case, ubiquitinated substrates (such as bacterial components or damaged membrane) are recognized by autophagy receptors, including p62 and NDP52, which direct formation of the autophagic membrane around the targeted pathogen (18–20). Autophagy-related (ATG) proteins also direct immunity-related GTPases (IRGs) and guanylate-binding proteins (GBPs) to pathogens (21). IRGs and GBPs belong to a family of GTPases that confer host cell resistance during infection by pathogens (22–24). IRGs cooperate with GBPs to target non-self vacuoles, trafficking nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, ATG proteins, and inflammasome complex assembly for host defense (25–27). Intracellular pathogens are also detected via nucleotide-binding oligomerization domain-like receptors (NLRs), a class of PRRs that reside in the cytosol (28). An important example is NLRP3, which acts as a scaffold protein for inflammasome assembly, leading to caspase-1 activation and maturation of pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18 (29, 30). These cytokines enhance the immune response and induce pathways leading to pyroptosis, a highly inflammatory type of programmed cell death (31).
A variety of different animal models have made important contributions to the study of macrophage-microbe interplay in vivo. Originally used for studying development, the zebrafish has many similarities with higher vertebrates (including mammals), which has led to their use for studying infection and immunity (32, 33). During the first 4 weeks of development, zebrafish lack adaptive immunity and rely on the innate immune response for host defense (34). This, together with ex utero development of embryos, necessitates the rapid development of innate immune cells, progenitors of which can be observed as early as 20 h postfertilization (hpf). Although the precise sites of development and maturation differ between zebrafish and human phagocytes, zebrafish macrophages retain close morphological and functional similarities with their mammalian counterparts (35). Primitive macrophages, originally identified by Philippe Herbomel and colleagues, use phagocytosis to control infection by Escherichia coli and Bacillus subtilis (36). Optical accessibility during early life stages make zebrafish larvae highly suited for non-invasive live microscopy. Studies on the zebrafish immune system identified macrophage-specific genes (including mpeg1 and csf1ra), discoveries that enabled the development of specific reporter lines (37, 38). By using transgenic lines that fluorescently label distinct leukocyte populations, studies have identified key roles for macrophages during infection control in vivo (6). Furthermore, the sequenced zebrafish genome and the ability to manipulate the immune system through chemical or genetic means (including morpholino oligonucleotides for transient depletion, or CRISPR/Cas9 for genome engineering) make the zebrafish a unique and powerful tool for studying host–pathogen interactions at the molecular, cellular, and whole-animal level (39–41).
In this review, we discuss novel aspects of host–pathogen interactions that have been recently revealed using bacterial (mycobacteria, Gram-positive, Gram-negative) and fungal zebrafish infection models, highlighting key roles for macrophages in host defense against a variety of important pathogens.
Zebrafish Macrophage–Bacteria Interactions In Vivo
Mycobacterium marinum is a natural pathogen of zebrafish, closely related to the causative agent of human tuberculosis (M. tuberculosis). Pioneering studies have shown that M. marinum infection leads to the aggregation of macrophages into granuloma-like structures that both contain and promote bacterial dissemination (Figure 1A) (42, 43). These structures are initiated by the ESX-1 secretion system, which induces expression of matrix metalloproteinase-9 in epithelial cells to recruit macrophages for bacterial phagocytosis (44, 45). Tissue-resident macrophages first responding to infection are microbicidal. Therefore, to create a replication niche, M. marinum induces chemokine (C-C motif) ligand 2 (CCL2) expression and recruits uninfected monocytes via the surface lipid phenolic glycolipid (PGL) (46). Moreover, M. marinum possess cell surface-associated phthiocerol dimycoceroserate lipids, which impede PAMP–TLR interactions and prevent the microbicidal response in newly recruited monocytes (47). Consistent with a protective niche, the granuloma supports bacterial growth, alteration of granuloma structure through disruption of E-cadherin increases immune cell accessibility, and reduces bacterial burden (48). Work has shown that macrophage deficiency leads to accelerated necrosis of the granuloma and increased susceptibility to infection (45). A balanced inflammatory response is crucial to prevent necrosis of the granuloma, as both low and high levels of tumor necrosis factor (TNF) can lead to increased bacterial replication (49). Supporting this, a forward genetic screen performed in zebrafish revealed that mutation of the lta4h locus (encoding leukotriene A4 hydrolase) can modulate production of anti- and pro-inflammatory lipid mediators and susceptibility to mycobacterial infection (50). Angiogenesis has also been implicated in granuloma expansion and bacterial dissemination via a mechanism that requires hypoxia-induced vascular endothelial growth factor expression and C-X-C chemokine receptor type 4 (CXCR4) signaling (51, 52). Collectively, these studies highlight a complex role for macrophages in granuloma formation and in host defense against mycobacteria.
