 Samuel Maldonado
Samuel Maldonado Patricia Fitzgerald-Bocarsly
Patricia Fitzgerald-Bocarsly- 1Rutgers School of Graduate Studies, Newark, NJ, United States
- 2Department of Pathology and Laboratory Medicine, New Jersey Medical School, Newark, NJ, United States
Due to the effectiveness of combined antiretroviral therapy, people living with HIV can control viral replication and live longer lifespans than ever. However, HIV-positive individuals still face challenges to their health and well-being, including dysregulation of the immune system resulting from years of chronic immune activation, as well as opportunistic infections from pathogenic fungi. This review focuses on one of the key players in HIV immunology, the plasmacytoid dendritic cell (pDC), which links the innate and adaptive immune response and is notable for being the body’s most potent producer of type-I interferons (IFNs). During chronic HIV infection, the pDC compartment is greatly dysregulated, experiencing a substantial depletion in number and compromise in function. This immune dysregulation may leave patients further susceptible to opportunistic infections. This is especially important when considering a new role for pDCs currently emerging in the literature: in addition to their role in antiviral immunity, recent studies suggest that pDCs also play an important role in antifungal immunity. Supporting this new role, pDCs express C-type lectin receptors including dectin-1, dectin-2, dectin-3, and mannose receptor, and toll-like receptors-4 and -9 that are involved in recognition, signaling, and response to a wide variety of fungal pathogens, including Aspergillus fumigatus, Cryptococcus neoformans, Candida albicans, and Pneumocystis jirovecii. Accordingly, pDCs have been demonstrated to recognize and respond to certain pathogenic fungi, measured via activation, cytokine production, and fungistatic activity in vitro, while in vivo mouse models indicated a strikingly vital role for pDCs in survival against pulmonary Aspergillus challenge. Here, we discuss the role of the pDC compartment and the dysregulation it undergoes during chronic HIV infection, as well as what is known so far about the role and mechanisms of pDC antifungal activity.
Introduction
An estimated 33 million people were living with HIV/AIDS worldwide in 2015 (1). Although the development of combined antiretroviral therapy has allowed HIV patients to suppress viral replication and live longer lifespans (2, 3), HIV evades the immune system and persists, causing chronic immune activation and inflammation (4). The persistence of infection and immune activation leads to significant detrimental changes that occur in the body, and pose major threats to patients’ well-being; in fact, in 2010, HIV/AIDS was still the fifth leading cause of disease burden worldwide (5).
Arguably the most significant pathological process faced by treated and untreated patients alike is the erosion of the immune system. HIV infection famously depletes CD4+ T-helper (TH) cells, first quickly in the acute phase of infection, followed by a near-total recovery in numbers, and finally a steady decline over a number of years (4) leaving the adaptive immune system at a tremendous disadvantage. T-cell depletion occurs via mechanisms such as direct infection, bystander effects due to chronic inflammation, and senescence (4, 6). The dysfunction of the immune system is also much broader; almost every known immune cell type has been associated with a dysfunction due to chronic HIV infection. Because HIV gains access to a cell via its interaction with CD4 and co-receptors CXCR4 and CCR5, it also has the capacity to infect other cells that express those receptors, including monocytes, macrophages, and plasmacytoid dendritic cells (pDCs) (4). In addition, the immune system may be further dysregulated due to chronic innate immune activation and the continued production of normally beneficial cytokines (7).
This erosion of the immune system leaves patients hyper-susceptible to a variety of opportunistic infections, including fungal infections not commonly seen in the general population. For example, Candida albicans infection is the most common fungal infection in HIV patients. While it usually presents as oropharyngeal thrush, it can span a broad spectrum of severity from asymptomatic to invasive candidiasis (8). The most common systemic fungal infection in the HIV population is cryptococcal meningitis, caused by Cryptococcus neoformans. Cryptococcal meningitis causes 181,000 deaths a year in HIV-positive patients, accounting for 15% of global AIDS-related deaths (9, 10). Aspergillus fumigatus is the causative agent of invasive Aspergillosis, which is less common in the context of HIV infection but is particularly aggressive and difficult to treat, resulting in a median survival of only 3 months after diagnosis (11, 12). These and other pathogenic fungi take advantage of an HIV patient’s impaired immune system. Studies early in the HIV epidemic suggested that HIV patients are much more likely to develop opportunistic infections when two events occur: absolute CD4+ cell counts drop below 250 cells/mm3, and interferon (IFN)-α production by virus-stimulated peripheral blood mononuclear cells (PBMCs) drops below 300 IU/ml (13). It is the latter that makes pDCs a cell type of great interest when studying HIV infection, as they are the body’s most potent producers of type-I IFNs. pDCs produce up to 100-fold more IFN-α than any other cell type in response to viral stimulation and serve as an important link between the innate and the adaptive branches of the immune system (14–16). pDC IFN production and influence over the rest of the immune response has made these cells the subject of extensive investigation in human and mouse models. Importantly, murine systems do not perfectly represent human ones, and studies on mouse pDC function must also be investigated in humans. For example, toll-like receptor (TLR) 9 is broadly expressed by murine myeloid cells, but is more limited to pDCs and B-cells in humans (17). Similarly, murine pDCs commonly produce IL-12 while human pDCs rarely, if ever, produce IL-12 (18).
Plasmacytoid dendritic cell dysregulation during chronic HIV infection has garnered attention due to the potentially far-reaching effects on patient outcome and well-being. However, an interesting development in the field of pDC research is the investigation into their role in fungal infection. Studies have demonstrated that pDCs have the machinery needed to recognize and respond to fungal stimulation, that they in fact do respond with certain cellular functions, and that they are necessary in vivo for a successful antifungal immune response. In this review, we outline the current paradigm concerning pDCs’ role in viral immunity and describe the dysregulation of the pDC compartment during chronic HIV infection. We then switch gears to the role of pDCs in antifungal immunity, where we highlight the components of fungal immunity that pDCs possess, and summarize what has been discovered to date concerning the mechanisms of pDC antifungal activity.
pDCs in Antiviral Immunity
Both the innate and the adaptive immune responses play active roles in combating viral infections such as HIV. The adaptive immune response includes a strong CD4+ and CD8+ T-cell response, as well as the production of neutralizing antibodies that begins during the acute phase of infection (19, 20). Mobilization of the adaptive immune response depends in part on pDCs, a lineage-negative subtype of dendritic cells that derives its name from a plasma-cell-like morphology. Although pDCs comprise only 0.2–0.8% of PBMC (14, 15, 21), their powerful immunomodulating capabilities make pDCs highly influential to the development of the immune response to HIV infection, and a lack of sufficient pDC numbers or function corresponds with high viral loads and opportunistic infections (22).
Human pDCs are generally found in peripheral blood and lymphoid tissue, so if an infection occurs in a different tissue (e.g., the lung or mucosa), they must migrate to sites of infection or inflammation by responding to chemokines. This occurs primarily via the chemokine receptors CMKLR1, CCR7, CXCR3, and CXCR4 (22–24) as well as through homing molecules, such as CD62L, which binds L-selectin and allows pDCs to track to high endothelial venules and the gut homing receptor α4β7 (25). Once at the site of infection, pDCs directly recognize and respond to viral stimulation. The initial interaction between pDCs and HIV virions is possible because pDCs express the surface molecules that are targeted by HIV: CD4, CCR4, and CXCR5, as reviewed in Ref. (16). The interaction between the virus and CD4 allows the pDC to endocytose the virus, which induces activation in a TLR-dependent or -independent manner.
In the TLR-dependent pathway, the endocytosed virus is delivered to endosomes in pDC that contain TLR-7 and 9. Acidification of the endosomes allows TLR-7 and 9 to access viral RNA or unmethylated CpG motifs, including that in viral DNA, respectively (26, 27). These TLRs have the capacity to recognize a host of viral, bacterial, and fungal nucleic acids, as well as small molecule artificial ligands such as imiquimod and resiquimod (28, 29). Ligation of TLR-7 or 9 induces a MyD88-dependent activation pathway that ultimately leads to pDC activation and cytokine production (30). Although less well-characterized than the TLR-dependent pathway, evidence exists for a TLR-independent activation pathway in pDCs. For example, one study found that IFN-α production by murine pDCs in response to Sendai virus was not abrogated in MyD88−/− mice and was not dependent on endosomal acidification, both of which are required for TLR9 signaling (31). Another study found that murine pDC IFN-α production in response to HSV-1 was not completely dependent on TLR9 (32). However, specific mechanisms of TLR-independent pDC activation and cytokine production are still under investigation.
Both the TLR-dependent and -independent signaling pathways converge on interferon regulatory factor (IRF) 7, the master regulator of IFN production which is constitutively expressed by pDCs at uniquely high levels (30, 33, 34). Phosphorylation of IRF7 causes it to translocate into the nucleus and induce the transcription of IFN genes, producing high concentrations of type-I IFN (35). TLR-7 and -9 ligation also lead to the activation of transcription factor NF-κB, which induces the production of inflammatory cytokines/chemokines and the upregulation of co-stimulatory molecules (21, 36). Activated pDCs can then produce type-I IFNs, type-III IFNs, and a network of other cytokines and chemokines. They also have the ability to become antigen-presenting cells, at which point they develop a morphology resembling conventional DCs (cDCs) (36–38).