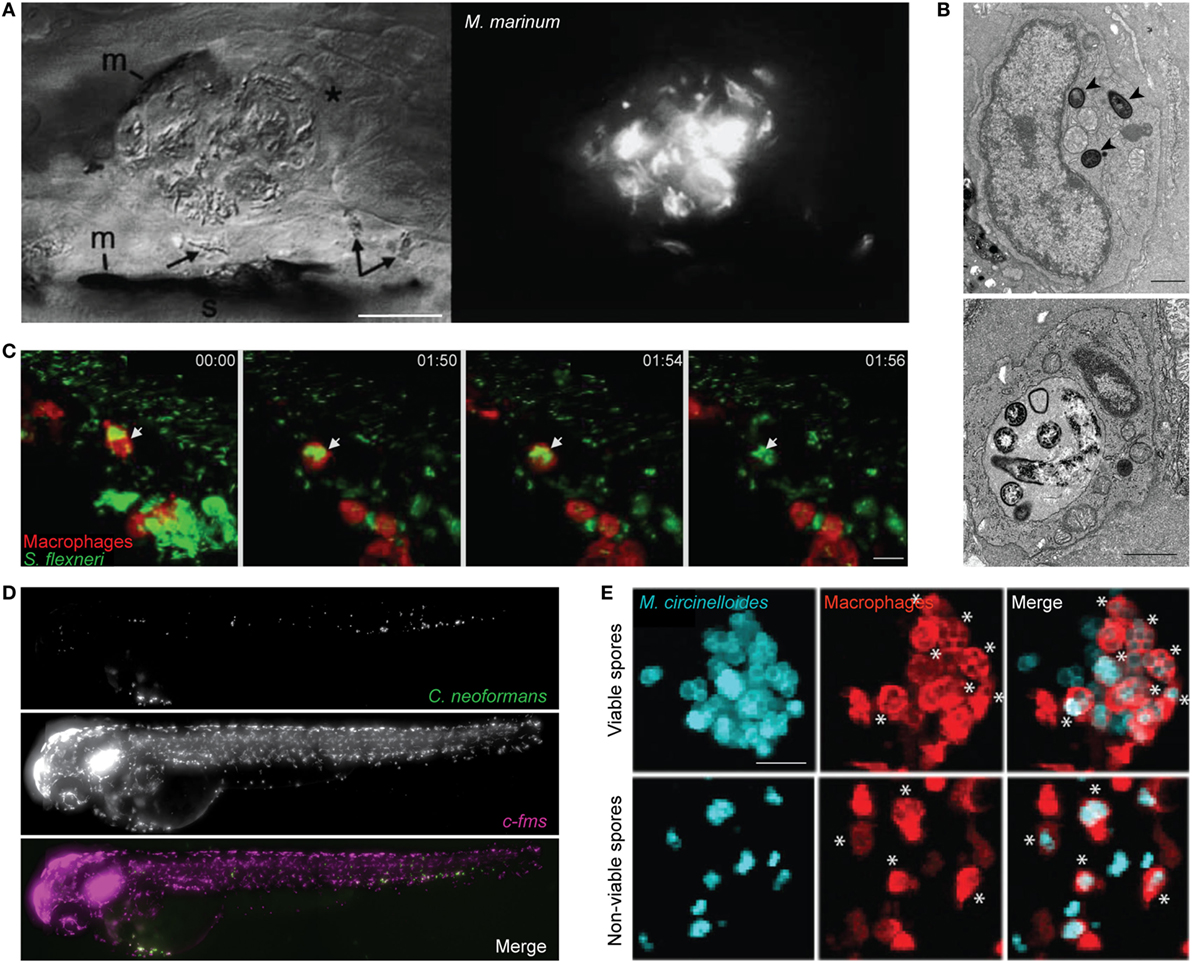
Figure 1. Zebrafish macrophage-microbe interactions in vivo. (A) Differential interference contrast (left) and fluorescent microscopy (right) image of macrophage aggregation to Mycobacterium marinum in the tail of wild-type AB larvae. Asterisk (*) indicates an infected macrophage at the aggregate; arrows indicate infected macrophages near the aggregate. m, melanocyte; s, striated muscle; scale bar 25 µm. Image adapted from Ref. (42). (B) Electron microscopy images of the caudal hematopoietic tissue of wild-type AB larvae injected intravenously with Listeria monocytogenes 3 h postinfection (hpi). Listeria in a macrophage cytosol (arrowheads; top image), and Listeria in a macrophage phagosome (bottom image). Scale bar 1 µm. Images adapted from Ref. (53). (C) Confocal time-lapse images of Tg(mpeg1:G/U:nfsb-mCherry) larva (red macrophages) infected with Shigella flexneri (green) by caudal vein injection, first frame at 20 min postinjection (mpi). White arrow depicts GFP-Shigella phagocytosed by a red macrophage, with a loss of red fluorescence at frame 01:56 indicating macrophage cell death. Maximum intensity projection of six planes every 2 µm, scale bar 10 µm. Images adapted from Ref. (54). (D) High content imaging of Tg(fms:Gal4.VP16)il86; Tg(UAS:nfsb.mCherry)il49 larvae harboring macrophages (middle) injected with Cryptococcus neoformans (top) into the yolk sac circulation valley; bottom panel showing a merged image of both macrophages (magenta) and C. neoformans (green). Maximum intensity projection of images obtained 2hpi. Images adapted from Ref. (55). (E) Hindbrain ventricle injection of viable (top row) or non-viable (bottom row) Mucor circinelloides spores (cyan) in Tg(mpeg1:G/U:nfsb-mCherry/mpx:GFP) larvae harboring red macrophages imaged at 10 h 45 min and 1 h 5 min, respectively. Asterisks (*) indicate spores inside macrophages (red). Z-stack of 15 sections every 7.3 µm; scale bar 20 µm. Images adapted from Ref. (56). All adapted images were used with the appropriate permissions from the copyright holders of this work.
Infection by Mycobacterium leprae, an ancient pathogen that causes leprosy, is restricted to humans and nine-banded armadillos (Dasypus novemcinctus) (57). M. leprae infection causes demyelination of peripheral nerves and axonal damage, which can lead to symptoms such as muscle weakness and numbness. Remarkably, new work has shown that zebrafish can be used to study M. leprae pathogenesis in vivo (58). Although M. leprae replication is not observed in zebrafish due to its long doubling time of 12–15 days, the macrophage response to bacteria is comparable to that observed during M. marinum infection. In the case of M. leprae, PGL-1 is responsible for mediating structural changes in myelin by inducing macrophage RNS production, which subsequently causes mitochondrial swelling, demyelination, and axonal damage.
Listeria monocytogenes, a Gram-positive foodborne pathogen, can cause listeriosis and meningitis in immunocompromised individuals, and spontaneous abortions during pregnancy (59, 60). Inside macrophages, L. monocytogenes can escape from the phagosome and proliferate in the cytosol (Figure 1B) (61). Bacterial escape from the phagosome to the cytosol is linked to expression of listeriolysin O (LLO), a pore-forming toxin that targets the phagosomal membrane (62). ActA, another major virulence factor of Listeria, enables actin tail polymerization and autophagy escape (59, 63, 64). In agreement with studies performed in vitro using tissue culture cells, virulence of Listeria in zebrafish is dependent on LLO and ActA (53). More recent work using zebrafish has shown that bacterial dissemination (via necrosis of infected macrophages and release of bacterial-containing blebs) is LLO-dependent (65). To counteract this, Gp96 (an endoplasmic reticulum chaperone) can protect the integrity of the host cell plasma membrane against pore-forming toxins. In a separate study, zebrafish infection with a Listeria strain ectopically expressing flagellin (called Lm-pyro) was shown to activate the inflammasome in macrophage and reduce infection (66). These results highlight the inflammasome as crucial for protection against Listeria.
Staphylococcus aureus is an opportunistic pathogen, which latently resides in one-third of humans. Invasive surgery or lesions can increase the risk of infection, and considering the emergence of antibiotic-resistant strains, S. aureus is recognized as a major human threat. While systemic S. aureus infections are controlled, zebrafish are susceptible to yolk sac infection (i.e., a site inaccessible to leukocytes), underscoring the importance of leukocytes for infection control (67). Experiments performed using larvae depleted of myeloid cells demonstrate a role for macrophages in restriction of S. aureus proliferation in the blood (67). Interestingly, the incomplete clearance of bacteria by leukocytes can result in an “immunological bottleneck,” viewed to select for persisting bacterial populations (68). The zebrafish can, therefore, be used to discover mechanisms used by S. aureus to evade destruction within leukocytes.
Colonization of humans by Burkholderia cenocepacia has severe consequences in cystic fibrosis (CF) patients and other immunocompromised individuals. Originally viewed to form a biofilm in CF patients, studies using clinical samples have shown that B. cenocepacia can reside in alveolar macrophages (69). More recently, a zebrafish infection model demonstrated that macrophages are crucial for B. cenocepacia replication (70). Consistent with this, depletion of macrophages from larvae restricts bacterial replication (71). During infection, macrophages express IL-1β and recruit uninfected cells to form cellular aggregates (70, 71). Paradoxically, the depletion of IL-1β (using morpholino oligonucleotide) during B. cenocepacia infection results in decreased survival, yet, inhibition of IL-1β signaling (using the IL-1 receptor antagonist anakinra) results in increased survival (71). Together, these experiments suggest the precise role of IL-1β during B. cenocepacia infection, and its manipulation for therapy, is complex.
Salmonella is a well-studied Gram-negative pathogen responsible for gastroenteritis, enteric fever, and bacteremia. Investigation of S. Typhimurium has made important contributions to macrophage biology (6, 11). During zebrafish infection, S. Typhimurium can replicate within macrophages and also extracellularly within the vasculature (72). A subpopulation of intracellular bacteria is lysed by mitochondrial-derived ROS produced by macrophages via a pathway dependent on immunoresponsive gene 1 (IRG1) (73). In addition, macrophages are responsible for the “fine-tuning” of the immune response to S. Typhimurium via secretion of granulocyte-colony stimulating factor (G-CSF), which in turn stimulates the transcription factor C/ebpβ and enhances neutrophil production by emergency granulopoiesis (74).
Shigella is a Gram-negative enteroinvasive pathogen classified by the WHO as a global threat due to its development of antibiotic resistance (75–77). Among the species of Shigella, S. flexneri is best recognized as a paradigm for studying macrophage cell death (78). In agreement with studies performed in vitro, S. flexneri can induce cell death in zebrafish macrophages in vivo (Figure 1C) (54). Despite this, macrophage-depleted transgenic zebrafish present increased mortality during infection (79). These results suggest that macrophages play an important role in the initial collection of injected bacteria, prior to the elimination of bacteria and cellular debris by neutrophils. The increasing risk of multidrug-resistant bacteria has driven the need for treatments that do not strictly rely on antibiotics. Injection of Shigella-infected zebrafish with predatory bacteria Bdellovibrio bacteriovorus revealed a synergy between predator–prey interactions with the host immune system to restrict multidrug-resistant infection (80). In this case, the reduction of Shigella burden by Bdellovibrio is beneficial for infection control by zebrafish leukocytes.