Perhaps the most significant contribution of pDCs to antiviral immunity is the production of massive amounts of type-I IFNs (mainly α and β) in response to viral stimulation. Most notably, pDCs can produce 100–1,000 times more type-I IFN than any other cell type (39–41). It has even been reported that as much as 60% of genes expressed in an activated pDC are dedicated to the production of type-I and type-III IFNs (42). Type-I IFNs all bind the same receptor, the IFN-α/β receptor, which is widely expressed by most nucleated cells in the body. The IFN-α/β receptor is composed of two subunits, IFNAR1 and IFNAR2, that form a heterodimer upon ligation of type-I IFN (43). IFNAR-associated Janus kinases then phosphorylate STAT1 and STAT2, which form a complex with IRF9 called the ISGF3 complex. The resulting signaling cascade activates gene transcription for factors that produce an antiviral state in surrounding tissues and mobilizes virtually all types of immune cells to combat viral infection (44, 45). For example, IFN-α acts as a survival, maturation, and cytokine/chemokine production factor for pDCs (46, 47) and cDCs (48), activates natural killer cells (26), biases the immune system toward a TH1 response (49, 50), primes CD8+ T-cells and induces memory CD8+ T-cells (51–53), promotes the development of regulatory T (Treg) cells (54), and enhances antibody production against soluble antigen (55). These effects, among others, are critical for the inhibition of viral replication and are why type-I IFNs are arguably the most important cytokines in antiviral immunity.
In addition to type-I IFNs, pDCs also produce type-III IFNs (IFN-λ1–3) after viral stimulation (38). The IFN-λ receptor is expressed on a much more limited scale than the IFN-α/β receptor—primarily epithelial cells and pDCs (38, 56)—suggesting that the biological activity of IFN-λ is more narrow than that of type-I IFN. However, type-I and type-III IFN share many common features: their signaling pathways include many of the same factors, including STAT1, STAT2, and the formation of the ISGF3 complex; they also activate several common genes (57, 58). Functionally, IFN-λ also has antiviral effects, especially for the protection of epithelial cells (59), the enhancement of TLR-mediated signaling in pDCs (56), development of Treg cells (60), and biasing toward a TH1 response by reducing IL-13 expression (61). However, it has also been reported that IFN-λ can be antagonistic to type-I IFN antiviral activity (62).
Other than IFNs, pDCs are able to produce several other cytokines and chemokines. NF-κB activation after viral stimulation induces pDCs to produce inflammatory cytokines and chemokines such as IL-12 (in murine but not human pDC), TNF-α, RANTES, IL-10, IP-10, IL-6, MIP-1α, and MIP-1β (36, 63). One study identified 12 chemokines that are produced in three distinct waves by pDCs in the hours following influenza virus infection (64). Furthermore, pDCs are also capable of other innate immune functions. Although resting pDCs are less efficient than myeloid DCs at capturing, processing, and presenting antigen (65, 66), activation of pDCs in response to influenza virus induces a change into a dendritic morphology and the upregulation of MHC-II, enhancing their ability to present antigen to naïve and memory T-cells (67, 68). In 2012, Tel et al. reported that pDCs could not only present tumor antigens but could also directly kill tumor cells. They termed these cells “killer pDCs” (69). Another interesting feature of the pDC is the expression of several surface receptors usually restricted to myeloid cells. On pDCs, these receptors include the C-type lectin receptors (CLRs) dectin-1, dectin-2, dectin-3, and mannose receptor (MR) (70–72), which will be discussed later. These receptors are of particular interest because they are involved in antifungal immunity.
pDC Dysregulation During Chronic HIV Infection
Despite the impressive antiviral functions of pDCs, HIV still manages to evade the immune system and persist in the body. The observation that HIV patient blood cells produced less IFN after stimulation led to the discovery of what were termed natural IFN-producing cells, which would later be known as pDCs (73–76). After the discovery of pDCs as the major IFN-α-producing cell, observations surfaced that showed chronic HIV infection leads to a decline in pDC number and function (77). The decline in absolute pDC numbers correlated directly with a decline in CD4+ T-cell count and increased viral load (22, 78), while pDC functional decline (percent of IFN-α-producing pDC after viral stimulation) correlated with an increase in patient viral load, but not CD4+ T-cell count (76). Although ART treatment results in a partial recovery of pDC numbers, they do not make a full recovery (79). The specific cause of this decline in pDC number is still under investigation. Studies on macaque models indicate that pDCs are recruited to lymph nodes where they die rapidly (80, 81); this was supported by a study that found higher numbers and accelerated pDC death in human lymph nodes (82). Other studies show that pDCs are susceptible to HIV infection, which can cause syncytia formation and cell death (83, 84). Other possible mechanisms include recruitment to mucosal tissues, bystander apoptosis similar to T-cells, and bone marrow suppression (85).
Along with lower numbers, HIV also causes functional differences in pDCs. One of the earliest observations in HIV patients was the high levels of circulating IFN-α seen in some untreated patients, which was associated with disease progression (13, 86). The pDCs that accumulate in HIV patient lymph nodes were also described as producing significantly higher levels of IFN-α than non-infected healthy controls (82). This suggests that at certain points during infection, pDCs are chronically activated and are continuously producing IFN-α. This chronic immune activation may be driving pathology, including the immune exhaustion of T-cell compartments (7, 87). Supporting this hypothesis, one study found that humanized pDC-depleted mice infected with HIV-1 show dramatically reduced cell death and immune cell depletion, a phenomenon they suggested stems from the lack of persistent pDC activation and IFN production (88). In addition, other pDC functions are found to be dysregulated in HIV patients: pDCs become less able to stimulate T-cell proliferation in mixed lymphocyte reactions (89), show a partial activation phenotype (90), and have a reduced ability to respond to TLR7 and -9 agonists (91).
Another area of research concerns the possibility that pDCs may be inducing the differentiation of human Treg cells that lead to tolerogenicity and suppression of T-cell proliferation during chronic HIV infection. Human pDCs have been shown to induce both CD8+ and CD4+ Treg cell development in certain circumstances. For example, pDCs stimulated with type B CpG oligodeoxynucleotides (CpGb) induced CD4+CD25− naïve T-cells to differentiate into CD4+CD25+ Treg cells, which suppressed T-cell proliferation and promoted tolerance (92). Another study found that pDCs activated with CD40L induced naïve CD8+ T-cells to proliferate, but the resulting CD8+ T-cells were poorly cytotoxic, had poor responses to secondary re-stimulation, and produced IL-10 (93). To that end, a study by Boasso et al. (94) found that T-cell proliferation in HIV-infected patients was being inhibited by pDCs through the expression of indoleamine-2,3-dioxygenase (IDO) (94). IDO is the first and rate-limiting enzyme in the catabolism of tryptophan, and its expression can be used by APCs as an immune modulator to limit T-cell proliferation (95). In the study, IDO mRNA expression in the blood of HIV-infected patients was directly correlated with viral load, and was responsible for impaired CD4+ T-cell proliferation. Using flow cytometric analysis, they found that pDCs were the main producers of IDO, and that pDC IDO expression was a direct response to HIV that did not require the presence of other leukocytes. This phenomenon may represent yet another way that pDCs contribute to the dysfunction of the immune system during chronic HIV infection. In addition, another study found that IDO expression by pDCs blocks T-cell differentiation into TH17 cells, which could further negatively impact adaptive immunity to fungal infection (96). This dysfunction, along with the dysregulation of virtually every immune cell type, leaves patients at higher risk of opportunistic infections and other comorbidities, including fungal infections with C. albicans and C. neoformans.
Select Components of the Immune Response to Fungal Infection
A discussion of HIV patient susceptibility to fungal infections requires an understanding of normal antifungal immunity. Although some fungi are commensal with the human host, this review focuses on pathogenic fungal infections. Fungal pathogens, such as A. fumigatus and C. neoformans, tend to be ubiquitous to the environment and are introduced into the human host through inhalation of spores or small yeast cells (97, 98). In the case of A. fumigatus and other sporulating fungi, spores (termed “resting conidia,” RC) that reach a suitable host begin to swell (termed “swollen conidia,” SC), and germinate (termed “germinating conidia,” GC), finally growing into filamentous hyphae (99). Fortunately, fungal pathogens express pathogen-associated molecular patterns (PAMPs), which are not found on mammalian cells and alert the immune system to the presence of non-self organisms (100). PAMPs can vary depending on the species, growth stage, and environment of the fungus (101, 102). Three significant PAMPs on fungal cell walls include β-glucans (polymers of glucose, typically with 1,3 linkages and occasional 1,6 branches), chitin (a polymer of N-acetylglucosamine), and mannans (polymers of mannose linked to fungal proteins) (97, 103). Fungi that make it into a human host can be recognized by innate immune cells through pattern recognition receptors (PRRs), such as TLRs and CLRs (100).
Once the PAMPs on fungi are recognized by cellular PRRs, a complex immune response begins that, incorporates both the innate and adaptive branches of the immune system (104). Phagocytes, such as monocytes, neutrophils, and macrophages, phagocytose and kill fungal pathogens directly (105), spread out over hyphae and kill them via oxidative and non-oxidative means, and in the case of neutrophils produce extracellular traps (106, 107). In addition to uptake and direct killing, cDCs also transport and present fungal antigen to T-cells and induce their differentiation into TH1, TH17, or Treg cells, depending on the environment and the fungus (108–110). While TH1 cells are thought to be the most protective, patients with defects in the TH1 compartment are not overly susceptible to certain fungal infections (97). This is because the TH17 compartment is also protective and sometimes essential to fungal clearance, depending on the species and route of infection (111). Conversely, inborn errors in IL-17 immunity can result in chronic mucocutaneous candidiasis in humans (112), and more broad susceptibility to C. albicans infection in mice, including cutaneous and oropharyngeal infection (113). Finally, the development of protective antibodies after vaccination of mice with C. albicans adhesins suggests that humoral immunity may also get involved during fungal infection, although in those studies the main effector cells responsible for vaccine efficacy were CD4+ T-cells, and neither B-cell transfer nor passive immunization with serum of vaccinated mice were protective to unvaccinated mice (114).
The TLRs involved in fungal recognition that are expressed on pDCs are TLR4 and TLR9 (17, 35). TLR4 (CD284) is mainly known for recognizing bacterial lipopolysaccharide, but it also appears to recognize mannan structures of fungal cell walls, leading to cytokine/chemokine production and recruitment of neutrophils. TLR4 is composed of a 608 residue extracellular domain, which contains 21 leucine-rich repeats and can be subdivided into N-, central, and C-terminal domains, and a 187-residue intracellular domain that signals via the TLR-associated adaptor MyD88 (115). Interruption of TLR4 signaling has been shown to increase susceptibility to C. albicans (116), A. fumigatus (117, 118), and Pneumocystis jirovecii (119) in mice. However, it is worth noting that TLR4 signaling did not induce cytokine production after recognizing mannans on C. neoformans cell walls (120), indicating fungal species-specific responses.
TLR9 (CD289) recognizes unmethylated CpG DNA sequences, which may allow it to recognize fungal DNA. As mentioned earlier, TLR9 is located in endosomes of pDCs, where it is known for responding to viral DNA in a MyD88-dependent manner. TLR9 has 25 intra-endosomal leucine-rich repeats, but a detailed structure for TLR9 has not yet been discovered (121). Studies show TLR9 induces IL-10 production by macrophages in response to C. albicans (120), pro-inflammatory cytokine secretion by mouse bone marrow-derived dendritic cells in response to A. fumigatus DNA (122), and IL-12p40 production and CD40 upregulation in DCs in response to C. neoformans (123). It is interesting to note that while MyD88 serves as a signaling adaptor for all of the TLRs except TLR3, patients with MyD88 deficiencies or mutations do not suffer from severe or recurrent fungal infections, suggesting that TLR signaling enhances but is not indispensable for antifungal immune responses (124).
C-type lectin receptors are a large family of receptors that contain C-type lectin-like domains (CTLDs), which, depending on the receptor, recognize a wide array of carbohydrates, proteins, or lipids (125). CLRs are a diverse family of receptors with a multitude of functions, but this review will focus on the four CLRs that have a role in fungal recognition and have been identified on human pDCs: dectin-1, dectin-2, dectin-3, and MR.
Dectin-1 (CLEC7A) recognizes β(1,3)- and β(1,6)-linked glucans, which are present on bacterial and fungal cell walls. B-glucans can comprise up to 50% of the cell walls of common pathogenic fungi, including A. fumigatus, C. neoformans, P. jirovecii, and C. albicans (126–130). Dectin-1 is composed of an extracellular CTLD, a single transmembrane domain, and an intracellular immunoreceptor tyrosine-based activation motif (ITAM) (126). The dimerization of dectin-1 and the phosphorylation of its intracellular ITAM recruits spleen tyrosine kinase (Syk), and induces a signaling cascade that includes the signaling adaptor caspase recruitment domain-containing protein 9 (CARD9). CARD9 is a major point of regulation within the signaling cascade of several CLRs, including dectin-1 and dectin-2, and is indispensable for the cell activation and cytokine production caused by dectin-1 and -2 ligation (131, 132). CARD9 deficiency has been linked to chronic and recurring fungal infections, especially by C. albicans (133, 134). In addition, pDCs from CARD9 knockout mice produced significantly less IL-6 and TNF-α than wild-type mice in response to influenza virus, while type-I IFN production was unaffected (135). Dectin-1/Syk/CARD9 signaling ultimately leads to the canonical activation and nuclear translocation of NF-κB subunits Rel-A (p65) and c-Rel. Dectin-1 also activates the NF-κB unit Rel-B through a non-canonical pathway, via Raf-1 instead of Syk (136). Dectin-1 has been shown to have an array of immune functions, including the recognition, uptake, and killing of fungi (137, 138), as well as cellular signaling leading to maturation, production of a respiratory burst, cytokine and chemokine production, enhanced survival and differentiation of TH cells that helps control fungal infection in the GI tract, and cytotoxic T-lymphocyte priming (128, 139–143).
Dectin-2 (CLEC6A) recognizes α-mannose structures present on the surface of many viruses, bacteria, and fungal pathogens. Dectin-2 has been shown to have a role in immunity to fungi, including C. albicans, A. fumigatus, and non-encapsulated C. neoformans, with a preference for hyphal as opposed to yeast or conidial forms of fungal cell walls (144–146). It is composed of an extracellular CTLD and a short cytoplasmic tail, which couples with FcRγ to initiate downstream signaling. The phosphorylation of FcRγ again recruits Syk and CARD9 (147, 148). Dectin-2 signaling also induces the activation of NF-κB, but in contrast to dectin-1, dectin-2 selectively activates the c-Rel subunit of NF-κB via MALT1. This selectivity suggests that dectin-2 is more specific for the production of cytokines that induce TH17 polarization (149). Dectin-2 signaling induces internalization of the fungus and the production of an array of cytokines, including TNF-α and IL-1β, 2, 6, 10, 12, and 23 (145, 147). Furthermore, blocking dectin-2 in mice abrogated the development of TH1 and TH17 cells, which left mice significantly more susceptible to C. albicans infection (150).
Similar to dectin-2, dectin-3 (CLEC4D, CLECSF8) also has a single extracellular CRD with a short cytoplasmic tail. The CRD of dectin-3 is highly homologous with that of dectin-2, and similarly recognizes α-mannans (151). In fact, dectin-3 was found to dimerize with dectin-2, and the resulting heterodimer was more adept at recognizing fungal PAMPs than the respective homodimers (152). Although the cytoplasmic tail of dectin-3 lacks a signaling motif and does not associate with any known signaling adaptor, including FcRγ, it does induce signaling via Syk and the CARD9/Bcl10/Malt1 complex and activates NF-κB (153). The cellular processes that are induced include phagocytosis, cytokine production, and a respiratory burst in macrophages (154). To illustrate its physiological relevance, murine bone marrow-derived macrophages pretreated with dectin-3-blocking monoclonal antibodies were inhibited from NF-κB nuclear translocation and cytokine production after C. albicans stimulation, and mice treated with dectin-3-blocking antibody succumbed rapidly to sub-lethal C. albicans IV challenge (152). Similarly, mice deficient in dectin-3 were more susceptible to DSS-induced colitis, and murine bone marrow-derived macrophages were impaired in phagocytic and fungicidal ability, NF-κB nuclear translocation, and cytokine production (155). Conversely, no role was found for dectin-3 in defense against C. neoformans, as dectin-3-deficient mice did not differ in pulmonary leukocyte recruitment, pulmonary cytokine profiles, or overall survival after pulmonary C. neoformans challenge. In addition, macrophages and DCs isolated from dectin-3 knockout mice did not differ in their phagocytic or fungicidal ability against C. neoformans (156).
Mannose receptor (CD206) is a highly endocytic receptor with three categories of extracellular binding domains, a single transmembrane domain, and a short cytoplasmic tail (157). The extracellular binding domains include an N-terminal cysteine-rich (CR) domain, which binds sulfated carbohydrates; a fibronectin type II (FNII) domain, which binds collagen; and eight CTLDs, which bind carbohydrates that terminate in d-mannose, l-fucose, or N-acetyl glucosamine, as reviewed in Ref. (158, 159). While the CR and FNII extracellular domains are involved in glycoprotein homeostasis (160, 161), it is the CTLDs that are responsible for recognizing carbohydrate entities on pathogens (159). For one particularly interesting example, MR binds the gp120 glycoprotein on HIV and mediates binding of the virus by dendritic cells (70) and macrophages (162). In the context of fungal infection, the MR has been shown to enhance uptake and clearance of pathogenic fungi, including A. fumigatus (163) C. albicans (164, 165), C. neoformans (166), and Pneumocystis carinii (167). However, MR seems to not be phagocytic in its own right; MR was unable to induce phagocytosis of its ligands when transfected onto Chinese hamster ovary cells (168). MR instead influences the signaling of other receptors. For example, Tachado et al. (169) discovered that human embryonic kidney 293 cells required transfection of both TLR2 and the MR in order to produce IL-8 in response to Pneumocystis. Similarly, in alveolar macrophages, Pneumocystis challenge led to direct interaction of TLR2 and MR, while blocking MR and TLR2 simultaneously abrogated IL-8 production (169). The downstream results of MR signaling are varied and dependent on many factors, including signaling from other receptors and the route of ligand delivery (158). A summary of the TLRs and CLRs discussed here can be found in Table 1.