Zebrafish Macrophage–Fungus Interactions In Vivo
Invasive fungal infections are a growing problem, causing significant morbidity and mortality in organ transplant patients. Immunosuppression using calcineurin inhibitors is a common strategy for the prevention of organ transplant rejection and increases the risk of infection by Aspergillus fumigatus (81). Alveolar macrophages and inflammatory monocytes in the murine lung have been described as critical for early antifungal immunity during Aspergillus infection (82). Real-time visualization using a zebrafish infection model revealed the inability of neutrophils to phagocytose fungal spores, and suggested macrophages as crucial for host defense against A. fumigatus (83, 84). In mouse models of A. fumigatus infection, treatment with calcineurin inhibitor FK506 leads to increased mortality (85). Consistent with this, studies using zebrafish infection showed a role for calcineurin in protection against Aspergillus (86). In this case, calcineurin activation leads to dephosphorylation of nuclear factor of activated T cells (NFAT), and FK506 treatment impairs neutrophil recruitment because of reduced TNF-α production by macrophages (86). A separate study revealed FK506 inhibits the calcineurin-dependent lateral transfer of A. fumigatus from necroptotic to naïve macrophages, allowing fungal escape and unrestricted growth (87). Collectively, these studies highlight the indispensable role of calcineurin in macrophages for Aspergillus control in vivo.
Candida albicans is an opportunistic fungal pathogen, which primarily affects immunocompromised individuals. Zebrafish infection models have been used to identify C. albicans virulence factors and indicate an important role for the filamentous (hyphal) form of C. albicans in pathogenesis (88–90). Strikingly, real-time microscopy of C. albicans infection showed the extrusion of hyphae from the zebrafish hindbrain (88). Fungal dissemination is observed by 24 h postinfection (hpi), followed by lethality resulting from uncontrolled hyphal growth (89). Here, macrophages can restrict germination (but not replication), and fungal killing by macrophages and neutrophils is a rare occurrence (89). Zebrafish infection has also demonstrated a new role for NADPH oxidase in controlling hyphal growth, helping to recruit macrophages through ROS and preventing germination (89, 91).
Another opportunistic fungal pathogen, Cryptococcus neoformans can be fatal in immunocompromised individuals and is responsible for over 600,000 deaths globally per annum (92). Although highly informative, mammalian and non-vertebrate infection models have limitations in visualizing fungus-leukocyte dynamics and the translatability to higher vertebrate models, respectively. Live imaging of zebrafish during Cryptococcus infection revealed macrophages are required for pathogen control (Figure 1D) (55, 93). In agreement with this, macrophage depletion prior to infection leads to uncontrolled fungal replication and increased zebrafish mortality (55, 93). Macrophage-depletion postinfection also leads to increased fungal burden (55). The Cryptococcus capsule is made of polysaccharides contributing immunosuppressive functions, including the inhibition of phagocytosis. Consistent with this, capsule enlargement that occurs during infection of zebrafish can prevent phagocytosis, resulting in fungal proliferation and zebrafish mortality (55). By tracking individual macrophages over time, the first in vivo observation of vomocytosis (the controlled non-lytic expulsion of pathogens from phagocytes) was captured (55). The precise role of vomocytosis in host defense is not yet known.
Mucor circinelloides is an emerging fungal pathogen in which the incidence of infection is increasingly associated with aging populations (94). M. circinelloides causes mucormycosis, a disease with a wide range of symptoms including fever and gastrointestinal bleeding (95). Treatment of mucormycosis in humans remains costly and unsuccessful, and fatalities are often linked to corticosteroid treatment and immune defects (96, 97). In agreement with this, immunosuppression by corticosteroid treatment results in increased zebrafish mortality (56). Moreover, macrophage-depleted zebrafish succumb to infection, highlighting a key role for macrophages in M. circinelloides control. Remarkably, macrophages accumulate around viable spores in a manner similar to the granuloma structures described for M. marinum infection (Figure 1E) (56). While the role of macrophage clusters during M. circinelloides infection is not fully known, the zebrafish infection model can provide a novel platform to study macrophage–fungal interplay during mucormycosis.
Discussion
Here, we describe recent mechanistic insights into the macrophage response to intracellular pathogens as revealed by zebrafish infection (Table 1). Although macrophage recruitment and phagocytosis is typically observed in response to infection, this is not always followed by pathogen restriction. Zebrafish infection has shown that, in some cases, macrophages can promote pathogenesis by shielding the pathogen from immune control or by providing a replicative niche. The zebrafish is a relatively new model for the study of human infectious disease. Therefore, a limitation of the system includes the lack of tools currently available, such as zebrafish antibodies and cell lines, which can impede in-depth mechanistic studies. On the other hand, the rapid development of transgenic zebrafish lines with fluorescently tagged proteins/cells, in combination with genome-editing technologies, compensate for these limitations. Considering advancements in RNAseq and high-resolution microscopy, we can expect that zebrafish infection will continue to illuminate fundamental aspects of host–pathogen interactions at the molecular, cellular, and whole animal level. The hope is that studying macrophage–microbe interactions in vivo using the zebrafish model can deliver therapeutic impact in humans.
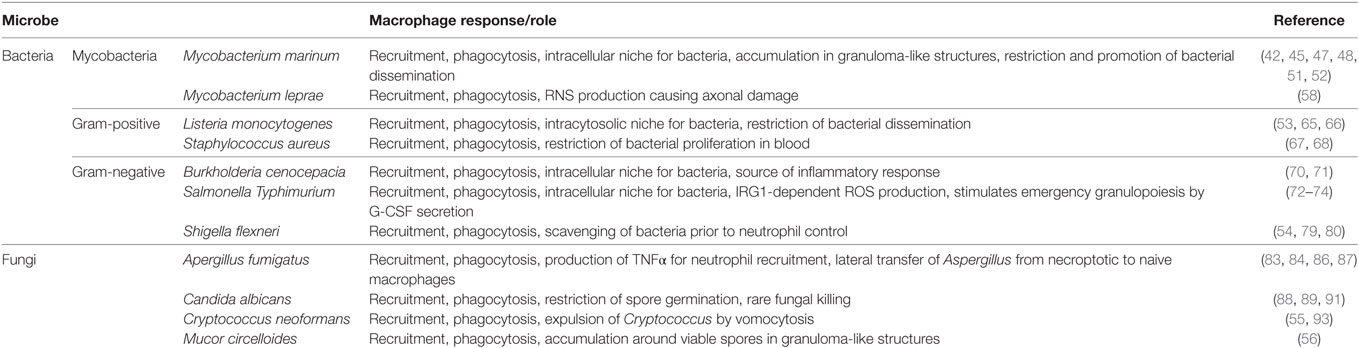
Table 1. The macrophage response/role during zebrafish infection.
Author Contributions
NY, E-MF, and SM jointly wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge Vincenzo Torraca, Gina Duggan, Alex Willis, and Joseph Wright for comments. NY is supported by the Joint Crick Ph.D. Programme with Imperial College London. Work in the Frickel laboratory is supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001076), the UK Medical Research Council (FC001076), and the Wellcome Trust (FC001076). Work in the Mostowy laboratory is supported by a Wellcome Trust Senior Research Fellowship (206444/Z/17/Z), Wellcome Trust Research Career Development Fellowship (WT097411MA), and the Lister Institute of Preventive Medicine.
References
1. Petrovski G, Zahuczky GB, Májai GN, Fésüs LS. Phagocytosis of cells dying through autophagy evokes a pro-inflammatory response in macrophages. Autophagy (2014) 3(5):508–10. doi:10.4161/auto.4731
2. Kapellos TS, Taylor L, Lee H, Cowley SA, James WS, Iqbal AJ, et al. A novel real time imaging platform to quantify macrophage phagocytosis. Biochem Pharmacol (2016) 116:107–19. doi:10.1016/j.bcp.2016.07.011
3. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumours and other ischemic tissues. Blood (2004) 104(8):2224–34. doi:10.1182/blood-2004-03-1109
4. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol (2014) 5(11):491. doi:10.3389/fimmu.2014.00491
5. Aderem A. Phagocytosis and the inflammatory response. J Infect Dis (2003) 187(Suppl2):S340–5. doi:10.1086/374747
6. Torraca V, Masud S, Spaink HP, Meijer AH. Macrophage-pathogen interactions in infectious diseases: new therapeutic insights from the zebrafish host model. Dis Model Mech (2014) 7(7):785–97. doi:10.1242/dmm.015594
7. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res (2010) 20(1):34–50. doi:10.1038/cr.2009.139
8. Yates RM, Hermetter A, Russell DG. The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic (2005) 6(5):413–20. doi:10.1111/j.1600-0854.2005.00284.x
9. Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol (2008) 9(10):781–95. doi:10.1038/nrm2515
10. Slauch JM. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol (2011) 80(3):580–3. doi:10.1111/j.1365-2958.2011.07612.x
11. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Ann Rev Immunol (1999) 17(1):593–623. doi:10.1146/annurev.immunol.17.1.593
12. Flannagan RS, Jaumouillé V, Grinstein S. The cell biology of phagocytosis. Ann Rev Pathol (2012) 7(1):61–98. doi:10.1146/annurev-pathol-011811-132445
13. Ray K, Marteyn B, Sansonetti PJ, Tang CM. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol (2009) 7(5):333–40. doi:10.1038/nrmicro2112
14. Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science (2013) 340(6133):701–6. doi:10.1126/science.1233028
15. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469(7330):323–35. doi:10.1038/nature09782
16. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi:10.1038/nri3532
17. Dikic I. Proteasomal and autophagic degradation systems. Ann Rev Biochem (2017) 86(1):193–224. doi:10.1146/annurev-biochem-061516-044908
18. Ivanov S, Roy CR. NDP52: the missing link between ubiquitinated bacteria and autophagy. Nat Immunol (2009) 10(11):1137–9. doi:10.1038/ni1109-1137
19. Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell (2009) 34(3):259–69. doi:10.1016/j.molcel.2009.04.026
20. Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell (2014) 54(2):224–33. doi:10.1016/j.molcel.2014.03.009
21. Clough B, Frickel E-M. The Toxoplasma parasitophorous vacuole: an evolving host-parasite frontier. Trends Parasitol (2017) 33(6):473–88. doi:10.1016/j.pt.2017.02.007
22. Meunier E, Broz P. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell Microbiol (2015) 18(2):168–80. doi:10.1111/cmi.12546
23. Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, et al. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature (2017) 551(7680):378–83. doi:10.1038/nature24467
24. Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, von der Malsburg A, et al. GBPs inhibit motility of Shigella flexneri but are targeted for degradation by the bacterial ubiquitin ligase IpaH9.8. Cell Host Microbe (2017) 22(4):507–18.e5. doi:10.1016/j.chom.2017.09.007
25. Kim B-H, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science (2011) 332(6030):717–21. doi:10.1126/science.1201711
26. Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science (2012) 336(6080):481–5. doi:10.1126/science.1217141
27. Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, et al. IRG and GBP host resistance factors target aberrant, "non-self" vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog (2013) 9(6):e1003414. doi:10.1371/journal.ppat.1003414
28. Franchi L, Warner N, Viani K, Nuñez G. Function of NOD-like receptors in microbial recognition and host defense. Immunol Rev (2009) 227(1):106–28. doi:10.1111/j.1600-065X.2008.00734.x
29. Kanneganti T-D, Lamkanfi M, Nuñez G. Intracellular NOD-like receptors in host defense and disease. Immunity (2007) 27(4):549–59. doi:10.1016/j.immuni.2007.10.002
30. Geddes K, Magalhães JG, Girardin SE. Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Discov (2009) 8(6):465–79. doi:10.1038/nrd2783
31. Sahoo M, Ceballos-Olvera I, del Barrio L, Re F. Role of the inflammasome, IL-1β, and IL-18 in bacterial infections. ScientificWorldJournal (2011) 11(2):2037–50. doi:10.1100/2011/212680
32. Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrate immunity. Dis Model Mech (2012) 5(1):38–47. doi:10.1242/dmm.007138
33. Novoa B, Figueras A. Zebrafish: model for the study of inflammation and the innate immune response to infectious diseases. Adv Exp Med Biol (2012) 946:253–75. doi:10.1007/978-1-4614-0106-3_15
34. Masud S, Torraca V, Meijer AH. Modelling infectious diseases in the context of a developing immune system. Curr Top Dev Biol (2017) 124:277–329. doi:10.1016/bs.ctdb.2016.10.006
35. Meeker ND, Trede NS. Immunology and zebrafish: spawning new models of human disease. Dev Comp Immunol (2008) 32(7):745–57. doi:10.1016/j.dci.2007.11.011
36. Herbomel P, Thisse B, Thisse C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development (1999) 126(17):3735–45.
37. Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood (2011) 117(4):e49–56. doi:10.1182/blood-2010-10-314120
38. Gray C, Loynes CA, Whyte MKB, Crossman DC, Renshaw SA, Chico TJA. Simultaneous intravital imaging of macrophage and neutrophil behaviour during inflammation using a novel transgenic zebrafish. Thromb Haemost (2011) 105(5):811–9. doi:10.1160/TH10-08-0525
39. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature (2013) 496(7446):498–503. doi:10.1038/nature12111
40. Blum M, De Robertis EM, Wallingford JB, Niehrs C. Morpholinos: antisense and sensibility. Dev Cell (2015) 35(2):145–9. doi:10.1016/j.devcel.2015.09.017
41. Li M, Zhao L, Page-McCaw PS, Chen W. Zebrafish genome engineering using the CRISPR-Cas9 system. Trends Genet (2016) 32(12):815–27. doi:10.1016/j.tig.2016.10.005
42. Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity (2002) 17(6):693–702. doi:10.1016/S1074-7613(02)00475-2
43. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell (2009) 136(1):37–49. doi:10.1016/j.cell.2008.11.014
44. Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science (2010) 327(5964):466–9. doi:10.1126/science.1179663
45. Pagán AJ, Yang CT, Cameron J, Swaim LE, Ellett F, Lieschke GJ, et al. Myeloid growth factors promote resistance to mycobacterial infection by curtailing granuloma necrosis through macrophage replenishment. Cell Host Microbe (2015) 18(1):15–26. doi:10.1016/j.chom.2015.06.008
46. Cambier CJ, O’Leary SM, O’Sullivan MP, Keane J, Ramakrishnan L. Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity (2017) 47(3):552–65.e4. doi:10.1016/j.immuni.2017.08.003
47. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature (2013) 505(7482):218–22. doi:10.1038/nature12799
48. Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity (2016) 45(4):861–76. doi:10.1016/j.immuni.2016.09.014
49. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell (2013) 153(3):521–34. doi:10.1016/j.cell.2013.03.022
50. Tobin DM, Vary JC, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell (2010) 140(5):717–30. doi:10.1016/j.cell.2010.02.013
51. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature (2015) 517(7536):612–5. doi:10.1038/nature13967
52. Torraca V, Tulotta C, Snaar-Jagalska BE, Meijer AH. The chemokine receptor CXCR4 promotes granuloma formation by sustaining a mycobacteria-induced angiogenesis programme. Sci Rep (2017) 7:45061. doi:10.1038/srep45061
53. Levraud J-P, Disson O, Kissa K, Bonne I, Cossart P, Herbomel P, et al. Real-time observation of Listeria monocytogenes-phagocyte interactions in living zebrafish larvae. Infect Immun (2009) 77(9):3651–60. doi:10.1128/IAI.00408-09