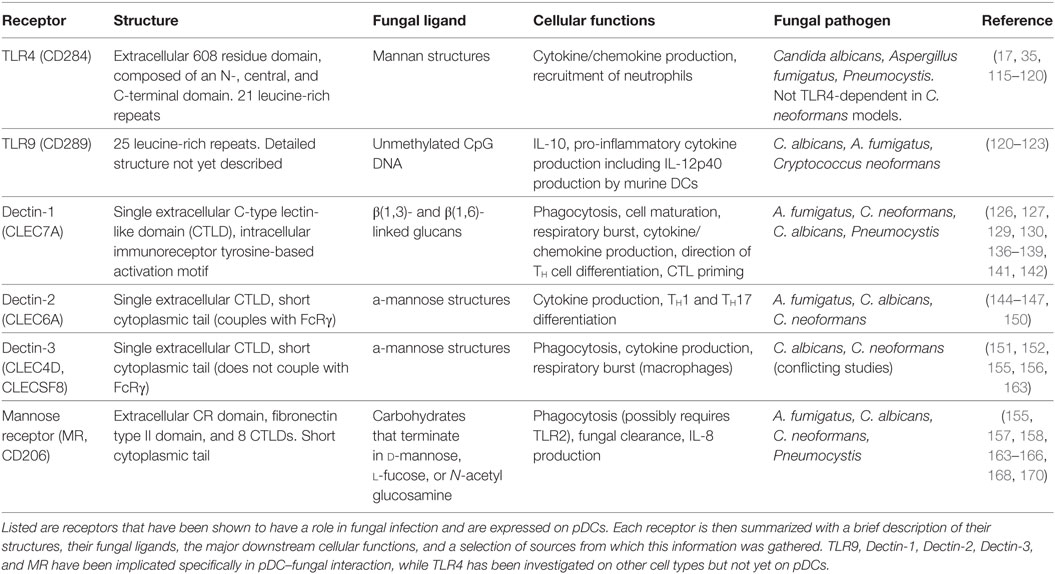
Table 1. A summary of the pattern recognition receptors (PRRs) on plasmacytoid dendritic cells (pDCs) that have a role in antifungal immunity.
pDCs in Antifungal Immunity
The involvement of pDCs in antifungal immunity is a topic of study currently emerging in the literature. An early indication came in 2004, when Romani et al. described how thymosin alpha 1, a peptide produced by the thymus, served to activate human cDCs and pDCs by enhancing their phagocytic capacity and IL-10 production in response to A. fumigatus stimulation (171). Later, Dan et al. found that mannoproteins from C. neoformans and CpG-containing DNA synergistically induced pDCs to produce greater amounts of pro-inflammatory cytokines, although stimulation with mannoproteins alone did not induce any cytokine production (170). This implied that pDCs were able to sense mannoproteins, which are classically recognized by CLRs, and that recognition of mannoproteins acted as an adjuvant for TLR9-dependent pDC activation by CpG. That same year, Ramirez-Ortiz et al. found that A. fumigatus DNA contained unmethylated CpG motifs, which triggered TLR9 in mouse and human pDCs and induced cytokine production in vitro (122). Perruccio et al. then confirmed that A. fumigatus DNA stimulated TLR-9 in pDCs and induced the production of IL-12p70, IL-10, and IFN-α (172).
The response of pDCs to fungal recognition is still under investigation, but a few studies have shed some light on mechanisms of pDC antifungal activity (summarized in Figure 1). Direct pDC antifungal activity was first demonstrated in 2011, when Ramirez-Ortiz et al. found that human pDCs directly inhibited growth of A. fumigatus hyphae and produced IFN-α and TNF-α in response to in vitro hyphal stimulation (173). They found that pDCs spread out over the surface of hyphae and inhibited hyphal viability by up to 80%. However, this strong antifungal activity was observed with a pDC:fungal ratio of 50:1, which seems highly unlikely to occur in a live organism. At a pDC-fungal ratio of 1:10, a 40% decrease in hyphal viability was still observed. At the same time, they found fungal stimulation lead to a large increase in pDC death, mediated partially by the production of gliotoxin by A. fumigatus. They also discovered that pDC lysates had the same antifungal activity as live cells, and implicated the release of calprotectin from dying pDCs as a contributor to the inhibition of fungal growth. Calprotectin has a documented role in antifungal immunity as a component of neutrophil extracellular traps (174). However, this was the first indication that pDCs may also use calprotectin. A later study by the same group indicated that pDC-Aspergillus interaction seemed to induce the production of pDC extracellular traps (pETs), similar to those extracellular traps laid by neutrophils (175). The production of extracellular traps by pDCs is a novel concept that has not been described elsewhere, and requires careful follow-up. Transcriptome analysis after A. fumigatus stimulation revealed the upregulation of several genes involved in activation, chemokine production, and antigen presentation, as well as genes associated with apoptosis. In vivo, mice depleted of pDCs were significantly more susceptible to Aspergillus-induced mortality, both after pulmonary and intravenous challenge (173). In addition, pulmonary challenge with A. fumigatus caused a six-fold increase in pDC number in the lungs. In a C. neoformans model, Hole et al. indicated that murine and human pDCs could phagocytose yeast cells and could directly inhibit C. neoformans growth through the use of reactive oxygen species (72).
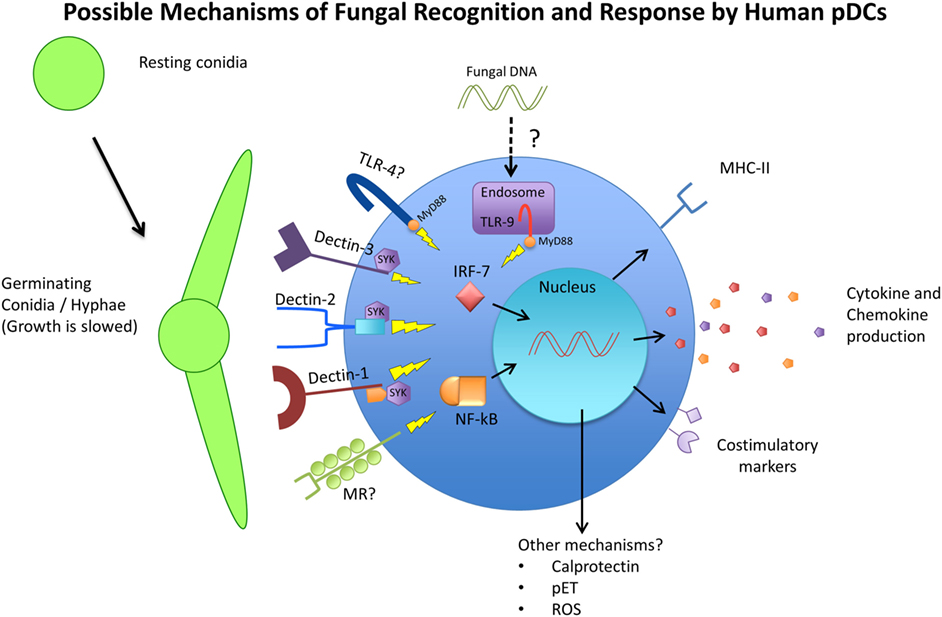
Figure 1. Schematic representing the available receptors and cellular responses of plasmacytoid dendritic cells (pDCs) to fungal pathogens. Resting conidia (RC) or yeast cells are less immunogenic than germinating and hyphal forms of fungi due to the increased availability of ligands such as beta-glucans and alpha-mannan structures of the cell wall. The receptors TLR4, TLR9, Dectin-1, Dectin-2, Dectin-3, and MR have been implicated in antifungal immunity in various models and studies and have also been observed to be expressed by pDCs. The cellular responses of pDCs to fungal stimulation have only begun to be characterized, but include phagocytosis, activation/maturation, cytokine and chemokine production, and MHC-II upregulation. Other pDC antifungal mechanisms that have been proposed include the release of calprotectin by apoptotic pDCs, the production of pDC extracellular traps (pETs), and the production of reactive oxygen species (ROS).
As mentioned above, pDCs express receptors that are known to be involved in fungal recognition (summarized on Figure 1). As early as 2002, Taylor et al. mentioned that murine pDCs express dectin-1, although the data were not shown. Seeds et al. (71) later confirmed that finding, and added that murine pDCs also expressed the CLRs dectin-2 and MR (71). Because murine pDCs are not perfect representations of human pDCs, these receptors also needed to be investigated on human pDCs. Studies on human pDCs observed the expression of dectin-1 (176), dectin-2 (177), and MR (70, 178). However, attempts to identify which pDC receptor is responsible for fungal recognition have delivered mixed results. One study singled out dectin-2 as the main receptor responsible for recognition of A. fumigatus hyphae by human pDCs (175). They used laminarin and mannan to block dectin-1 and dectin-2, respectively, and found that while both blocking agents decreased the ability of pDCs to associate with fungal hyphae, only mannan’s effect reached statistical significance. However, the definition and use of mannan as a dectin-2 blocker may be problematic, as it can be the ligand for other CLRs, including dectin-3 and MR. The study did follow-up by blocking with specific antibodies against dectin-1 and dectin-2, which recapitulated their results that dectin-2 was more responsible for pDC–hyphal association. Hole et al. observed the presence of dectin-3 on murine pDCs, and demonstrated that blocking dectin-3 on human pDCs resulted in a blunted reaction to C. neoformans stimulation (72). In their study, pDCs from dectin-3 knockout mice experienced the greatest reduction in fungal uptake and growth inhibition, greater than dectin-1, dectin-2, and MR knockout mice. Accordingly, pDCs from dectin-3 knockout mice were greatly deficient in their ability to phagocytose C. neoformans yeast cells. It is interesting to note that these findings stand in contrast to the aforementioned study where dectin-3 was not required for protection against C. neoformans (156). Nevertheless, the data again indicated that while dectin-3 knockout mice were the most affected in their antifungal activity and the only ones to achieve significance, knocking out dectin-1, dectin-2, and MR also resulted in varying degrees of reduced antifungal activity. This allows the possibility that pDCs use a combination of receptors to recognize and respond to fungal pathogens and that these PRRs have a measure of redundancy.