54. Mostowy S, Boucontet L, Mazon Moya MJ, Sirianni A, Boudinot P, Hollinshead M, et al. The zebrafish as a new model for the in vivo study of Shigella flexneri interaction with phagocytes and bacterial autophagy. PLoS Pathog (2013) 9(9):e1003588. doi:10.1371/journal.ppat.1003588
55. Bojarczuk A, Miller KA, Hotham R, Lewis A, Ogryzko NV, Kamuyango AA, et al. Cryptococcus neoformans intracellular proliferation and capsule size determines early macrophage control of infection. Sci Rep (2016) 6:21489. doi:10.1038/srep21489
56. Voelz K, Gratacap RL, Wheeler RT. A zebrafish larval model reveals early tissue-specific innate immune responses to Mucor circinelloides. Dis Model Mech (2015) 8(11):1375–88. doi:10.1242/dmm.019992
57. Balamayooran G, Pena M, Sharma R, Truman RW. The armadillo as an animal model and reservoir host for Mycobacterium leprae. Clin Dermatol (2015) 33(1):108–15. doi:10.1016/j.clindermatol.2014.07.001
58. Madigan CA, Cambier CJ, Kelly-Scumpia KM, Scumpia PO, Cheng TY, Zailaa J, et al. A macrophage response to Mycobacterium leprae phenolic glycolipid initiates nerve damage in leprosy. Cell (2017) 170(5):973–85.e10. doi:10.1016/j.cell.2017.07.030
59. Cossart P. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci U S A (2011) 108(49):19484–91. doi:10.1073/pnas.1112371108
60. David DJV, Cossart P. Recent advances in understanding Listeria monocytogenes infection: the importance of subcellular and physiological context. F1000Res (2017) 6:1126. doi:10.12688/f1000research.11363.1
61. Chen GY, Pensinger DA, Sauer J-D. Listeria monocytogenes cytosolic metabolism promotes replication, survival and evasion of innate immunity. Cell Microbiol (2017) 19(10):e12762. doi:10.1111/cmi.12762
62. Seveau S. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Subcell Biochem (2014) 80:161–95. doi:10.1007/978-94-017-8881-6_9
63. Birmingham CL, Higgins DE, Brumell JH. Avoiding death by autophagy: interactions of Listeria monocytogenes with the macrophage autophagy system. Autophagy (2008) 4(3):368–71. doi:10.4161/auto.5594
64. Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol (2009) 11(10):1233–40. doi:10.1038/ncb1967
65. Mesquita FS, Brito CU, Mazon Moya MJ, Pinheiro JC, Mostowy S, Cabanes D, et al. Endoplasmic reticulum chaperone Gp96 controls actomyosin dynamics and protects against pore-forming toxins. EMBO Rep (2017) 18(2):303–18. doi:10.15252/embr.201642833
66. Vincent WJB, Freisinger CM, Lam PY, Huttenlocher A, Sauer JD. Macrophages mediate flagellin induced inflammasome activation and host defense in zebrafish. Cell Microbiol (2015) 18(4):591–604. doi:10.1111/cmi.12536
67. Prajsnar TK, Cunliffe VT, Foster SJ, Renshaw SA. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell Microbiol (2008) 10(11):2312–25. doi:10.1111/j.1462-5822.2008.01213.x
68. Prajsnar TK, Hamilton R, Garcia-Lara J, McVicker G, Williams A, Boots M, et al. A privileged intraphagocyte niche is responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cell Microbiol (2012) 14(10):1600–19. doi:10.1111/j.1462-5822.2012.01826.x
69. Sajjan U, Corey M, Humar A, Tullis E, Cutz E, Ackerley C, et al. Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J Med Microbiol (2001) 50(6):535–46. doi:10.1099/0022-1317-50-6-535
70. Vergunst AC, Meijer AH, Renshaw SA, O’Callaghan D. Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect Immun (2010) 78(4):1495–508. doi:10.1128/IAI.00743-09
71. Mesureur J, Feliciano JR, Wagner N, Gomes MC, Zhang L, Blanco-Gonzalez M, et al. Macrophages, but not neutrophils, are critical for proliferation of Burkholderia cenocepacia and ensuing host-damaging inflammation. PLoS Pathog (2017) 13(6):e1006437. doi:10.1371/journal.ppat.1006437
72. van der Sar AM, Musters RJP, van Eeden FJM, Appelmelk BJ, Vandenbroucke-Grauls CMJE, Bitter W. Zebrafish embryos as a model host for the real time analysis of Salmonella typhimurium infections. Cell Microbiol (2003) 5(9):601–11. doi:10.1046/j.1462-5822.2003.00303.x
73. Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, et al. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating β-oxidation-dependent mitochondrial ROS production. Cell Metab (2013) 18(2):265–78. doi:10.1016/j.cmet.2013.06.018
74. Hall CJ, Flores MV, Oehlers SH, Sanderson LE, Lam EY, Crosier KE, et al. Infection-responsive expansion of the hematopoietic stem and progenitor cell compartment in zebrafish is dependent upon inducible nitric oxide. Cell Stem Cell (2012) 10(2):198–209. doi:10.1016/j.stem.2012.01.007
75. Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, et al. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ (1999) 77(8):651–66.