Considered together, this evidence strongly indicates that pDCs play an important role in antifungal immunity. However, there remains much to be discovered concerning the mechanisms of pDC–fungal interaction, cellular responses to recognition, and the role of pDCs in the broader landscape of antifungal immunity.
Concluding Remarks
One of the most insidious threats to the health and well-being of immunocompromised individuals, including those living with HIV, is the threat of opportunistic infections. Opportunistic fungal infections can be particularly fast, lethal, and difficult to treat. The main contributing factor for increased susceptibility to fungal infection in immunocompromised patients is the depletion and dysregulation of many components of the immune system; even when infection is well-controlled with antiretroviral medication, over time this dysregulation is measurable in individuals chronically infected with HIV. The pDC is no exception; HIV patients have a decline in number and functionality of their pDC compartment. This is especially problematic when considering the role of pDCs in fungal infection that is currently emerging in the literature. Studies have shown that pDCs have the machinery to recognize and respond to fungal infection, and do so in a manner that indicates pDCs are involved in a successful antifungal immune response. Therefore, dysregulation of the pDC compartment may be a contributing factor to the increased susceptibility of fungal infections that afflict HIV patients and other immunocompromised populations.
Author Contributions
SM performed all literature searches and writing of this review. PF-B provided focused expertise and guidance on the field of plasmacytoid dendritic cell and HIV biology, extensive revision of this manuscript, and final approval of the version for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the contributions of fellow lab members PD, HH, ZR, and HD as well as past members JD, VS, TS, MP, MN, and DD.
Funding
SM is supported by National Institutes of Health Integrated T32 Training Program in Infection, Immunity and Inflammation (Grant # 5T32AI125185) and the MD/PhD program at Rutgers New Jersey Medical School. PF-B is funded by NIH grants AI026806 and AI106125.
References
1. WHO. Global Health Sector Strategy on HIV 2016–2021. Geneva, Switzerland: WHO Press (2016). 60 p.
2. Zaidi J, Grapsa E, Tanser F, Newell ML, Barnighausen T. Dramatic increase in HIV prevalence after scale-up of antiretroviral treatment. AIDS (2013) 27(14):2301–5. doi:10.1097/QAD.0b013e328362e832
3. May MT, Gompels M, Delpech V, Porter K, Orkin C, Kegg S, et al. Impact on life expectancy of HIV-1 positive individuals of CD4+ cell count and viral load response to antiretroviral therapy. AIDS (2014) 28(8):1193–202. doi:10.1097/QAD.0000000000000243
4. Maartens G, Celum C, Lewin SR. HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet (2014) 384(9939):258–71. doi:10.1016/S0140-6736(14)60164-1
5. Ortblad KF, Lozano R, Murray CJ. The burden of HIV: insights from the Global Burden of Disease Study 2010. AIDS (2013) 27(13):2003–17. doi:10.1097/QAD.0b013e328362ba67
6. Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature (2013) 498(7454):376–9. doi:10.1038/nature12274
7. Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol (2008) 126(3):235–42. doi:10.1016/j.clim.2007.08.015
8. Anwar KP, Malik A, Subhan KH. Profile of candidiasis in HIV infected patients. Iran J Microbiol (2012) 4(4):204–9.
9. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS (2009) 23(4):525–30. doi:10.1097/QAD.0b013e328322ffac
10. Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis (2017) 17(8):873–81. doi:10.1016/S1473-3099(17)30243-8
11. Denning DW, Follansbee SE, Scolaro M, Norris S, Edelstein H, Stevens DA. Pulmonary aspergillosis in the acquired immunodeficiency syndrome. N Engl J Med (1991) 324(10):654–62. doi:10.1056/NEJM199103073241003
12. Holding KJ, Dworkin MS, Wan PC, Hanson DL, Klevens RM, Jones JL, et al. Aspergillosis among people infected with human immunodeficiency virus: incidence and survival. Adult and Adolescent Spectrum of HIV Disease Project. Clin Infect Dis (2000) 31(5):1253–7. doi:10.1086/317452
13. Lopez C, Fitzgerald PA, Siegal FP. Severe acquired immune deficiency syndrome in male homosexuals: diminished capacity to make interferon-alpha in vitro associated with severe opportunistic infections. J Infect Dis (1983) 148(6):962–6. doi:10.1093/infdis/148.6.962
14. McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol (2005) 79(1):17–27. doi:10.1128/JVI.79.1.17-27.2005
15. Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev (2008) 19(1):3–19. doi:10.1016/j.cytogfr.2007.10.006
16. Fitzgerald-Bocarsly P, Jacobs ES. Plasmacytoid dendritic cells in HIV infection: striking a delicate balance. J Leukoc Biol (2010) 87(4):609–20. doi:10.1189/jlb.0909635
17. Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med (2001) 194(6):863–9. doi:10.1084/jem.194.6.863
18. Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol (2001) 31(10):3026–37. doi:10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H
19. McMichael AJ, Rowland-Jones SL. Cellular immune responses to HIV. Nature (2001) 410(6831):980–7. doi:10.1038/35073658
20. Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol (2003) 21:265–304. doi:10.1146/annurev.immunol.21.120601.141053
21. O’Brien M, Manches O, Bhardwaj N. Plasmacytoid dendritic cells in HIV infection. Adv Exp Med Biol (2013) 762:71–107. doi:10.1007/978-1-4614-4433-6_3
22. Schmidt B, Fujimura SH, Martin JN, Levy JA. Variations in plasmacytoid dendritic cell (PDC) and myeloid dendritic cell (MDC) levels in HIV-infected subjects on and off antiretroviral therapy. J Clin Immunol (2006) 26(1):55–64. doi:10.1007/s10875-006-8401-3
23. Zabel BA, Silverio AM, Butcher EC. Chemokine-like receptor 1 expression and chemerin-directed chemotaxis distinguish plasmacytoid from myeloid dendritic cells in human blood. J Immunol (2005) 174(1):244–51. doi:10.4049/jimmunol.174.1.244
24. Lande R, Gilliet M. Plasmacytoid dendritic cells: key players in the initiation and regulation of immune responses. Ann N Y Acad Sci (2010) 1183:89–103. doi:10.1111/j.1749-6632.2009.05152.x
25. Bargatze RF, Jutila MA, Butcher EC. Distinct roles of L-selectin and integrins alpha 4 beta 7 and LFA-1 in lymphocyte homing to Peyer’s patch-HEV in situ: the multistep model confirmed and refined. Immunity (1995) 3(1):99–108. doi:10.1016/1074-7613(95)90162-0
26. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity (2004) 21(1):107–19. doi:10.1016/j.immuni.2004.06.007
27. Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via toll-like receptor-viral RNA interactions. J Clin Invest (2005) 115(11):3265–75. doi:10.1172/JCI26032
28. Gibson SJ, Lindh JM, Riter TR, Gleason RM, Rogers LM, Fuller AE, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol (2002) 218(1–2):74–86. doi:10.1016/S0008-8749(02)00517-8
29. Colak E, Leslie A, Zausmer K, Khatamzas E, Kubarenko AV, Pichulik T, et al. RNA and imidazoquinolines are sensed by distinct TLR7/8 ectodomain sites resulting in functionally disparate signaling events. J Immunol (2014) 192(12):5963–73. doi:10.4049/jimmunol.1303058
30. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi:10.1038/nature03464
31. Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann KK, et al. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol (2004) 173(10):5935–43. doi:10.4049/jimmunol.173.10.5935
32. Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A (2004) 101(31):11416–21. doi:10.1073/pnas.0403555101
33. Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, et al. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J Leukoc Biol (2003) 74(6):1125–38. doi:10.1189/jlb.0603255
34. O’Brien M, Manches O, Sabado RL, Baranda SJ, Wang Y, Marie I, et al. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-alpha-producing and partially matured phenotype. J Clin Invest (2011) 121(3):1088–101. doi:10.1172/JCI44960
35. Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol (2004) 173(3):1535–48. doi:10.4049/jimmunol.173.3.1535
36. Fuchsberger M, Hochrein H, O’Keeffe M. Activation of plasmacytoid dendritic cells. Immunol Cell Biol (2005) 83(5):571–7. doi:10.1111/j.1440-1711.2005.01392.x
37. Decalf J, Fernandes S, Longman R, Ahloulay M, Audat F, Lefrerre F, et al. Plasmacytoid dendritic cells initiate a complex chemokine and cytokine network and are a viable drug target in chronic HCV patients. J Exp Med (2007) 204(10):2423–37. doi:10.1084/jem.20070814
38. Yin Z, Dai J, Deng J, Sheikh F, Natalia M, Shih T, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol (2012) 189(6):2735–45. doi:10.4049/jimmunol.1102038
39. Feldman M, Fitzgerald-Bocarsly P. Sequential enrichment and immunocytochemical visualization of human interferon-alpha-producing cells. J Interferon Res (1990) 10(4):435–46. doi:10.1089/jir.1990.10.435
40. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284(5421):1835–7. doi:10.1126/science.284.5421.1835
41. Liu Y-J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23:275–306. doi:10.1146/annurev.immunol.23.021704.115633
42. Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood (2006) 107(6):2423–31. doi:10.1182/blood-2005-07-2709
43. Novick D, Cohen B, Rubinstein M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell (1994) 77(3):391–400. doi:10.1016/0092-8674(94)90154-6
44. Biron CA. Role of early cytokines, including alpha and beta interferons (IFN-a/b), in innate and adaptive immune responses to viral infections. Semin Immunol (1998) 10:383–90. doi:10.1006/smim.1998.0138
45. Fitzgerald-Bocarsly P, Feng D. The role of type I interferon production by dendritic cells in host defense. Biochimie (2007) 89(6–7):843–55. doi:10.1016/j.biochi.2007.04.018
46. Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol (2001) 166(5):2961–9. doi:10.4049/jimmunol.166.5.2961
47. Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology (2005) 343(2):256–66. doi:10.1016/j.virol.2005.09.059
48. Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, et al. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A (2003) 100(19):10872–7. doi:10.1073/pnas.1934678100
49. Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med (1993) 178(5):1655–63. doi:10.1084/jem.178.5.1655
50. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol (2000) 1(4):305–10. doi:10.1038/79747
51. Rothenfusser S, Hornung V, Ayyoub M, Britsch S, Towarowski A, Krug A, et al. CpG-A and CpG-B oligonucleotides differentially enhance human peptide-specific primary and memory CD8+ T-cell responses in vitro. Blood (2004) 103(6):2162–9. doi:10.1182/blood-2003-04-1091
52. Yoneyama H, Matsuno K, Toda E, Nishiwaki T, Matsuo N, Nakano A, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med (2005) 202(3):425–35. doi:10.1084/jem.20041961
53. Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation (2006) 114(23):2482–9. doi:10.1161/CIRCULATIONAHA.106.642801
54. Dikopoulos N, Bertoletti A, Kroger A, Hauser H, Schirmbeck R, Reimann J. Type I IFN negatively regulates CD8+ T cell responses through IL-10-producing CD4+ T regulatory 1 cells. J Immunol (2005) 174(1):99–109. doi:10.4049/jimmunol.174.1.99
55. Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14(4):461–70. doi:10.1016/S1074-7613(01)00126-1
56. Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol (2008) 180(4):2474–85. doi:10.4049/jimmunol.180.4.2474
57. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol (2006) 80(9):4501–9. doi:10.1128/JVI.80.9.4501-4509.2006
58. Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem (2007) 282(10):7576–81. doi:10.1074/jbc.M608618200
59. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog (2008) 4(3):e1000017. doi:10.1371/journal.ppat.1000017
60. Mennechet FJ, Uze G. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood (2006) 107(11):4417–23. doi:10.1182/blood-2005-10-4129
61. Jordan WJ, Eskdale J, Srinivas S, Pekarek V, Kelner D, Rodia M, et al. Human interferon lambda-1 (IFN-lambda1/IL-29) modulates the Th1/Th2 response. Genes Immun (2007) 8(3):254–61. doi:10.1038/sj.gene.6364382
62. Bordi L, Lalle E, Lapa D, Caglioti C, Quartu S, Capobianchi MR, et al. Type III interferon (IFN-lambda) antagonizes the antiviral activity of interferon-alpha in vitro. J Biol Regul Homeost Agents (2013) 27(4):1001–9.
63. Megjugorac NJ, Young HA, Amrute SB, Olshalsky SL, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J Leukoc Biol (2004) 75(3):504–14. doi:10.1189/jlb.0603291
64. Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood (2006) 107(7):2613–8. doi:10.1182/blood-2005-07-2965
65. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol (2004) 5(12):1219–26. doi:10.1038/ni1141
66. Fanning SL, George TC, Feng D, Feldman SB, Megjugorac NJ, Izaguirre AG, et al. Receptor cross-linking on human plasmacytoid dendritic cells leads to the regulation of IFN-alpha production. J Immunol (2006) 177(9):5829–39. doi:10.4049/jimmunol.177.9.5829
67. Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity (2008) 29(3):352–61. doi:10.1016/j.immuni.2008.09.002
68. Sadaka C, Marloie-Provost MA, Soumelis V, Benaroch P. Developmental regulation of MHC II expression and transport in human plasmacytoid-derived dendritic cells. Blood (2009) 113(10):2127–35. doi:10.1182/blood-2008-10-178152
69. Tel J, Smits EL, Anguille S, Joshi RN, Figdor CG, de Vries IJ. Human plasmacytoid dendritic cells are equipped with antigen-presenting and tumoricidal capacities. Blood (2012) 120(19):3936–44. doi:10.1182/blood-2012-06-435941
70. Milone MC, Fitzgerald-Bocarsly P. The mannose receptor mediates induction of IFN-alpha in peripheral blood dendritic cells by enveloped RNA and DNA viruses. J Immunol (1998) 161(5):2391–9.
71. Seeds RE, Gordon S, Miller JL. Characterisation of myeloid receptor expression and interferon alpha/beta production in murine plasmacytoid dendritic cells by flow cytomtery. J Immunol Methods (2009) 350(1–2):106–17. doi:10.1016/j.jim.2009.07.016
72. Hole CR, Leopold Wager CM, Mendiola AS, Wozniak KL, Campuzano A, Lin X, et al. Antifungal activity of plasmacytoid dendritic cells against Cryptococcus neoformans in vitro requires expression of dectin-3 (CLEC4D) and reactive oxygen species. Infect Immun (2016) 84(9):2493–504. doi:10.1128/IAI.00103-16
73. Siegal FP, Lopez C, Fitzgerald PA, Shah K, Baron P, Leiderman IZ, et al. Opportunistic infections in acquired immune deficiency syndrome result from synergistic defects of both the natural and adaptive components of cellular immunity. J Clin Invest (1986) 78(1):115–23. doi:10.1172/JCI112539
74. Howell DM, Feldman SB, Kloser P, Fitzgerald-Bocarsly P. Decreased frequency of functional natural interferon-producing cells in peripheral blood of patients with the acquired immune deficiency syndrome. Clin Immunol Immunopathol (1994) 71(2):223–30. doi:10.1006/clin.1994.1076
75. Feldman SB, Milone MC, Kloser P, Fitzgerald-Bocarsly P. Functional deficiencies in two distinct interferon alpha-producing cell populations in peripheral blood mononuclear cells from human immunodeficiency virus seropositive patients. J Leukoc Biol (1995) 57(2):214–20.
76. Feldman S, Stein D, Amrute S, Denny T, Garcia Z, Kloser P, et al. Decreased interferon-alpha production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin Immunol (2001) 101(2):201–10. doi:10.1006/clim.2001.5111
77. Soumelis V, Scott I, Gheyas F, Bouhour D, Cozon G, Cotte L, et al. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood (2001) 98(4):906–12. doi:10.1182/blood.V98.4.906
78. Donaghy H, Pozniak A, Gazzard B, Qazi N, Gilmour J, Gotch F, et al. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood (2001) 98(8):2574–6. doi:10.1182/blood.V98.8.2574
79. Chehimi J, Azzoni L, Farabaugh M, Creer SA, Tomescu C, Hancock A, et al. Baseline viral load and immune activation determine the extent of reconstitution of innate immune effectors in HIV-1-infected subjects undergoing antiretroviral treatment. J Immunol (2007) 179(4):2642–50. doi:10.4049/jimmunol.179.4.2642
80. Malleret B, Maneglier B, Karlsson I, Lebon P, Nascimbeni M, Perie L, et al. Primary infection with simian immunodeficiency virus: plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood (2008) 112(12):4598–608. doi:10.1182/blood-2008-06-162651
81. Brown KN, Wijewardana V, Liu X, Barratt-Boyes SM. Rapid influx and death of plasmacytoid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog (2009) 5(5):e1000413. doi:10.1371/journal.ppat.1000413
82. Lehmann C, Lafferty M, Garzino-Demo A, Jung N, Hartmann P, Fatkenheuer G, et al. Plasmacytoid dendritic cells accumulate and secrete interferon alpha in lymph nodes of HIV-1 patients. PLoS One (2010) 5(6):e11110. doi:10.1371/journal.pone.0011110
83. Patterson S, Rae A, Hockey N, Gilmour J, Gotch F. Plasmacytoid dendritic cells are highly susceptible to human immunodeficiency virus type 1 infection and release infectious virus. J Virol (2001) 75(14):6710–3. doi:10.1128/JVI.75.14.6710-6713.2001
84. Schmidt B, Scott I, Whitmore RG, Foster H, Fujimura S, Schmitz J, et al. Low-level HIV infection of plasmacytoid dendritic cells: onset of cytopathic effects and cell death after PDC maturation. Virology (2004) 329(2):280–8. doi:10.1016/j.virol.2004.08.016
85. Wijewardana V, Brown KN, Barratt-Boyes SM. Studies of plasmacytoid dendritic cell dynamics in simian immunodeficiency virus infection of nonhuman primates provide insights into HIV pathogenesis. Curr HIV Res (2009) 7(1):23–9. doi:10.2174/157016209787048483