76. Jennison AV, Verma NK. Shigella flexneri infection: pathogenesis and vaccine development. FEMS Microbiol Rev (2004) 28(1):43–58. doi:10.1016/j.femsre.2003.07.002
77. Puzari M, Sharma M, Chetia P. Emergence of antibiotic resistant Shigella species: a matter of concern. J Infect Public Health (2017). doi:10.1016/j.jiph.2017.09.025
78. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358(6382):167–9. doi:10.1038/358167a0
79. Mazon Moya MJ, Willis AR, Torraca V, Boucontet L, Shenoy AR, Colucci-Guyon E, et al. Septins restrict inflammation and protect zebrafish larvae from Shigella infection. PLoS Pathog (2017) 13(6):e1006467. doi:10.1371/journal.ppat.1006467
80. Willis AR, Moore C, Mazon-Moya M, Krokowski S, Lambert C, Till R, et al. Injections of predatory bacteria work alongside host immune cells to treat Shigella infection in zebrafish larvae. Curr Biol (2016) 26(24):3343–51. doi:10.1016/j.cub.2016.09.067
81. Sugui JA, Kwon-Chung KJ, Juvvadi PR, Latgé J-P, Steinbach WJ. Aspergillus fumigatus and related species. Cold Spring Harb Perspect Med (2014) 5(2):a019786. doi:10.1101/cshperspect.a019786
82. Espinosa V, Jhingran A, Dutta O, Kasahara S, Donnelly R, Du P, et al. Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog (2014) 10(2):e1003940. doi:10.1371/journal.ppat.1003940
83. Knox BP, Deng Q, Rood M, Eickhoff JC, Keller NP, Huttenlocher A. Distinct innate immune phagocyte responses to Aspergillus fumigatus conidia and hyphae in zebrafish larvae. Eukaryot Cell (2014) 13(10):1266–77. doi:10.1128/EC.00080-14
84. Knox BP, Huttenlocher A, Keller NP. Real-time visualization of immune cell clearance of Aspergillus fumigatus spores and hyphae. Fungal Genet Biol (2017) 105:52–4. doi:10.1016/j.fgb.2017.05.005
85. Herbst S, Shah A, Carby M, Chusney G, Kikkeri N, Dorling A, et al. A new and clinically relevant murine model of solid-organ transplant aspergillosis. Dis Model & Mech (2013) 6(3):643–51. doi:10.1242/dmm.010330
86. Herbst S, Shah A, Mazon-Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med (2015) 7(3):240–58. doi:10.15252/emmm.201404556
87. Shah A, Kannambath S, Herbst S, Rogers A, Soresi S, Carby M, et al. Calcineurin orchestrates lateral transfer of Aspergillus fumigatus during macrophage cell death. Am J Respir Crit Care Med (2016) 194(9):1127–39. doi:10.1164/rccm.201601-0070OC
88. Chao CC, Hsu PC, Jen CF, Chen IH, Wang CH, Chan HC, et al. Zebrafish as a model host for Candida albicans infection. Infect Immun (2010) 78(6):2512–21. doi:10.1128/IAI.01293-09
89. Brothers KM, Newman ZR, Wheeler RT. Live imaging of disseminated candidiasis in zebrafish reveals role of phagocyte oxidase in limiting filamentous growth. Eukaryot Cell (2011) 10(7):932–44. doi:10.1128/EC.05005-11
90. Chen YZ, Yang YL, Chu WL, You MS, Lo HJ. Zebrafish egg infection model for studying Candida albicans adhesion factors. PLoS One (2015) 10(11):e0143048. doi:10.1371/journal.pone.0143048
91. Brothers KM, Gratacap RL, Barker SE, Newman ZR, Norum A, Wheeler RT. NADPH oxidase-driven phagocyte recruitment controls Candida albicans filamentous growth and prevents mortality. PLoS Pathog (2013) 9(10):e1003634. doi:10.1371/journal.ppat.1003634
92. Mada PK, Alam MU. Cryptococcus (Cryptococcosis). Treasure Island, FL: StatPearls Publishing (2017).
93. Tenor JL, Oehlers SH, Yang JL, Tobin DM, Perfect JR. Live imaging of host-parasite interactions in a zebrafish infection model reveals cryptococcal determinants of virulence and central nervous system invasion. MBio (2015) 6(5):e01425–15. doi:10.1128/mBio.01425-15
94. Ibrahim AS, Kontoyiannis DP. Update on mucormycosis pathogenesis. Curr Opin Infect Dis (2013) 26(6):508–15. doi:10.1097/QCO.0000000000000008
95. Pak J, Tucci VT, Vincent AL, Sandin RL, Greene JN. Mucormycosis in immunochallenged patients. J Emerg Trauma Shock (2008) 1(2):106–13. doi:10.4103/0974-2700.42203
96. Waldorf AR, Levitz SM, Diamond RD. In vivo bronchoalveolar macrophage defense against Rhizopus oryzae and Aspergillus fumigatus. J Infect Dis (1984) 150(5):752–60. doi:10.1093/infdis/150.5.752
Keywords: host–pathogen interactions, infection, inflammation, macrophage, zebrafish
Citation: Yoshida N, Frickel E-M and Mostowy S (2017) Macrophage–Microbe Interactions: Lessons from the Zebrafish Model. Front. Immunol. 8:1703. doi: 10.3389/fimmu.2017.01703
Received: 19 October 2017; Accepted: 20 November 2017;
Published: 01 December 2017
Edited by:
Etienne Meunier, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Jessica Lynn Humann, Florida A&M University, United StatesMatthias Hauptmann, Research Center Borstel, Germany
Copyright: © 2017 Yoshida, Frickel and Mostowy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva-Maria Frickel, ZXZhLmZyaWNrZWxAY3JpY2suYWMudWs=;
Serge Mostowy, cy5tb3N0b3d5QGltcGVyaWFsLmFjLnVr