86. Barron MA, Blyveis N, Palmer BE, MaWhinney S, Wilson CC. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals. J Infect Dis (2003) 187(1):26–37. doi:10.1086/345957
87. Khaitan A, Unutmaz D. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep (2011) 8(1):4–11. doi:10.1007/s11904-010-0066-0
88. Li G, Cheng M, Nunoya J, Cheng L, Guo H, Yu H, et al. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog (2014) 10(7):e1004291. doi:10.1371/journal.ppat.1004291
89. Donaghy H, Gazzard B, Gotch F, Patterson S. Dysfunction and infection of freshly isolated blood myeloid and plasmacytoid dendritic cells in patients infected with HIV-1. Blood (2003) 101(11):4505–11. doi:10.1182/blood-2002-10-3189
90. Dillon SM, Robertson KB, Pan SC, Mawhinney S, Meditz AL, Folkvord JM, et al. Plasmacytoid and myeloid dendritic cells with a partial activation phenotype accumulate in lymphoid tissue during asymptomatic chronic HIV-1 infection. J Acquir Immune Defic Syndr (2008) 48(1):1–12. doi:10.1097/QAI.0b013e3181664b60
91. Martinson JA, Roman-Gonzalez A, Tenorio AR, Montoya CJ, Gichinga CN, Rugeles MT, et al. Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell Immunol (2007) 250(1–2):75–84. doi:10.1016/j.cellimm.2008.01.007
92. Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, et al. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J Immunol (2004) 173(7):4433–42. doi:10.4049/jimmunol.173.7.4433
93. Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med (2002) 195(6):695–704. doi:10.1084/jem.20011603
94. Boasso A, Herbeuval JP, Hardy AW, Anderson SA, Dolan MJ, Fuchs D, et al. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood (2007) 109(8):3351–9. doi:10.1182/blood-2006-07-034785
95. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189(9):1363–72. doi:10.1084/jem.189.9.1363
96. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol (2009) 183(4):2475–83. doi:10.4049/jimmunol.0900986
98. Vallabhaneni S, Mody RK, Walker T, Chiller T. The global burden of fungal diseases. Infect Dis Clin North Am (2016) 30(1):1–11. doi:10.1016/j.idc.2015.10.004
99. Tronchin G, Bouchara JP, Ferron M, Larcher G, Chabasse D. Cell surface properties of Aspergillus fumigatus conidia: correlation between adherence, agglutination, and rearrangements of the cell wall. Can J Microbiol (1995) 41(8):714–21. doi:10.1139/m95-098
100. van de Veerdonk FL, Kullberg BJ, van der Meer JW, Gow NA, Netea MG. Host-microbe interactions: innate pattern recognition of fungal pathogens. Curr Opin Microbiol (2008) 11(4):305–12. doi:10.1016/j.mib.2008.06.002
101. Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, et al. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog (2005) 1(3):e30. doi:10.1371/journal.ppat.0010030
102. Latge JP. Tasting the fungal cell wall. Cell Microbiol (2010) 12(7):863–72. doi:10.1111/j.1462-5822.2010.01474.x
103. Levitz SM. Innate recognition of fungal cell walls. PLoS Pathog (2010) 6(4):e1000758. doi:10.1371/journal.ppat.1000758
104. Hamad M. Innate and adaptive antifungal immune responses: partners on an equal footing. Mycoses (2011) 55:205–17. doi:10.1111/j.1439-0507.2011.02078.x
105. Brown GD. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol (2011) 29:1–21. doi:10.1146/annurev-immunol-030409-101229
106. Hohl TM, Feldmesser M. Aspergillus fumigatus: principles of pathogenesis and host defense. Eukaryot Cell (2007) 6(11):1953–63. doi:10.1128/EC.00274-07
107. Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog (2010) 6(4):e1000873. doi:10.1371/journal.ppat.1000873
108. Bozza S, Gaziano R, Spreca A, Bacci A, Montagnoli C, di Francesco P, et al. Dendritic cells transport conidia and hyphae of Aspergillus fumigatus from the airways to the draining lymph nodes and initiate disparate Th responses to the fungus. J Immunol (2002) 168(3):1362–71. doi:10.4049/jimmunol.168.3.1362
109. Huang H, Ostroff GR, Lee CK, Wang JP, Specht CA, Levitz SM. Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and toll-like receptor agonists. Infect Immun (2009) 77(5):1774–81. doi:10.1128/IAI.00086-09
110. Roy RM, Klein BS. Dendritic cells in antifungal immunity and vaccine design. Cell Host Microbe (2012) 11(5):436–46. doi:10.1016/j.chom.2012.04.005
111. Peck A, Mellins ED. Precarious balance: Th17 cells in host defense. Infect Immun (2010) 78(1):32–8. doi:10.1128/IAI.00929-09
112. Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol (2012) 12(6):616–22. doi:10.1097/ACI.0b013e328358cc0b
113. Ling Y, Puel A. IL-17 and infections. Actas Dermosifiliogr (2014) 105(Suppl 1):34–40. doi:10.1016/S0001-7310(14)70016-X
114. Spellberg B, Ibrahim AS, Lin L, Avanesian V, Fu Y, Lipke P, et al. Antibody titer threshold predicts anti-candidal vaccine efficacy even though the mechanism of protection is induction of cell-mediated immunity. J Infect Dis (2008) 197(7):967–71. doi:10.1086/529204
115. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi:10.1038/41131
116. Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis (2002) 185(10):1483–9. doi:10.1086/340511
117. Mambula SS, Sau K, Henneke P, Golenbock DT, Levitz SM. Toll-like receptor (TLR) signaling in response to Aspergillus fumigatus. J Biol Chem (2002) 277(42):39320–6. doi:10.1074/jbc.M201683200
118. Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, et al. The contribution of the toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol (2004) 172(5):3059–69. doi:10.4049/jimmunol.172.5.3059
119. Ding K, Shibui A, Wang Y, Takamoto M, Matsuguchi T, Sugane K. Impaired recognition by toll-like receptor 4 is responsible for exacerbated murine Pneumocystis pneumonia. Microbes Infect (2005) 7(2):195–203. doi:10.1016/j.micinf.2004.10.010
120. Nakamura K, Miyagi K, Koguchi Y, Kinjo Y, Uezu K, Kinjo T, et al. Limited contribution of toll-like receptor 2 and 4 to the host response to a fungal infectious pathogen, Cryptococcus neoformans. FEMS Immunol Med Microbiol (2006) 47(1):148–54. doi:10.1111/j.1574-695X.2006.00078.x
121. Botos I, Segal DM, Davies DR. The structural biology of toll-like receptors. Structure (2011) 19(4):447–59. doi:10.1016/j.str.2011.02.004
122. Ramirez-Ortiz ZG, Specht CA, Wang JP, Lee CK, Bartholomeu DC, Gazzinelli RT, et al. Toll-like receptor 9-dependent immune activation by unmethylated CpG motifs in Aspergillus fumigatus DNA. Infect Immun (2008) 76(5):2123–9. doi:10.1128/IAI.00047-08
123. Nakamura K, Miyazato A, Xiao G, Hatta M, Inden K, Aoyagi T, et al. Deoxynucleic acids from Cryptococcus neoformans activate myeloid dendritic cells via a TLR9-dependent pathway. J Immunol (2008) 180(6):4067–74. doi:10.4049/jimmunol.180.6.4067
124. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) (2010) 89(6):403–25. doi:10.1097/MD.0b013e3181fd8ec3
125. Vautier S, Sousa Mda G, Brown GD. C-type lectins, fungi and Th17 responses. Cytokine Growth Factor Rev (2010) 21(6):405–12. doi:10.1016/j.cytogfr.2010.10.001
126. Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature (2001) 413(6851):36–7. doi:10.1038/35092620
127. Odabasi Z, Paetznick VL, Rodriguez JR, Chen E, McGinnis MR, Ostrosky-Zeichner L. Differences in beta-glucan levels in culture supernatants of a variety of fungi. Med Mycol (2006) 44(3):267–72. doi:10.1080/13693780500474327
128. Dennehy KM, Brown GD. The role of the beta-glucan receptor Dectin-1 in control of fungal infection. J Leukoc Biol (2007) 82(2):253–8. doi:10.1189/jlb.1206753
129. Seeds RE, Mukhopadhyay S, Jones IM, Gordon S, Miller JL. The role of myeloid receptors on murine plasmacytoid dendritic cells in induction of type I interferon. Int Immunopharmacol (2011) 11(7):794–801. doi:10.1016/j.intimp.2011.01.013
130. Rhein J, Bahr NC, Morawski BM, Schutz C, Zhang Y, Finkelman M, et al. Detection of high cerebrospinal fluid levels of (1 – >3)-beta-d-Glucan in cryptococcal meningitis. Open Forum Infect Dis (2014) 1(3):ofu105. doi:10.1093/ofid/ofu052.1014
131. Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature (2006) 442(7103):651–6. doi:10.1038/nature04926
132. Drummond RA, Lionakis MS. Mechanistic insights into the role of C-type lectin receptor/CARD9 signaling in human antifungal immunity. Front Cell Infect Microbiol (2016) 6:39. doi:10.3389/fcimb.2016.00039
133. Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood (2013) 121(13):2385–92. doi:10.1182/blood-2012-08-450551
134. Alves de Medeiros AK, Lodewick E, Bogaert DJ, Haerynck F, Daele S. Van, Lambrecht B, et al. Chronic and invasive fungal infections in a family with CARD9 deficiency. J Clin Immunol (2016) 36(3):204–9. doi:10.1007/s10875-016-0255-8
135. Uematsu T, Iizasa E, Kobayashi N, Yoshida H, Hara H. Loss of CARD9-mediated innate activation attenuates severe influenza pneumonia without compromising host viral immunity. Sci Rep (2015) 5:17577. doi:10.1038/srep17577
136. Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol (2009) 10(2):203–13. doi:10.1038/ni.1692
137. Herre J, Marshall AS, Caron E, Edwards AD, Williams DL, Schweighoffer E, et al. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood (2004) 104(13):4038–45. doi:10.1182/blood-2004-03-1140
138. Hefter M, Lother J, Weiss E, Schmitt AL, Fliesser M, Einsele H, et al. Human primary myeloid dendritic cells interact with the opportunistic fungal pathogen Aspergillus fumigatus via the C-type lectin receptor Dectin-1. Med Mycol (2017) 55(5):573–8. doi:10.1093/mmy/myw105
139. Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J (2005) 24(6):1277–86. doi:10.1038/sj.emboj.7600594
140. Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol (2006) 6:33–43. doi:10.1038/nri1745
141. Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, et al. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol (2009) 182(8):4938–46. doi:10.4049/jimmunol.0804250
142. Rivera A, Hohl TM, Collins N, Leiner I, Gallegos A, Saijo S, et al. Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J Exp Med (2011) 208(2):369–81. doi:10.1084/jem.20100906
143. Drummond RA, Dambuza IM, Vautier S, Taylor JA, Reid DM, Bain CC, et al. CD4(+) T-cell survival in the GI tract requires dectin-1 during fungal infection. Mucosal Immunol (2016) 9(2):492–502. doi:10.1038/mi.2015.79
144. McGreal EP, Rosas M, Brown GD, Zamze S, Wong SY, Gordon S, et al. The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology (2006) 16(5):422–30. doi:10.1093/glycob/cwj077
145. Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem (2006) 281(50):38854–66. doi:10.1074/jbc.M606542200
146. Kerscher B, Willment JA, Brown GD. The Dectin-2 family of C-type lectin-like receptors: an update. Int Immunol (2013) 25(5):271–7. doi:10.1093/intimm/dxt006
147. Bi L, Gojestani S, Wu W, Hsu YM, Zhu J, Ariizumi K, et al. CARD9 mediates dectin-2-induced IkappaBalpha kinase ubiquitination leading to activation of NF-kappaB in response to stimulation by the hyphal form of Candida albicans. J Biol Chem (2010) 285(34):25969–77. doi:10.1074/jbc.M110.131300
148. Drummond RA, Saijo S, Iwakura Y, Brown GD. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur J Immunol (2011) 41(2):276–81. doi:10.1002/eji.201041252
149. Gringhuis SI, Wevers BA, Kaptein TM, van Capel TM, Theelen B, Boekhout T, et al. Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog (2011) 7(1):e1001259. doi:10.1371/journal.ppat.1001259
150. Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med (2009) 206(9):2037–51. doi:10.1084/jem.20082818
151. Arce I, Martinez-Munoz L, Roda-Navarro P, Fernandez-Ruiz E. The human C-type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur J Immunol (2004) 34(1):210–20. doi:10.1002/eji.200324230
152. Zhu LL, Zhao XQ, Jiang C, You Y, Chen XP, Jiang YY, et al. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity (2013) 39(2):324–34. doi:10.1016/j.immuni.2013.05.017
153. Zhao XQ, Zhu LL, Chang Q, Jiang C, You Y, Luo T, et al. C-type lectin receptor dectin-3 mediates trehalose 6,6’-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-kappaB activation. J Biol Chem (2014) 289(43):30052–62. doi:10.1074/jbc.M114.588574
154. Graham LM, Gupta V, Schafer G, Reid DM, Kimberg M, Dennehy KM, et al. The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J Biol Chem (2012) 287(31):25964–74. doi:10.1074/jbc.M112.384164
155. Wang T, Pan D, Zhou Z, You Y, Jiang C, Zhao X, et al. Dectin-3 deficiency promotes colitis development due to impaired antifungal innate immune responses in the Gut. PLoS Pathog (2016) 12(6):e1005662. doi:10.1371/journal.ppat.1005662
156. Campuzano A, Castro-Lopez N, Wozniak KL, Leopold Wager CM, Wormley FL Jr. Dectin-3 is not required for protection against Cryptococcus neoformans infection. PLoS One (2017) 12(1):e0169347. doi:10.1371/journal.pone.0169347
157. Ezekowitz RA, Sastry K, Bailly P, Warner A. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J Exp Med (1990) 172(6):1785–94. doi:10.1084/jem.172.6.1785
158. Gazi U, Martinez-Pomares L. Influence of the mannose receptor in host immune responses. Immunobiology (2009) 214(7):554–61. doi:10.1016/j.imbio.2008.11.004
159. Martinez-Pomares L. The mannose receptor. J Leukoc Biol (2012) 92(6):1177–86. doi:10.1189/jlb.0512231
160. Lee SJ, Evers S, Roeder D, Parlow AF, Risteli J, Risteli L, et al. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science (2002) 295(5561):1898–901. doi:10.1126/science.1069540
161. Napper CE, Drickamer K, Taylor ME. Collagen binding by the mannose receptor mediated through the fibronectin type II domain. Biochem J (2006) 395(3):579–86. doi:10.1042/BJ20052027
162. Nguyen DG, Hildreth JE. Involvement of macrophage mannose receptor in the binding and transmission of HIV by macrophages. Eur J Immunol (2003) 33(2):483–93. doi:10.1002/immu.200310024
163. Wang Q, Zhao G, Lin J, Li C, Jiang N, Xu Q, et al. Role of the mannose receptor during Aspergillus fumigatus infection and interaction with dectin-1 in corneal epithelial cells. Cornea (2016) 35(2):267–73. doi:10.1097/ICO.0000000000000710
164. Marodi L, Korchak HM, Johnston RB Jr. Mechanisms of host defense against Candida species. I. Phagocytosis by monocytes and monocyte-derived macrophages. J Immunol (1991) 146(8):2783–9.
165. Porcaro I, Vidal M, Jouvert S, Stahl PD, Giaimis J. Mannose receptor contribution to Candida albicans phagocytosis by murine E-clone J774 macrophages. J Leukoc Biol (2003) 74(2):206–15. doi:10.1189/jlb.1202608
166. Dan JM, Kelly RM, Lee CK, Levitz SM. Role of the mannose receptor in a murine model of Cryptococcus neoformans infection. Infect Immun (2008) 76(6):2362–7. doi:10.1128/IAI.00095-08
167. Ezekowitz RA, Williams DJ, Koziel H, Armstrong MY, Warner A, Richards FF, et al. Uptake of Pneumocystis carinii mediated by the macrophage mannose receptor. Nature (1991) 351(6322):155–8. doi:10.1038/351155a0
168. Le Cabec V, Emorine LJ, Toesca I, Cougoule C, Maridonneau-Parini I. The human macrophage mannose receptor is not a professional phagocytic receptor. J Leukoc Biol (2005) 77(6):934–43. doi:10.1189/jlb.1204705
169. Tachado SD, Zhang J, Zhu J, Patel N, Cushion M, Koziel H. Pneumocystis-mediated IL-8 release by macrophages requires coexpression of mannose receptors and TLR2. J Leukoc Biol (2007) 81(1):205–11. doi:10.1189/jlb.1005580
170. Dan JM, Wang JP, Lee CK, Levitz SM. Cooperative stimulation of dendritic cells by Cryptococcus neoformans mannoproteins and CpG oligodeoxynucleotides. PLoS One (2008) 3(4):e2046. doi:10.1371/journal.pone.0002046
171. Romani L, Bistoni F, Gaziano R, Bozza S, Montagnoli C, Perruccio K, et al. Thymosin alpha 1 activates dendritic cells for antifungal Th1 resistance through toll-like receptor signaling. Blood (2004) 103(11):4232–9. doi:10.1182/blood-2003-11-4036
172. Perruccio K, Bozza S, Montagnoli C, Bellocchio S, Aversa F, Martelli M, et al. Prospects for dendritic cell vaccination against fungal infections in hematopoietic transplantation. Blood Cells Mol Dis (2004) 33(3):248–55. doi:10.1016/j.bcmd.2004.08.011
173. Ramirez-Ortiz ZG, Lee CK, Wang JP, Boon L, Specht CA, Levitz SM. A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe (2011) 9(5):415–24. doi:10.1016/j.chom.2011.04.007
174. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog (2009) 5(10):e1000639. doi:10.1371/journal.ppat.1000639
175. Loures FV, Rohm M, Lee CK, Santos E, Wang JP, Specht CA, et al. Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PLoS Pathog (2015) 11(2):e1004643. doi:10.1371/journal.ppat.1004643
176. Joo H, Upchurch K, Zhang W, Ni L, Li D, Xue Y, et al. Opposing roles of dectin-1 expressed on human plasmacytoid dendritic cells and myeloid dendritic cells in Th2 polarization. J Immunol (2015) 195(4):1723–31. doi:10.4049/jimmunol.1402276
177. Gavino AC, Chung JS, Sato K, Ariizumi K, Cruz PD Jr. Identification and expression profiling of a human C-type lectin, structurally homologous to mouse dectin-2. Exp Dermatol (2005) 14(4):281–8. doi:10.1111/j.0906-6705.2005.00312.x
Keywords: plasmacytoid dendritic cells, C-type lectin receptors, toll-like receptors, innate antifungal immunity, HIV
Citation: Maldonado S and Fitzgerald-Bocarsly P (2017) Antifungal Activity of Plasmacytoid Dendritic Cells and the Impact of Chronic HIV Infection. Front. Immunol. 8:1705. doi: 10.3389/fimmu.2017.01705
Received: 02 October 2017; Accepted: 20 November 2017;
Published: 04 December 2017
Edited by:
Steven Templeton, Indiana University School of Medicine, Terre Haute, United StatesReviewed by:
Rebecca Drummond, National Institutes of Health (NIH), United StatesPaolo Puccetti, University of Perugia, Italy
Copyright: © 2017 Maldonado and Fitzgerald-Bocarsly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Fitzgerald-Bocarsly, Qm9jYXJzbHlAbmptcy5ydXRnZXJzLmVkdQ==